
Insights into the Structure and Function of the Pex1/Pex6 AAA-ATPase in Peroxisome Homeostasis


Abstract
:1. Introduction
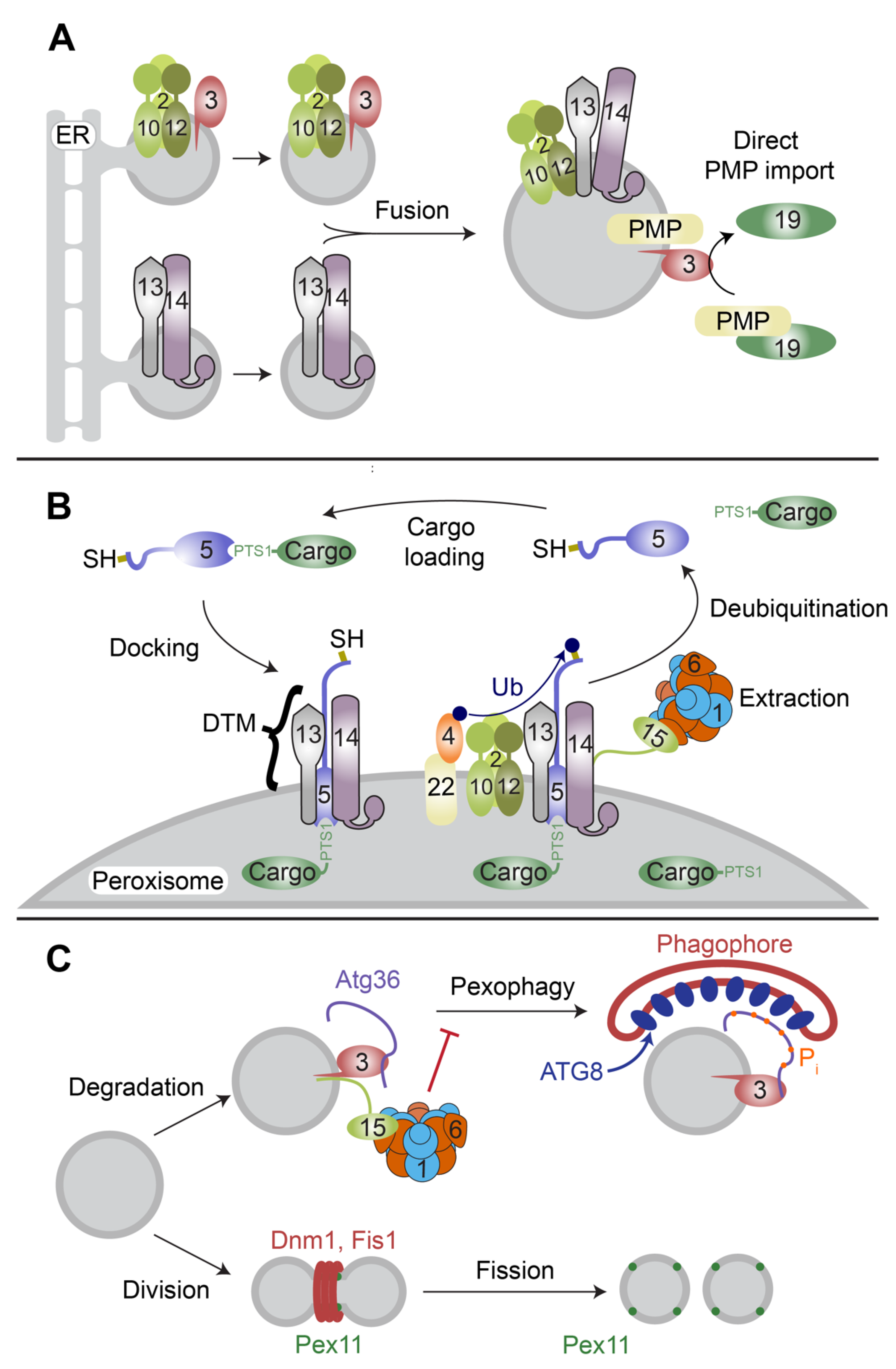
2. Peroxisome Membrane Biogenesis
3. Matrix Protein Import
4. Proposed Roles for Pex1 and Pex6 in Peroxisome Biogenesis and Maintenance
5. Pex1/Pex6 Structure and Threading Mechanism
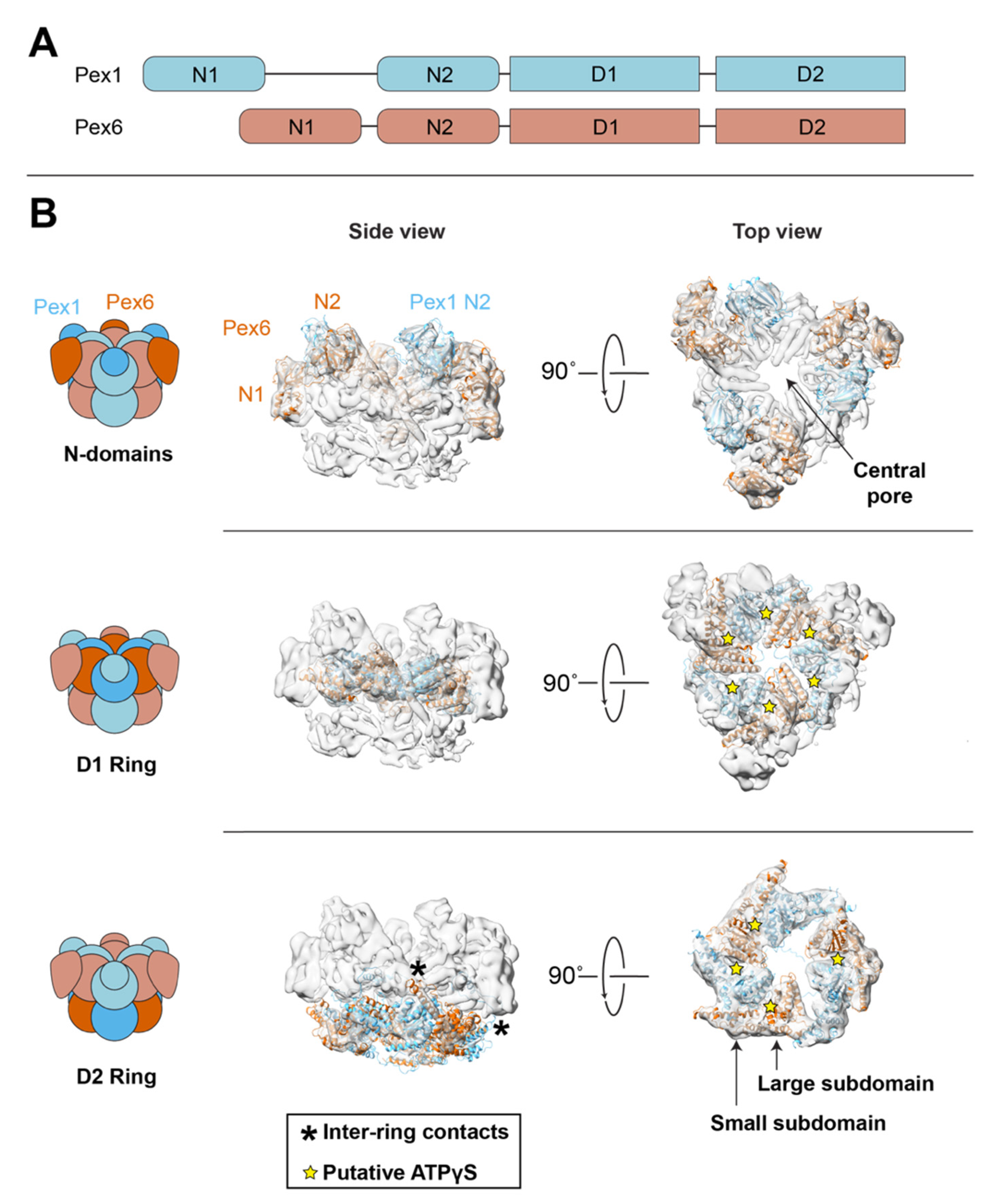
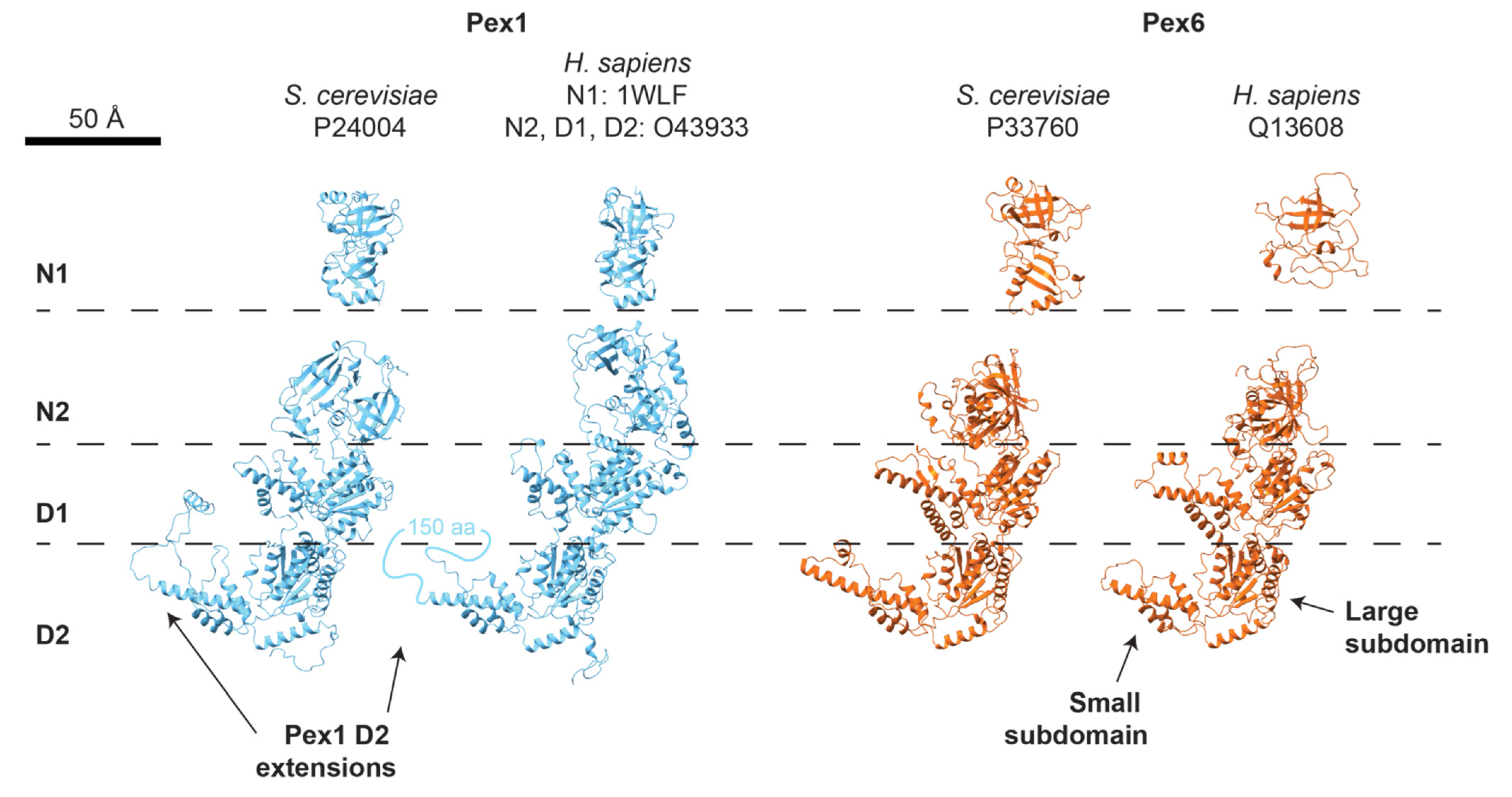
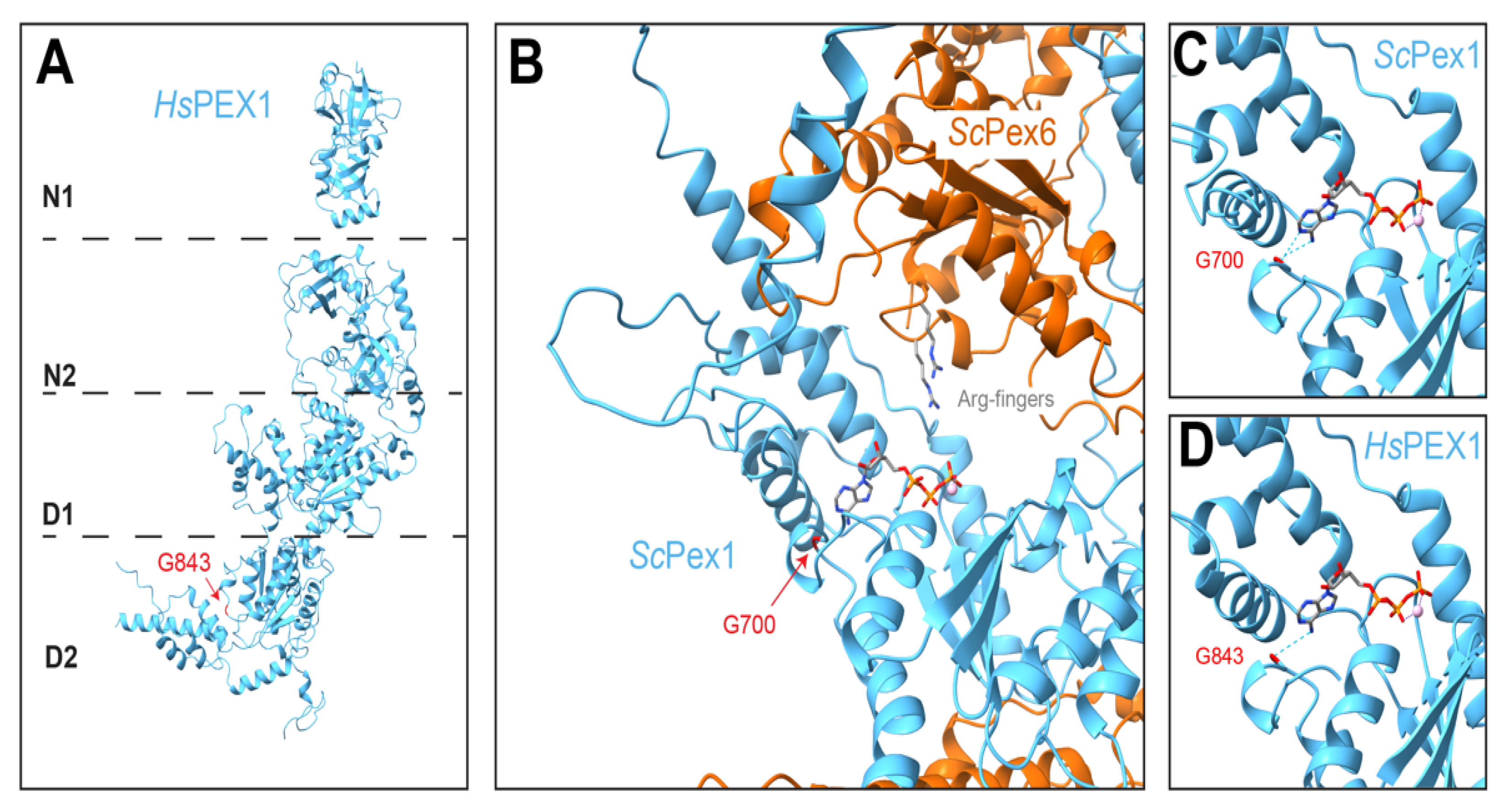
5.1. Pex1/Pex6 Architecture
5.2. Pex1/Pex6 Substrate Processing
5.3. Coordination between ATPase Rings
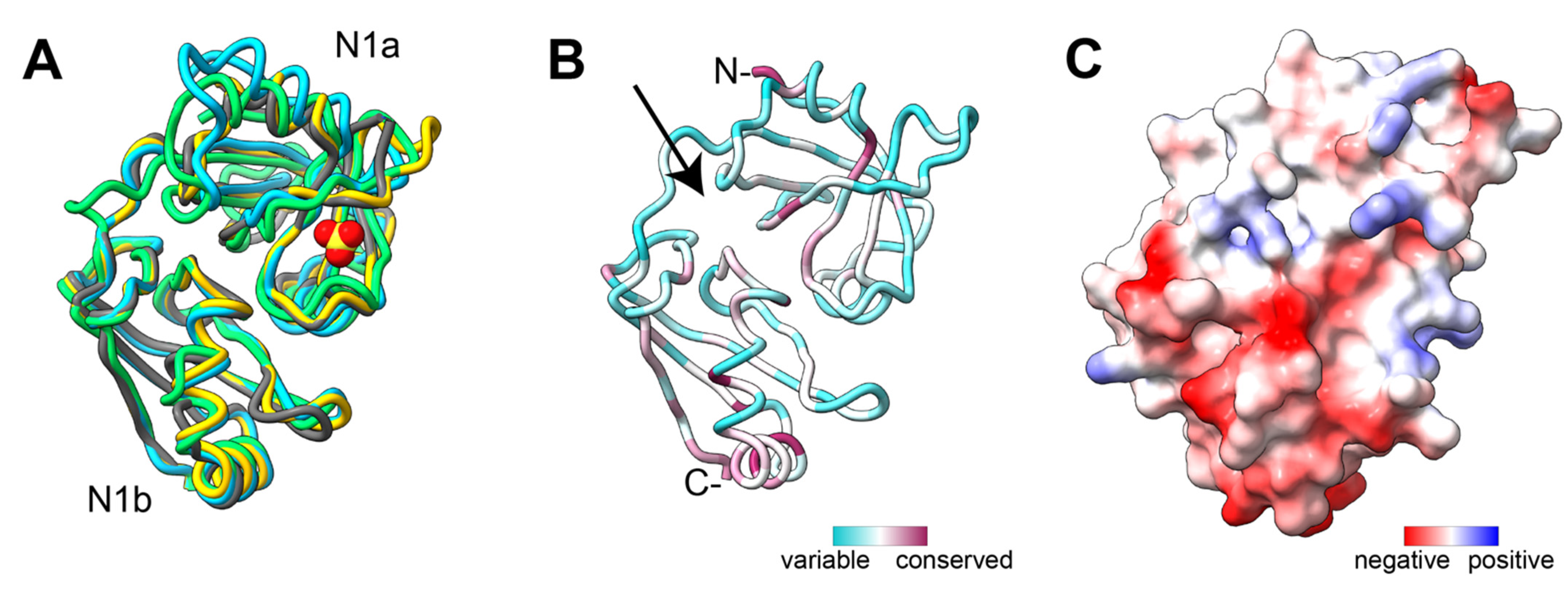
5.4. Domains for Substrate Engagement
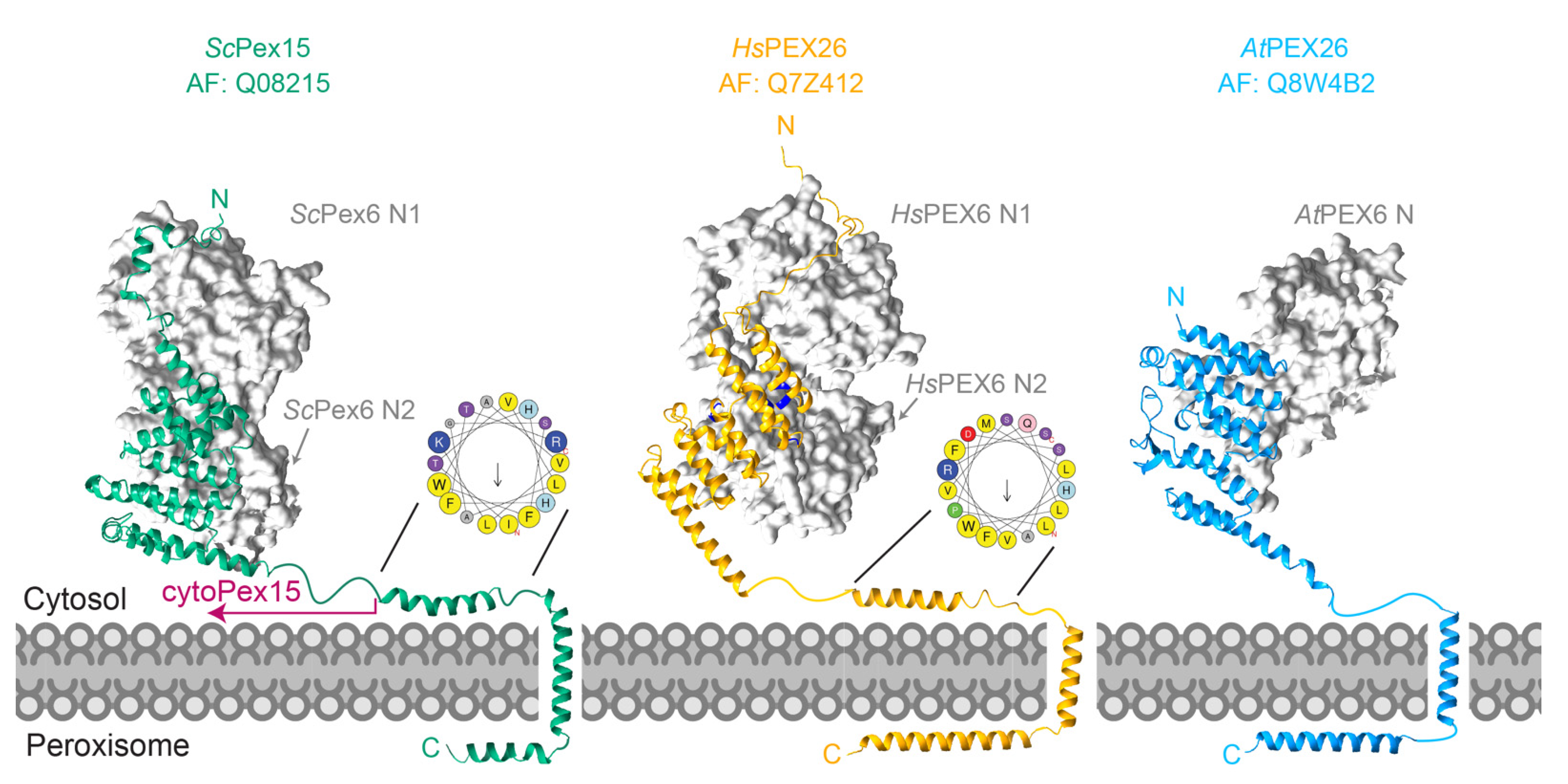
5.5. Comparison to Human PEX1/PEX6
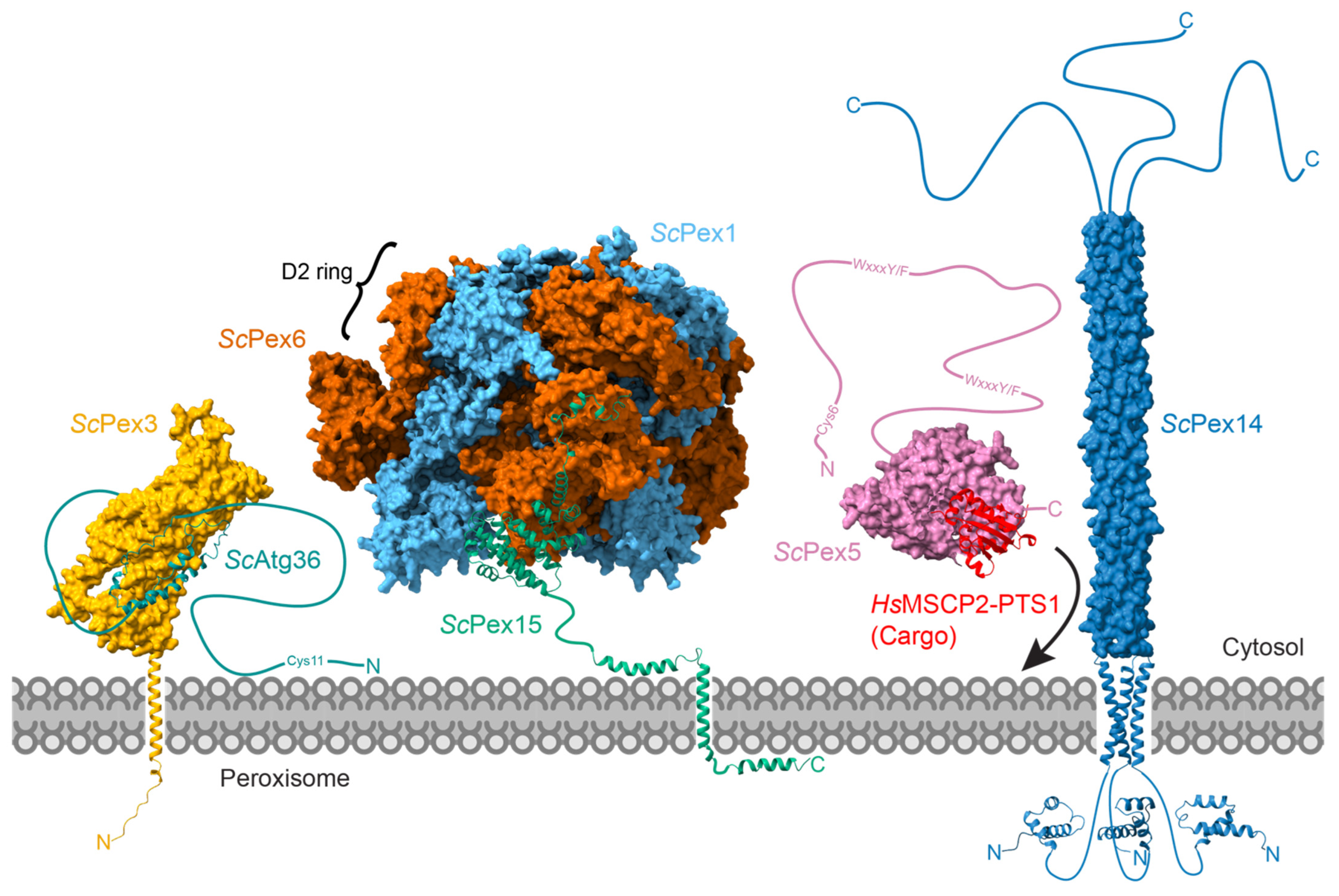
6. Pex15/PEX26 as a Partner and Substrate
7. Potential Substrates of Pex1/Pex6
8. Peroxisomes and Pex1/Pex6 in Disease
9. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gabaldón, T. Peroxisome Diversity and Evolution. Philos. Trans. R. Soc. B Biol. Sci. 2010, 365, 765–773. [Google Scholar] [CrossRef] [Green Version]
- Jansen, R.L.M.; Santana Molina, C.; Van Den Noort, M.; Devos, D.P.; Van Der Klei, I.J. Comparative Genomics of Peroxisome Biogenesis Proteins: Making Sense of the PEX Proteins. Front. Cell Dev. Biol. 2021, 9, 654163. [Google Scholar] [CrossRef]
- Nordgren, M.; Fransen, M. Peroxisomal Metabolism and Oxidative Stress. Biochimie 2014, 98, 56–62. [Google Scholar] [CrossRef] [Green Version]
- Goldman, B.M.; Blobel, G. Biogenesis of Peroxisomes: Intracellular Site of Synthesis of Catalase and Uricase. Proc. Natl. Acad. Sci. USA 1978, 75, 5066–5070. [Google Scholar] [CrossRef] [Green Version]
- Gurvitz, A.; Rottensteiner, H. The Biochemistry of Oleate Induction: Transcriptional Upregulation and Peroxisome Proliferation. Biochim. Biophys. Acta BBA-Mol. Cell Res. 2006, 1763, 1392–1402. [Google Scholar] [CrossRef]
- Schrader, M.; Costello, J.L.; Godinho, L.F.; Azadi, A.S.; Islinger, M. Proliferation and Fission of Peroxisomes—An Update. Biochim. Biophys. Acta BBA-Mol. Cell Res. 2016, 1863, 971–983. [Google Scholar] [CrossRef]
- Gould, S.J.; McCollum, D.; Spong, A.P.; Heyman, J.A.; Subramani, S. Development of the Yeast Pichia Pastoris as a Model Organism for a Genetic and Molecular Analysis of Peroxisome Assembly. Yeast 1992, 8, 613–628. [Google Scholar] [CrossRef]
- Mukherji, S.; O’Shea, E.K. Mechanisms of Organelle Biogenesis Govern Stochastic Fluctuations in Organelle Abundance. eLife 2014, 3, e02678. [Google Scholar] [CrossRef]
- Saffian, D.; Grimm, I.; Girzalsky, W.; Erdmann, R. ATP-Dependent Assembly of the Heteromeric Pex1p-Pex6p-Complex of the Peroxisomal Matrix Protein Import Machinery. J. Struct. Biol. 2012, 179, 126–132. [Google Scholar] [CrossRef]
- Birschmann, I.; Rosenkranz, K.; Erdmann, R.; Kunau, W.-H. Structural and Functional Analysis of the Interaction of the AAA-Peroxins Pex1p and Pex6p. FEBS J. 2005, 272, 47–58. [Google Scholar] [CrossRef]
- Tamura, S.; Yasutake, S.; Matsumoto, N.; Fujiki, Y. Dynamic and Functional Assembly of the AAA Peroxins, Pex1p and Pex6p, and Their Membrane Receptor Pex26p. J. Biol. Chem. 2006, 281, 27693–27704. [Google Scholar] [CrossRef] [Green Version]
- Matsumoto, N.; Tamura, S.; Fujiki, Y. The Pathogenic Peroxin Pex26p Recruits the Pex1p–Pex6p AAA ATPase Complexes to Peroxisomes. Nat. Cell Biol. 2003, 5, 454–460. [Google Scholar] [CrossRef]
- Kiel, J.A.K.W.; Veenhuis, M.; van der Klei, I.J. PEX Genes in Fungal Genomes: Common, Rare or Redundant. Traffic 2006, 7, 1291–1303. [Google Scholar] [CrossRef] [Green Version]
- Oliveira, M.E.; Gouveia, A.M.; Pinto, R.A.; Sá-Miranda, C.; Azevedo, J.E. The Energetics of Pex5p-Mediated Peroxisomal Protein Import. J. Biol. Chem. 2003, 278, 39483–39488. [Google Scholar] [CrossRef] [Green Version]
- Romano, F.B.; Blok, N.B.; Rapoport, T.A. Peroxisome Protein Import Recapitulated in Xenopus Egg Extracts. J. Cell Biol. 2019, 218, 2021–2034. [Google Scholar] [CrossRef] [Green Version]
- Law, K.B.; Bronte-Tinkew, D.; Di Pietro, E.; Snowden, A.; Jones, R.O.; Moser, A.; Brumell, J.H.; Braverman, N.; Kim, P.K. The Peroxisomal AAA ATPase Complex Prevents Pexophagy and Development of Peroxisome Biogenesis Disorders. Autophagy 2017, 13, 868–884. [Google Scholar] [CrossRef]
- Nuttall, J.M.; Motley, A.M.; Hettema, E.H. Deficiency of the Exportomer Components Pex1, Pex6, and Pex15 Causes Enhanced Pexophagy in Saccharomyces Cerevisiae. Autophagy 2014, 10, 835–845. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.; Kamber, R.A.; Denic, V. The Peroxisomal Exportomer Directly Inhibits Phosphoactivation of the Pexophagy Receptor Atg36 to Suppress Pexophagy in Yeast. eLife 2022, 11, e74531. [Google Scholar] [CrossRef]
- Waterham, H.R.; Ebberink, M.S. Genetics and Molecular Basis of Human Peroxisome Biogenesis Disorders. Biochim. Biophys. Acta BBA-Mol. Basis Dis. 2012, 1822, 1430–1441. [Google Scholar] [CrossRef] [Green Version]
- Waterham, H.R.; Ferdinandusse, S.; Wanders, R.J.A. Human Disorders of Peroxisome Metabolism and Biogenesis. Biochim. Biophys. Acta BBA-Mol. Cell Res. 2016, 1863, 922–933. [Google Scholar] [CrossRef]
- Kim, P.K.; Mullen, R.T.; Schumann, U.; Lippincott-Schwartz, J. The Origin and Maintenance of Mammalian Peroxisomes Involves a de Novo PEX16-Dependent Pathway from the ER. J. Cell Biol. 2006, 173, 521–532. [Google Scholar] [CrossRef] [Green Version]
- Motley, A.M.; Hettema, E.H. Yeast Peroxisomes Multiply by Growth and Division. J. Cell Biol. 2007, 178, 399–410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoepfner, D.; Schildknegt, D.; Braakman, I.; Philippsen, P.; Tabak, H.F. Contribution of the Endoplasmic Reticulum to Peroxisome Formation. Cell 2005, 122, 85–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thoms, S.; Harms, I.; Kalies, K.-U.; Gärtner, J. Peroxisome Formation Requires the Endoplasmic Reticulum Channel Protein Sec61: ER Translocon Requirement for Peroxisome Formation. Traffic 2012, 13, 599–609. [Google Scholar] [CrossRef] [PubMed]
- Schuldiner, M.; Metz, J.; Schmid, V.; Denic, V.; Rakwalska, M.; Schmitt, H.D.; Schwappach, B.; Weissman, J.S. The GET Complex Mediates Insertion of Tail-Anchored Proteins into the ER Membrane. Cell 2008, 134, 634–645. [Google Scholar] [CrossRef]
- van der Zand, A.; Braakman, I.; Tabak, H.F. Peroxisomal Membrane Proteins Insert into the Endoplasmic Reticulum. Mol. Biol. Cell 2010, 21, 2057–2065. [Google Scholar] [CrossRef] [Green Version]
- Lam, S.K.; Yoda, N.; Schekman, R. A Vesicle Carrier That Mediates Peroxisome Protein Traffic from the Endoplasmic Reticulum. Proc. Natl. Acad. Sci. USA 2010, 107, 21523–21528. [Google Scholar] [CrossRef] [Green Version]
- Agrawal, G.; Fassas, S.N.; Xia, Z.-J.; Subramani, S. Distinct Requirements for Intra-ER Sorting and Budding of Peroxisomal Membrane Proteins from the ER. J. Cell Biol. 2016, 212, 335–348. [Google Scholar] [CrossRef] [Green Version]
- van der Zand, A.; Gent, J.; Braakman, I.; Tabak, H.F. Biochemically Distinct Vesicles from the Endoplasmic Reticulum Fuse to Form Peroxisomes. Cell 2012, 149, 397–409. [Google Scholar] [CrossRef] [Green Version]
- Sugiura, A.; Mattie, S.; Prudent, J.; McBride, H.M. Newly Born Peroxisomes Are a Hybrid of Mitochondrial and ER-Derived Pre-Peroxisomes. Nature 2017, 542, 251–254. [Google Scholar] [CrossRef]
- Motley, A.M.; Galvin, P.C.; Ekal, L.; Nuttall, J.M.; Hettema, E.H. Reevaluation of the Role of Pex1 and Dynamin-Related Proteins in Peroxisome Membrane Biogenesis. J. Cell Biol. 2015, 211, 1041–1056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knoops, K.; de Boer, R.; Kram, A.; van der Klei, I.J. Yeast Pex1 Cells Contain Peroxisomal Ghosts That Import Matrix Proteins upon Reintroduction of Pex1. J. Cell Biol. 2015, 211, 955–962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raychaudhuri, S.; Prinz, W.A. Nonvesicular Phospholipid Transfer between Peroxisomes and the Endoplasmic Reticulum. Proc. Natl. Acad. Sci. USA 2008, 105, 15785–15790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farré, J.-C.; Mahalingam, S.S.; Proietto, M.; Subramani, S. Peroxisome Biogenesis, Membrane Contact Sites, and Quality Control. EMBO Rep. 2019, 20, e46864. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.M.; Morrell, J.C.; Gould, S.J. Multiple Distinct Targeting Signals in Integral Peroxisomal Membrane Proteins. J. Cell Biol. 2001, 153, 1141–1150. [Google Scholar] [CrossRef]
- Rottensteiner, H.; Kramer, A.; Lorenzen, S.; Stein, K.; Landgraf, C.; Volkmer-Engert, R.; Erdmann, R. Peroxisomal Membrane Proteins Contain Common Pex19p-Binding Sites That Are an Integral Part of Their Targeting Signals. Mol. Biol. Cell 2004, 15, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hettema, E.H.; Girzalsky, W.; van den Berg, M.; Erdmann, R.; Distel, B. Saccharomyces Cerevisiae Pex3p and Pex19p Are Required for Proper Localization and Stability of Peroxisomal Membrane Proteins. EMBO J. 2000, 19, 223–233. [Google Scholar] [CrossRef] [Green Version]
- Sacksteder, K.A.; Jones, J.M.; South, S.T.; Li, X.; Liu, Y.; Gould, S.J. Pex19 Binds Multiple Peroxisomal Membrane Proteins, Is Predominantly Cytoplasmic, and Is Required for Peroxisome Membrane Synthesis. J. Cell Biol. 2000, 148, 931–944. [Google Scholar] [CrossRef] [Green Version]
- Fang, Y.; Morrell, J.C.; Jones, J.M.; Gould, S.J. PEX3 Functions as a PEX19 Docking Factor in the Import of Class I Peroxisomal Membrane Proteins. J. Cell Biol. 2004, 164, 863–875. [Google Scholar] [CrossRef] [Green Version]
- Jones, J.M.; Morrell, J.C.; Gould, S.J. PEX19 Is a Predominantly Cytosolic Chaperone and Import Receptor for Class 1 Peroxisomal Membrane Proteins. J. Cell Biol. 2004, 164, 57–67. [Google Scholar] [CrossRef] [Green Version]
- Shibata, H.; Kashiwayama, Y.; Imanaka, T.; Kato, H. Domain Architecture and Activity of Human Pex19p, a Chaperone-like Protein for Intracellular Trafficking of Peroxisomal Membrane Proteins. J. Biol. Chem. 2004, 279, 38486–38494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, Y.; Shibata, H.; Nakatsu, T.; Nakano, H.; Kashiwayama, Y.; Imanaka, T.; Kato, H. Structural Basis for Docking of Peroxisomal Membrane Protein Carrier Pex19p onto Its Receptor Pex3p. EMBO J. 2010, 29, 4083–4093. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Yagita, Y.; Fujiki, Y. Assembly of Peroxisomal Membrane Proteins via the Direct Pex19p-Pex3p Pathway. Traffic 2016, 17, 433–455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Pieuchot, L.; Loh, R.A.; Yang, J.; Kari, T.M.A.; Wong, J.Y.; Jedd, G. Hydrophobic Handoff for Direct Delivery of Peroxisome Tail-Anchored Proteins. Nat. Commun. 2014, 5, 5790. [Google Scholar] [CrossRef] [Green Version]
- Motley, A.M.; Ward, G.P.; Hettema, E.H. Dnm1p-Dependent Peroxisome Fission Requires Caf4p, Mdv1p and Fis1p. J. Cell Sci. 2008, 121, 1633–1640. [Google Scholar] [CrossRef] [Green Version]
- Schrader, M.; Reuber, B.E.; Morrell, J.C.; Jimenez-Sanchez, G.; Obie, C.; Stroh, T.A.; Valle, D.; Schroer, T.A.; Gould, S.J. Expression of PEX11β Mediates Peroxisome Proliferation in the Absence of Extracellular Stimuli. J. Biol. Chem. 1998, 273, 29607–29614. [Google Scholar] [CrossRef] [Green Version]
- Koch, A.; Yoon, Y.; Bonekamp, N.A.; McNiven, M.A.; Schrader, M. A Role for Fis1 in Both Mitochondrial and Peroxisomal Fission in Mammalian Cells. Mol. Biol. Cell 2005, 16, 5077–5086. [Google Scholar] [CrossRef]
- Koch, J.; Brocard, C. PEX11 Proteins Attract Mff and HFis1 to Coordinate Peroxisomal Fission. J. Cell Sci. 2012, 125 Pt 16, 3813–3826. [Google Scholar] [CrossRef] [Green Version]
- Opaliński, Ł.; Kiel, J.A.K.W.; Williams, C.; Veenhuis, M.; van der Klei, I.J. Membrane Curvature during Peroxisome Fission Requires Pex11. EMBO J. 2011, 30, 5–16. [Google Scholar] [CrossRef] [Green Version]
- Mahalingam, S.S.; Shukla, N.; Farré, J.-C.; Zientara-Rytter, K.; Subramani, S. Balancing the Opposing Principles That Govern Peroxisome Homeostasis. Trends Biochem. Sci. 2021, 46, 200–212. [Google Scholar] [CrossRef]
- Walton, P.A.; Hill, P.E.; Subramani, S. Import of Stably Folded Proteins into Peroxisomes. Mol. Biol. Cell 1995, 6, 675–683. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Purdue, P.E.; Lazarow, P.B. Eci1p Uses a PTS1 to Enter Peroxisomes: Either Its Own or That of a Partner, Dci1p. Eur. J. Cell Biol. 2001, 80, 126–138. [Google Scholar] [CrossRef] [PubMed]
- DeLoache, W.C.; Russ, Z.N.; Dueber, J.E. Towards Repurposing the Yeast Peroxisome for Compartmentalizing Heterologous Metabolic Pathways. Nat. Commun. 2016, 7, 11152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antonenkov, V.D.; Sormunen, R.T.; Hiltunen, J.K. The Rat Liver Peroxisomal Membrane Forms a Permeability Barrier for Cofactors but Not for Small Metabolites in Vitro. J. Cell Sci. 2004, 117, 5633–5642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nötzel, C.; Lingner, T.; Klingenberg, H.; Thoms, S. Identification of New Fungal Peroxisomal Matrix Proteins and Revision of the PTS1 Consensus: Peroxisomal Targeting Signal Type 1 (PTS1) in Yeast. Traffic 2016, 17, 1110–1124. [Google Scholar] [CrossRef]
- Fung, K.; Clayton, C. Recognition of a Peroxisomal Tripeptide Entry Signal by the Glycosomes of Trypanosoma Brucei. Mol. Biochem. Parasitol. 1991, 45, 261–264. [Google Scholar] [CrossRef]
- Dodt, G.; Braverman, N.; Wong, C.; Moser, A.; Moser, H.W.; Watkins, P.; Valle, D.; Gould, S.J. Mutations in the PTS1 Receptor Gene, PXR1, Define Complementation Group 2 of the Peroxisome Biogenesis Disorders. Nat. Genet. 1995, 9, 115–125. [Google Scholar] [CrossRef]
- Terlecky, S.R.; Nuttley, W.M.; McCollum, D.; Sock, E.; Subramani, S. The Pichia Pastoris Peroxisomal Protein PAS8p Is the Receptor for the C-Terminal Tripeptide Peroxisomal Targeting Signal. EMBO J. 1995, 14, 3627–3634. [Google Scholar] [CrossRef]
- Stanley, W.A.; Filipp, F.V.; Kursula, P.; Schüller, N.; Erdmann, R.; Schliebs, W.; Sattler, M.; Wilmanns, M. Recognition of a Functional Peroxisome Type 1 Target by the Dynamic Import Receptor Pex5p. Mol. Cell 2006, 24, 653–663. [Google Scholar] [CrossRef] [Green Version]
- Carvalho, A.F.; Costa-Rodrigues, J.; Correia, I.; Costa Pessoa, J.; Faria, T.Q.; Martins, C.L.; Fransen, M.; Sá-Miranda, C.; Azevedo, J.E. The N-Terminal Half of the Peroxisomal Cycling Receptor Pex5p Is a Natively Unfolded Domain. J. Mol. Biol. 2006, 356, 864–875. [Google Scholar] [CrossRef] [Green Version]
- Gaussmann, S.; Gopalswamy, M.; Eberhardt, M.; Reuter, M.; Zou, P.; Schliebs, W.; Erdmann, R.; Sattler, M. Membrane Interactions of the Peroxisomal Proteins PEX5 and PEX14. Front. Cell Dev. Biol. 2021, 9, 651449. [Google Scholar] [CrossRef] [PubMed]
- Schäfer, A.; Kerssen, D.; Veenhuis, M.; Kunau, W.-H.; Schliebs, W. Functional Similarity between the Peroxisomal PTS2 Receptor Binding Protein Pex18p and the N-Terminal Half of the PTS1 Receptor Pex5p. Mol. Cell. Biol. 2004, 24, 8895–8906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klein, A.T.J.; Berg, M.; van den Bottger, G.; Tabak, H.F.; Distel, B. Saccharomyces Cerevisiae Acyl-CoA Oxidase Follows a Novel, Non-PTS1, Import Pathway into Peroxisomes That Is Dependent on Pex5p. J. Biol. Chem. 2002, 277, 25011–25019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gunkel, K.; van Dijk, R.; Veenhuis, M.; van der Klei, I.J. Routing of Hansenula Polymorpha Alcohol Oxidase: An Alternative Peroxisomal Protein-Sorting Machinery. Mol. Biol. Cell 2004, 15, 1347–1355. [Google Scholar] [CrossRef] [Green Version]
- Meinecke, M.; Cizmowski, C.; Schliebs, W.; Krüger, V.; Beck, S.; Wagner, R.; Erdmann, R. The Peroxisomal Importomer Constitutes a Large and Highly Dynamic Pore. Nat. Cell Biol. 2010, 12, 273–277. [Google Scholar] [CrossRef]
- Niederhoff, K.; Meindl-Beinker, N.M.; Kerssen, D.; Perband, U.; Schäfer, A.; Schliebs, W.; Kunau, W.-H. Yeast Pex14p Possesses Two Functionally Distinct Pex5p and One Pex7p Binding Sites. J. Biol. Chem. 2005, 280, 35571–35578. [Google Scholar] [CrossRef] [Green Version]
- Dias, A.F.; Rodrigues, T.A.; Pedrosa, A.G.; Barros-Barbosa, A.; Francisco, T.; Azevedo, J.E. The Peroxisomal Matrix Protein Translocon Is a Large Cavity-Forming Protein Assembly into Which PEX5 Protein Enters to Release Its Cargo. J. Biol. Chem. 2017, 292, 15287–15300. [Google Scholar] [CrossRef] [Green Version]
- Gould, S.J.; Kalish, J.E.; Morrell, J.C.; Bjorkman, J.; Urquhart, I.A.J.; CraneH, D.I. Pex13p Is an SH3 Protein of the Peroxisome Membrane and a Docking Factor for the Predominantly Cytoplasmic PTSl Receptor. J. Cell Biol. 1996, 135, 11. [Google Scholar] [CrossRef] [Green Version]
- Urquhart, A.J.; Kennedy, D.; Gould, S.J.; Crane, D.I. Interaction of Pex5p, the Type 1 Peroxisome Targeting Signal Receptor, with the Peroxisomal Membrane Proteins Pex14p and Pex13p. J. Biol. Chem. 2000, 275, 4127–4136. [Google Scholar] [CrossRef] [Green Version]
- Deckers, M.; Emmrich, K.; Girzalsky, W.; Awa, W.L.; Kunau, W.-H.; Erdmann, R. Targeting of Pex8p to the Peroxisomal Importomer. Eur. J. Cell Biol. 2010, 89, 924–931. [Google Scholar] [CrossRef]
- Otera, H.; Setoguchi, K.; Hamasaki, M.; Kumashiro, T.; Shimizu, N.; Fujiki, Y. Peroxisomal Targeting Signal Receptor Pex5p Interacts with Cargoes and Import Machinery Components in a Spatiotemporally Differentiated Manner: Conserved Pex5p WXXXF/Y Motifs Are Critical for Matrix Protein Import. Mol. Cell. Biol. 2002, 22, 1639–1655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agne, B.; Meindl, N.M.; Niederhoff, K.; Einwächter, H.; Rehling, P.; Sickmann, A.; Meyer, H.E.; Girzalsky, W.; Kunau, W.-H. Pex8p: An Intraperoxisomal Organizer of the Peroxisomal Import Machinery. Mol. Cell 2003, 11, 635–646. [Google Scholar] [CrossRef]
- Reguenga, C.; Oliveira, M.E.M.; Gouveia, A.M.M.; Sá-Miranda, C.; Azevedo, J.E. Characterization of the Mammalian Peroxisomal Import Machinery: Pex2p, Pex5p, Pex12p, AND Pex14p are subunits of the same protein assembly. J. Biol. Chem. 2001, 276, 29935–29942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montilla-Martinez, M.; Beck, S.; Klümper, J.; Meinecke, M.; Schliebs, W.; Wagner, R.; Erdmann, R. Distinct Pores for Peroxisomal Import of PTS1 and PTS2 Proteins. Cell Rep. 2015, 13, 2126–2134. [Google Scholar] [CrossRef] [Green Version]
- Shiozawa, K.; Konarev, P.V.; Neufeld, C.; Wilmanns, M.; Svergun, D.I. Solution Structure of Human Pex5·Pex14·PTS1 Protein Complexes Obtained by Small Angle X-ray Scattering. J. Biol. Chem. 2009, 284, 25334–25342. [Google Scholar] [CrossRef] [Green Version]
- Williams, C.; van den Berg, M.; Distel, B. Saccharomyces Cerevisiae Pex14p Contains Two Independent Pex5p Binding Sites, Which Are Both Essential for PTS1 Protein Import. FEBS Lett. 2005, 579, 3416–3420. [Google Scholar] [CrossRef] [Green Version]
- Pires, J.R.; Hong, X.; Brockmann, C.; Volkmer-Engert, R.; Schneider-Mergener, J.; Oschkinat, H.; Erdmann, R. The ScPex13p SH3 Domain Exposes Two Distinct Binding Sites for Pex5p and Pex14p. J. Mol. Biol. 2003, 326, 1427–1435. [Google Scholar] [CrossRef]
- Girzalsky, W.; Rehling, P.; Stein, K.; Kipper, J.; Blank, L.; Kunau, W.-H.; Erdmann, R. Involvement of Pex13p in Pex14p Localization and Peroxisomal Targeting Signal 2–Dependent Protein Import into Peroxisomes. J. Cell Biol. 1999, 144, 1151–1162. [Google Scholar] [CrossRef]
- Oliveira, M.E.M.; Reguenga, C.; Gouveia, A.M.M.; Guimarães, C.P.; Schliebs, W.; Kunau, W.-H.; Silva, M.T.; Sá-Miranda, C.; Azevedo, J.E. Mammalian Pex14p: Membrane Topology and Characterisation of the Pex14p–Pex14p Interaction. Biochim. Biophys. Acta BBA-Biomembr. 2002, 1567, 13–22. [Google Scholar] [CrossRef] [Green Version]
- Shimizu, N.; Itoh, R.; Hirono, Y.; Otera, H.; Ghaedi, K.; Tateishi, K.; Tamura, S.; Okumoto, K.; Harano, T.; Mukai, S.; et al. The Peroxin Pex14p. J. Biol. Chem. 1999, 274, 12593–12604. [Google Scholar] [CrossRef] [Green Version]
- Barros-Barbosa, A.; Ferreira, M.J.; Rodrigues, T.A.; Pedrosa, A.G.; Grou, C.P.; Pinto, M.P.; Fransen, M.; Francisco, T.; Azevedo, J.E. Membrane Topologies of PEX 13 and PEX 14 Provide New Insights on the Mechanism of Protein Import into Peroxisomes. FEBS J. 2019, 286, 205–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skowyra, M.; Rapoport, T.A. Mechanism of PEX5-Mediated Protein Import into Peroxisomes. bioRxiv 2022. [Google Scholar] [CrossRef]
- Kerssen, D.; Hambruch, E.; Klaas, W.; Platta, H.W.; de Kruijff, B.; Erdmann, R.; Kunau, W.-H.; Schliebs, W. Membrane Association of the Cycling Peroxisome Import Receptor Pex5p. J. Biol. Chem. 2006, 281, 27003–27015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenkranz, K.; Birschmann, I.; Grunau, S.; Girzalsky, W.; Kunau, W.-H.; Erdmann, R. Functional Association of the AAA Complex and the Peroxisomal Importomer. FEBS J. 2006, 273, 3804–3815. [Google Scholar] [CrossRef]
- Marzioch, M.; Erdmann, R.; Veenhuis, M.; Kunau, W.H. PAS7 Encodes a Novel Yeast Member of the WD-40 Protein Family Essential for Import of 3-Oxoacyl-CoA Thiolase, a PTS2-Containing Protein, into Peroxisomes. EMBO J. 1994, 13, 4908–4918. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.W.; Lazarow, P.B. PEB1 (PAS7) in Saccharomyces Cerevisiae Encodes a Hydrophilic, Intra-Peroxisomal Protein That Is a Member of the WD Repeat Family and Is Essential for the Import of Thiolase into Peroxisomes. J. Cell Biol. 1995, 129, 65–80. [Google Scholar] [CrossRef]
- Swinkels, B.W.; Gould, S.J.; Bodnar, A.G.; Rachubinski, R.A.; Subramani, S. A Novel, Cleavable Peroxisomal Targeting Signal at the Amino-Terminus of the Rat 3-Ketoacyl-CoA Thiolase. EMBO J. 1991, 10, 3255–3262. [Google Scholar] [CrossRef]
- Braverman, N.; Steel, G.; Obie, C.; Moser, A.; Moser, H.; Gould, S.J.; Valle, D. Human PEXl Encodes the Peroxisomal PTS2 Receptor and Is Responsible for Rhizomelic Chondrodysplasia Punctata. Nat. Genet. 1997, 15, 369–376. [Google Scholar] [CrossRef]
- Braverman, N. An Isoform of Pex5p, the Human PTS1 Receptor, Is Required for the Import of PTS2 Proteins into Peroxisomes. Hum. Mol. Genet. 1998, 7, 1195–1205. [Google Scholar] [CrossRef] [Green Version]
- Matsumura, T.; Otera, H.; Fujiki, Y. Disruption of the Interaction of the Longer Isoform of Pex5p, Pex5pL, with Pex7p Abolishes Peroxisome Targeting Signal Type 2 Protein Import in Mammals. J. Biol. Chem. 2000, 275, 21715–21721. [Google Scholar] [CrossRef] [Green Version]
- Dodt, G.; Warren, D.; Becker, E.; Rehling, P.; Gould, S.J. Domain Mapping of Human PEX5 Reveals Functional and Structural Similarities to Saccharomyces Cerevisiae Pex18p and Pex21p. J. Biol. Chem. 2001, 276, 41769–41781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Purdue, P.E.; Yang, X.; Lazarow, P.B. Pex18p and Pex21p, a Novel Pair of Related Peroxins Essential for Peroxisomal Targeting by the PTS2 Pathway. J. Cell Biol. 1998, 143, 1859–1869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukai, S.; Fujiki, Y. Molecular Mechanisms of Import of Peroxisome-Targeting Signal Type 2 (PTS2) Proteins by PTS2 Receptor Pex7p and PTS1 Receptor Pex5pL. J. Biol. Chem. 2006, 281, 37311–37320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, C.; Distel, B. Pex13p: Docking or Cargo Handling Protein? Biochim. Biophys. Acta BBA-Mol. Cell Res. 2006, 1763, 1585–1591. [Google Scholar] [CrossRef] [Green Version]
- Freitas, M.O.; Francisco, T.; Rodrigues, T.A.; Alencastre, I.S.; Pinto, M.P.; Grou, C.P.; Carvalho, A.F.; Fransen, M.; Sá-Miranda, C.; Azevedo, J.E. PEX5 Protein Binds Monomeric Catalase Blocking Its Tetramerization and Releases It upon Binding the N-Terminal Domain of PEX14. J. Biol. Chem. 2011, 286, 40509–40519. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Visser, N.V.; Veenhuis, M.; van der Klei, I.J. Physical Interactions of the Peroxisomal Targeting Signal 1 Receptor Pex5p, Studied by Fluorescence Correlation Spectroscopy. J. Biol. Chem. 2003, 278, 43340–43345. [Google Scholar] [CrossRef] [Green Version]
- Rodrigues, T.A.; Alencastre, I.S.; Francisco, T.; Brites, P.; Fransen, M.; Grou, C.P.; Azevedo, J.E. A PEX7-Centered Perspective on the Peroxisomal Targeting Signal Type 2-Mediated Protein Import Pathway. Mol. Cell. Biol. 2014, 34, 2917–2928. [Google Scholar] [CrossRef] [Green Version]
- Pedrosa, A.G.; Francisco, T.; Bicho, D.; Dias, A.F.; Barros-Barbosa, A.; Hagmann, V.; Dodt, G.; Rodrigues, T.A.; Azevedo, J.E. Peroxisomal Monoubiquitinated PEX5 Interacts with the AAA ATPases PEX1 and PEX6 and Is Unfolded during Its Dislocation into the Cytosol. J. Biol. Chem. 2018, 293, 11553–11563. [Google Scholar] [CrossRef] [Green Version]
- Hensel, A.; Beck, S.; El Magraoui, F.; Platta, H.W.; Girzalsky, W.; Erdmann, R. Cysteine-Dependent Ubiquitination of Pex18p Is Linked to Cargo Translocation across the Peroxisomal Membrane. J. Biol. Chem. 2011, 286, 43495–43505. [Google Scholar] [CrossRef] [Green Version]
- Léon, S.; Subramani, S. A Conserved Cysteine Residue of Pichia Pastoris Pex20p Is Essential for Its Recycling from the Peroxisome to the Cytosol. J. Biol. Chem. 2007, 282, 7424–7430. [Google Scholar] [CrossRef] [Green Version]
- Platta, H.W.; Girzalsky, W.; Erdmann, R. Ubiquitination of the Peroxisomal Import Receptor Pex5p. Biochem. J. 2004, 384, 37–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Platta, H.W.; Grunau, S.; Rosenkranz, K.; Girzalsky, W.; Erdmann, R. Functional Role of the AAA Peroxins in Dislocation of the Cycling PTS1 Receptor Back to the Cytosol. Nat. Cell Biol. 2005, 7, 817–822. [Google Scholar] [CrossRef] [PubMed]
- Kragt, A.; Voorn-Brouwer, T.; van den Berg, M.; Distel, B. The Saccharomyces Cerevisiae Peroxisomal Import Receptor Pex5p Is Monoubiquitinated in Wild Type Cells. J. Biol. Chem. 2005, 280, 7867–7874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Platta, H.W.; El Magraoui, F.; Bäumer, B.E.; Schlee, D.; Girzalsky, W.; Erdmann, R. Pex2 and Pex12 Function as Protein-Ubiquitin Ligases in Peroxisomal Protein Import. Mol. Cell. Biol. 2009, 29, 5505–5516. [Google Scholar] [CrossRef] [Green Version]
- Wiebel, F.F.; Kunau, W.-H. The Pas2 Protein Essential for Peroxisome Biogenesis Is Related to Ubiquitin-Conjugating Enzymes. Nature 1992, 359, 73–76. [Google Scholar] [CrossRef]
- Grou, C.P.; Carvalho, A.F.; Pinto, M.P.; Wiese, S.; Piechura, H.; Meyer, H.E.; Warscheid, B.; Sá-Miranda, C.; Azevedo, J.E. Members of the E2D (UbcH5) Family Mediate the Ubiquitination of the Conserved Cysteine of Pex5p, the Peroxisomal Import Receptor. J. Biol. Chem. 2008, 283, 14190–14197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, C.; van den Berg, M.; Sprenger, R.R.; Distel, B. A Conserved Cysteine Is Essential for Pex4p-Dependent Ubiquitination of the Peroxisomal Import Receptor Pex5p. J. Biol. Chem. 2007, 282, 22534–22543. [Google Scholar] [CrossRef] [Green Version]
- Carvalho, A.F.; Pinto, M.P.; Grou, C.P.; Alencastre, I.S.; Fransen, M.; Sá-Miranda, C.; Azevedo, J.E. Ubiquitination of Mammalian Pex5p, the Peroxisomal Import Receptor. J. Biol. Chem. 2007, 282, 31267–31272. [Google Scholar] [CrossRef] [Green Version]
- McClellan, A.J.; Laugesen, S.H.; Ellgaard, L. Cellular Functions and Molecular Mechanisms of Non-Lysine Ubiquitination. Open Biol. 2019, 9, 190147. [Google Scholar] [CrossRef] [Green Version]
- Apanasets, O.; Grou, C.P.; Van Veldhoven, P.P.; Brees, C.; Wang, B.; Nordgren, M.; Dodt, G.; Azevedo, J.E.; Fransen, M. PEX5, the Shuttling Import Receptor for Peroxisomal Matrix Proteins, Is a Redox-Sensitive Protein. Traffic Cph. Den. 2014, 15, 94–103. [Google Scholar] [CrossRef] [Green Version]
- Ma, C.; Hagstrom, D.; Polley, S.G.; Subramani, S. Redox-Regulated Cargo Binding and Release by the Peroxisomal Targeting Signal Receptor, Pex5. J. Biol. Chem. 2013, 288, 27220–27231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, C.; Bener Aksam, E.; Gunkel, K.; Veenhuis, M.; van der Klei, I.J. The Relevance of the Non-Canonical PTS1 of Peroxisomal Catalase. Biochim. Biophys. Acta BBA-Mol. Cell Res. 2012, 1823, 1133–1141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujiki, Y.; Bassik, M.C. A New Paradigm in Catalase Research. Trends Cell Biol. 2021, 31, 148–151. [Google Scholar] [CrossRef] [PubMed]
- Schliebs, W.; Girzalsky, W.; Erdmann, R. Peroxisomal Protein Import and ERAD: Variations on a Common Theme. Nat. Rev. Mol. Cell Biol. 2010, 11, 885–890. [Google Scholar] [CrossRef]
- Debelyy, M.O.; Platta, H.W.; Saffian, D.; Hensel, A.; Thoms, S.; Meyer, H.E.; Warscheid, B.; Girzalsky, W.; Erdmann, R. Ubp15p, a Ubiquitin Hydrolase Associated with the Peroxisomal Export Machinery. J. Biol. Chem. 2011, 286, 28223–28234. [Google Scholar] [CrossRef] [Green Version]
- Grou, C.P.; Francisco, T.; Rodrigues, T.A.; Freitas, M.O.; Pinto, M.P.; Carvalho, A.F.; Domingues, P.; Wood, S.A.; Rodríguez-Borges, J.E.; Sá-Miranda, C.; et al. Identification of Ubiquitin-Specific Protease 9X (USP9X) as a Deubiquitinase Acting on Ubiquitin-Peroxin 5 (PEX5) Thioester Conjugate. J. Biol. Chem. 2012, 287, 12815–12827. [Google Scholar] [CrossRef] [Green Version]
- Zhao, C.; Slevin, J.T.; Whiteheart, S.W. Cellular Functions of NSF: Not Just SNAPs and SNAREs. FEBS Lett. 2007, 581, 2140–2149. [Google Scholar] [CrossRef] [Green Version]
- Yedidi, R.S.; Wendler, P.; Enenkel, C. AAA-ATPases in Protein Degradation. Front. Mol. Biosci. 2017, 4, 42. [Google Scholar] [CrossRef] [Green Version]
- Monroe, N.; Hill, C.P. Meiotic Clade AAA ATPases: Protein Polymer Disassembly Machines. J. Mol. Biol. 2016, 428, 1897–1911. [Google Scholar] [CrossRef] [Green Version]
- Kappel, L.; Loibl, M.; Zisser, G.; Klein, I.; Fruhmann, G.; Gruber, C.; Unterweger, S.; Rechberger, G.; Pertschy, B.; Bergler, H. Rlp24 Activates the AAA-ATPase Drg1 to Initiate Cytoplasmic Pre-60S Maturation. J. Cell Biol. 2012, 199, 771–782. [Google Scholar] [CrossRef] [Green Version]
- Latterich, M.; Fröhlich, K.-U.; Schekman, R. Membrane Fusion and the Cell Cycle: Cdc48p Participates in the Fusion of ER Membranes. Cell 1995, 82, 885–893. [Google Scholar] [CrossRef] [Green Version]
- Erdmann, R.; Veenhuis, M.; Mertens, D.; Kunau, W.H. Isolation of Peroxisome-Deficient Mutants of Saccharomyces Cerevisiae. Proc. Natl. Acad. Sci. USA 1989, 86, 5419–5423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erdmann, R.; Wiebel, F.R.; Flessau, A.; Rytka, J.; Beyer, A.; Fr, K.-U. PAS1, a Yeast Gene Required for Peroxisome Biogenesis, Encodes a Member of a Novel Family of Putative ATPases. Cell 1991, 64, 499–510. [Google Scholar] [CrossRef]
- Voorn-Brouwer, T.; van der Leij, I.; Hemrika, W.; Distel, B.; Tabak, H.F. Sequence of the PAS8 Gene, the Product of Which Is Essential for Biogenesis of Peroxisomes in Saccharomyces Cerevisiae. Biochim. Biophys. Acta BBA-Gene Struct. Expr. 1993, 1216, 325–328. [Google Scholar] [CrossRef]
- Tsukamoto, T.; Miura, S.; Nakai, T.; Yokota, S.; Shimozawa, N.; Suzuki, Y.; Orii, T.; Fujiki, Y.; Sakai, F.; Bogaki, A.; et al. Peroxisome Assembly Factor–2, a Putative ATPase Cloned by Functional Complementation on a Peroxisome–Deficient Mammalian Cell Mutant. Nat. Genet. 1995, 11, 395–401. [Google Scholar] [CrossRef]
- Reuber, B.E.; Germain-Lee, E.; Collins, C.S.; Morrell, J.C.; Ameritunga, R.; Moser, H.W.; Valle, D.; Gould, S.J. Mutations in PEX1 Are the Most Common Cause of Peroxisome Biogenesis Disorders. Nat. Genet. 1997, 17, 445–448. [Google Scholar] [CrossRef]
- Portsteffen, H.; Beyer, A.; Becker, E.; Epplen, C.; Pawlak, A.; Kunau, W.-H.; Dodt, G. Human PEX1 Is Mutated in Complementation Group 1 of the Peroxisome Biogenesis Disorders. Nat. Genet. 1997, 17, 449–452. [Google Scholar] [CrossRef]
- Meyer, H.; Bug, M.; Bremer, S. Emerging Functions of the VCP/P97 AAA-ATPase in the Ubiquitin System. Nat. Cell Biol. 2012, 14, 117–123. [Google Scholar] [CrossRef]
- Titorenko, V.I.; Rachubinski, R.A. Peroxisomal Membrane Fusion Requires Two Aaa Family Atpases, Pex1p and Pex6p. J. Cell Biol. 2000, 150, 881–886. [Google Scholar] [CrossRef] [Green Version]
- Titorenko, V.I.; Chan, H.; Rachubinski, R.A. Fusion of Small Peroxisomal Vesicles in Vitro Reconstructs an Early Step in the in Vivo Multistep Peroxisome Assembly Pathway of Yarrowia Lipolytica. J. Cell Biol. 2000, 148, 29–44. [Google Scholar] [CrossRef] [Green Version]
- Collins, C.S.; Kalish, J.E.; Morrell, J.C.; McCaffery, J.M.; Gould, S.J. The Peroxisome Biogenesis Factors Pex4p, Pex22p, Pex1p, and Pex6p Act in the Terminal Steps of Peroxisomal Matrix Protein Import. Mol. Cell. Biol. 2000, 20, 7516–7526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Wang, W. Mechanisms and Functions of Pexophagy in Mammalian Cells. Cells 2021, 10, 1094. [Google Scholar] [CrossRef] [PubMed]
- Kim, P.K.; Hailey, D.W.; Mullen, R.T.; Lippincott-Schwartz, J. Ubiquitin Signals Autophagic Degradation of Cytosolic Proteins and Peroxisomes. Proc. Natl. Acad. Sci. USA 2008, 105, 20567–20574. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Han, H.; Zhou, M.-T.; Yang, B.; Ta, A.P.; Li, N.; Chen, J.; Wang, W. Proteomic Analysis of the Human Tankyrase Protein Interaction Network Reveals Its Role in Pexophagy. Cell Rep. 2017, 20, 737–749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deosaran, E.; Larsen, K.B.; Hua, R.; Sargent, G.; Wang, Y.; Kim, S.; Lamark, T.; Jauregui, M.; Law, K.; Lippincott-Schwartz, J.; et al. NBR1 Acts as an Autophagy Receptor for Peroxisomes. J. Cell Sci. 2012, 126 Pt 4, 939–952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sargent, G.; van Zutphen, T.; Shatseva, T.; Zhang, L.; Di Giovanni, V.; Bandsma, R.; Kim, P.K. PEX2 Is the E3 Ubiquitin Ligase Required for Pexophagy during Starvation. J. Cell Biol. 2016, 214, 677–690. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Tripathi, D.N.; Jing, J.; Alexander, A.; Kim, J.; Powell, R.T.; Dere, R.; Tait-Mulder, J.; Lee, J.-H.; Paull, T.T.; et al. ATM Functions at the Peroxisome to Induce Pexophagy in Response to ROS. Nat. Cell Biol. 2015, 17, 1259–1269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nordgren, M.; Francisco, T.; Lismont, C.; Hennebel, L.; Brees, C.; Wang, B.; Van Veldhoven, P.P.; Azevedo, J.E.; Fransen, M. Export-Deficient Monoubiquitinated PEX5 Triggers Peroxisome Removal in SV40 Large T Antigen-Transformed Mouse Embryonic Fibroblasts. Autophagy 2015, 11, 1326–1340. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez, K.L.; Ratzel, S.E.; Burks, K.H.; Danan, C.H.; Wages, J.M.; Zolman, B.K.; Bartel, B. A Pex1 Missense Mutation Improves Peroxisome Function in a Subset of Arabidopsis Pex6 Mutants without Restoring PEX5 Recycling. Proc. Natl. Acad. Sci. USA 2018, 115, E3163–E3172. [Google Scholar] [CrossRef] [Green Version]
- Tamura, S.; Matsumoto, N.; Takeba, R.; Fujiki, Y. AAA Peroxins and Their Recruiter Pex26p Modulate the Interactions of Peroxins Involved in Peroxisomal Protein Import. J. Biol. Chem. 2014, 289, 24336–24346. [Google Scholar] [CrossRef] [Green Version]
- Seo, J.-G.; Lai, C.-Y.; Miceli, M.V.; Jazwinski, S.M. A Novel Role of Peroxin PEX6: Suppression of Aging Defects in Mitochondria. Aging Cell 2007, 6, 405–413. [Google Scholar] [CrossRef] [PubMed]
- Gardner, B.M.; Chowdhury, S.; Lander, G.C.; Martin, A. The Pex1/Pex6 Complex Is a Heterohexameric AAA + Motor with Alternating and Highly Coordinated Subunits. J. Mol. Biol. 2015, 427, 1375–1388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blok, N.B.; Tan, D.; Wang, R.Y.-R.; Penczek, P.A.; Baker, D.; DiMaio, F.; Rapoport, T.A.; Walz, T. Unique Double-Ring Structure of the Peroxisomal Pex1/Pex6 ATPase Complex Revealed by Cryo-Electron Microscopy. Proc. Natl. Acad. Sci. USA 2015, 112, E4017–E4025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciniawsky, S.; Grimm, I.; Saffian, D.; Girzalsky, W.; Erdmann, R.; Wendler, P. Molecular Snapshots of the Pex1/6 AAA+ Complex in Action. Nat. Commun. 2015, 6, 7331. [Google Scholar] [CrossRef] [Green Version]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly Accurate Protein Structure Prediction with AlphaFold. Nature 2021, 596, 590–596. [Google Scholar] [CrossRef]
- Varadi, M.; Anyango, S.; Deshpande, M.; Nair, S.; Natassia, C.; Yordanova, G.; Yuan, D.; Stroe, O.; Wood, G.; Laydon, A.; et al. AlphaFold Protein Structure Database: Massively Expanding the Structural Coverage of Protein-Sequence Space with High-Accuracy Models. Nucleic Acids Res. 2022, 50, D439–D444. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Meng, E.C.; Couch, G.S.; Croll, T.I.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Structure Visualization for Researchers, Educators, and Developers. Protein Sci. 2021, 30, 70–82. [Google Scholar] [CrossRef]
- Shiozawa, K.; Maita, N.; Tomii, K.; Seto, A.; Goda, N.; Akiyama, Y.; Shimizu, T.; Shirakawa, M.; Hiroaki, H. Structure of the N-Terminal Domain of PEX1 AAA-ATPase: Characterization of a putative adaptor-binding domain. J. Biol. Chem. 2004, 279, 50060–50068. [Google Scholar] [CrossRef] [Green Version]
- Gates, S.N.; Martin, A. Stairway to Translocation: AAA+ Motor Structures Reveal the Mechanisms of ATP-dependent Substrate Translocation. Protein Sci. 2020, 29, 407–419. [Google Scholar] [CrossRef]
- Puchades, C.; Sandate, C.R.; Lander, G.C. The Molecular Principles Governing the Activity and Functional Diversity of AAA+ Proteins. Nat. Rev. Mol. Cell Biol. 2020, 21, 43–58. [Google Scholar] [CrossRef]
- Glynn, S.E.; Martin, A.; Nager, A.R.; Baker, T.A.; Sauer, R.T. Structures of Asymmetric ClpX Hexamers Reveal Nucleotide-Dependent Motions in a AAA+ Protein-Unfolding Machine. Cell 2009, 139, 744–756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wendler, P.; Ciniawsky, S.; Kock, M.; Kube, S. Structure and Function of the AAA+ Nucleotide Binding Pocket. Biochim. Biophys. Acta 2012, 1823, 2–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bodnar, N.; Rapoport, T. Toward an Understanding of the Cdc48/P97 ATPase. F1000Research 2017, 6, 1318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matveeva, E.A.; He, P.; Whiteheart, S.W. N-Ethylmaleimide-Sensitive Fusion Protein Contains High and Low Affinity ATP-Binding Sites That Are Functionally Distinct. J. Biol. Chem. 1997, 272, 26413–26418. [Google Scholar] [CrossRef] [Green Version]
- Konagurthu, A.S.; Whisstock, J.C.; Stuckey, P.J.; Lesk, A.M. MUSTANG: A Multiple Structural Alignment Algorithm. Proteins Struct. Funct. Bioinform. 2006, 64, 559–574. [Google Scholar] [CrossRef]
- Zhao, M.; Wu, S.; Zhou, Q.; Vivona, S.; Cipriano, D.J.; Cheng, Y.; Brunger, A.T. Mechanistic Insights into the Recycling Machine of the SNARE Complex. Nature 2015, 518, 61–67. [Google Scholar] [CrossRef] [Green Version]
- Bodnar, N.O.; Kim, K.H.; Ji, Z.; Wales, T.E.; Svetlov, V.; Nudler, E.; Engen, J.R.; Walz, T.; Rapoport, T.A. Structure of the Cdc48 ATPase with Its Ubiquitin-Binding Cofactor Ufd1–Npl4. Nat. Struct. Mol. Biol. 2018, 25, 616–622. [Google Scholar] [CrossRef]
- Gardner, B.M.; Castanzo, D.T.; Chowdhury, S.; Stjepanovic, G.; Stefely, M.S.; Hurley, J.H.; Lander, G.C.; Martin, A. The Peroxisomal AAA-ATPase Pex1/Pex6 Unfolds Substrates by Processive Threading. Nat. Commun. 2018, 9, 135. [Google Scholar] [CrossRef] [Green Version]
- Pan, M.; Yu, Y.; Ai, H.; Zheng, Q.; Xie, Y.; Liu, L.; Zhao, M. Mechanistic Insight into Substrate Processing and Allosteric Inhibition of Human P97. Nat. Struct. Mol. Biol. 2021, 28, 614–625. [Google Scholar] [CrossRef]
- Cooney, I.; Han, H.; Stewart, M.G.; Carson, R.H.; Hansen, D.T.; Iwasa, J.H.; Price, J.C.; Hill, C.P.; Shen, P.S. Structure of the Cdc48 Segregase in the Act of Unfolding an Authentic Substrate. Science 2019, 365, 502–505. [Google Scholar] [CrossRef]
- Han, H.; Monroe, N.; Sundquist, W.I.; Shen, P.S.; Hill, C.P. The AAA ATPase Vps4 Binds ESCRT-III Substrates through a Repeating Array of Dipeptide-Binding Pockets. eLife 2017, 6, e31324. [Google Scholar] [CrossRef] [PubMed]
- de la Peña, A.H.; Goodall, E.A.; Gates, S.N.; Lander, G.C.; Martin, A. Substrate-Engaged 26 S Proteasome Structures Reveal Mechanisms for ATP-Hydrolysis–Driven Translocation. Science 2018, 362, eaav0725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puchades, C.; Rampello, A.J.; Shin, M.; Giuliano, C.J.; Wiseman, R.L.; Glynn, S.E.; Lander, G.C. Structure of the Mitochondrial Inner Membrane AAA+ Protease YME1 Gives Insight into Substrate Processing. Science 2017, 358, eaao0464. [Google Scholar] [CrossRef] [Green Version]
- Lo, Y.-H.; Sobhany, M.; Hsu, A.L.; Ford, B.L.; Krahn, J.M.; Borgnia, M.J.; Stanley, R.E. Cryo-EM Structure of the Essential Ribosome Assembly AAA-ATPase Rix7. Nat. Commun. 2019, 10, 513. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.; Lupoli, T.J.; Kovach, A.; Meng, X.; Zhao, G.; Nathan, C.F.; Li, H. ATP Hydrolysis-Coupled Peptide Translocation Mechanism of Mycobacterium Tuberculosis ClpB. Proc. Natl. Acad. Sci. USA 2018, 115, E9560–E9569. [Google Scholar] [CrossRef] [Green Version]
- Dong, Y.; Zhang, S.; Wu, Z.; Li, X.; Wang, W.L.; Zhu, Y.; Stoilova-McPhie, S.; Lu, Y.; Finley, D.; Mao, Y. Cryo-EM Structures and Dynamics of Substrate-Engaged Human 26S Proteasome. Nature 2019, 565, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Ripstein, Z.A.; Huang, R.; Augustyniak, R.; Kay, L.E.; Rubinstein, J.L. Structure of a AAA+ Unfoldase in the Process of Unfolding Substrate. eLife 2017, 6, e25754. [Google Scholar] [CrossRef]
- Martin, A.; Baker, T.A.; Sauer, R.T. Rebuilt AAA + Motors Reveal Operating Principles for ATP-Fuelled Machines. Nature 2005, 437, 1115–1120. [Google Scholar] [CrossRef]
- Cordova, J.C.; Olivares, A.O.; Shin, Y.; Stinson, B.M.; Calmat, S.; Schmitz, K.R.; Aubin-Tam, M.-E.; Baker, T.A.; Lang, M.J.; Sauer, R.T. Stochastic but Highly Coordinated Protein Unfolding and Translocation by the ClpXP Proteolytic Machine. Cell 2014, 158, 647–658. [Google Scholar] [CrossRef] [Green Version]
- Beckwith, R.; Estrin, E.; Worden, E.J.; Martin, A. Reconstitution of the 26S Proteasome Reveals Functional Asymmetries in Its AAA+ Unfoldase. Nat. Struct. Mol. Biol. 2013, 20, 1164–1172. [Google Scholar] [CrossRef] [Green Version]
- Sauer, R.T.; Fei, X.; Bell, T.A.; Baker, T.A. Structure and Function of ClpXP, a AAA+ Proteolytic Machine Powered by Probabilistic ATP Hydrolysis. Crit. Rev. Biochem. Mol. Biol. 2022, 57, 188–204. [Google Scholar] [CrossRef] [PubMed]
- Augustin, S.; Gerdes, F.; Lee, S.; Tsai, F.T.F.; Langer, T.; Tatsuta, T. An Intersubunit Signaling Network Coordinates ATP Hydrolysis by M-AAA Proteases. Mol. Cell 2009, 35, 574–585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazal, H.; Iljina, M.; Riven, I.; Haran, G. Ultrafast Pore-Loop Dynamics in a AAA+ Machine Point to a Brownian-Ratchet Mechanism for Protein Translocation. Sci. Adv. 2021, 7, eabg4674. [Google Scholar] [CrossRef] [PubMed]
- Olivares, A.O.; Nager, A.R.; Iosefson, O.; Sauer, R.T.; Baker, T.A. Mechanochemical Basis of Protein Degradation by a Double-Ring AAA+ Machine. Nat. Struct. Mol. Biol. 2014, 21, 871–875. [Google Scholar] [CrossRef] [Green Version]
- Lopez, K.E.; Rizo, A.N.; Tse, E.; Lin, J.; Scull, N.W.; Thwin, A.C.; Lucius, A.L.; Shorter, J.; Southworth, D.R. Conformational Plasticity of the ClpAP AAA+ Protease Couples Protein Unfolding and Proteolysis. Nat. Struct. Mol. Biol. 2020, 27, 406–416. [Google Scholar] [CrossRef] [PubMed]
- Ripstein, Z.A.; Vahidi, S.; Houry, W.A.; Rubinstein, J.L.; Kay, L.E. A Processive Rotary Mechanism Couples Substrate Unfolding and Proteolysis in the ClpXP Degradation Machinery. eLife 2020, 9, e52158. [Google Scholar] [CrossRef] [PubMed]
- Fei, X.; Bell, T.A.; Jenni, S.; Stinson, B.M.; Baker, T.A.; Harrison, S.C.; Sauer, R.T. Structures of the ATP-Fueled ClpXP Proteolytic Machine Bound to Protein Substrate. eLife 2020, 9, e52774. [Google Scholar] [CrossRef]
- Aubin-Tam, M.-E.; Olivares, A.O.; Sauer, R.T.; Baker, T.A.; Lang, M.J. Single-Molecule Protein Unfolding and Translocation by an ATP-Fueled Proteolytic Machine. Cell 2011, 145, 257–267. [Google Scholar] [CrossRef] [Green Version]
- Avellaneda, M.J.; Franke, K.B.; Sunderlikova, V.; Bukau, B.; Mogk, A.; Tans, S.J. Processive Extrusion of Polypeptide Loops by a Hsp100 Disaggregase. Nature 2020, 578, 317–320. [Google Scholar] [CrossRef]
- Han, H.; Fulcher, J.M.; Dandey, V.P.; Iwasa, J.H.; Sundquist, W.I.; Kay, M.S.; Shen, P.S.; Hill, C.P. Structure of Vps4 with Circular Peptides and Implications for Translocation of Two Polypeptide Chains by AAA+ ATPases. eLife 2019, 8, e44071. [Google Scholar] [CrossRef]
- Sen, M.; Maillard, R.A.; Nyquist, K.; Rodriguez-Aliaga, P.; Pressé, S.; Martin, A.; Bustamante, C. The ClpXP Protease Unfolds Substrates Using a Constant Rate of Pulling but Different Gears. Cell 2013, 155, 636–646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, D.; Blok, N.B.; Rapoport, T.A.; Walz, T. Structures of the Double-Ring AAA ATPase Pex1–Pex6 Involved in Peroxisome Biogenesis. FEBS J. 2016, 283, 986–992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, R.C.; Jahn, R.; Brunger, A.T. NSF N-Terminal Domain Crystal Structure: Models of NSF Function. Mol. Cell 1999, 4, 97–107. [Google Scholar] [CrossRef]
- Hänzelmann, P.; Schindelin, H. The Interplay of Cofactor Interactions and Post-Translational Modifications in the Regulation of the AAA+ ATPase P97. Front. Mol. Biosci. 2017, 4, 21. [Google Scholar] [CrossRef]
- Rosenzweig, R.; Farber, P.; Velyvis, A.; Rennella, E.; Latham, M.P.; Kay, L.E. ClpB N-Terminal Domain Plays a Regulatory Role in Protein Disaggregation. Proc. Natl. Acad. Sci. USA 2015, 112, E6872–E6881. [Google Scholar] [CrossRef] [Green Version]
- Tzeng, S.-R.; Tseng, Y.-C.; Lin, C.-C.; Hsu, C.-Y.; Huang, S.-J.; Kuo, Y.-T.; Chang, C.-I. Molecular Insights into Substrate Recognition and Discrimination by the N-Terminal Domain of Lon AAA+ Protease. eLife 2021, 10, e64056. [Google Scholar] [CrossRef]
- Birschmann, I.; Stroobants, A.K.; van den Berg, M.; Schäfer, A.; Rosenkranz, K.; Kunau, W.-H.; Tabak, H.F. Pex15p of Saccharomyces Cerevisiae Provides a Molecular Basis for Recruitment of the AAA Peroxin Pex6p to Peroxisomal Membranes. Mol. Biol. Cell 2003, 14, 2226–2236. [Google Scholar] [CrossRef] [Green Version]
- Grimm, I.; Saffian, D.; Girzalsky, W.; Erdmann, R. Nucleotide-Dependent Assembly of the Peroxisomal Receptor Export Complex. Sci. Rep. 2016, 6, 19838. [Google Scholar] [CrossRef] [Green Version]
- Schieferdecker, A.; Wendler, P. Structural Mapping of Missense Mutations in the Pex1/Pex6 Complex. Int. J. Mol. Sci. 2019, 20, 3756. [Google Scholar] [CrossRef] [Green Version]
- Elgersma, Y. Overexpression of Pex15p, a Phosphorylated Peroxisomal Integral Membrane Protein Required for Peroxisome Assembly in S.Cerevisiae, Causes Proliferation of the Endoplasmic Reticulum Membrane. EMBO J. 1997, 16, 7326–7341. [Google Scholar] [CrossRef]
- Goto, S.; Mano, S.; Nakamori, C.; Nishimura, M. Arabidopsis ABERRANT PEROXISOME MORPHOLOGY9 Is a Peroxin That Recruits the PEX1-PEX6 Complex to Peroxisomes. Plant Cell 2011, 23, 1573–1587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyata, N.; Fujiki, Y. Shuttling Mechanism of Peroxisome Targeting Signal Type 1 Receptor Pex5: ATP-Independent Import and ATP-Dependent Export. Mol. Cell. Biol. 2005, 25, 10822–10832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weller, S.; Cajigas, I.; Morrell, J.; Obie, C.; Steel, G.; Gould, S.J.; Valle, D. Alternative Splicing Suggests Extended Function of PEX26 in Peroxisome Biogenesis. Am. J. Hum. Genet. 2005, 76, 987–1007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lotz-Havla, A.S.; Woidy, M.; Guder, P.; Schmiesing, J.; Erdmann, R.; Waterham, H.R.; Muntau, A.C.; Gersting, S.W. Edgetic Perturbations Contribute to Phenotypic Variability in PEX26 Deficiency. Front. Genet. 2021, 12, 726174. [Google Scholar] [CrossRef]
- Guder, P.; Lotz-Havla, A.S.; Woidy, M.; Reiß, D.D.; Danecka, M.K.; Schatz, U.A.; Becker, M.; Ensenauer, R.; Pagel, P.; Büttner, L.; et al. Isoform-Specific Domain Organization Determines Conformation and Function of the Peroxisomal Biogenesis Factor PEX26. Biochim. Biophys. Acta BBA-Mol. Cell Res. 2019, 1866, 518–531. [Google Scholar] [CrossRef]
- Evans, R.; O’Neill, M.; Pritzel, A.; Antropova, N.; Senior, A.; Green, T.; Žídek, A.; Bates, R.; Blackwell, S.; Yim, J.; et al. Protein Complex Prediction with AlphaFold-Multimer. bioRxiv 2022. [Google Scholar] [CrossRef]
- Furuki, S.; Tamura, S.; Matsumoto, N.; Miyata, N.; Moser, A.; Moser, H.W.; Fujiki, Y. Mutations in the Peroxin Pex26p Responsible for Peroxisome Biogenesis Disorders of Complementation Group 8 Impair Its Stability, Peroxisomal Localization, and Interaction with the Pex1p·Pex6p Complex. J. Biol. Chem. 2006, 281, 1317–1323. [Google Scholar] [CrossRef] [Green Version]
- Halbach, A.; Landgraf, C.; Lorenzen, S.; Rosenkranz, K.; Volkmer-Engert, R.; Erdmann, R.; Rottensteiner, H. Targeting of the Tail-Anchored Peroxisomal Membrane Proteins PEX26 and PEX15 Occurs through C-Terminal PEX19-Binding Sites. J. Cell Sci. 2006, 119, 2508–2517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fokkema, I.F.A.C.; Taschner, P.E.M.; Schaafsma, G.C.P.; Celli, J.; Laros, J.F.J.; den Dunnen, J.T. LOVD v.2.0: The next Generation in Gene Variant Databases. Hum. Mutat. 2011, 32, 557–563. [Google Scholar] [CrossRef]
- Krogh, A.; Larsson, B.; von Heijne, G.; Sonnhammer, E.L.L. Predicting Transmembrane Protein Topology with a Hidden Markov Model: Application to Complete Genomes11Edited by F. Cohen. J. Mol. Biol. 2001, 305, 567–580. [Google Scholar] [CrossRef] [Green Version]
- Gautier, R.; Douguet, D.; Antonny, B.; Drin, G. HELIQUEST: A Web Server to Screen Sequences with Specific -Helical Properties. Bioinformatics 2008, 24, 2101–2102. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wigley, D.B. The “glutamate Switch” Provides a Link between ATPase Activity and Ligand Binding in AAA+ Proteins. Nat. Struct. Mol. Biol. 2008, 15, 1223–1227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weibezahn, J.; Schlieker, C.; Bukau, B.; Mogk, A. Characterization of a Trap Mutant of the AAA+ Chaperone ClpB. J. Biol. Chem. 2003, 278, 32608–32617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costello, J.L.; Castro, I.G.; Camões, F.; Schrader, T.A.; McNeall, D.; Yang, J.; Giannopoulou, E.-A.; Gomes, S.; Pogenberg, V.; Bonekamp, N.A.; et al. Predicting the Targeting of Tail-Anchored Proteins to Subcellular Compartments in Mammalian Cells. J. Cell Sci. 2017, 130, 1675–1687. [Google Scholar] [CrossRef] [Green Version]
- Matsumoto, N.; Tamura, S.; Furuki, S.; Miyata, N.; Moser, A.; Shimozawa, N.; Moser, H.W.; Suzuki, Y.; Kondo, N.; Fujiki, Y. Mutations in Novel Peroxin Gene PEX26 That Cause Peroxisome-Biogenesis Disorders of Complementation Group 8 Provide a Genotype-Phenotype Correlation. Am. J. Hum. Genet. 2003, 73, 233–246. [Google Scholar] [CrossRef] [Green Version]
- Dammai, V.; Subramani, S. The Human Peroxisomal Targeting Signal Receptor, Pex5p, Is Translocated into the Peroxisomal Matrix and Recycled to the Cytosol. Cell 2001, 105, 187–196. [Google Scholar] [CrossRef] [Green Version]
- Berner, N.; Reutter, K.-R.; Wolf, D.H. Protein Quality Control of the Endoplasmic Reticulum and Ubiquitin–Proteasome-Triggered Degradation of Aberrant Proteins: Yeast Pioneers the Path. Annu. Rev. Biochem. 2018, 87, 751–782. [Google Scholar] [CrossRef]
- Schwerter, D.; Grimm, I.; Girzalsky, W.; Erdmann, R. Receptor Recognition by the Peroxisomal AAA Complex Depends on the Presence of the Ubiquitin Moiety and Is Mediated by Pex1p. J. Biol. Chem. 2018, 293, 15458–15470. [Google Scholar] [CrossRef] [Green Version]
- Hagmann, V.; Sommer, S.; Fabian, P.; Bierlmeier, J.; van Treel, N.; Mootz, H.D.; Schwarzer, D.; Azevedo, J.E.; Dodt, G. Chemically Monoubiquitinated PEX5 Binds to the Components of the Peroxisomal Docking and Export Machinery. Sci. Rep. 2018, 8, 16014. [Google Scholar] [CrossRef]
- Miyata, N.; Okumoto, K.; Mukai, S.; Noguchi, M.; Fujiki, Y. AWP1/ZFAND6 Functions in Pex5 Export by Interacting with Cys-Monoubiquitinated Pex5 and Pex6 AAA ATPase. Traffic 2012, 13, 168–183. [Google Scholar] [CrossRef]
- Pedrosa, A.G.; Francisco, T.; Ferreira, M.J.; Rodrigues, T.A.; Barros-Barbosa, A.; Azevedo, J.E. A Mechanistic Perspective on PEX1 and PEX6, Two AAA+ Proteins of the Peroxisomal Protein Import Machinery. Int. J. Mol. Sci. 2019, 20, 5246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, Z.; Li, H.; Peterle, D.; Paulo, J.A.; Ficarro, S.B.; Wales, T.E.; Marto, J.A.; Gygi, S.P.; Engen, J.R.; Rapoport, T.A. Translocation of Polyubiquitinated Protein Substrates by the Hexameric Cdc48 ATPase. Mol. Cell 2022, 82, 570–584.e8. [Google Scholar] [CrossRef] [PubMed]
- Haslberger, T.; Zdanowicz, A.; Brand, I.; Kirstein, J.; Turgay, K.; Mogk, A.; Bukau, B. Protein Disaggregation by the AAA+ Chaperone ClpB Involves Partial Threading of Looped Polypeptide Segments. Nat. Struct. Mol. Biol. 2008, 15, 641–650. [Google Scholar] [CrossRef]
- Monroe, N.; Han, H.; Shen, P.S.; Sundquist, W.I.; Hill, C.P. Structural Basis of Protein Translocation by the Vps4-Vta1 AAA ATPase. eLife 2017, 6, e24487. [Google Scholar] [CrossRef] [PubMed]
- Yifrach, E.; Chuartzman, S.G.; Dahan, N.; Maskit, S.; Zada, L.; Weill, U.; Yofe, I.; Olender, T.; Schuldiner, M.; Zalckvar, E. Characterization of Proteome Dynamics in Oleate Reveals a Novel Peroxisome Targeting Receptor. J. Cell Sci. 2016, 129, 4067–4075. [Google Scholar] [CrossRef] [Green Version]
- Effelsberg, D.; Cruz-Zaragoza, L.D.; Schliebs, W.; Erdmann, R. Pex9p Is a Novel Yeast Peroxisomal Import Receptor for PTS1-Proteins. J. Cell Sci. 2016, 129, 4057–4066. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, C.; Tan, L.-J.; Mochida, K.; Kirisako, H.; Koizumi, M.; Asai, E.; Sakoh-Nakatogawa, M.; Ohsumi, Y.; Nakatogawa, H. Hrr25 Triggers Selective Autophagy–Related Pathways by Phosphorylating Receptor Proteins. J. Cell Biol. 2014, 207, 91–105. [Google Scholar] [CrossRef] [Green Version]
- Motley, A.M.; Nuttall, J.M.; Hettema, E.H. Pex3-Anchored Atg36 Tags Peroxisomes for Degradation in Saccharomyces Cerevisiae. EMBO J. 2012, 31, 2852–2868. [Google Scholar] [CrossRef] [Green Version]
- Steinberg, S.J.; Dodt, G.; Raymond, G.V.; Braverman, N.E.; Moser, A.B.; Moser, H.W. Peroxisome Biogenesis Disorders. Biochim. Biophys. Acta BBA-Mol. Cell Res. 2006, 1763, 1733–1748. [Google Scholar] [CrossRef] [Green Version]
- Santos, M.J.; Hoefler, S.; Moser, A.B.; Moser, H.W.; Lazarow, P.B. Peroxisome assembly mutations in humans: Structural heterogeneity in Zellweger syndrome. J. Cell. Physiol. 1992, 151, 103–112. [Google Scholar] [CrossRef]
- Santos, M.J.; Imanaka, T.; Shio, H.; Lazarow, P.B. Peroxisomal Integral Membrane Proteins in Control and Zellweger Fibroblasts. J. Biol. Chem. 1988, 263, 10502–10509. [Google Scholar] [CrossRef]
- Soliman, K.; Göttfert, F.; Rosewich, H.; Thoms, S.; Gärtner, J. Super-Resolution Imaging Reveals the Sub-Diffraction Phenotype of Zellweger Syndrome Ghosts and Wild-Type Peroxisomes. Sci. Rep. 2018, 8, 7809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- South, S.T.; Gould, S.J. Peroxisome Synthesis in the Absence of Preexisting Peroxisomes. J. Cell Biol. 1999, 144, 255–266. [Google Scholar] [CrossRef] [PubMed]
- Schrader, M.; Costello, J.; Godinho, L.F.; Islinger, M. Peroxisome-Mitochondria Interplay and Disease. J. Inherit. Metab. Dis. 2015, 38, 681–702. [Google Scholar] [CrossRef]
- Kleinecke, S.; Richert, S.; de Hoz, L.; Brügger, B.; Kungl, T.; Asadollahi, E.; Quintes, S.; Blanz, J.; McGonigal, R.; Naseri, K.; et al. Peroxisomal Dysfunctions Cause Lysosomal Storage and Axonal Kv1 Channel Redistribution in Peripheral Neuropathy. eLife 2017, 6, e23332. [Google Scholar] [CrossRef] [Green Version]
- Berger, J.; Dorninger, F.; Forss-Petter, S.; Kunze, M. Peroxisomes in Brain Development and Function. Biochim. Biophys. Acta BBA-Mol. Cell Res. 2016, 1863, 934–955. [Google Scholar] [CrossRef] [Green Version]
- Geisbrecht, B.V.; Collins, C.S.; Reuber, B.E.; Gould, S.J. Disruption of a PEX1–PEX6 Interaction Is the Most Common Cause of the Neurologic Disorders Zellweger Syndrome, Neonatal Adrenoleukodystrophy, and Infantile Refsum Disease. Proc. Natl. Acad. Sci. USA 1998, 95, 8630–8635. [Google Scholar] [CrossRef] [Green Version]
- Ratbi, I.; Falkenberg, K.D.; Sommen, M.; Al-Sheqaih, N.; Guaoua, S.; Vandeweyer, G.; Urquhart, J.E.; Chandler, K.E.; Williams, S.G.; Roberts, N.A.; et al. Heimler Syndrome Is Caused by Hypomorphic Mutations in the Peroxisome-Biogenesis Genes PEX1 and PEX6. Am. J. Hum. Genet. 2015, 97, 535–545. [Google Scholar] [CrossRef]
- Klouwer, F.C.C.; Falkenberg, K.D.; Ofman, R.; Koster, J.; van Gent, D.; Ferdinandusse, S.; Wanders, R.J.A.; Waterham, H.R. Autophagy Inhibitors Do Not Restore Peroxisomal Functions in Cells With the Most Common Peroxisome Biogenesis Defect. Front. Cell Dev. Biol. 2021, 9, 661298. [Google Scholar] [CrossRef]
- Nazarko, T.Y. Pexophagy Is Responsible for 65% of Cases of Peroxisome Biogenesis Disorders. Autophagy 2017, 13, 991–994. [Google Scholar] [CrossRef] [Green Version]
- Ebberink, M.S.; Mooijer, P.A.W.; Gootjes, J.; Koster, J.; Wanders, R.J.A.; Waterham, H.R. Genetic Classification and Mutational Spectrum of More than 600 Patients with a Zellweger Syndrome Spectrum Disorder. Hum. Mutat. 2011, 32, 59–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walter, C.; Gootjes, J.; Mooijer, P.A.; Portsteffen, H.; Klein, C.; Waterham, H.R.; Barth, P.G.; Epplen, J.T.; Kunau, W.-H.; Wanders, R.J.A.; et al. Disorders of Peroxisome Biogenesis Due to Mutations in PEX1: Phenotypes and PEX1 Protein Levels. Am. J. Hum. Genet. 2001, 69, 35–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maxwell, M.A.; Allen, T.; Solly, P.B.; Svingen, T.; Paton, B.C.; Crane, D.I. Novel PEX1 Mutations and Genotype–Phenotype Correlations in Australasian Peroxisome Biogenesis Disorder Patients. Hum. Mutat. 2002, 20, 342–351. [Google Scholar] [CrossRef] [PubMed]
- Argyriou, C.; Polosa, A.; Cecyre, B.; Hsieh, M.; Di Pietro, E.; Cui, W.; Bouchard, J.-F.; Lachapelle, P.; Braverman, N. A Longitudinal Study of Retinopathy in the PEX1-Gly844Asp Mouse Model for Mild Zellweger Spectrum Disorder. Exp. Eye Res. 2019, 186, 107713. [Google Scholar] [CrossRef]
- Twomey, E.C.; Ji, Z.; Wales, T.E.; Bodnar, N.O.; Ficarro, S.B.; Marto, J.A.; Engen, J.R.; Rapoport, T.A. Substrate Processing by the Cdc48 ATPase Complex Is Initiated by Ubiquitin Unfolding. Science 2019, 365, eaax1033. [Google Scholar] [CrossRef]
- Nashiro, C.; Kashiwagi, A.; Matsuzaki, T.; Tamura, S.; Fujiki, Y. Recruiting Mechanism of the AAA Peroxins, Pex1p and Pex6p, to Pex26p on the Peroxisomal Membrane. Traffic 2011, 12, 774–788. [Google Scholar] [CrossRef]
- Zhang, R.; Chen, L.; Jiralerspong, S.; Snowden, A.; Steinberg, S.; Braverman, N. Recovery of PEX1-Gly843Asp Peroxisome Dysfunction by Small-Molecule Compounds. Proc. Natl. Acad. Sci. USA 2010, 107, 5569–5574. [Google Scholar] [CrossRef] [Green Version]
- MacLean, G.E.; Argyriou, C.; Di Pietro, E.; Sun, X.; Birjandian, S.; Saberian, P.; Hacia, J.G.; Braverman, N.E. Zellweger Spectrum Disorder Patient–Derived Fibroblasts with the PEX1-Gly843Asp Allele Recover Peroxisome Functions in Response to Flavonoids. J. Cell. Biochem. 2019, 120, 3243–3258. [Google Scholar] [CrossRef]
- Banerjee, S.; Bartesaghi, A.; Merk, A.; Rao, P.; Bulfer, S.L.; Yan, Y.; Green, N.; Mroczkowski, B.; Neitz, R.J.; Wipf, P.; et al. 2.3 Å Resolution Cryo-EM Structure of Human P97 and Mechanism of Allosteric Inhibition. Science 2016, 351, 871–875. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Hu, Y.; Hau, R.; Musharrafieh, R.; Ma, C.; Zhou, X.; Chen, Y.; Wang, J. Identification of NMS-873, an Allosteric and Specific P97 Inhibitor, as a Broad Antiviral against Both Influenza A and B Viruses. Eur. J. Pharm. Sci. 2019, 133, 86–94. [Google Scholar] [CrossRef]
- Zhou, H.-J.; Wang, J.; Yao, B.; Wong, S.; Djakovic, S.; Kumar, B.; Rice, J.; Valle, E.; Soriano, F.; Menon, M.-K.; et al. Discovery of a First-in-Class, Potent, Selective, and Orally Bioavailable Inhibitor of the P97 AAA ATPase (CB-5083). J. Med. Chem. 2015, 58, 9480–9497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dephoure, N.; Zhou, C.; Villén, J.; Beausoleil, S.A.; Bakalarski, C.E.; Elledge, S.J.; Gygi, S.P. A Quantitative Atlas of Mitotic Phosphorylation. Proc. Natl. Acad. Sci. USA 2008, 105, 10762–10767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuoka, S.; Ballif, B.A.; Smogorzewska, A.; McDonald, E.R.; Hurov, K.E.; Luo, J.; Bakalarski, C.E.; Zhao, Z.; Solimini, N.; Lerenthal, Y.; et al. ATM and ATR Substrate Analysis Reveals Extensive Protein Networks Responsive to DNA Damage. Science 2007, 316, 1160–1166. [Google Scholar] [CrossRef] [Green Version]
- Dinkel, H.; Chica, C.; Via, A.; Gould, C.M.; Jensen, L.J.; Gibson, T.J.; Diella, F. Phospho.ELM: A Database of Phosphorylation Sites--Update 2011. Nucleic Acids Res. 2011, 39, D261–D267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, N.Y.; Jo, D.S.; Park, S.J.; Lee, H.; Bae, J.-E.; Hong, Y.; Kim, J.B.; Kim, Y.H.; Park, H.J.; Choi, J.Y.; et al. Depletion of HNRNPA1 Induces Peroxisomal Autophagy by Regulating PEX1 Expression. Biochem. Biophys. Res. Commun. 2021, 545, 69–74. [Google Scholar] [CrossRef]
- Tomko, R.J.; Hochstrasser, M. Order of the Proteasomal ATPases and Eukaryotic Proteasome Assembly. Cell Biochem. Biophys. 2011, 60, 13–20. [Google Scholar] [CrossRef] [Green Version]
- Zavodszky, E.; Peak-Chew, S.-Y.; Juszkiewicz, S.; Narvaez, A.J.; Hegde, R.S. Identification of a Quality-Control Factor That Monitors Failures during Proteasome Assembly. Science 2021, 373, 998–1004. [Google Scholar] [CrossRef]
- Narayan, V.; Ly, T.; Pourkarimi, E.; Murillo, A.B.; Gartner, A.; Lamond, A.I.; Kenyon, C. Deep Proteome Analysis Identifies Age-Related Processes in C. Elegans. Cell Syst. 2016, 3, 144–159. [Google Scholar] [CrossRef] [Green Version]
- Uzor, N.-E.; Scheihing, D.M.; Kim, G.S.; Moruno-Manchon, J.F.; Zhu, L.; Reynolds, C.R.; Stephenson, J.M.; Holmes, A.; McCullough, L.D.; Tsvetkov, A.S. Aging Lowers PEX5 Levels in Cortical Neurons in Male and Female Mouse Brains. Mol. Cell. Neurosci. 2020, 107, 103536. [Google Scholar] [CrossRef]
- Huang, K.; Chen, W.; Zhu, F.; Li, P.W.-L.; Kapahi, P.; Bai, H. RiboTag Translatomic Profiling of Drosophila Oenocytes under Aging and Induced Oxidative Stress. BMC Genom. 2019, 20, 50. [Google Scholar] [CrossRef] [Green Version]
- Huang, K.; Kim, J.; Vo, P.; Miao, T.; Bai, H. Peroxisome Import Stress Impairs Ribosome Biogenesis and Induces Integrative Stress Response through EIF2α Phosphorylation. bioRxiv 2020. [Google Scholar] [CrossRef]
- Dixit, E.; Boulant, S.; Zhang, Y.; Lee, A.S.Y.; Odendall, C.; Shum, B.; Hacohen, N.; Chen, Z.J.; Whelan, S.P.; Fransen, M.; et al. Peroxisomes Are Signaling Platforms for Antiviral Innate Immunity. Cell 2010, 141, 668–681. [Google Scholar] [CrossRef] [Green Version]
- Grewal, P.S.; Samson, J.A.; Baker, J.J.; Choi, B.; Dueber, J.E. Peroxisome Compartmentalization of a Toxic Enzyme Improves Alkaloid Production. Nat. Chem. Biol. 2021, 17, 96–103. [Google Scholar] [CrossRef]
- Dusséaux, S.; Wajn, W.T.; Liu, Y.; Ignea, C.; Kampranis, S.C. Transforming Yeast Peroxisomes into Microfactories for the Efficient Production of High-Value Isoprenoids. Proc. Natl. Acad. Sci. USA 2020, 117, 31789–31799. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.-S.; Li, T.; Zhou, W.; Jiang, M.; Tao, X.-Y.; Liu, M.; Zhao, M.; Ren, Y.-H.; Gao, B.; Wang, F.-Q.; et al. The Yeast Peroxisome: A Dynamic Storage Depot and Subcellular Factory for Squalene Overproduction. Metab. Eng. 2020, 57, 151–161. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Walker A | Pore Loop 1 | Walker B | ISS | Arg Finger | ||
|---|---|---|---|---|---|---|
| ATP Binding | Substrate Binding | ATP Hydrolysis | Inter-Subunit Signaling | Assembly, ATP Hydrolysis | ||
| Consensus | GxxGxGKT | +ΩΦx- | ΦΦΦΦDE | DGF | ALLRPGR | |
| ScPex1 | GKQGIGKT | CETLHE-TSNLDKTQ | LIVLDN | QVTKI | LLFDKHF | |
| HsPEX1 | GGKGSGKS | CKALR--GKRLENIQ | VVLLDD | MIKEF | LLV…VHI | |
| AtPEX1 | GPPGSGKT | CSTLA--LEKVQHIH | VIILDD | VIDDY | TLSSSGR | |
| D1 | ScPex6 | S…NNVGKA | CLSLTSNSRQLDSTS | VIFLAH | LLDDF | SFRS--H |
| HsPEX6 | GPPGCGKT | CSSLC--AESSGAVE | VLLLTA | LLLNE | DVQ--TA | |
| AtPEX6 | GIPGCGKR | CHSLL--ASSERKTS | ILLLRH | VIREL | TIR--RC | |
| ScPex1 | GYPGCGKT | GPEIL--NKFIGASE | ILFFDE | QMDGA | ALLRPGR | |
| HsPEX1 | GPPGTGKT | GPELL--SKYIGASE | ILFFDE | QLDGV | ALLRPGR | |
| D2 | AtPEX1 | GPPGCGKT | GPELL--NKYIGASE | ILFFDE | ELDGV | ALLRPGR |
| ScPex6 | GPPGTGKT | GPELL--NMYIGESE | VIFFDE | ELDGM | ALLRPGR | |
| HsPEX6 | GPPGTGKT | SPELI--NMYVGQSE | IIFFDE | ELDGL | ALLRPGR | |
| AtPEX6 | GPPGTGKT | GPELI--NMYIGESE | VIFFDE | EMDGM | ALLRPGR | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Judy, R.M.; Sheedy, C.J.; Gardner, B.M. Insights into the Structure and Function of the Pex1/Pex6 AAA-ATPase in Peroxisome Homeostasis. Cells 2022, 11, 2067. https://doi.org/10.3390/cells11132067
Judy RM, Sheedy CJ, Gardner BM. Insights into the Structure and Function of the Pex1/Pex6 AAA-ATPase in Peroxisome Homeostasis. Cells. 2022; 11(13):2067. https://doi.org/10.3390/cells11132067
Chicago/Turabian StyleJudy, Ryan M., Connor J. Sheedy, and Brooke M. Gardner. 2022. "Insights into the Structure and Function of the Pex1/Pex6 AAA-ATPase in Peroxisome Homeostasis" Cells 11, no. 13: 2067. https://doi.org/10.3390/cells11132067
APA StyleJudy, R. M., Sheedy, C. J., & Gardner, B. M. (2022). Insights into the Structure and Function of the Pex1/Pex6 AAA-ATPase in Peroxisome Homeostasis. Cells, 11(13), 2067. https://doi.org/10.3390/cells11132067