
Lysosomal Proteomics Links Disturbances in Lipid Homeostasis and Sphingolipid Metabolism to CLN5 Disease

 , , , , , , , , , , and
, , , , , , , , , , and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Disease Models and Standard Methodologies
2.2. Subcellular Fractionation of Cells and Tissues for Lysosome Enrichment
2.3. Proteomic Analysis
2.4. Development of a Scoring System for Lysosomal Proteins and Data Confidence
2.5. Detection and Quantification of Intracellular Lipids by Fluorescence Microscopy
2.6. Cell Treatment
2.7. Functional Studies on Zebrafish
3. Results
3.1. Proteomic Analysis and Bioinformatic Categorization
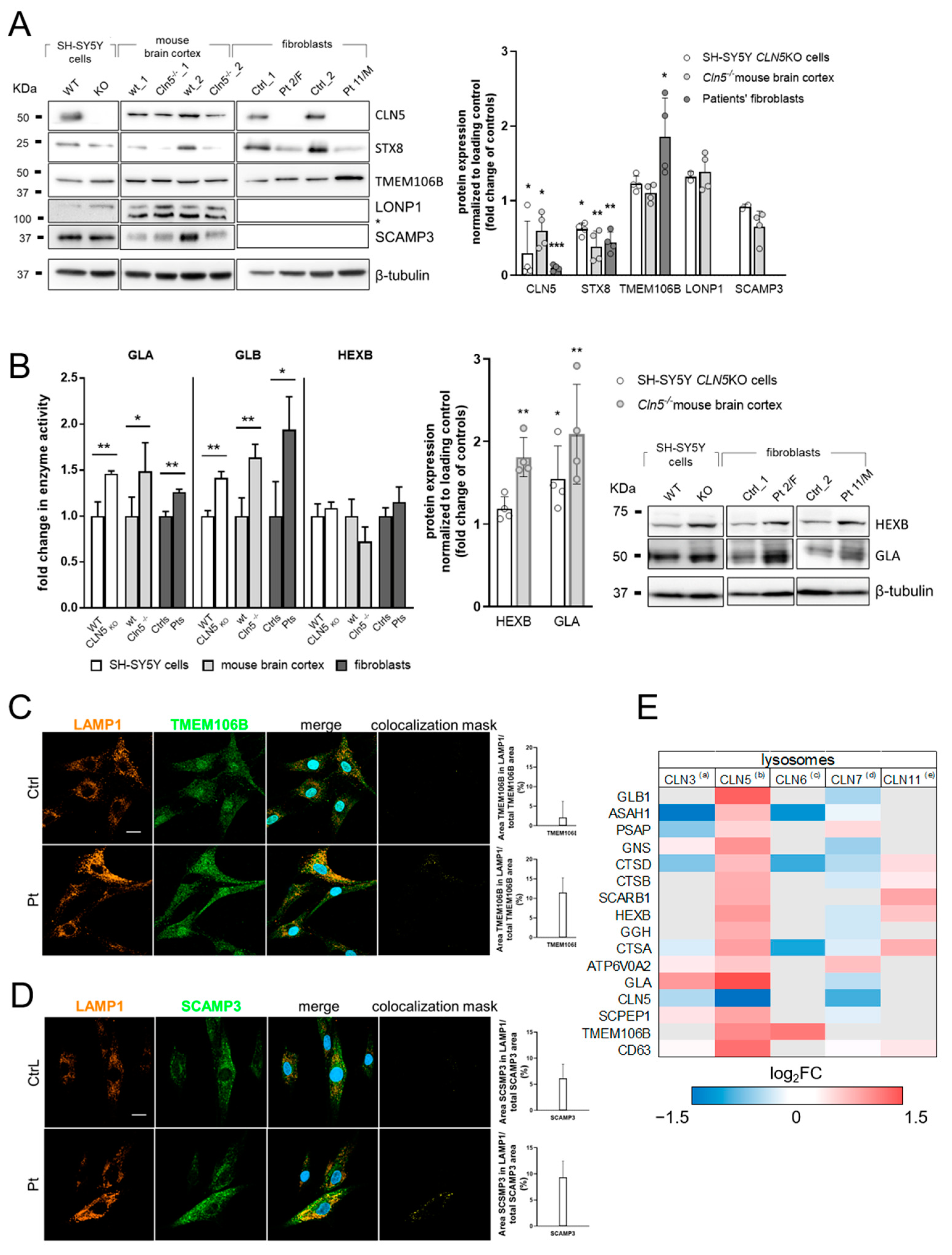
3.2. Validation of Selected DEP
3.3. Characterization of the Zebrafish cln5 Knockdown Model
3.4. In-Vitro Modulation of ROS Overproduction
3.5. Modulation of Locomotor Phenotype in the Zebrafish Model
3.6. Modulation of Lipid Overproduction
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Schulz, A.; Kohlschütter, A. NCL Disorders: Frequent Causes of Childhood Dementia. Iran. J. Child Neurol. 2013, 7, 1–8. [Google Scholar] [PubMed]
- Schulz, A.; Ajayi, T.; Specchio, N.; de Los Reyes, E.; Gissen, P.; Ballon, D.; Dyke, J.P.; Cahan, H.; Slasor, P.; Jacoby, D.; et al. Study of Intraventricular Cerliponase Alfa for CLN2 Disease. N. Engl. J. Med. 2018, 378, 1898–1907. [Google Scholar] [CrossRef] [PubMed]
- Cherukuri, A.; Cahan, H.; de Hart, G.; Van Tuyl, A.; Slasor, P.; Bray, L.; Henshaw, J.; Ajayi, T.; Jacoby, D.; O’Neill, C.A.; et al. Immunogenicity to cerliponase alfa intracerebroventricular enzyme replacement therapy for CLN2 disease: Results from a Phase 1/2 study. Clin. Immunol. 2018, 197, 68–76. [Google Scholar] [CrossRef] [PubMed]
- Lyly, A.; von Schantz, C.; Heine, C.; Schmiedt, M.-L.; Sipilä, T.; Jalanko, A.; Kyttälä, A. Novel interactions of CLN5 support molecular networking between Neuronal Ceroid Lipofuscinosis proteins. BMC Cell Biol. 2009, 10, 83. [Google Scholar] [CrossRef] [Green Version]
- Blom, T.; Schmiedt, M.-L.; Wong, A.M.; Kyttala, A.; Soronen, J.; Jauhiainen, M.; Tyynela, J.; Cooper, J.D.; Jalanko, A. Exacerbated neuronal ceroid lipofuscinosis phenotype in Cln1/5 double-knockout mice. Dis. Model. Mech. 2013, 6, 342–357. [Google Scholar] [CrossRef] [Green Version]
- Scifo, E.; Szwajda, A.; Debski, J.; Uusi-Rauva, K.; Kesti, T.; Dadlez, M.; Gingras, A.C.; Tyynelä, J.; Baumann, M.H.; Jalanko, A.; et al. Drafting the CLN3 protein interactome in SH-SY5Y human neuroblastoma cells: A label-free quantitative proteomics approach. J. Proteome Res. 2013, 12, 2101–2115. [Google Scholar] [CrossRef] [Green Version]
- Vesa, J.; Chin, M.H.; Oelgeschläger, K.; Isosomppi, J.; DellAngelica, E.C.; Jalanko, A.; Peltonen, L. Neuronal ceroid lipofuscinoses are connected at molecular; CLN3, interaction of C. protein with C. and Neuronal Ceroid Lipofuscinoses Are Connected at Molecular Level: Interaction of CLN5 Protein with CLN2 and CLN3. Mol. Biol. Cell 2002, 13, 2410–2420. [Google Scholar] [CrossRef] [Green Version]
- Huber, R.J.; Mathavarajah, S. Cln5 is secreted and functions as a glycoside hydrolase in Dictyostelium. Cell. Signal 2018, 42, 236–248. [Google Scholar] [CrossRef]
- Mamo, A.; Jules, F.; Dumaresq-Doiron, K.; Costantino, S.; Lefrancois, S. The Role of Ceroid Lipofuscinosis Neuronal Protein 5 (CLN5) in Endosomal Sorting. Mol. Cell. Biol. 2012, 32, 1855–1866. [Google Scholar] [CrossRef] [Green Version]
- Adams, J.; Feuerborn, M.; Molina, J.A.; Wilden, A.R.; Adhikari, B.; Budden, T.; Lee, S.Y. Autophagy–lysosome pathway alterations and alpha-synuclein up-regulation in the subtype of neuronal ceroid lipofuscinosis, CLN5 disease. Sci. Rep. 2019, 9, 151. [Google Scholar] [CrossRef] [Green Version]
- Doccini, S.; Morani, F.; Nesti, C.; Pezzini, F.; Calza, G.; Soliymani, R.; Signore, G.; Rocchiccioli, S.; Kanninen, K.M.; Huuskonen, M.T.; et al. Proteomic and functional analyses in disease models reveal CLN5 protein involvement in mitochondrial dysfunction. Cell Death Discov. 2020, 6, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klein, Z.A.; Takahashi, H.; Ma, M.; Stagi, M.; Zhou, M.; Lam, T.T.; Strittmatter, S.M. Loss of TMEM106B Ameliorates Lysosomal and Frontotemporal Dementia-Related Phenotypes in Progranulin-Deficient Mice. Neuron 2017, 95, 281–296.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Danyukova, T.; Ariunbat, K.; Thelen, M.; Brocke-Ahmadinejad, N.; Mole, S.E.; Storch, S. Loss of CLN7 results in depletion of soluble lysosomal proteins and impaired mTOR reactivation. Hum. Mol. Genet. 2018, 27, 1711–1722. [Google Scholar] [CrossRef] [PubMed]
- Tuermer, A.; Mausbach, S.; Kaade, E.; Damme, M.; Sylvester, M.; Gieselmann, V.; Thelen, M. CLN6 deficiency causes selective changes in the lysosomal protein composition. Proteomics 2021, 21, 2100043. [Google Scholar] [CrossRef] [PubMed]
- Schmidtke, C.; Tiede, S.; Thelen, M.; Käkelä, R.; Jabs, S.; Makrypidi, G.; Sylvester, M.; Schweizer, M.; Braren, I.; Brocke-Ahmadinejad, N.; et al. Lysosomal proteome analysis reveals that CLN3-defective cells have multiple enzyme deficiencies associated with changes in intracellular trafficking. J. Biol. Chem. 2019, 294, 9592–9604. [Google Scholar] [CrossRef]
- Faller, K.M.E.; Gutierrez-Quintana, R.; Mohammed, A.; Rahim, A.A.; Tuxworth, R.I.; Wager, K.; Bond, M. The neuronal ceroid lipofuscinoses: Opportunities from model systems. Biochim. Biophys. Acta Mol. Basis Dis. 2015, 1852, 2267–2278. [Google Scholar] [CrossRef] [Green Version]
- Palmer, D.N.; Barry, L.A.; Tyynelä, J.; Cooper, J.D. NCL disease mechanisms. Biochim. Biophys. Acta Mol. Basis Dis. 2013, 1832, 1882–1893. [Google Scholar] [CrossRef]
- Mahmood, F.; Fu, S.; Cooke, J.; Wilson, S.W.; Cooper, J.D.; Russell, C. A zebrafish model of CLN2 disease is deficient in tripeptidyl peptidase 1 and displays progressive neurodegeneration accompanied by a reduction in proliferation. Brain 2013, 136, 1488–1507. [Google Scholar] [CrossRef] [Green Version]
- Luo, C.; Zhao, S.; Dai, W.; Zheng, N.; Wang, J. Proteomic Analysis of Lysosomal Membrane Proteins in Bovine Mammary Epithelial Cells Illuminates Potential Novel Lysosome Functions in Lactation. J. Agric. Food Chem. 2018, 66, 13041–13049. [Google Scholar] [CrossRef]
- Chapel, A.; Kieffer-Jaquinod, S.; Sagné, C.; Verdon, Q.; Ivaldi, C.; Mellal, M.; Thirion, J.; Jadot, M.; Bruley, C.; Garin, J.; et al. An Extended Proteome Map of the Lysosomal Membrane Reveals Novel Potential Transporters. Mol. Cell. Proteom. 2013, 12, 1572–1588. [Google Scholar] [CrossRef] [Green Version]
- hLGDB v.1.2. Available online: http://lysosome.unipg.it/ (accessed on 26 April 2022).
- Brozzi, A.; Urbanelli, L.; Luc Germain, P.; Magini, A.; Emiliani, C. hLGDB: A database of human lysosomal genes and their regulation. Database 2013, 2013, bat024. [Google Scholar] [CrossRef] [PubMed]
- Bateman, A.; Martin, M.-J.; Orchard, S.; Magrane, M.; Agivetova, R.; Ahmad, S.; Alpi, E.; Bowler-Barnett, E.H.; Britto, R.; Bursteinas, B.; et al. UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. [Google Scholar] [CrossRef]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Nesti, C.; Meschini, M.C.; Meunier, B.; Sacchini, M.; Doccini, S.; Romano, A.; Petrillo, S.; Pezzini, I.; Seddiki, N.; Rubegni, A.; et al. Additive effect of nuclear and mitochondrial mutations in a patient with mitochondrial encephalomyopathy. Hum. Mol. Genet. 2015, 24, 3248–3256. [Google Scholar] [CrossRef] [Green Version]
- Sarkar, S.; Davies, J.E.; Huang, Z.; Tunnacliffe, A.; Rubinsztein, D.C. Trehalose, a Novel mTOR-independent Autophagy Enhancer, Accelerates the Clearance of Mutant Huntingtin and α-Synuclein. J. Biol. Chem. 2007, 282, 5641–5652. [Google Scholar] [CrossRef] [Green Version]
- Colaco, A.; Kaya, E.; Adriaenssens, E.; Davis, L.C.; Zampieri, S.; Fernández-Suárez, M.E.; Tan, C.Y.; Deegan, P.B.; Porter, F.D.; Galione, A.; et al. Mechanistic convergence and shared therapeutic targets in Niemann-Pick disease. J. Inherit. Metab. Dis. 2020, 43, 574–585. [Google Scholar] [CrossRef] [Green Version]
- Qiu, B.; Simon, M. BODIPY 493/503 Staining of Neutral Lipid Droplets for Microscopy and Quantification by Flow Cytometry. Bio-Protocol 2016, 6, e1912. [Google Scholar] [CrossRef] [Green Version]
- Marchese, M.; Pappalardo, A.; Baldacci, J.; Verri, T.; Doccini, S.; Cassandrini, D.; Bruno, C.; Fiorillo, C.; Garcia-Gil, M.; Bertini, E.; et al. Dolichol-phosphate mannose synthase depletion in zebrafish leads to dystrophic muscle with hypoglycosylated α-dystroglycan. Biochem. Biophys. Res. Commun. 2016, 477, 137–143. [Google Scholar] [CrossRef]
- D’Amore, A.; Tessa, A.; Naef, V.; Bassi, M.T.; Citterio, A.; Romaniello, R.; Fichi, G.; Galatolo, D.; Mero, S.; Battini, R.; et al. Loss of ap4s1 in zebrafish leads to neurodevelopmental defects resembling spastic paraplegia 52. Ann. Clin. Transl. Neurol. 2020, 7, 584–589. [Google Scholar] [CrossRef] [Green Version]
- Lalowski, M.M.; Björk, S.; Finckenberg, P.; Soliymani, R.; Tarkia, M.; Calza, G.; Blokhina, D.; Tulokas, S.; Kankainen, M.; Lakkisto, P.; et al. Characterizing the Key Metabolic Pathways of the Neonatal Mouse Heart Using a Quantitative Combinatorial Omics Approach. Front. Physiol. 2018, 9, 365. [Google Scholar] [CrossRef]
- Sharma, M.; Burre, J.; Sudhof, T.C. Proteasome Inhibition Alleviates SNARE-Dependent Neurodegeneration. Sci. Transl. Med. 2012, 4, 147ra113. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-J.; Zhang, Z.; Sarkar, C.; Tsai, P.-C.; Lee, Y.-C.; Dye, L.; Mukherjee, A.B. Palmitoyl protein thioesterase-1 deficiency impairs synaptic vesicle recycling at nerve terminals, contributing to neuropathology in humans and mice. J. Clin. Investig. 2008, 118, 3075–3086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Settembre, C.; Ballabio, A. Lysosome: Regulator of lipid degradation pathways. Trends Cell Biol. 2014, 24, 743–750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmiedt, M.-L.; Blom, T.; Blom, T.; Kopra, O.; Wong, A.; von Schantz-Fant, C.; Ikonen, E.; Kuronen, M.; Jauhiainen, M.; Cooper, J.D.; et al. Cln5-deficiency in mice leads to microglial activation, defective myelination and changes in lipid metabolism. Neurobiol. Dis. 2012, 46, 19–29. [Google Scholar] [CrossRef] [PubMed]
- Haddad, S.E.; Khoury, M.; Daoud, M.; Kantar, R.; Harati, H.; Mousallem, T.; Alzate, O.; Meyer, B.; Boustany, R.-M. CLN5 and CLN8 protein association with ceramide synthase: Biochemical and proteomic approaches. Electrophoresis 2012, 33, 3798–3809. [Google Scholar] [CrossRef] [PubMed]
- Li, P.-L.; Gulbins, E. Bioactive Lipids and Redox Signaling: Molecular Mechanism and Disease Pathogenesis. Antioxid. Redox Signal. 2018, 28, 911–915. [Google Scholar] [CrossRef]
- Simonati, A.; Williams, R.E.; Nardocci, N.; Laine, M.; Battini, R.; Schulz, A.; Garavaglia, B.; Moro, F.; Pezzini, F.; Santorelli, F.M. Phenotype and natural history of variant late infantile ceroid-lipofuscinosis 5. Dev. Med. Child Neurol. 2017, 59, 815–822. [Google Scholar] [CrossRef] [Green Version]
- Kopra, O. A mouse model for Finnish variant late infantile neuronal ceroid lipofuscinosis, CLN5, reveals neuropathology associated with early aging. Hum. Mol. Genet. 2004, 13, 2893–2906. [Google Scholar] [CrossRef] [Green Version]
- Wong, C.O.; Palmieri, M.; Li, J.; Akhmedov, D.; Chao, Y.; Broadhead, G.T.; Zhu, M.X.; Berdeaux, R.; Collins, C.A.; Sardiello, M.; et al. Diminished MTORC1-Dependent JNK Activation Underlies the Neurodevelopmental Defects Associated with Lysosomal Dysfunction. Cell Rep. 2015, 12, 2009–2020. [Google Scholar] [CrossRef] [Green Version]
- Palmieri, M.; Pal, R.; Nelvagal, H.R.; Lotfi, P.; Stinnett, G.R.; Seymour, M.L.; Chaudhury, A.; Bajaj, L.; Bondar, V.V.; Bremner, L.; et al. mTORC1-independent TFEB activation via Akt inhibition promotes cellular clearance in neurodegenerative storage diseases. Nat. Commun. 2017, 8, 14338. [Google Scholar] [CrossRef]
- Kline, R.A.; Wishart, T.M.; Mills, K.; Heywood, W.E. Applying modern Omic technologies to the Neuronal Ceroid Lipofuscinoses. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165498. [Google Scholar] [CrossRef] [PubMed]
- Jabs, S.; Quitsch, A.; Käkelä, R.; Koch, B.; Tyynelä, J.; Brade, H.; Glatzel, M.; Walkley, S.; Saftig, P.; Vanier, M.T.; et al. Accumulation of bis(monoacylglycero)phosphate and gangliosides in mouse models of neuronal ceroid lipofuscinosis. J. Neurochem. 2008, 106, 1415–1425. [Google Scholar] [CrossRef] [PubMed]
- Ben-David, O.; Futerman, A.H. The role of the ceramide acyl chain length in neurodegeneration: Involvement of ceramide synthases. Neuromol. Med. 2010, 12, 341–350. [Google Scholar] [CrossRef] [PubMed]
- Patwardhan, G.A.; Beverly, L.J.; Siskind, L.J. Sphingolipids and mitochondrial apoptosis. J. Bioenerg. Biomembr. 2016, 48, 153–168. [Google Scholar] [CrossRef] [PubMed]
- Nikolova-Karakashian, M.N.; Reid, M.B. Sphingolipid Metabolism, Oxidant Signaling, and Contractile Function of Skeletal Muscle. Antioxid. Redox Signal. 2011, 15, 2501–2517. [Google Scholar] [CrossRef] [Green Version]
- De Faria Poloni, J.; Chapola, H.; Feltes, B.C.; Bonatto, D. The importance of sphingolipids and reactive oxygen species in cardiovascular development. Biol. Cell 2014, 106, 167–181. [Google Scholar] [CrossRef]
- Shacka, J.J.; Roth, K.A. Cathepsin D Deficiency and NCL/Batten Disease: There’s More to Death than Apoptosis. Autophagy 2007, 3, 474–476. [Google Scholar] [CrossRef] [Green Version]
- Misrani, A.; Tabassum, S.; Yang, L. Mitochondrial Dysfunction and Oxidative Stress in Alzheimer’s Disease. Front. Aging Neurosci. 2021, 13, 7588. [Google Scholar] [CrossRef]
- Esteras, N.; Kopach, O.; Maiolino, M.; Lariccia, V.; Amoroso, S.; Qamar, S.; Wray, S.; Rusakov, D.A.; Jaganjac, M.; Abramov, A.Y. Mitochondrial ROS control neuronal excitability and cell fate in frontotemporal dementia. Alzheimer’s Dement. 2021, 18, 318–338. [Google Scholar] [CrossRef]
- Nimmo, G.A.M.; Venkatesh, S.; Pandey, A.K.; Marshall, C.R.; Hazrati, L.-N.; Blaser, S.; Ahmed, S.; Cameron, J.; Singh, K.; Ray, P.N.; et al. Bi-allelic mutations of LONP1 encoding the mitochondrial LonP1 protease cause pyruvate dehydrogenase deficiency and profound neurodegeneration with progressive cerebellar atrophy. Hum. Mol. Genet. 2019, 28, 290–306. [Google Scholar] [CrossRef]
- Aoh, Q.L.; Castle, A.M.; Hubbard, C.H.; Katsumata, O.; Castle, J.D. SCAMP3 Negatively Regulates Epidermal Growth Factor Receptor Degradation and Promotes Receptor Recycling. Mol. Biol. Cell 2009, 20, 1816–1832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iftikhar, M.; Lu, Y.; Zhou, M. An overview of therapeutic potential of N-alkylated 1-deoxynojirimycin congeners. Carbohydr. Res. 2021, 504, 108317. [Google Scholar] [CrossRef] [PubMed]
- Miyawaki, I. Application of zebrafish to safety evaluation in drug discovery. J. Toxicol. Pathol. 2020, 33, 197–210. [Google Scholar] [CrossRef] [PubMed]
- Rupprecht, P.; Prendergast, A.; Wyart, C.; Friedrich, R.W. Remote z-scanning with a macroscopic voice coil motor for fast 3D multiphoton laser scanning microscopy. Biomed. Opt. Express 2016, 7, 1656. [Google Scholar] [CrossRef]
- Westerfield, M. The Zebrafish Book. A Guide for the Laboratory Use of Zebrafish (Danio rerio), 4th ed.; Univ. of Oregon Press: Eugene, OR, USA, 2000; ISBN 0 19 963808X. [Google Scholar]
- Thisse, C.; Thisse, B. High-resolution in situ hybridization to whole-mount zebrafish embryos. Nat. Protoc. 2008, 3, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Scifo, E.; Szwajda, A.; Soliymani, R.; Pezzini, F.; Bianchi, M.; Dapkunas, A.; Debski, J.; Uusi-Rauva, K.; Dadlez, M.; Gingras, A.C.; et al. Proteomic analysis of the palmitoyl protein thioesterase 1 interactome in SH-SY5Y human neuroblastoma cells. J. Proteomics 2015, 123, 42–53. [Google Scholar] [CrossRef]
- Laakkonen, E.K.; Soliymani, R.; Karvinen, S.; Kaprio, J.; Kujala, U.M.; Baumann, M.; Sipilä, S.; Kovanen, V.; Lalowski, M. Estrogenic regulation of skeletal muscle proteome: A study of premenopausal women and postmenopausal MZ cotwins discordant for hormonal therapy. Aging Cell 2017, 16, 1276–1287. [Google Scholar] [CrossRef]
- Santi, M.; Finamore, F.; Cecchettini, A.; Santorelli, F.M.; Doccini, S.; Rocchiccioli, S.; Signore, G. Protein Delivery by Peptide-Based Stealth Liposomes: A Biomolecular Insight into Enzyme Replacement Therapy. Mol. Pharm. 2020, 17, 4510–4521. [Google Scholar] [CrossRef]
- Tajima, Y.; Kawashima, I.; Tsukimura, T.; Sugawara, K.; Kuroda, M.; Suzuki, T.; Togawa, T.; Chiba, Y.; Jigami, Y.; Ohno, K.; et al. Use of a Modified α-N-Acetylgalactosaminidase in the Development of Enzyme Replacement Therapy for Fabry Disease. Am. J. Hum. Genet. 2009, 85, 569–580. [Google Scholar] [CrossRef] [Green Version]
- Cromwell, O.; Bennett, J.P.; Hide, I.; Kay, A.B.; Gomperts, B.D. Mechanisms of granule enzyme secretion from permeabilized guinea pig eosinophils. Dependence on Ca2+ and guanine nucleotides. J. Immunol. 1991, 147, 1905–1911. [Google Scholar]
- Naef, V.; Marchese, M.; Ogi, A.; Fichi, G.; Galatolo, D.; Licitra, R.; Doccini, S.; Verri, T.; Argenton, F.; Morani, F.; et al. Efficient Neuroprotective Rescue of Sacsin-Related Disease Phenotypes in Zebrafish. Int. J. Mol. Sci. 2021, 22, 8401. [Google Scholar] [CrossRef] [PubMed]
- Cox, A.G.; Tsomides, A.; Yimlamai, D.; Hwang, K.L.; Miesfeld, J.; Galli, G.G.; Fowl, B.H.; Fort, M.; Ma, K.Y.; Sullivan, M.R.; et al. Yap regulates glucose utilization and sustains nucleotide synthesis to enable organ growth. EMBO J. 2018, 37, e100294. [Google Scholar] [CrossRef] [PubMed]
- Brogi, L.; Marchese, M.; Cellerino, A.; Licitra, R.; Naef, V.; Mero, S.; Bibbiani, C.; Fronte, B. β-Glucans as Dietary Supplement to Improve Locomotion and Mitochondrial Respiration in a Model of Duchenne Muscular Dystrophy. Nutrients 2021, 13, 1619. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Doccini, S.; Marchese, M.; Morani, F.; Gammaldi, N.; Mero, S.; Pezzini, F.; Soliymani, R.; Santi, M.; Signore, G.; Ogi, A.; et al. Lysosomal Proteomics Links Disturbances in Lipid Homeostasis and Sphingolipid Metabolism to CLN5 Disease. Cells 2022, 11, 1840. https://doi.org/10.3390/cells11111840
Doccini S, Marchese M, Morani F, Gammaldi N, Mero S, Pezzini F, Soliymani R, Santi M, Signore G, Ogi A, et al. Lysosomal Proteomics Links Disturbances in Lipid Homeostasis and Sphingolipid Metabolism to CLN5 Disease. Cells. 2022; 11(11):1840. https://doi.org/10.3390/cells11111840
Chicago/Turabian StyleDoccini, Stefano, Maria Marchese, Federica Morani, Nicola Gammaldi, Serena Mero, Francesco Pezzini, Rabah Soliymani, Melissa Santi, Giovanni Signore, Asahi Ogi, and et al. 2022. "Lysosomal Proteomics Links Disturbances in Lipid Homeostasis and Sphingolipid Metabolism to CLN5 Disease" Cells 11, no. 11: 1840. https://doi.org/10.3390/cells11111840