
Therapeutic Validation of GEF-H1 Using a De Novo Designed Inhibitor in Models of Retinal Disease

 , , ,
, , ,  , , ,
, , , 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. GEF-H1 Inhibitors
2.2. RhoA Binding Assay
2.3. Cell Culture and RPE Cell Isolation
2.4. Cell Transfection Methods
2.5. Cell Cytokine Treatment
2.6. Immunoblotting
2.7. Immunofluorescence
2.8. Gene Reporter Assays
2.9. Migration Assay
2.10. Paracellular Permeability
2.11. Toxicity Assays
2.12. Experimental Autoimmune Uveitis (EAU)
2.13. Flow Cytometry
2.14. Histology
2.15. Statistical Methods
3. Results
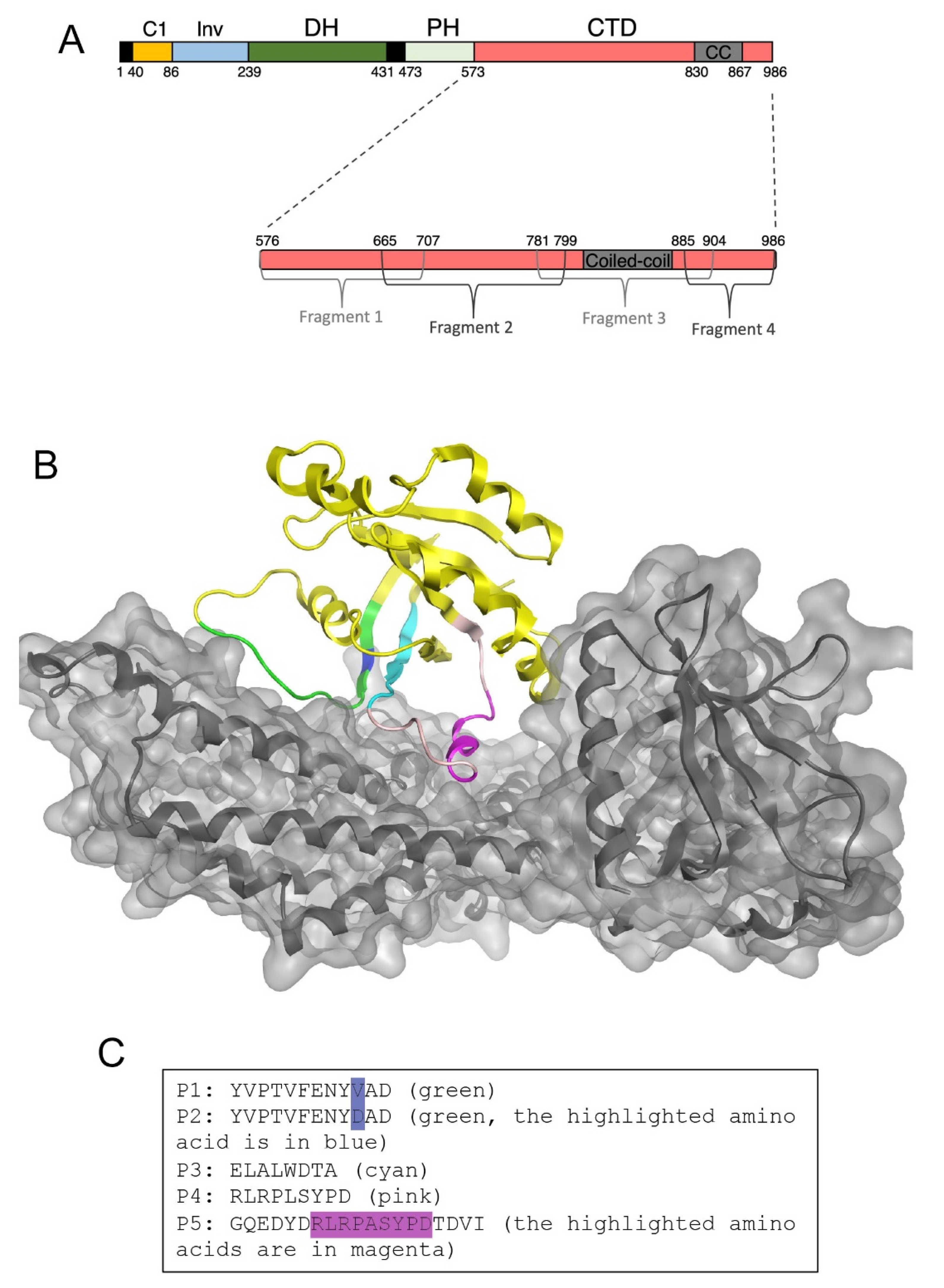
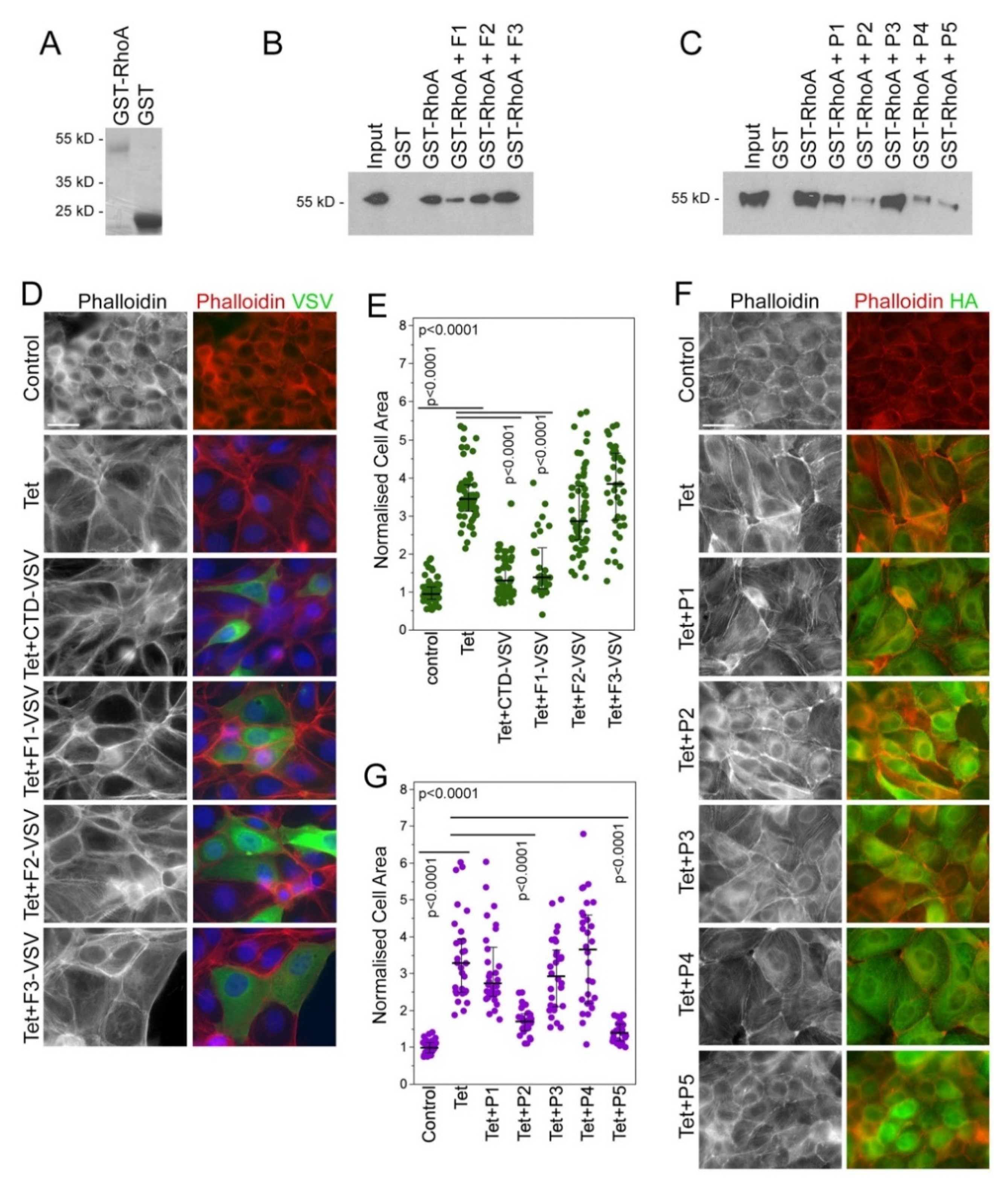
3.1. Identification of Peptide Inhibitors of GEF-H1
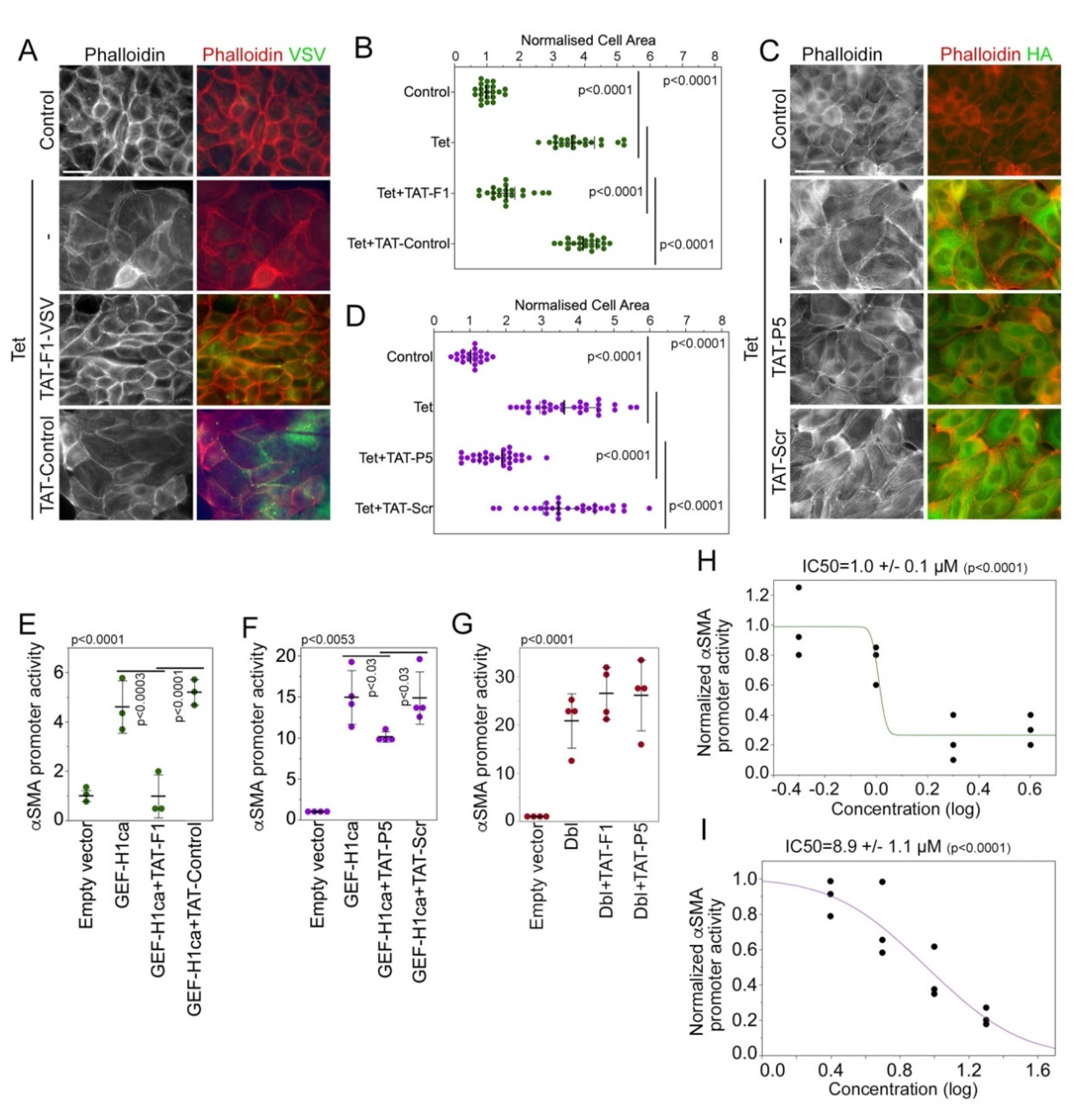
3.2. Characterization of Membrane-Permeable GEF-H1 Inhibitors
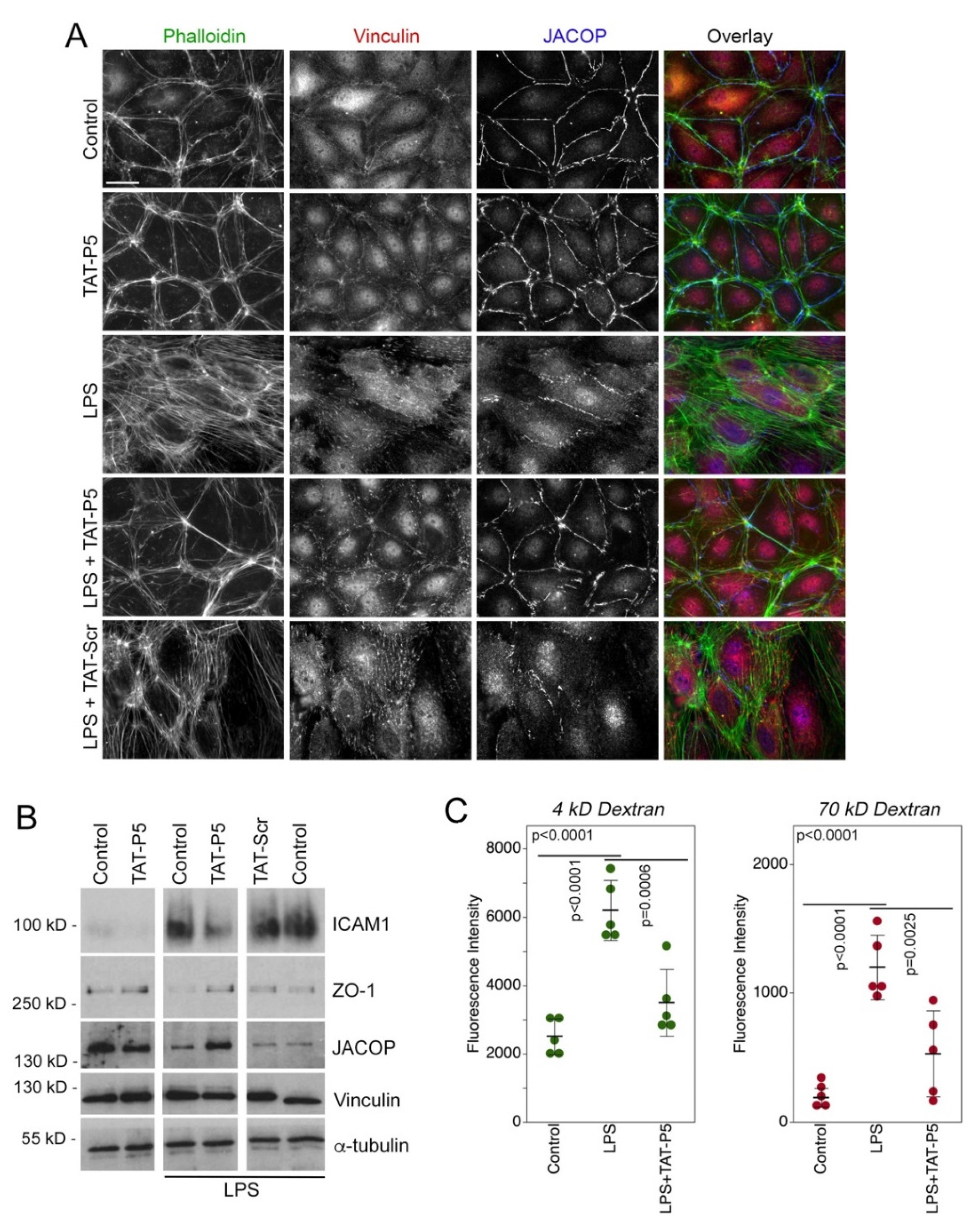
3.3. GEF-H1 Inhibitors Antagonize TGFβ and LPS Signaling in Cellular Disease Models
3.4. GEF-H1 Inhibitors Ameliorate Retinal Autoimmune Disease
4. Discussion
5. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- O’Leary, F.; Campbell, M. The blood-retina barrier in health and disease. FEBS J. 2021. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Chen, M.; Forrester, J.V. Para-inflammation in the aging retina. Prog. Retin. Eye Res. 2009, 28, 348–368. [Google Scholar] [CrossRef] [PubMed]
- Mudhar, H.S. A brief review of the histopathology of proliferative vitreoretinopathy (PVR). Eye 2020, 34, 246–250. [Google Scholar] [CrossRef] [PubMed]
- Willermain, F.; Libert, S.; Motulsky, E.; Salik, D.; Caspers, L.; Perret, J.; Delporte, C. Origins and consequences of hyperosmolar stress in retinal pigmented epithelial cells. Front. Physiol. 2014, 5, 199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rohrer, B. Anaphylatoxin Signaling in Retinal Pigment and Choroidal Endothelial Cells: Characteristics and Relevance to Age-Related Macular Degeneration. Adv. Exp. Med. Biol. 2018, 1074, 45–51. [Google Scholar] [CrossRef]
- Ren, Y.; Li, R.; Zheng, Y.; Busch, H. Cloning and characterization of GEF-H1, a microtubule-associated guanine nucleotide exchange factor for Rac and Rho GTPases. J. Biol. Chem. 1998, 273, 34954–34960. [Google Scholar] [CrossRef] [Green Version]
- Glaven, J.A.; Whitehead, I.; Bagrodia, S.; Kay, R.; Cerione, R.A. The Dbl-related protein, Lfc, localizes to microtubules and mediates the activation of Rac signaling pathways in cells. J. Biol. Chem. 1999, 274, 2279–2285. [Google Scholar] [CrossRef] [Green Version]
- Bakal, C.J.; Finan, D.; LaRose, J.; Wells, C.D.; Gish, G.; Kulkarni, S.; DeSepulveda, P.; Wilde, A.; Rottapel, R. The Rho GTP exchange factor Lfc promotes spindle assembly in early mitosis. Proc. Natl. Acad. Sci. USA 2005, 102, 9529–9534. [Google Scholar] [CrossRef] [Green Version]
- Krendel, M.; Zenke, F.T.; Bokoch, G.M. Nucleotide exchange factor GEF-H1 mediates cross-talk between microtubules and the actin cytoskeleton. Nat. Cell Biol. 2002, 4, 294–301. [Google Scholar] [CrossRef]
- Benais-Pont, G.; Punn, A.; Flores-Maldonado, C.; Eckert, J.; Raposo, G.; Fleming, T.P.; Cereijido, M.; Balda, M.S.; Matter, K. Identification of a tight junction-associated guanine nucleotide exchange factor that activates Rho and regulates paracellular permeability. J. Cell Biol. 2003, 160, 729–740. [Google Scholar] [CrossRef] [Green Version]
- Samarin, S.N.; Ivanov, A.I.; Flatau, G.; Parkos, C.A.; Nusrat, A. Rho/Rho-associated kinase-II signaling mediates disassembly of epithelial apical junctions. Mol. Biol. Cell 2007, 18, 3429–3439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Birkenfeld, J.; Nalbant, P.; Bohl, B.P.; Pertz, O.; Hahn, K.M.; Bokoch, G.M. GEF-H1 modulates localized RhoA activation during cytokinesis under the control of mitotic kinases. Dev. Cell 2007, 12, 699–712. [Google Scholar] [CrossRef] [PubMed]
- Aijaz, S.; D’Atri, F.; Citi, S.; Balda, M.S.; Matter, K. Binding of GEF-H1 to the tight junction-associated adaptor cingulin results in inhibition of Rho signaling and G1/S phase transition. Dev. Cell 2005, 8, 777–786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nalbant, P.; Chang, Y.C.; Birkenfeld, J.; Chang, Z.F.; Bokoch, G.M. Guanine nucleotide exchange factor-H1 regulates cell migration via localized activation of RhoA at the leading edge. Mol. Biol. Cell 2009, 20, 4070–4082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Birukova, A.A.; Adyshev, D.; Gorshkov, B.; Bokoch, G.M.; Birukov, K.G.; Verin, A.D. GEF-H1 is involved in agonist-induced human pulmonary endothelial barrier dysfunction. Am. J. Physiol. Lung Cell. Mol. Physiol. 2006, 290, L540–L548. [Google Scholar] [CrossRef] [Green Version]
- Numaga-Tomita, T.; Kitajima, N.; Kuroda, T.; Nishimura, A.; Miyano, K.; Yasuda, S.; Kuwahara, K.; Sato, Y.; Ide, T.; Birnbaumer, L.; et al. TRPC3-GEF-H1 axis mediates pressure overload-induced cardiac fibrosis. Sci. Rep. 2016, 6, 39383. [Google Scholar] [CrossRef]
- Tsapara, A.; Luthert, P.; Greenwood, J.; Hill, C.S.; Matter, K.; Balda, M.S. The RhoA activator GEF-H1/Lfc is a transforming growth factor-beta target gene and effector that regulates alpha-smooth muscle actin expression and cell migration. Mol. Biol. Cell 2010, 21, 860–870. [Google Scholar] [CrossRef] [Green Version]
- Nie, M.; Balda, M.S.; Matter, K. Stress- and Rho-activated ZO-1-associated nucleic acid binding protein binding to p21 mRNA mediates stabilization, translation, and cell survival. Proc. Natl. Acad. Sci. USA 2012, 109, 10897–10902. [Google Scholar] [CrossRef] [Green Version]
- Mambetsariev, I.; Tian, Y.; Wu, T.; Lavoie, T.; Solway, J.; Birukov, K.G.; Birukova, A.A. Stiffness-activated GEF-H1 expression exacerbates LPS-induced lung inflammation. PLoS ONE 2014, 9, e92670. [Google Scholar] [CrossRef]
- Fine, N.; Dimitriou, I.D.; Rullo, J.; Sandi, M.J.; Petri, B.; Haitsma, J.; Ibrahim, H.; La Rose, J.; Glogauer, M.; Kubes, P.; et al. GEF-H1 is necessary for neutrophil shear stress-induced migration during inflammation. J. Cell Biol. 2016, 215, 107–119. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Wang, J.; Yang, J.; Yang, X.; He, J.; Wang, R.; Liu, S.; Zhou, L.; Ma, L. Guanine nucleotide exchange factor -H1 promotes inflammatory cytokine production and intracellular mycobacterial elimination in macrophages. Cell Cycle 2017, 16, 1695–1704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dan, Q.; Shi, Y.; Rabani, R.; Venugopal, S.; Xiao, J.; Anwer, S.; Ding, M.; Speight, P.; Pan, W.; Alexander, R.T.; et al. Claudin-2 suppresses GEF-H1, RHOA, and MRTF, thereby impacting proliferation and profibrotic phenotype of tubular cells. J. Biol. Chem. 2019, 294, 15446–15465. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wu, S. Effects of fasudil on pulmonary hypertension in clinical practice. Pulm. Pharmacol. Ther. 2017, 46, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Ahmadieh, H.; Nourinia, R.; Hafezi-Moghadam, A.; Sabbaghi, H.; Nakao, S.; Zandi, S.; Yaseri, M.; Tofighi, Z.; Akbarian, S. Intravitreal injection of a Rho-kinase inhibitor (fasudil) combined with bevacizumab versus bevacizumab monotherapy for diabetic macular oedema: A pilot randomised clinical trial. Br. J. Ophthalmol. 2019, 103, 922–927. [Google Scholar] [CrossRef]
- Koch, J.C.; Kuttler, J.; Maass, F.; Lengenfeld, T.; Zielke, E.; Bahr, M.; Lingor, P. Compassionate Use of the ROCK Inhibitor Fasudil in Three Patients With Amyotrophic Lateral Sclerosis. Front. Neurol. 2020, 11, 173. [Google Scholar] [CrossRef]
- Defert, O.; Boland, S. Rho kinase inhibitors: A patent review (2014–2016). Expert Opin. Ther. Pat. 2017, 27, 507–515. [Google Scholar] [CrossRef]
- Tanihara, H.; Inoue, T.; Yamamoto, T.; Kuwayama, Y.; Abe, H.; Suganami, H.; Araie, M.; Group, K.C.S. Intra-ocular pressure-lowering effects of a Rho kinase inhibitor, ripasudil (K-115), over 24 hours in primary open-angle glaucoma and ocular hypertension: A randomized, open-label, crossover study. Acta Ophthalmol. 2015, 93, e254–e260. [Google Scholar] [CrossRef]
- Terry, S.J.; Zihni, C.; Elbediwy, A.; Vitiello, E.; Leefa Chong San, I.V.; Balda, M.S.; Matter, K. Spatially restricted activation of RhoA signalling at epithelial junctions by p114RhoGEF drives junction formation and morphogenesis. Nat. Cell Biol. 2011, 13, 159–166. [Google Scholar] [CrossRef] [Green Version]
- Terry, S.; Nie, M.; Matter, K.; Balda, M.S. Rho signaling and tight junction functions. Physiology 2010, 25, 16–26. [Google Scholar] [CrossRef]
- Joo, E.; Olson, M.F. Regulation and functions of the RhoA regulatory guanine nucleotide exchange factor GEF-H1. Small GTPases 2020, 12, 358–371. [Google Scholar] [CrossRef]
- Martin-Martin, N.; Dan, Q.; Amoozadeh, Y.; Waheed, F.; McMorrow, T.; Ryan, M.P.; Szaszi, K. RhoA and Rho kinase mediate cyclosporine A and sirolimus-induced barrier tightening in renal proximal tubular cells. Int. J. Biochem. Cell Biol. 2012, 44, 178–188. [Google Scholar] [CrossRef] [PubMed]
- Mu, S.; Liu, Y.; Jiang, J.; Ding, R.; Li, X.; Li, X.; Ma, X. Unfractionated heparin ameliorates pulmonary microvascular endothelial barrier dysfunction via microtubule stabilization in acute lung injury. Respir. Res. 2018, 19, 220. [Google Scholar] [CrossRef] [PubMed]
- Osborne, L.D.; Li, G.Z.; How, T.; O’Brien, E.T.; Blobe, G.C.; Superfine, R.; Mythreye, K. TGF-beta regulates LARG and GEF-H1 during EMT to affect stiffening response to force and cell invasion. Mol. Biol. Cell 2014, 25, 3528–3540. [Google Scholar] [CrossRef] [PubMed]
- Keller, M.; Dubois, F.; Teulier, S.; Martin, A.P.J.; Levallet, J.; Maille, E.; Brosseau, S.; Elie, N.; Hergovich, A.; Bergot, E.; et al. NDR2 kinase contributes to cell invasion and cytokinesis defects induced by the inactivation of RASSF1A tumor-suppressor gene in lung cancer cells. J. Exp. Clin. Cancer Res. 2019, 38, 158. [Google Scholar] [CrossRef] [Green Version]
- Sun, Z.; Zhang, H.; Zhang, Y.; Liao, L.; Zhou, W.; Zhang, F.; Lian, F.; Huang, J.; Xu, P.; Zhang, R.; et al. Covalent Inhibitors Allosterically Block the Activation of Rho Family Proteins and Suppress Cancer Cell Invasion. Adv. Sci. 2020, 7, 2000098. [Google Scholar] [CrossRef]
- Biondini, M.; Duclos, G.; Meyer-Schaller, N.; Silberzan, P.; Camonis, J.; Parrini, M.C. RalB regulates contractility-driven cancer dissemination upon TGFbeta stimulation via the RhoGEF GEF-H1. Sci. Rep. 2015, 5, 11759. [Google Scholar] [CrossRef] [Green Version]
- Cullis, J.; Meiri, D.; Sandi, M.J.; Radulovich, N.; Kent, O.A.; Medrano, M.; Mokady, D.; Normand, J.; Larose, J.; Marcotte, R.; et al. The RhoGEF GEF-H1 is required for oncogenic RAS signaling via KSR-1. Cancer Cell 2014, 25, 181–195. [Google Scholar] [CrossRef] [Green Version]
- Birkenfeld, J.; Nalbant, P.; Yoon, S.H.; Bokoch, G.M. Cellular functions of GEF-H1, a microtubule-regulated Rho-GEF: Is altered GEF-H1 activity a crucial determinant of disease pathogenesis? Trends Cell Biol. 2008, 18, 210–219. [Google Scholar] [CrossRef]
- Guo, F.; Tang, J.; Zhou, Z.; Dou, Y.; Van Lonkhuyzen, D.; Gao, C.; Huan, J. GEF-H1-RhoA signaling pathway mediates LPS-induced NF-kappaB transactivation and IL-8 synthesis in endothelial cells. Mol. Immunol. 2012, 50, 98–107. [Google Scholar] [CrossRef] [Green Version]
- Tornavaca, O.; Chia, M.; Dufton, N.; Almagro, L.O.; Conway, D.E.; Randi, A.M.; Schwartz, M.A.; Matter, K.; Balda, M.S. ZO-1 controls endothelial adherens junctions, cell-cell tension, angiogenesis, and barrier formation. J. Cell Biol. 2015, 208, 821–838. [Google Scholar] [CrossRef] [Green Version]
- Kreis, T.E. Microtubules containing detyrosinated tubulin are less dynamic. EMBO J. 1987, 6, 2597–2606. [Google Scholar] [CrossRef] [PubMed]
- Daro, E.; van der Sluijs, P.; Galli, T.; Mellman, I. Rab4 and cellubrevin define different early endosome populations on the pathway of transferrin receptor recycling. Proc. Natl. Acad. Sci. USA 1996, 93, 9559–9564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terry, S.J.; Elbediwy, A.; Zihni, C.; Harris, A.R.; Bailly, M.; Charras, G.T.; Balda, M.S.; Matter, K. Stimulation of cortical myosin phosphorylation by p114RhoGEF drives cell migration and tumor cell invasion. PLoS ONE 2012, 7, e50188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agarwal, R.K.; Silver, P.B.; Caspi, R.R. Rodent models of experimental autoimmune uveitis. Methods Mol. Biol. 2012, 900, 443–469. [Google Scholar] [CrossRef] [Green Version]
- Xu, H.; Koch, P.; Chen, M.; Lau, A.; Reid, D.M.; Forrester, J.V. A clinical grading system for retinal inflammation in the chronic model of experimental autoimmune uveoretinitis using digital fundus images. Exp. Eye Res. 2008, 87, 319–326. [Google Scholar] [CrossRef]
- Copland, D.A.; Liu, J.; Schewitz-Bowers, L.P.; Brinkmann, V.; Anderson, K.; Nicholson, L.B.; Dick, A.D. Therapeutic dosing of fingolimod (FTY720) prevents cell infiltration, rapidly suppresses ocular inflammation, and maintains the blood-ocular barrier. Am. J. Pathol. 2012, 180, 672–681. [Google Scholar] [CrossRef] [Green Version]
- Kallenberg, D.; Tripathi, V.; Javaid, F.; Pilotti, C.; George, J.; Davis, S.; Blackburn, J.W.; O’Connor, M.; Dowsett, L.; Bowers, C.E.; et al. A Humanized Antibody against LRG1 that Inhibits Angiogenesis and Reduces Retinal Vascular Leakage. bioRxiv 2020. [Google Scholar] [CrossRef]
- Abramoff, M.D.; Garvin, M.K.; Sonka, M. Retinal imaging and image analysis. IEEE Rev. Biomed. Eng. 2010, 3, 169–208. [Google Scholar] [CrossRef] [Green Version]
- Garvin, M.K.; Abramoff, M.D.; Wu, X.; Russell, S.R.; Burns, T.L.; Sonka, M. Automated 3-D intraretinal layer segmentation of macular spectral-domain optical coherence tomography images. IEEE Trans. Med. Imaging 2009, 28, 1436–1447. [Google Scholar] [CrossRef] [Green Version]
- Antony, B.; Abramoff, M.D.; Tang, L.; Ramdas, W.D.; Vingerling, J.R.; Jansonius, N.M.; Lee, K.; Kwon, Y.H.; Sonka, M.; Garvin, M.K. Automated 3-D method for the correction of axial artifacts in spectral-domain optical coherence tomography images. Biomed. Opt. Express 2011, 2, 2403–2416. [Google Scholar] [CrossRef]
- Harry, R.; Gegg, M.; Hankey, D.; Zambarakji, H.; Pryce, G.; Baker, D.; Calder, V.; Adamson, P.; Greenwood, J. Suppression of autoimmune retinal disease by lovastatin does not require Th2 cytokine induction. J. Immunol. 2005, 174, 2327–2335. [Google Scholar] [CrossRef] [PubMed]
- Derewenda, U.; Oleksy, A.; Stevenson, A.S.; Korczynska, J.; Dauter, Z.; Somlyo, A.P.; Otlewski, J.; Somlyo, A.V.; Derewenda, Z.S. The crystal structure of RhoA in complex with the DH/PH fragment of PDZRhoGEF, an activator of the Ca2+ sensitization pathway in smooth muscle. Structure 2004, 12, 1955–1965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cimini, S.; Sclip, A.; Mancini, S.; Colombo, L.; Messa, M.; Cagnotto, A.; Di Fede, G.; Tagliavini, F.; Salmona, M.; Borsello, T. The cell-permeable Abeta1-6A2VTAT(D) peptide reverts synaptopathy induced by Abeta1-42wt. Neurobiol. Dis. 2016, 89, 101–111. [Google Scholar] [CrossRef] [PubMed]
- Heck, J.N.; Ponik, S.M.; Garcia-Mendoza, M.G.; Pehlke, C.A.; Inman, D.R.; Eliceiri, K.W.; Keely, P.J. Microtubules regulate GEF-H1 in response to extracellular matrix stiffness. Mol. Biol. Cell 2012, 23, 2583–2592. [Google Scholar] [CrossRef]
- Kratzer, E.; Tian, Y.; Sarich, N.; Wu, T.; Meliton, A.; Leff, A.; Birukova, A.A. Oxidative stress contributes to lung injury and barrier dysfunction via microtubule destabilization. Am. J. Respir. Cell Mol. Biol. 2012, 47, 688–697. [Google Scholar] [CrossRef] [Green Version]
- Tian, Y.; Gawlak, G.; O’Donnell, J.J., 3rd; Mambetsariev, I.; Birukova, A.A. Modulation of Endothelial Inflammation by Low and High Magnitude Cyclic Stretch. PLoS ONE 2016, 11, e0153387. [Google Scholar] [CrossRef] [Green Version]
- Dick, A.D.; Forrester, J.V.; Liversidge, J.; Cope, A.P. The role of tumour necrosis factor (TNF-alpha) in experimental autoimmune uveoretinitis (EAU). Prog. Retin. Eye Res. 2004, 23, 617–637. [Google Scholar] [CrossRef]
- Chen, J.; Caspi, R.R. Clinical and Functional Evaluation of Ocular Inflammatory Disease Using the Model of Experimental Autoimmune Uveitis. Methods Mol. Biol. 2019, 1899, 211–227. [Google Scholar] [CrossRef]
- Gardner, P.J.; Yazid, S.; Chu, C.J.; Copland, D.A.; Adamson, P.; Dick, A.D.; Calder, V.L. TNFalpha Regulates SIRT1 Cleavage during Ocular Autoimmune Disease. Am. J. Pathol. 2015, 185, 1324–1333. [Google Scholar] [CrossRef]
- Eskandarpour, M.; Alexander, R.; Adamson, P.; Calder, V.L. Pharmacological Inhibition of Bromodomain Proteins Suppresses Retinal Inflammatory Disease and Downregulates Retinal Th17 Cells. J. Immunol. 2017, 198, 1093–1103. [Google Scholar] [CrossRef] [Green Version]
- Waldrep, J.C.; Ramanadham, S.; Wood, J.D.; Donoso, L.A. Temporal analysis of retinal function in IRBP peptide-induced experimental autoimmune uveoretinitis (EAU). Reg. Immunol. 1990, 3, 247–253. [Google Scholar] [PubMed]
- Li, X.; Wang, Y.; Yang, C.; Shi, S.; Jin, L.; Luo, Z.; Yu, J.; Zhang, Z.; Yang, Z.; Chen, H. Supramolecular nanofibers of triamcinolone acetonide for uveitis therapy. Nanoscale 2014, 6, 14488–14494. [Google Scholar] [CrossRef] [PubMed]
- Kakiashvili, E.; Dan, Q.; Vandermeer, M.; Zhang, Y.; Waheed, F.; Pham, M.; Szaszi, K. The epidermal growth factor receptor mediates tumor necrosis factor-alpha-induced activation of the ERK/GEF-H1/RhoA pathway in tubular epithelium. J. Biol. Chem. 2011, 286, 9268–9279. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.H.; Eskandarpour, M.; Zhang, X.; Galatowicz, G.; Greenwood, J.; Lightman, S.; Calder, V. Small-molecule antagonist of VLA-4 (GW559090) attenuated neuro-inflammation by targeting Th17 cell trafficking across the blood-retinal barrier in experimental autoimmune uveitis. J. Neuroinflamm. 2021, 18, 49. [Google Scholar] [CrossRef]
- Pepple, K.L.; Wilson, L.; Van Gelder, R.N.; Kovaleva, M.; Ubah, O.C.; Steven, J.; Barelle, C.J.; Porter, A. Uveitis Therapy With Shark Variable Novel Antigen Receptor Domains Targeting Tumor Necrosis Factor Alpha or Inducible T-Cell Costimulatory Ligand. Transl. Vis. Sci. Technol. 2019, 8, 11. [Google Scholar] [CrossRef]
- Lieb, W.S.; Lungu, C.; Tamas, R.; Berreth, H.; Rathert, P.; Storz, P.; Olayioye, M.A.; Hausser, A. The GEF-H1/PKD3 signaling pathway promotes the maintenance of triple-negative breast cancer stem cells. Int. J. Cancer 2020, 146, 3423–3434. [Google Scholar] [CrossRef]
- Cao, J.; Yang, T.; Tang, D.; Zhou, F.; Qian, Y.; Zou, X. Increased expression of GEF-H1 promotes colon cancer progression by RhoA signaling. Pathol. Res. Pract. 2019, 215, 1012–1019. [Google Scholar] [CrossRef]
- Cheng, I.K.; Tsang, B.C.; Lai, K.P.; Ching, A.K.; Chan, A.W.; To, K.F.; Lai, P.B.; Wong, N. GEF-H1 over-expression in hepatocellular carcinoma promotes cell motility via activation of RhoA signalling. J. Pathol. 2012, 228, 575–585. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mills, C.; Hemkemeyer, S.A.; Alimajstorovic, Z.; Bowers, C.; Eskandarpour, M.; Greenwood, J.; Calder, V.; Chan, A.W.E.; Gane, P.J.; Selwood, D.L.; et al. Therapeutic Validation of GEF-H1 Using a De Novo Designed Inhibitor in Models of Retinal Disease. Cells 2022, 11, 1733. https://doi.org/10.3390/cells11111733
Mills C, Hemkemeyer SA, Alimajstorovic Z, Bowers C, Eskandarpour M, Greenwood J, Calder V, Chan AWE, Gane PJ, Selwood DL, et al. Therapeutic Validation of GEF-H1 Using a De Novo Designed Inhibitor in Models of Retinal Disease. Cells. 2022; 11(11):1733. https://doi.org/10.3390/cells11111733
Chicago/Turabian StyleMills, Clare, Sandra A. Hemkemeyer, Zerin Alimajstorovic, Chantelle Bowers, Malihe Eskandarpour, John Greenwood, Virginia Calder, A. W. Edith Chan, Paul J. Gane, David L. Selwood, and et al. 2022. "Therapeutic Validation of GEF-H1 Using a De Novo Designed Inhibitor in Models of Retinal Disease" Cells 11, no. 11: 1733. https://doi.org/10.3390/cells11111733
APA StyleMills, C., Hemkemeyer, S. A., Alimajstorovic, Z., Bowers, C., Eskandarpour, M., Greenwood, J., Calder, V., Chan, A. W. E., Gane, P. J., Selwood, D. L., Matter, K., & Balda, M. S. (2022). Therapeutic Validation of GEF-H1 Using a De Novo Designed Inhibitor in Models of Retinal Disease. Cells, 11(11), 1733. https://doi.org/10.3390/cells11111733