
Aberrant Synaptic Pruning in CNS Diseases: A Critical Player in HIV-Associated Neurological Dysfunction?

Abstract
1. Introduction
2. Synaptic Degeneration Associated with HAND and HIV-Associated Pain
2.1. Synaptic Degeneration and HAND
2.2. Synaptic Degeneration and HIV-Associated Pain
2.3. What Is Causing Synaptic Degeneration in HIV Patients?
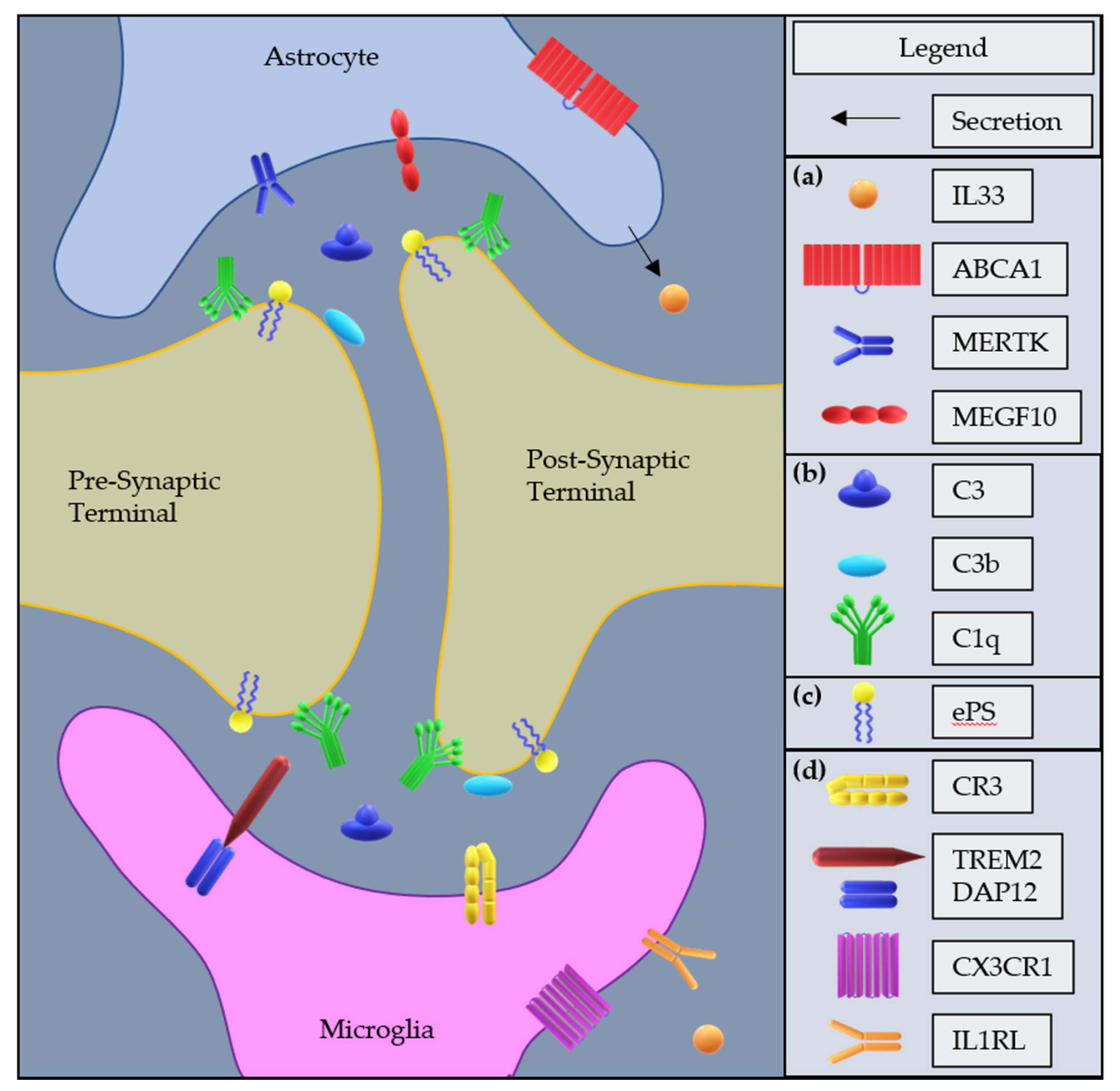
3. Glia-Mediated Synaptic Pruning in the Normal CNS
3.1. Discovery and Overview of Glia-Mediated Synaptic Pruning
3.2. Microglial Mechanisms of Synaptic Pruning
3.3. Astrocyte Mechanisms of Synaptic Pruning
3.4. Interactions between Microglia and Astrocytes Mediating Synaptic Pruning
4. Glia-Mediated Synaptic Pruning in CNS Diseases
4.1. The Role of Glia in Alzheimer’s Disease
4.1.1. Microglia-Mediated Synaptic Pruning in AD
4.1.2. Astrocyte-Mediated Synaptic Pruning in AD
4.2. The Role of Glia in West Nile Virus CNS Infection
Microglia-Mediated Synaptic Pruning in WNV
4.3. The Role of Glia in Zika Virus CNS Infection
4.3.1. Microglia-Mediated Synaptic Pruning in ZIKV
4.3.2. Astrocyte-Mediated Synaptic Pruning in ZKV
4.4. Overview of Glia-Mediated Synaptic Pruning during Disease
5. Does Dysregulated Synaptic Pruning Contribute to HIV-Associated Neurological Disorders?
5.1. The Role of Glia in HIV CNS Infection and Their Potential Role in Synaptic Pruning
5.2. Potential Molecular Mechanisms of HIV-Induced Glia-Mediated Synaptic Pruning
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Wang, Y.; Liu, M.; Lu, Q.; Farrell, M.; Lappin, J.M.; Shi, J.; Lu, L.; Bao, Y. Global prevalence and burden of HIV-associated neurocognitive disorder. Neurology 2020, 95, e2610–e2621. [Google Scholar] [CrossRef] [PubMed]
- Parker, R.; Stein, D.J.; Jelsma, J. Pain in people living with HIV/AIDS: A systematic review. J. Int. AIDS Soc. 2014, 17, 18719. [Google Scholar] [CrossRef] [PubMed]
- Smail, R.C.; Brew, B.J. HIV-associated neurocognitive disorder. Handb. Clin. Neurol. 2018, 152, 75–97. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.-J.; Fu, Y.-Y.; Wei, Q.-Q.; Zhang, Z.-J. Neuroinflammation in HIV-Related Neuropathic Pain. Front. Pharmacol. 2021, 12, 653852. [Google Scholar] [CrossRef] [PubMed]
- Israel, S.M.; Hassanzadeh-Behbahani, S.; Turkeltaub, P.E.; Moore, D.J.; Ellis, R.J.; Jiang, X. Different roles of frontal versus striatal atrophy in HIV-associated neurocognitive disorders. Hum. Brain Mapp. 2019, 40, 3010–3026. [Google Scholar] [CrossRef]
- Moore, D.J.; Masliah, E.; Rippeth, J.D.; Gonzalez, R.; Carey, C.L.; Cherner, M.; Ellis, R.J.; Achim, C.L.; Marcotte, T.D.; Heaton, R.K.; et al. Cortical and subcortical neurodegeneration is associated with HIV neurocognitive impairment. AIDS 2006, 20, 879–887. [Google Scholar] [CrossRef]
- Robinson-Papp, J.; Schütz, S.G. HIV-related neuropathy: Current perspectives. HIV/AIDS—Res. Palliat. Care 2013, 5, 243–251. [Google Scholar] [CrossRef]
- Tagliati, M.; Grinnell, J.; Godbold, J.; Simpson, D.M. Peripheral Nerve Function in HIV Infection. Arch. Neurol. 1999, 56, 84–89. [Google Scholar] [CrossRef]
- Ma, L.; Yue, L.; Zhang, Y.; Wang, Y.; Han, B.; Cui, S.; Liu, F.-Y.; Wan, Y.; Yi, M. Spontaneous Pain Disrupts Ventral Hippocampal CA1-Infralimbic Cortex Connectivity and Modulates Pain Progression in Rats with Peripheral Inflammation. Cell Rep. 2019, 29, 1579–1593.e6. [Google Scholar] [CrossRef]
- Chang, P.-C.; Pollema-Mays, S.L.; Centeno, M.V.; Procissi, D.; Contini, M.; Baria, A.T.; Martina, M.; Apkarian, A.V. Role of nucleus accumbens in neuropathic pain: Linked multi-scale evidence in the rat transitioning to neuropathic pain. Pain 2014, 155, 1128–1139. [Google Scholar] [CrossRef]
- Saylor, D.; Dickens, A.; Sacktor, N.; Haughey, N.; Slusher, B.; Pletnikov, M.; Mankowski, J.L.; Brown, A.; Volsky, D.J.; McArthur, J.C. Erratum: HIV-associated neurocognitive disorder—pathogenesis and prospects for treatment. Nat. Rev. Neurol. 2016, 12, 309. [Google Scholar] [CrossRef] [PubMed]
- Gelman, B.B.; Chen, T.; Lisinicchia, J.G.; Soukup, V.M.; Carmical, J.R.; Starkey, J.; Masliah, E.; Commins, D.L.; Brandt, D.; Grant, I.; et al. The National NeuroAIDS Tissue Consortium Brain Gene Array: Two Types of HIV-Associated Neurocognitive Impairment. PLoS ONE 2012, 7, e46178. [Google Scholar] [CrossRef] [PubMed]
- Ru, W.; Tang, S.-J. HIV-associated synaptic degeneration. Mol. Brain 2017, 10, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Yuan, S.-B.; Shi, Y.; Chen, J.; Zhou, X.; Li, G.; Gelman, B.B.; Bs, J.G.L.; Carlton, S.M.; Ferguson, M.R.; Tan, A.; et al. Gp120 in the pathogenesis of human immunodeficiency virus-associated pain. Ann. Neurol. 2014, 75, 837–850. [Google Scholar] [CrossRef] [PubMed]
- Everall, I.P.; Heaton, R.K.; Marcotte, T.D.; Ellis, R.; A McCutchan, J.; Atkinson, J.H.; Grant, I.; Mallory, M.; Masliah, E.; HNRC Group. Cortical Synaptic Density is Reduced in Mild to Moderate Human Immunodeficiency Virus Neurocognitive Disorder. Brain Pathol. 2006, 9, 209–217. [Google Scholar] [CrossRef]
- Buzhdygan, T.; Lisinicchia, J.; Patel, V.; Johnson, K.; Neugebauer, V.; Paessler, S.; Jennings, K.; Gelman, B. Neuropsychological, Neurovirological and Neuroimmune Aspects of Abnormal GABAergic Transmission in HIV Infection. J. Neuroimmune Pharmacol. 2016, 11, 279–293. [Google Scholar] [CrossRef]
- Abidin, A.Z.; Dsouza, A.M.; Schifitto, G.; Wismüller, A. Detecting cognitive impairment in HIV-infected individuals using mutual connectivity analysis of resting state functional MRI. J. NeuroVirology 2020, 26, 188–200. [Google Scholar] [CrossRef]
- Terry, R.D.; Masliah, E.; Salmon, D.P.; Butters, N.; DeTeresa, R.; Hill, R.; Hansen, L.A.; Katzman, R. Physical basis of cognitive alterations in alzheimer’s disease: Synapse loss is the major correlate of cognitive impairment. Ann. Neurol. 1991, 30, 572–580. [Google Scholar] [CrossRef]
- Cheng, H.-C.; Ulane, C.M.; Burke, R. Clinical progression in Parkinson disease and the neurobiology of axons. Ann. Neurol. 2010, 67, 715–725. [Google Scholar] [CrossRef]
- Milnerwood, A.J.; Raymond, L.A. Early synaptic pathophysiology in neurodegeneration: Insights from Huntington’s disease. Trends Neurosci. 2010, 33, 513–523. [Google Scholar] [CrossRef]
- Sadek, J.R.; Pergam, S.A.; Harrington, J.A.; Echevarria, L.A.; Davis, L.E.; Goade, D.; Harnar, J.; Nofchissey, R.A.; Sewell, C.M.; Ettestad, P.; et al. Persistent neuropsychological impairment associated with West Nile virus infection. J. Clin. Exp. Neuropsychol. 2010, 32, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Braz, J.; Solorzano, C.; Wang, X.; Basbaum, A.I. Transmitting Pain and Itch Messages: A Contemporary View of the Spinal Cord Circuits that Generate Gate Control. Neuron 2014, 82, 522–536. [Google Scholar] [CrossRef] [PubMed]
- Kuner, R.; Flor, H. Erratum: Structural plasticity and reorganisation in chronic pain. Nat. Rev. Neurosci. 2017, 18, 113. [Google Scholar] [CrossRef] [PubMed]
- Woolf, C.J. Central sensitization: Implications for the diagnosis and treatment of pain. Pain 2011, 152, S2–S15. [Google Scholar] [CrossRef] [PubMed]
- Tashima, R.; Koga, K.; Yoshikawa, Y.; Sekine, M.; Watanabe, M.; Tozaki-Saitoh, H.; Furue, H.; Yasaka, T.; Tsuda, M. A subset of spinal dorsal horn interneurons crucial for gating touch-evoked pain-like behavior. Proc. Natl. Acad. Sci. USA 2021, 118, e2021220118. [Google Scholar] [CrossRef] [PubMed]
- Bezprozvanny, I.; Hiesinger, P.R. The synaptic maintenance problem: Membrane recycling, Ca2+ homeostasis and late onset degeneration. Mol. Neurodegener. 2013, 8, 23. [Google Scholar] [CrossRef]
- Trachtenberg, J.T.; Chen, B.E.; Knott, G.W.; Feng, G.; Sanes, J.R.; Welker, E.; Svoboda, K. Long-term in vivo imaging of experience-dependent synaptic plasticity in adult cortex. Nature 2002, 420, 788–794. [Google Scholar] [CrossRef]
- Ertürk, A.; Wang, Y.; Sheng, M. Local Pruning of Dendrites and Spines by Caspase-3-Dependent and Proteasome-Limited Mechanisms. J. Neurosci. 2014, 34, 1672–1688. [Google Scholar] [CrossRef]
- Green, M.V.; Raybuck, J.D.; Zhang, X.; Wu, M.M.; Thayer, S.A. Scaling Synapses in the Presence of HIV. Neurochem. Res. 2018, 44, 234–246. [Google Scholar] [CrossRef]
- Deshpande, M.; Zheng, J.; Borgmann, K.; Persidsky, R.; Wu, L.; Schellpeper, C.; Ghorpade, A. Role of activated astrocytes in neuronal damage: Potential links to HIV-1-associated dementia. Neurotox. Res. 2005, 7, 183–192. [Google Scholar] [CrossRef]
- Budka, H. Neuropathology of Human Immunodeficiency Virus Infection. Brain Pathol. 1991, 1, 163–175. [Google Scholar] [CrossRef] [PubMed]
- Hua, J.Y.; Smith, S.J. Neural activity and the dynamics of central nervous system development. Nat. Neurosci. 2004, 7, 327–332. [Google Scholar] [CrossRef] [PubMed]
- Andoh, M.; Koyama, R. Microglia regulate synaptic development and plasticity. Dev. Neurobiol. 2021, 81, 568–590. [Google Scholar] [CrossRef] [PubMed]
- Paolicelli, R.C.; Bolasco, G.; Pagani, F.; Maggi, L.; Scianni, M.; Panzanelli, P.; Giustetto, M.; Ferreira, T.A.; Guiducci, E.; Dumas, L.; et al. Synaptic Pruning by Microglia Is Necessary for Normal Brain Development. Science 2011, 333, 1456–1458. [Google Scholar] [CrossRef] [PubMed]
- Schafer, D.P.; Lehrman, E.K.; Kautzman, A.G.; Koyama, R.; Mardinly, A.R.; Yamasaki, R.; Ransohoff, R.M.; Greenberg, M.E.; Barres, B.A.; Stevens, B. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron 2012, 74, 691–705. [Google Scholar] [CrossRef]
- Chung, W.-S.; Clarke, L.E.; Wang, G.X.; Stafford, B.K.; Sher, A.; Chakraborty, C.; Joung, J.; Foo, L.C.; Thompson, A.; Chen, C.; et al. Astrocytes mediate synapse elimination through MEGF10 and MERTK pathways. Nature 2013, 504, 394–400. [Google Scholar] [CrossRef]
- Schafer, D.P.; Lehrman, E.K.; Heller, C.T.; Stevens, B. An Engulfment Assay: A Protocol to Assess Interactions between CNS Phagocytes and Neurons. J. Vis. Exp. 2014, 51482. [Google Scholar] [CrossRef]
- Wilton, D.K.; Dissing-Olesen, L.; Stevens, B. Neuron-Glia Signaling in Synapse Elimination. Annu. Rev. Neurosci. 2019, 42, 107–127. [Google Scholar] [CrossRef]
- Wang, C.; Yue, H.; Hu, Z.; Shen, Y.; Ma, J.; Li, J.; Wang, X.-D.; Wang, L.; Sun, B.; Shi, P.; et al. Microglia mediate forgetting via complement-dependent synaptic elimination. Science 2020, 367, 688–694. [Google Scholar] [CrossRef]
- Simonetti, M.; Hagenston, A.M.; Vardeh, D.; Freitag, H.E.; Mauceri, D.; Lu, J.; Satagopam, V.; Schneider, R.; Costigan, M.; Bading, H.; et al. Nuclear Calcium Signaling in Spinal Neurons Drives a Genomic Program Required for Persistent Inflammatory Pain. Neuron 2013, 77, 43–57. [Google Scholar] [CrossRef]
- Vainchtein, I.D.; Chin, G.; Cho, F.S.; Kelley, K.W.; Miller, J.G.; Chien, E.C.; Liddelow, S.A.; Nguyen, P.T.; Nakao-Inoue, H.; Dorman, L.C.; et al. Astrocyte-derived interleukin-33 promotes microglial synapse engulfment and neural circuit development. Science 2018, 359, 1269–1273. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.; Dissing-Olesen, L.; Stevens, B. New insights on the role of microglia in synaptic pruning in health and disease. Curr. Opin. Neurobiol. 2015, 36, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Merle, N.S.; Church, S.E.; Fremeaux-Bacchi, V.; Roumenina, L.T. Complement System Part I—Molecular Mechanisms of Activation and Regulation. Front. Immunol. 2015, 6, 262. [Google Scholar] [CrossRef] [PubMed]
- Stevens, B.; Allen, N.J.; Vazquez, L.E.; Howell, G.R.; Christopherson, K.S.; Nouri, N.; Micheva, K.; Mehalow, A.; Huberman, A.D.; Stafford, B.; et al. The Classical Complement Cascade Mediates CNS Synapse Elimination. Cell 2007, 131, 1164–1178. [Google Scholar] [CrossRef] [PubMed]
- Hoshiko, M.; Arnoux, I.; Avignone, E.; Yamamoto, N.; Audinat, E. Deficiency of the Microglial Receptor CX3CR1 Impairs Postnatal Functional Development of Thalamocortical Synapses in the Barrel Cortex. J. Neurosci. 2012, 32, 15106–15111. [Google Scholar] [CrossRef] [PubMed]
- Gunner, G.; Cheadle, L.; Johnson, K.M.; Ayata, P.; Badimon, A.; Mondo, E.; Nagy, M.A.; Liu, L.; Bemiller, S.M.; Kim, K.-W.; et al. Sensory lesioning induces microglial synapse elimination via ADAM10 and fractalkine signaling. Nat. Neurosci. 2019, 22, 1075–1088. [Google Scholar] [CrossRef] [PubMed]
- Scott-Hewitt, N.; Perrucci, F.; Morini, R.; Erreni, M.; Mahoney, M.; Witkowska, A.; Carey, A.; Faggiani, E.; Schuetz, L.T.; Mason, S.; et al. Local externalization of phosphatidylserine mediates developmental synaptic pruning by microglia. EMBO J. 2020, 39, e105380. [Google Scholar] [CrossRef]
- Yeh, F.L.; Hansen, D.; Sheng, M. TREM2, Microglia, and Neurodegenerative Diseases. Trends Mol. Med. 2017, 23, 512–533. [Google Scholar] [CrossRef]
- Filipello, F.; Morini, R.; Corradini, I.; Zerbi, V.; Canzi, A.; Michalski, B.; Erreni, M.; Markicevic, M.; Starvaggi-Cucuzza, C.; Otero, K.; et al. The Microglial Innate Immune Receptor TREM2 Is Required for Synapse Elimination and Normal Brain Connectivity. Immunity 2018, 48, 979–991.e8. [Google Scholar] [CrossRef]
- Takahashi, K.; Rochford, C.D.; Neumann, H. Clearance of apoptotic neurons without inflammation by microglial triggering receptor expressed on myeloid cells-2. J. Exp. Med. 2005, 201, 647–657. [Google Scholar] [CrossRef]
- Kobayashi, M.; Konishi, H.; Sayo, A.; Takai, T.; Kiyama, H. TREM2/DAP12 Signal Elicits Proinflammatory Response in Microglia and Exacerbates Neuropathic Pain. J. Neurosci. 2016, 36, 11138–11150. [Google Scholar] [CrossRef] [PubMed]
- Zhong, L.; Chen, X.-F.; Wang, T.; Wang, Z.; Liao, C.; Wang, Z.; Huang, R.; Wang, D.; Li, X.; Wu, L.; et al. Soluble TREM2 induces inflammatory responses and enhances microglial survival. J. Exp. Med. 2017, 214, 597–607. [Google Scholar] [CrossRef] [PubMed]
- Morizawa, Y.M.; Hirayama, Y.; Ohno, N.; Shibata, S.; Shigetomi, E.; Sui, Y.; Nabekura, J.; Sato, K.; Okajima, F.; Takebayashi, H.; et al. Reactive astrocytes function as phagocytes after brain ischemia via ABCA1-mediated pathway. Nat. Commun. 2017, 8, 28. [Google Scholar] [CrossRef] [PubMed]
- Iram, T.; Ramirez-Ortiz, Z.; Byrne, M.H.; Coleman, U.A.; Kingery, N.D.; Means, T.K.; Frenkel, D.; El Khoury, J. Megf10 Is a Receptor for C1Q That Mediates Clearance of Apoptotic Cells by Astrocytes. J. Neurosci. 2016, 36, 5185–5192. [Google Scholar] [CrossRef] [PubMed]
- Hamon, Y.; Trompier, R.; Ma, Z.; Venegas, V.; Pophillat, M.; Mignotte, V.; Zhou, Z.; Chimini, G. Cooperation between Engulfment Receptors: The Case of ABCA1 and MEGF10. PLoS ONE 2006, 1, e120. [Google Scholar] [CrossRef] [PubMed]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Münch, A.E.; Chung, W.-S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef]
- Jay, T.R.; Von Saucken, V.E.; Munoz, B.; Codocedo, J.F.; Atwood, B.K.; Lamb, B.T.; Landreth, G.E. TREM2 is required for microglial instruction of astrocytic synaptic engulfment in neurodevelopment. Glia 2019, 67, 1873–1892. [Google Scholar] [CrossRef]
- Selkoe, D.J. Alzheimer’s Disease Is a Synaptic Failure. Science 2002, 298, 789–791. [Google Scholar] [CrossRef]
- Montero-Crespo, M.; Domínguez-Álvaro, M.; Alonso-Nanclares, L.; DeFelipe, J.; Blazquez-Llorca, L. Three-dimensional analysis of synaptic organization in the hippocampal CA1 field in Alzheimer’s disease. Brain 2021, 144, 553–573. [Google Scholar] [CrossRef]
- Kim, S.-M.; Mun, B.-R.; Lee, S.-J.; Joh, Y.; Lee, H.-Y.; Ji, K.-Y.; Choi, H.-R.; Lee, E.-H.; Kim, E.-M.; Jang, J.-H.; et al. TREM2 promotes Aβ phagocytosis by upregulating C/EBPα-dependent CD36 expression in microglia. Sci. Rep. 2017, 7, 1–12. [Google Scholar] [CrossRef]
- Wang, Y.; Ulland, T.K.; Ulrich, J.D.; Song, W.; Tzaferis, J.A.; Hole, J.T.; Yuan, P.; Mahan, T.E.; Shi, Y.; Gilfillan, S.; et al. TREM2-mediated early microglial response limits diffusion and toxicity of amyloid plaques. J. Exp. Med. 2016, 213, 667–675. [Google Scholar] [CrossRef] [PubMed]
- Osborn, L.M.; Kamphuis, W.; Wadman, W.J.; Hol, E.M. Astrogliosis: An integral player in the pathogenesis of Alzheimer’s disease. Prog. Neurobiol. 2016, 144, 121–141. [Google Scholar] [CrossRef] [PubMed]
- Gratuze, M.; Leyns, C.E.G.; Holtzman, D.M. New insights into the role of TREM2 in Alzheimer’s disease. Mol. Neurodegener. 2018, 13, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Gratuze, M.; Leyns, C.E.; Sauerbeck, A.D.; St-Pierre, M.-K.; Xiong, M.; Kim, N.; Serrano, J.R.; Tremblay, M.; Kummer, T.T.; Colonna, M.; et al. Impact of TREM2R47H variant on tau pathology–induced gliosis and neurodegeneration. J. Clin. Investig. 2020, 130, 4954–4968. [Google Scholar] [CrossRef]
- Hong, S.; Beja-Glasser, V.F.; Nfonoyim, B.M.; Frouin, A.; Li, S.; Ramakrishnan, S.; Merry, K.M.; Shi, Q.; Rosenthal, A.; Barres, B.A.; et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 2016, 352, 712–716. [Google Scholar] [CrossRef]
- Ferreira, S.T.; Lourenco, M.V.; Oliveira, M.M.; De Felice, F.G. Soluble amyloid-β oligomers as synaptotoxins leading to cognitive impairment in Alzheimer’s disease. Front. Cell. Neurosci. 2015, 9, 191. [Google Scholar] [CrossRef]
- Corder, E.H.; Saunders, A.M.; Risch, N.J.; Strittmatter, W.J.; Schmechel, D.E.; Gaskell, P.C., Jr.; Rimmler, J.B.; Locke, P.A.; Conneally, P.M.; Schmader, K.E.; et al. Protective effect of apolipoprotein E type 2 allele for late onset Alzheimer disease. Nat. Genet. 1994, 7, 180–184. [Google Scholar] [CrossRef]
- Corder, E.H.; Saunders, A.M.; Strittmatter, W.J.; Schmechel, D.E.; Gaskell, P.C.; Small, G.W.; Roses, A.D.; Haines, J.L.; Pericak-Vance, M.A. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 1993, 261, 921–923. [Google Scholar] [CrossRef]
- Chung, W.-S.; Verghese, P.B.; Chakraborty, C.; Joung, J.; Hyman, B.T.; Ulrich, J.D.; Holtzman, D.M.; Barres, B.A. Novel allele-dependent role for APOE in controlling the rate of synapse pruning by astrocytes. Proc. Natl. Acad. Sci. USA 2016, 113, 10186–10191. [Google Scholar] [CrossRef]
- Wang, Y.; Balaji, V.; Kaniyappan, S.; Krüger, L.; Irsen, S.; Tepper, K.; Chandupatla, R.; Maetzler, W.; Schneider, A.; Mandelkow, E.; et al. The release and trans-synaptic transmission of Tau via exosomes. Mol. Neurodegener. 2017, 12, 1–25. [Google Scholar] [CrossRef]
- Ghag, G.; Bhatt, N.; Cantu, D.V.; Guerrero-Munoz, M.J.; Ellsworth, A.; Sengupta, U.; Kayed, R. Soluble tau aggregates, not large fibrils, are the toxic species that display seeding and cross-seeding behavior. Protein Sci. 2018, 27, 1901–1909. [Google Scholar] [CrossRef] [PubMed]
- Winkelmann, E.R.; Luo, H.; Wang, T. West Nile Virus Infection in the Central Nervous System. F1000Research 2016, 5, 105. [Google Scholar] [CrossRef] [PubMed]
- Peng, B.-H.; Wang, T. West Nile Virus Induced Cell Death in the Central Nervous System. Pathogens 2019, 8, 215. [Google Scholar] [CrossRef] [PubMed]
- Seitz, S.; Clarke, P.; Tyler, K.L. Pharmacologic Depletion of Microglia Increases Viral Load in the Brain and Enhances Mortality in Murine Models of Flavivirus-Induced Encephalitis. J. Virol. 2018, 92, e00525-18. [Google Scholar] [CrossRef]
- Vasek, M.J.; Garber, C.; Dorsey, D.; Durrant, D.M.; Bollman, B.; Soung, A.; Yu, J.; Perez-Torres, C.; Frouin, A.; Wilton, D.K.; et al. A complement–microglial axis drives synapse loss during virus-induced memory impairment. Nature 2016, 534, 538–543. [Google Scholar] [CrossRef]
- Garber, C.; Soung, A.; Vollmer, L.L.; Kanmogne, M.; Last, A.; Brown, J.; Klein, R.S. T cells promote microglia-mediated synaptic elimination and cognitive dysfunction during recovery from neuropathogenic flaviviruses. Nat. Neurosci. 2019, 22, 1276–1288. [Google Scholar] [CrossRef]
- Maximova, O.A.; Bernbaum, J.G.; Pletnev, A.G. West Nile Virus Spreads Transsynaptically within the Pathways of Motor Control: Anatomical and Ultrastructural Mapping of Neuronal Virus Infection in the Primate Central Nervous System. PLOS Neglected Trop. Dis. 2016, 10, e0004980. [Google Scholar] [CrossRef]
- Figueiredo, C.P.; Barros-Aragão, F.G.Q.; Neris, R.L.S.; Frost, P.S.; Soares, C.; Souza, I.N.O.; Zeidler, J.D.; Zamberlan, D.C.; De Sousa, V.L.; Souza, A.S.; et al. Zika virus replicates in adult human brain tissue and impairs synapses and memory in mice. Nat. Commun. 2019, 10, 1–16. [Google Scholar] [CrossRef]
- Potokar, M.; Jorgačevski, J.; Zorec, R. Astrocytes in Flavivirus Infections. Int. J. Mol. Sci. 2019, 20, 691. [Google Scholar] [CrossRef]
- Enlow, W.; Bordeleau, M.; Piret, J.; Ibáñez, F.G.; Uyar, O.; Venable, M.-C.; Goyette, N.; Carbonneau, J.; Tremblay, M.-E.; Boivin, G. Microglia are involved in phagocytosis and extracellular digestion during Zika virus encephalitis in young adult immunodeficient mice. J. Neuroinflamm. 2021, 18, 1–24. [Google Scholar] [CrossRef]
- Lall, D.; Lorenzini, I.; Mota, T.A.; Bell, S.; Mahan, T.E.; Ulrich, J.D.; Davtyan, H.; Rexach, J.E.; Muhammad, A.G.; Shelest, O.; et al. C9orf72 deficiency promotes microglial-mediated synaptic loss in aging and amyloid accumulation. Neuron 2021, 109, 2275–2291.e8. [Google Scholar] [CrossRef] [PubMed]
- Aono, H.; Choudhury, M.E.; Higaki, H.; Miyanishi, K.; Kigami, Y.; Fujita, K.; Akiyama, J.-I.; Takahashi, H.; Yano, H.; Kubo, M.; et al. Microglia may compensate for dopaminergic neuron loss in experimental Parkinsonism through selective elimination of glutamatergic synapses from the subthalamic nucleus. Glia 2017, 65, 1833–1847. [Google Scholar] [CrossRef] [PubMed]
- Werneburg, S.; Jung, J.; Kunjamma, R.B.; Ha, S.-K.; Luciano, N.J.; Willis, C.M.; Gao, G.; Biscola, N.P.; Havton, L.; Crocker, S.J.; et al. Targeted Complement Inhibition at Synapses Prevents Microglial Synaptic Engulfment and Synapse Loss in Demyelinating Disease. Immunity 2019, 52, 167–182.e7. [Google Scholar] [CrossRef]
- Churchill, M.J.; Wesselingh, S.L.; Cowley, D.; Pardo, C.A.; McArthur, J.C.; Brew, B.J.; Gorry, P.R. Extensive astrocyte infection is prominent in human immunodeficiency virus-associated dementia. Ann. Neurol. 2009, 66, 253–258. [Google Scholar] [CrossRef] [PubMed]
- Patters, B.J.; Kumar, S. The role of exosomal transport of viral agents in persistent HIV pathogenesis. Retrovirology 2018, 15, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Albert, S.M. The Neurology of Aids, Third Edition. Neurology 2012, 79, 835. [Google Scholar] [CrossRef]
- Liu, Y.; Jones, M.; Hingtgen, C.M.; Bu, G.; Laribee, R.N.; Tanzi, R.E.; Moir, R.; Nath, A.; He, J.J. Uptake of HIV-1 Tat protein mediated by low-density lipoprotein receptor-related protein disrupts the neuronal metabolic balance of the receptor ligands. Nat. Med. 2000, 6, 1380–1387. [Google Scholar] [CrossRef]
- Ru, W.; Tang, S.-J. HIV-1 gp120Bal down-Regulates Phosphorylated NMDA Receptor Subunit 1 in Cortical Neurons via Activation of Glutamate and Chemokine Receptors. J. Neuroimmune Pharmacol. 2015, 11, 182–191. [Google Scholar] [CrossRef]
- Bonaviaab, R.; Bajettoab, A.; Barberoab, S.; Albini, A.; Noonan, D.; Schettini, G. HIV-1 Tat Causes Apoptotic Death and Calcium Homeostasis Alterations in Rat Neurons. Biochem. Biophys. Res. Commun. 2001, 288, 301–308. [Google Scholar] [CrossRef]
- Fontana, G.; Valenti, L.; Raiteri, M. Gp120 can revert antagonism at the glycine site of NMDA receptors mediating GABA release from cultured hippocampal neurons. J. Neurosci. Res. 1997, 49, 732–738. [Google Scholar] [CrossRef]
- Nakanishi, N.; Kang, Y.-J.; Tu, S.; McKercher, S.R.; Masliah, E.; Lipton, S.A. Differential Effects of Pharmacologic and Genetic Modulation of NMDA Receptor Activity on HIV/gp120-Induced Neuronal Damage in an In Vivo Mouse Model. J. Mol. Neurosci. 2015, 58, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Thaney, V.E.; Sanchez, A.B.; Fields, J.A.; Minassian, A.; Young, J.W.; Maung, R.; Kaul, M. Transgenic mice expressing HIV-1 envelope protein gp120 in the brain as an animal model in neuroAIDS research. J. NeuroVirology 2017, 24, 156–167. [Google Scholar] [CrossRef] [PubMed]
- Hammond, J.W.; Qiu, W.Q.; Marker, D.F.; Chamberlain, J.M.; Greaves-Tunnell, W.; Bellizzi, M.J.; Lu, S.-M.; Gelbard, H.A. HIV Tat causes synapse loss in a mouse model of HIV-associated neurocognitive disorder that is independent of the classical complement cascade component C1q. Glia 2018, 66, 2563–2574. [Google Scholar] [CrossRef] [PubMed]
- Ru, W.; Liu, X.; Bae, C.; Shi, Y.; Walikonis, R.; Chung, J.M.; Tang, S.-J. Microglia Mediate HIV-1 gp120-Induced Synaptic Degeneration in Spinal Pain Neural Circuits. J. Neurosci. 2019, 39, 8408–8421. [Google Scholar] [CrossRef]
- Raybuck, J.D.; Hargus, N.J.; Thayer, S.A. A GluN2B-Selective NMDAR Antagonist Reverses Synapse Loss and Cognitive Impairment Produced by the HIV-1 Protein Tat. J. Neurosci. 2017, 37, 7837–7847. [Google Scholar] [CrossRef]
- Tremblay, M.; Marker, D.; Puccini, J.M.; Muly, E.C.; Lu, S.-M.; A Gelbard, H. Ultrastructure of microglia-synapse interactions in the HIV-1 Tat-injected murine central nervous system. Commun. Integr. Biol. 2013, 6, e27670. [Google Scholar] [CrossRef]
- Kedzierska, K.; Azzam, R.; Ellery, P.; Mak, J.; Jaworowski, A.; Crowe, S.M. Defective phagocytosis by human monocyte/macrophages following HIV-1 infection: Underlying mechanisms and modulation by adjunctive cytokine therapy. J. Clin. Virol. 2002, 26, 247–263. [Google Scholar] [CrossRef]
- McGuire, J.L.; CNS HIV Antiretroviral Therapy Effects Research (CHARTER) Group; Gill, A.J.; Douglas, S.D.; Kolson, D.L. The complement system, neuronal injury, and cognitive function in horizontally-acquired HIV-infected youth. J. NeuroVirology 2016, 22, 823–830. [Google Scholar] [CrossRef]
- Nitkiewicz, J.; Borjabad, A.; Morgello, S.; Murray, J.; Chao, W.; Emdad, L.; Fisher, P.B.; Potash, M.J.; Volsky, D.J. HIV induces expression of complement component C3 in astrocytes by NF-κB-dependent activation of interleukin-6 synthesis. J. Neuroinflammation 2017, 14, 1–15. [Google Scholar] [CrossRef]
- Shapshak, P.; Duncan, R.; Minagar, A.; de la Vega, P.R.; Stewart, R.V.; Goodkin, K. Elevated expression OF IFN-gamma in the HIV-1 infected brain. Front. Biosci. 2004, 9, 1073–1081. [Google Scholar] [CrossRef]
- Schrier, R.D.; Hong, S.; Crescini, M.; Ellis, R.; Pérez-Santiago, J.; Spina, C.; Letendre, S.; HNRP Group. Cerebrospinal Fluid (CSF) CD8+ T-Cells That Express Interferon-Gamma Contribute to HIV Associated Neurocognitive Disorders (HAND). PLoS ONE 2015, 10, e0116526. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Diseases | Microglia | Astrocytes |
|---|---|---|
| AD | C1q, C3, CR3 [65] 1 | APOE [69] 2 |
| TREM2 [64] 1 | ||
| WNV | C3, C3aR [75] 1 | No reported pruning activity |
| IFN-γ [76] 1 | ||
| ZIKV | C3, C1q [78] | Phagocytosis of debris was reported, but no mechanism was reported [80] |
| TNF-α [78] | ||
| IFN-γ [76] 1 | ||
| Amyotrophic lateral sclerosis (ALS) | C1q implicated [81] | No reported pruning activity |
| Parkinson’s Disease (PD) | Pruning was reported, but no mechanism was reported [82] 1 | Neuronal Phagocytosis, no mechanism reported [82] |
| Multiple sclerosis (MS) | C3 [83] 1 | No reported pruning activity |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Watson, Z.; Tang, S.-J. Aberrant Synaptic Pruning in CNS Diseases: A Critical Player in HIV-Associated Neurological Dysfunction? Cells 2022, 11, 1943. https://doi.org/10.3390/cells11121943
Watson Z, Tang S-J. Aberrant Synaptic Pruning in CNS Diseases: A Critical Player in HIV-Associated Neurological Dysfunction? Cells. 2022; 11(12):1943. https://doi.org/10.3390/cells11121943
Chicago/Turabian StyleWatson, Zachary, and Shao-Jun Tang. 2022. "Aberrant Synaptic Pruning in CNS Diseases: A Critical Player in HIV-Associated Neurological Dysfunction?" Cells 11, no. 12: 1943. https://doi.org/10.3390/cells11121943
APA StyleWatson, Z., & Tang, S.-J. (2022). Aberrant Synaptic Pruning in CNS Diseases: A Critical Player in HIV-Associated Neurological Dysfunction? Cells, 11(12), 1943. https://doi.org/10.3390/cells11121943