
Inhibition of ATR Reverses a Mitochondrial Respiratory Insufficiency


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methods
2.1. Nematode Strains and Maintenance
2.2. Bacterial Feeding RNAi
2.3. Microscopy
2.4. mRNA and mtDNA Quantitation
2.5. Mutation Screening Assays
2.6. Nucleotide Extraction and Mass Spectrometry
2.7. R-Loop Quantification
2.8. Spliceosome Reporter Assays
2.9. Nematode Oxygen Consumption
2.10. Polysome Profiling
2.11. Western Analysis
2.12. DNA Damage Response (DDR) Screen
3. Results
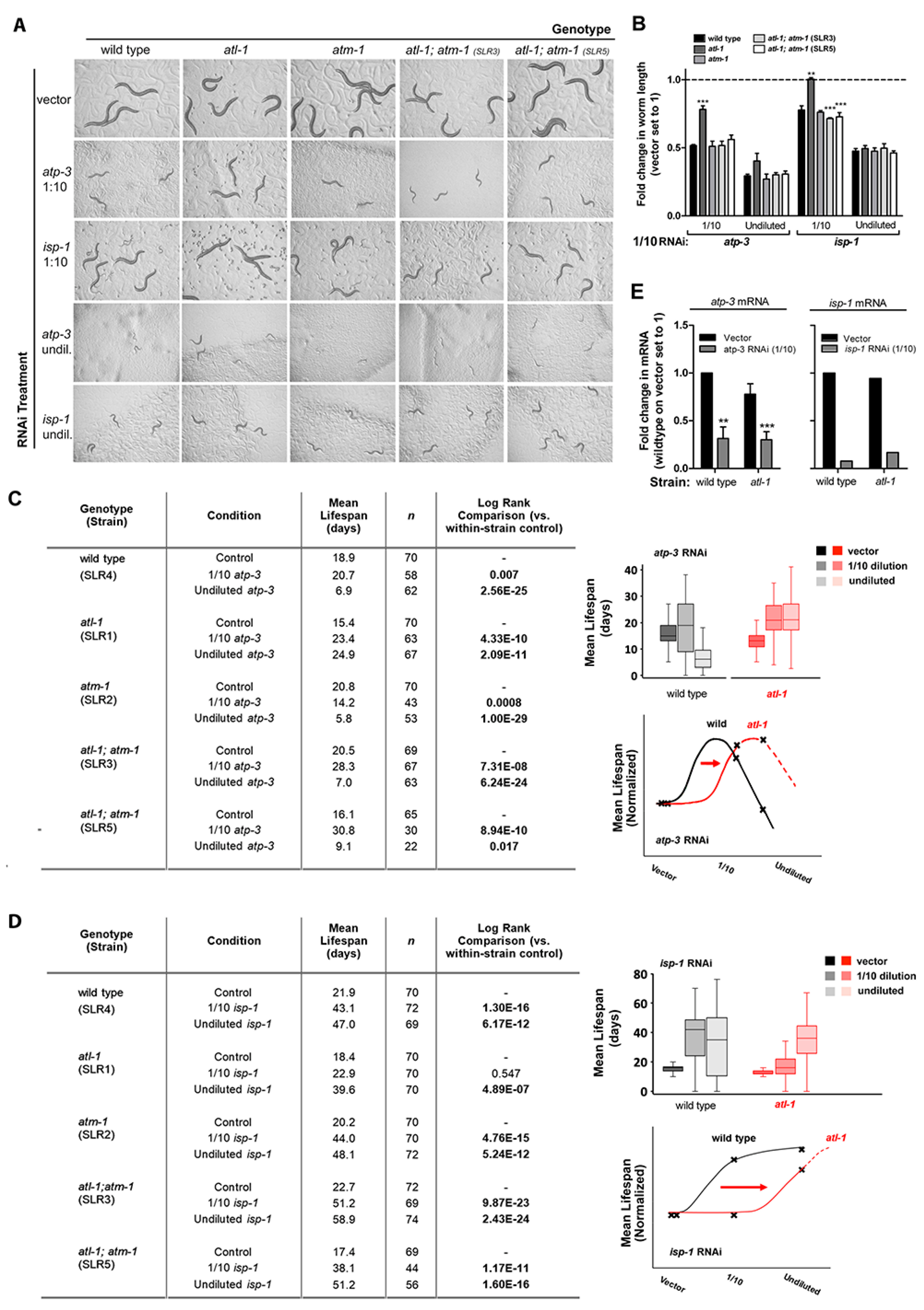
3.1. Loss of ATL-1/ATR Desensitizes Worms to Mitochondrial ETC Stress
3.2. MAK-1 and MAK-2 Participate in Mitochondrial Retrograde Response Signaling
3.3. Knockdown of DNA Pol α Reverses the Small Phenotype of atm-1(gk186); atl-1(tm853) Mutants
3.4. No Evidence for Permanent Nuclear DNA Damage Following Mitochondrial ETC Disruption
3.5. Ribonucleotide and Deoxyribonucleotide Pools Are Disrupted by Mitochondrial ETC Dysfunction
3.6. Evidence for RNA Polymerase Stalling Following Mitochondrial ETC Disruption
3.7. RNA Splicing Is Not Altered by Mitochondrial ETC Disruption
3.8. Reduced ATL-1 Activity Does Not Enhance Hormetic Stress Response Activation
3.9. Oxygen Consumption Remains Surprisingly Unaltered in atl-1 Mutants Exposed to ETC Disruption
3.10. Loss of ATR Leads to Recovery of Translational Activity in Worms Exposed to ETC Disruption
4. Discussion
4.1. Mode of ATL-1 Activation Following Mitochondrial ETC Disruption
4.2. Downstream Targets of ATL-1 That Modulate Mitochondrial ETC Activity
4.3. Mitochondrial Dysfunction in Mammals
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ATM | ataxia telangiectasia mutated |
| ATFS-1 | activating transcription factor associated with stress |
| ATL-1 | ataxia telangiectasia mutated-like (C. elegans ortholog of ATR) |
| ATR | ataxia telangiectasia mutated and Rad3-related |
| DDR | DNA damage response |
| DHODH | dihydroorotate dehydrogenase |
| DNA pol α | DNA polymerase alpha |
| DNA polA1 | DNA polymerase alpha catalytic subunit |
| ETC | electron transport chain |
| mtDNA | mitochondrial DNA |
| MAPKAP | p38 MAPK-activated protein kinase |
| mTOR | mechanistic target of rapamycin |
| PIKK | phosphoinositide 3-kinase-related kinase |
| R-loop | DNA::RNA hybrid |
| RNA pol II | RNA polymerase II |
| TUNEL | terminal deoxynucleotidyl transferase dUTP nick end labeling |
References
- Schaefer, A.M.; Taylor, R.W.; Turnbull, D.M.; Chinnery, P.F. The epidemiology of mitochondrial disorders—Past, present and future. Biochim. Biophys. Acta (BBA)-Bioenerg. 2004, 1659, 115–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.X.; Dewson, G. Mitochondria and apoptosis: Emerging concepts. F1000Prime Rep. 2015, 7, 42. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Samuel, B.S.; Breen, P.C.; Ruvkun, G. Caenorhabditis elegans pathways that surveil and defend mitochondria. Nature 2014, 508, 406–410. [Google Scholar] [CrossRef] [Green Version]
- Khutornenko, A.A.; Roudko, V.V.; Chernyak, B.V.; Vartapetian, A.B.; Chumakov, P.M.; Evstafieva, A.G. Pyrimidine biosynthesis links mitochondrial respiration to the p53 pathway. Proc. Natl. Acad. Sci. USA 2010, 107, 12828–12833. [Google Scholar] [CrossRef] [Green Version]
- Wallace, D.C. Mitochondrial DNA mutations in disease and aging. Environ. Mol. Mutagen. 2010, 51, 440–450. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.C. Mitochondria and cancer. Nat. Rev. Cancer 2012, 12, 685–698. [Google Scholar] [CrossRef] [Green Version]
- Lane, R.K.; Hilsabeck, T.; Rea, S.L. The role of mitochondrial dysfunction in age-related diseases. Biochim. Biophys. Acta (BBA)-Bioenerg. 2015, 1847, 1387–1400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossignol, R.; Faustin, B.; Rocher, C.; Malgat, M.; Mazat, J.P.; Letellier, T. Mitochondrial threshold effects. Biochem. J. 2003, 370, 751–762. [Google Scholar] [CrossRef] [Green Version]
- Munkácsy, E.; Rea, S.L. The paradox of mitochondrial dysfunction and extended longevity. Exp. Gerontol. 2014, 56, 221–233. [Google Scholar] [CrossRef] [Green Version]
- Arnould, T.; Michel, S.; Renard, P. Mitochondria Retrograde Signaling and the UPR(mt): Where Are We in Mammals? Int. J. Mol. Sci. 2015, 16, 18224–18251. [Google Scholar] [CrossRef] [Green Version]
- Nargund, A.M.; Pellegrino, M.W.; Fiorese, C.J.; Baker, B.M.; Haynes, C.M. Mitochondrial import efficiency of ATFS-1 regulates mitochondrial UPR activation. Science 2012, 337, 587–590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biswas, G.; Adebanjo, O.A.; Freedman, B.D.; Anandatheerthavarada, H.K.; Vijayasarathy, C.; Zaidi, M.; Kotlikoff, M.; Avadhani, N.G. Retrograde Ca2+ signaling in C2C12 skeletal myocytes in response to mitochondrial genetic and metabolic stress: A novel mode of inter-organelle crosstalk. EMBO J. 1999, 18, 522–533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munkacsy, E.; Khan, M.H.; Lane, R.K.; Borror, M.B.; Park, J.H.; Bokov, A.F.; Fisher, A.L.; Link, C.D.; Rea, S.L. DLK-1, SEK-3 and PMK-3 Are Required for the Life Extension Induced by Mitochondrial Bioenergetic Disruption in C. elegans. PLoS Genet. 2016, 12, e1006133. [Google Scholar] [CrossRef] [PubMed]
- Heo, J.M.; Livnat-Levanon, N.; Taylor, E.B.; Jones, K.T.; Dephoure, N.; Ring, J.; Xie, J.; Brodsky, J.L.; Madeo, F.; Gygi, S.P.; et al. A stress-responsive system for mitochondrial protein degradation. Mol. Cell 2010, 40, 465–480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, P.; Jin, X.; Peng, Y.; Wang, M.; Liu, H.; Liu, X.; Zhang, Z.; Ji, Y.; Zhang, J.; Liang, M.; et al. The exome sequencing identified the mutation in YARS2 encoding the mitochondrial tyrosyl-tRNA synthetase as a nuclear modifier for the phenotypic manifestation of Leber’s hereditary optic neuropathy-associated mitochondrial DNA mutation. Hum. Mol. Genet. 2016, 25, 584–596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huyen, Y.; Jeffrey, P.D.; Derry, W.B.; Rothman, J.H.; Pavletich, N.P.; Stavridi, E.S.; Halazonetis, T.D. Structural Differences in the DNA Binding Domains of Human p53 and Its C. elegans Ortholog Cep-1. Structure 2004, 12, 1237–1243. [Google Scholar] [CrossRef] [Green Version]
- Ventura, N.; Rea, S.L.; Schiavi, A.; Torgovnick, A.; Testi, R.; Johnson, T.E. p53/CEP-1 increases or decreases lifespan, depending on level of mitochondrial bioenergetic stress. Aging Cell 2009, 8, 380–393. [Google Scholar] [CrossRef] [Green Version]
- Schiavi, A.; Torgovnick, A.; Kell, A.; Megalou, E.; Castelein, N.; Guccini, I.; Marzocchella, L.; Gelino, S.; Hansen, M.; Malisan, F.; et al. Autophagy induction extends lifespan and reduces lipid content in response to frataxin silencing in C. elegans. Exp. Gerontol. 2013, 48, 191–201. [Google Scholar] [CrossRef] [Green Version]
- Lovejoy, C.A.; Cortez, D. Common mechanisms of PIKK regulation. DNA Repair 2009, 8, 1004–1008. [Google Scholar] [CrossRef] [Green Version]
- Matsuoka, S.; Ballif, B.A.; Smogorzewska, A.; McDonald, E.R., III; Hurov, K.E.; Luo, J.; Bakalarski, C.E.; Zhao, Z.; Solimini, N.; Lerenthal, Y.; et al. ATM and ATR Substrate Analysis Reveals Extensive Protein Networks Responsive to DNA Damage. Science 2007, 316, 1160–1166. [Google Scholar] [CrossRef] [Green Version]
- Marechal, A.; Zou, L. DNA damage sensing by the ATM and ATR kinases. Cold Spring Harb. Perspect. Biol. 2013, 5, a012716. [Google Scholar] [CrossRef] [PubMed]
- Vousden, K.H.; Lane, D.P. p53 in health and disease. Nat. Rev. Mol. Cell Biol. 2007, 8, 275–283. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.Q.; Kastan, M.B. Participation of ATM in insulin signalling through phosphorylation of eIF-4E-binding protein 1. Nat. Cell Biol. 2000, 2, 893–898. [Google Scholar] [CrossRef] [PubMed]
- Schneider, J.G.; Finck, B.N.; Ren, J.; Standley, K.N.; Takagi, M.; Maclean, K.H.; Bernal-Mizrachi, C.; Muslin, A.J.; Kastan, M.B.; Semenkovich, C.F. ATM-dependent suppression of stress signaling reduces vascular disease in metabolic syndrome. Cell Metab. 2006, 4, 377–389. [Google Scholar] [CrossRef] [Green Version]
- Cosentino, C.; Grieco, D.; Costanzo, V. ATM activates the pentose phosphate pathway promoting anti-oxidant defence and DNA repair. EMBO J. 2011, 30, 546–555. [Google Scholar] [CrossRef] [Green Version]
- Schroeder, E.A.; Raimundo, N.; Shadel, G.S. Epigenetic silencing mediates mitochondria stress-induced longevity. Cell Metab. 2013, 17, 954–964. [Google Scholar] [CrossRef] [Green Version]
- Fang, E.F.; Bohr, V.A. NAD+: The convergence of DNA repair and mitophagy. Autophagy 2017, 13, 442–443. [Google Scholar] [CrossRef] [Green Version]
- Chiolo, I.; Minoda, A.; Colmenares, S.U.; Polyzos, A.; Costes, S.V.; Karpen, G.H. Double-strand breaks in heterochromatin move outside of a dynamic HP1a domain to complete recombinational repair. Cell 2011, 144, 732–744. [Google Scholar] [CrossRef] [Green Version]
- Gupta, A.; Sharma, S.; Reichenbach, P.; Marjavaara, L.; Nilsson, A.K.; Lingner, J.; Chabes, A.; Rothstein, R.; Chang, M. Telomere length homeostasis responds to changes in intracellular dNTP pools. Genetics 2013, 193, 1095–1105. [Google Scholar] [CrossRef] [Green Version]
- Kumar, A.; Mazzanti, M.; Mistrik, M.; Kosar, M.; Beznoussenko, G.V.; Mironov, A.A.; Garre, M.; Parazzoli, D.; Shivashankar, G.V.; Scita, G.; et al. ATR mediates a checkpoint at the nuclear envelope in response to mechanical stress. Cell 2014, 158, 633–646. [Google Scholar] [CrossRef] [Green Version]
- Hilton, B.A.; Li, Z.; Musich, P.R.; Wang, H.; Cartwright, B.M.; Serrano, M.; Zhou, X.Z.; Lu, K.P.; Zou, Y. ATR Plays a Direct Antiapoptotic Role at Mitochondria, which Is Regulated by Prolyl Isomerase Pin1. Mol. Cell 2015, 60, 35–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yi, C.; Tong, J.; Lu, P.; Wang, Y.; Zhang, J.; Sun, C.; Yuan, K.; Xue, R.; Zou, B.; Li, N.; et al. Formation of a Snf1-Mec1-Atg1 Module on Mitochondria Governs Energy Deprivation-Induced Autophagy by Regulating Mitochondrial Respiration. Dev. Cell 2017, 41, 59–71.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wood, W.B. (Ed.) The Nematode Caenorhabditis elegans; Cold Spring Harbor Laboratory: New York, NY, USA, 1988; p. 667. [Google Scholar]
- Timmons, L.; Fire, A. Specific interference by ingested dsRNA. Nature 1998, 395, 854. [Google Scholar] [CrossRef] [PubMed]
- Hoogewijs, D.; Houthoofd, K.; Matthijssens, F.; Vandesompele, J.; Vanfleteren, J.R. Selection and validation of a set of reliable reference genes for quantitative sod gene expression analysis in C. elegans. BMC Mol. Biol. 2008, 9, 9. [Google Scholar] [CrossRef] [Green Version]
- Sugimoto, T.; Mori, C.; Takanami, T.; Sasagawa, Y.; Saito, R.; Ichiishi, E.; Higashitani, A. Caenorhabditis elegans par2.1/mtssb-1 is essential for mitochondrial DNA replication and its defect causes comprehensive transcriptional alterations including a hypoxia response. Exp. Cell Res. 2008, 314, 103–114. [Google Scholar] [CrossRef]
- Rooney, J.P.; Ryde, I.T.; Sanders, L.H.; Howlett, E.H.; Colton, M.D.; Germ, K.E.; Mayer, G.D.; Greenamyre, J.T.; Meyer, J.N. PCR based determination of mitochondrial DNA copy number in multiple species. Methods Mol. Biol. 2015, 1241, 23–38. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.Z.; Pannunzio, N.R.; Hsieh, C.L.; Yu, K.; Lieber, M.R. Complexities due to single-stranded RNA during antibody detection of genomic RNA:DNA hybrids. BMC Res. Notes 2015, 8, 127. [Google Scholar] [CrossRef] [Green Version]
- Kuroyanagi, H.; Watanabe, Y.; Suzuki, Y.; Hagiwara, M. Position-dependent and neuron-specific splicing regulation by the CELF family RNA-binding protein UNC-75 in Caenorhabditis elegans. Nucleic Acids Res. 2013, 41, 4015–4025. [Google Scholar] [CrossRef] [Green Version]
- Kuroyanagi, H.; Ohno, G.; Sakane, H.; Maruoka, H.; Hagiwara, M. Visualization and genetic analysis of alternative splicing regulation in vivo using fluorescence reporters in transgenic Caenorhabditis elegans. Nat. Protoc. 2010, 5, 1495–1517. [Google Scholar] [CrossRef]
- Luz, A.L.; Rooney, J.P.; Kubik, L.L.; Gonzalez, C.P.; Song, D.H.; Meyer, J.N. Mitochondrial Morphology and Fundamental Parameters of the Mitochondrial Respiratory Chain Are Altered in Caenorhabditis elegans Strains Deficient in Mitochondrial Dynamics and Homeostasis Processes. PLoS ONE 2015, 10, e0130940. [Google Scholar] [CrossRef]
- Steffen, K.K.; MacKay, V.L.; Kerr, E.O.; Tsuchiya, M.; Hu, D.; Fox, L.A.; Dang, N.; Johnston, E.D.; Oakes, J.A.; Tchao, B.N.; et al. Yeast life span extension by depletion of 60s ribosomal subunits is mediated by Gcn4. Cell 2008, 133, 292–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torgovnick, A.; Schiavi, A.; Shaik, A.; Kassahun, H.; Maglioni, S.; Rea, S.L.; Johnson, T.E.; Reinhardt, H.C.; Honnen, S.; Schumacher, B.; et al. BRCA1 and BARD1 mediate apoptotic resistance but not longevity upon mitochondrial stress in Caenorhabditis elegans. EMBO Rep. 2018, 19, e45856. [Google Scholar] [CrossRef] [PubMed]
- Rea, S.L.; Ventura, N.; Johnson, T.E. Relationship between mitochondrial electron transport chain dysfunction, development, and life extension in Caenorhabditis elegans. PLoS Biol. 2007, 5, e259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Satsuka, A.; Mehta, K.; Laimins, L. p38MAPK and MK2 pathways are important for the differentiation-dependent human papillomavirus life cycle. J. Virol. 2015, 89, 1919–1924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reinhardt, H.C.; Aslanian, A.S.; Lees, J.A.; Yaffe, M.B. p53-Deficient Cells Rely on ATM- and ATR-Mediated Checkpoint Signaling through the p38MAPK/MK2 Pathway for Survival after DNA Damage. Cancer Cell 2007, 11, 175–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manke, I.A.; Nguyen, A.; Lim, D.; Stewart, M.Q.; Elia, A.E.; Yaffe, M.B. MAPKAP kinase-2 is a cell cycle checkpoint kinase that regulates the G2/M transition and S phase progression in response to UV irradiation. Mol. Cell 2005, 17, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Muse, T.; Boulton, S.J. Distinct modes of ATR activation after replication stress and DNA double-strand breaks in Caenorhabditis elegans. EMBO J. 2005, 24, 4345–4355. [Google Scholar] [CrossRef] [Green Version]
- Dmitrieva, N.I.; Celeste, A.; Nussenzweig, A.; Burg, M.B. Ku86 preserves chromatin integrity in cells adapted to high NaCl. Proc. Natl. Acad. Sci. USA 2005, 102, 10730–10735. [Google Scholar] [CrossRef] [Green Version]
- Desler, C.; Lykke, A.; Rasmussen, L.J. The effect of mitochondrial dysfunction on cytosolic nucleotide metabolism. J. Nucleic Acids 2010, 2010, 701518. [Google Scholar] [CrossRef] [Green Version]
- Kuchta, R.D.; Stengel, G. Mechanism and evolution of DNA primases. Biochim. Biophys. Acta (BBA)-Bioenerg. 2010, 1804, 1180–1189. [Google Scholar] [CrossRef] [Green Version]
- Guilliam, T.A.; Keen, B.A.; Brissett, N.C.; Doherty, A.J. Primase-polymerases are a functionally diverse superfamily of replication and repair enzymes. Nucleic Acids Res. 2015, 43, 6651–6664. [Google Scholar] [CrossRef] [Green Version]
- Zou, L.; Elledge, S.J. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science 2003, 300, 1542–1548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsang, W.Y.; Lemire, B.D. Mitochondrial Genome Content Is Regulated during Nematode Development. Biochem. Biophys. Res. Commun. 2002, 291, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Derheimer, F.A.; O’Hagan, H.M.; Krueger, H.M.; Hanasoge, S.; Paulsen, M.T.; Ljungman, M. RPA and ATR link transcriptional stress to p53. Proc. Natl. Acad. Sci. USA 2007, 104, 12778–12783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamperl, S.; Cimprich, K.A. The contribution of co-transcriptional RNA:DNA hybrid structures to DNA damage and genome instability. DNA Repair 2014, 19, 84–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sollier, J.; Cimprich, K.A. Breaking bad: R-loops and genome integrity. Trends Cell Biol. 2015, 25, 514–522. [Google Scholar] [CrossRef] [Green Version]
- Proudfoot, N.J.; Furger, A.; Dye, M.J. Integrating mRNA processing with transcription. Cell 2002, 108, 501–512. [Google Scholar] [CrossRef] [Green Version]
- Heintz, C.; Doktor, T.K.; Lanjuin, A.; Escoubas, C.; Zhang, Y.; Weir, H.J.; Dutta, S.; Silva-Garcia, C.G.; Bruun, G.H.; Morantte, I.; et al. Splicing factor 1 modulates dietary restriction and TORC1 pathway longevity in C. elegans. Nature 2017, 541, 102–106. [Google Scholar] [CrossRef]
- Kuroyanagi, H.; Kobayashi, T.; Mitani, S.; Hagiwara, M. Transgenic alternative-splicing reporters reveal tissue-specific expression profiles and regulation mechanisms in vivo. Nat. Methods 2006, 3, 909–915. [Google Scholar] [CrossRef]
- Ventura, N.; Rea, S.L. Caenorhabditis elegans mitochondrial mutants as an investigative tool to study human neurodegenerative diseases associated with mitochondrial dysfunction. Biotechnol. J. 2007, 2, 584–595. [Google Scholar] [CrossRef]
- Mori, C.; Takanami, T.; Higashitani, A. Maintenance of mitochondrial DNA by the Caenorhabditis elegans ATR checkpoint protein ATL-1. Genetics 2008, 180, 681–686. [Google Scholar] [CrossRef] [Green Version]
- Henderson, S.T.; Bonafe, M.; Johnson, T.E. daf-16 protects the nematode Caenorhabditis elegans during food deprivation. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2006, 61, 444–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bloss, T.A.; Witze, E.S.; Rothman, J.H. Suppression of CED-3-independent apoptosis by mitochondrial βNAC in Caenorhabditis elegans. Nature 2003, 424, 1066–1071. [Google Scholar] [CrossRef] [PubMed]
- Arsenovic, P.T.; Maldonado, A.T.; Colleluori, V.D.; Bloss, T.A. Depletion of the C. elegans NAC engages the unfolded protein response, resulting in increased chaperone expression and apoptosis. PLoS ONE 2012, 7, e44038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.K.; Lund, J.; Kiraly, M.; Duke, K.; Jiang, M.; Stuart, J.M.; Eizinger, A.; Wylie, B.N.; Davidson, G.S. A gene expression map for Caenorhabditis elegans. Science 2001, 293, 2087–2092. [Google Scholar] [CrossRef] [Green Version]
- Houtkooper, R.H.; Mouchiroud, L.; Ryu, D.; Moullan, N.; Katsyuba, E.; Knott, G.; Williams, R.W.; Auwerx, J. Mitonuclear protein imbalance as a conserved longevity mechanism. Nature 2013, 497, 451–457. [Google Scholar] [CrossRef] [Green Version]
- West, A.P.; Khoury-Hanold, W.; Staron, M.; Tal, M.C.; Pineda, C.M.; Lang, S.M.; Bestwick, M.; Duguay, B.A.; Raimundo, N.; MacDuff, D.A.; et al. Mitochondrial DNA stress primes the antiviral innate immune response. Nature 2015, 520, 553–557. [Google Scholar] [CrossRef] [Green Version]
- Lewis, S.C.; Joers, P.; Willcox, S.; Griffith, J.D.; Jacobs, H.T.; Hyman, B.C. A rolling circle replication mechanism produces multimeric lariats of mitochondrial DNA in Caenorhabditis elegans. PLoS Genet. 2015, 11, e1004985. [Google Scholar] [CrossRef] [Green Version]
- Starokadomskyy, P.; Gemelli, T.; Rios, J.J.; Xing, C.; Wang, R.C.; Li, H.; Pokatayev, V.; Dozmorov, I.; Khan, S.; Miyata, N.; et al. DNA polymerase-alpha regulates the activation of type I interferons through cytosolic RNA:DNA synthesis. Nat. Immunol. 2016, 17, 495–504. [Google Scholar] [CrossRef] [Green Version]
- Milenkovic, D.; Blaza, J.N.; Larsson, N.G.; Hirst, J. The Enigma of the Respiratory Chain Supercomplex. Cell Metab. 2017, 25, 765–776. [Google Scholar] [CrossRef] [Green Version]
- George, R.; Beddoe, T.; Landl, K.; Lithgow, T. The yeast nascent polypeptide-associated complex initiates protein targeting to mitochondria in vivo. Proc. Natl. Acad. Sci. USA 1998, 95, 2296–2301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- George, R.; Walsh, P.; Beddoe, T.; Lithgow, T. The nascent polypeptide-associated complex (NAC) promotes interaction of ribosomes with the mitochondrial surface in vivo. FEBS Lett. 2002, 516, 213–216. [Google Scholar] [CrossRef] [Green Version]
- Yogev, O.; Karniely, S.; Pines, O. Translation-coupled translocation of yeast fumarase into mitochondria in vivo. J. Biol. Chem. 2007, 282, 29222–29229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, K.-S.; Kim, H.-S.; Jin, U.-H.; Lee, S.; Park, J.-A.; Lim, Y.; Pai, H.-S. Silencing of NbBTF3 results in developmental defects and disturbed gene expression in chloroplasts and mitochondria of higher plants. Planta 2007, 225, 1459–1469. [Google Scholar] [CrossRef]
- del Alamo, M.; Hogan, D.J.; Pechmann, S.; Albanese, V.; Brown, P.O.; Frydman, J. Defining the specificity of cotranslationally acting chaperones by systematic analysis of mRNAs associated with ribosome-nascent chain complexes. PLoS Biol. 2011, 9, e1001100. [Google Scholar] [CrossRef] [Green Version]
- Amanchy, R.; Periaswamy, B.; Mathivanan, S.; Reddy, R.; Tattikota, S.G.; Pandey, A. A curated compendium of phosphorylation motifs. Nat. Biotechnol. 2007, 25, 285–286. [Google Scholar] [CrossRef]
- Johnson, S.C.; Yanos, M.E.; Kayser, E.B.; Quintana, A.; Sangesland, M.; Castanza, A.; Uhde, L.; Hui, J.; Wall, V.Z.; Gagnidze, A.; et al. mTOR inhibition alleviates mitochondrial disease in a mouse model of Leigh syndrome. Science 2013, 342, 1524–1528. [Google Scholar] [CrossRef] [Green Version]
- Zid, B.M.; Rogers, A.N.; Katewa, S.D.; Vargas, M.A.; Kolipinski, M.C.; Lu, T.A.; Benzer, S.; Kapahi, P. 4E-BP extends lifespan upon dietary restriction by enhancing mitochondrial activity in Drosophila. Cell 2009, 139, 149–160. [Google Scholar] [CrossRef] [Green Version]
- Bonawitz, N.D.; Chatenay-Lapointe, M.; Pan, Y.; Shadel, G.S. Reduced TOR Signaling Extends Chronological Life Span via Increased Respiration and Upregulation of Mitochondrial Gene Expression. Cell Metab. 2007, 5, 265–277. [Google Scholar] [CrossRef] [Green Version]
- Garelick, M.G.; Mackay, V.L.; Yanagida, A.; Academia, E.C.; Schreiber, K.H.; Ladiges, W.C.; Kennedy, B.K. Chronic rapamycin treatment or lack of S6K1 does not reduce ribosome activity in vivo. Cell Cycle 2013, 12, 2493–2504. [Google Scholar] [CrossRef] [Green Version]
- Jones, M.R.; Huang, J.C.; Chua, S.Y.; Baillie, D.L.; Rose, A.M. The atm-1 gene is required for genome stability in Caenorhabditis elegans. Mol. Genet. Genom. 2012, 287, 325–335. [Google Scholar] [CrossRef] [Green Version]
- Kamath, R.S.; Ahringer, J. Genome-wide RNAi screening in Caenorhabditis elegans. Methods 2003, 30, 313–321. [Google Scholar] [CrossRef]
- Butler, J.A.; Ventura, N.; Johnson, T.E.; Rea, S.L. Long-lived mitochondrial (Mit) mutants of Caenorhabditis elegans utilize a novel metabolism. FASEB J. 2010, 24, 4977–4988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Pothof, J.; van Haaften, G.; Thijssen, K.; Kamath, R.S.; Fraser, A.G.; Ahringer, J.; Plasterk, R.H.; Tijsterman, M. Identification of genes that protect the C. elegans genome against mutations by genome-wide RNAi. Genes Dev. 2003, 17, 443–448. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.-C.; Stanfield, G.M.; Horvitz, H.R. NUC-1, a Caenorhabditis elegans DNase II homolog, functions in an intermediate step of DNA degradation during apoptosis. Genes Dev. 2000, 14, 536–548. [Google Scholar] [CrossRef] [PubMed]
- Bhaskaran, S.; Butler, J.A.; Becerra, S.; Fassio, V.; Girotti, M.; Rea, S.L. Breaking Caenorhabditis elegans the easy way using the Balch homogenizer: An old tool for a new application. Anal. Biochem. 2011, 413, 123–132. [Google Scholar] [CrossRef] [Green Version]
- Huang, D.; Zhang, Y.; Chen, X. Analysis of intracellular nucleoside triphosphate levels in normal and tumor cell lines by high-performance liquid chromatography. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2003, 784, 101–109. [Google Scholar] [CrossRef]
- Hoaglin, D.C.; Mosteller, F.; Tukey, J.W. Understanding Robust and Exploratory Data Analysis; Wiley: New York, NY, USA, 1983. [Google Scholar]
- Dupont, W.D.; Plummer, W.D., Jr. Power and sample size calculations. A review and computer program. Control. Clin. Trials 1990, 11, 116–128. [Google Scholar] [CrossRef]
- Dupont, W.D.; Plummer, W.D., Jr. Power and sample size calculations for studies involving linear regression. Control. Clin. Trials 1998, 19, 589–601. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Borror, M.B.; Girotti, M.; Kar, A.; Cain, M.K.; Gao, X.; MacKay, V.L.; Herron, B.; Bhaskaran, S.; Becerra, S.; Novy, N.; et al. Inhibition of ATR Reverses a Mitochondrial Respiratory Insufficiency. Cells 2022, 11, 1731. https://doi.org/10.3390/cells11111731
Borror MB, Girotti M, Kar A, Cain MK, Gao X, MacKay VL, Herron B, Bhaskaran S, Becerra S, Novy N, et al. Inhibition of ATR Reverses a Mitochondrial Respiratory Insufficiency. Cells. 2022; 11(11):1731. https://doi.org/10.3390/cells11111731
Chicago/Turabian StyleBorror, Megan B., Milena Girotti, Adwitiya Kar, Meghan K. Cain, Xiaoli Gao, Vivian L. MacKay, Brent Herron, Shylesh Bhaskaran, Sandra Becerra, Nathan Novy, and et al. 2022. "Inhibition of ATR Reverses a Mitochondrial Respiratory Insufficiency" Cells 11, no. 11: 1731. https://doi.org/10.3390/cells11111731
APA StyleBorror, M. B., Girotti, M., Kar, A., Cain, M. K., Gao, X., MacKay, V. L., Herron, B., Bhaskaran, S., Becerra, S., Novy, N., Ventura, N., Johnson, T. E., Kennedy, B. K., & Rea, S. L. (2022). Inhibition of ATR Reverses a Mitochondrial Respiratory Insufficiency. Cells, 11(11), 1731. https://doi.org/10.3390/cells11111731