
The Role of Microglia in Neuroinflammation of the Spinal Cord after Peripheral Nerve Injury

 , , ,
, , ,
Abstract
: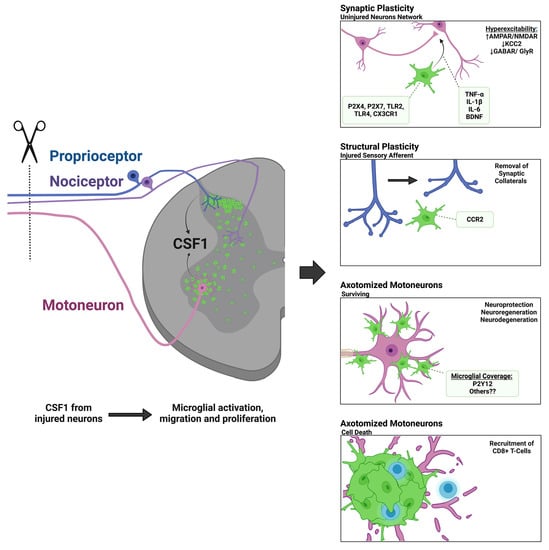
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
1.1. Peripheral Nerve Injuries
1.2. Microgliosis around Axotomized Motoneurons
1.3. Microgliosis in the Dorsal Horn around the Central Projections of Sensory Afferents Axotomized in the Periphery
1.4. Scope of the Present Review
2. Mechanisms of Spinal Microglial Activation, Motility, and Recruitment
2.1. Microglia Activation Dynamics in the Spinal Cord after Peripheral Nerve Injury
2.2. Colony Stimulating Factor 1 (CSF1) and Other DNAX Activating Protein of 12kDa (DAP12) Activating Pathways
2.3. C-C Chemokine Receptor Type 2 (CCR2) and Fractalkine Receptor (CX3CR1)
2.4. Toll-Like Receptors (TLRs)
2.5. Activation of Microglia by Purinergic Receptors
3. Theories about the Function of Microglia around Axotomized Motoneurons
3.1. Neuroprotection or Neurodegeneration
3.2. Synaptic Plasticity around Axotomized Motoneurons
3.3. Microglia and Immune System Responses around Axotomized Motoneurons
3.4. Motor Axon Regeneration
4. Theories behind Microgliosis around the Central Branches of Sensory Afferents Injured in the Peripheral Nerve
Complement as a Targeting Mechanism for Synaptic Pruning
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ADAM10 | metalloproteinase domain-containing protein |
| ADP | Adenosine Diphosphate |
| AKT | protein kinase B |
| AR | P1 Adenosine Receptors |
| ATP | Adenosine Triphosphate |
| BAX | bcl-2-like protein 4 |
| BCL-2 | B-cell lymphoma-2 |
| BDNF | Brain-Derived Neurotrophic Factor |
| C1q | Complement component 1q |
| CSF1 | Colony Stimulating Factor 1 |
| CSF1-R | CSF1 Receptor |
| CCI | Chronic Constriction Injuries |
| CCL2 | C-C Chemokine Ligand type 2 |
| CCR2 | C-C Chemokine Receptor type 2 |
| ChAT | Choline Acetyltransferase |
| CNS | Central Nervous System |
| CREB | cAMP Response Element-Binding protein |
| CX3CL1 | Fractalkine |
| CX3CR1 | Fractalkine receptor |
| DAMPs | damage-associated molecular patterns |
| DAP12 | DNAX Activating Protein of 12kDa |
| DRG | Dorsal Root Ganglion |
| E/I | excitatory/inhibitory |
| FAS | fas cell surface death receptor |
| GFAP | Glial fibrillary acidic protein |
| GFP | Green Fluorescent Protein |
| HGMB1 | High Mobility Group Box 1 protein |
| IBA1 | Ionized calcium Binding Adaptor molecule 1 |
| ICE | interleukin-1b-converting enzyme |
| IL | Interleukins |
| NO | Nitric Oxide |
| NOS | NO Synthase |
| iNOS | inducible NOS |
| IP3 | Inositol Triphosphate |
| IRF8 | Interferon Regulatory Factor 8 |
| ITAMs | Immunoreceptor Tyrosine-based Activation Motifs |
| KCC2 | chloride potassium transporter isoform 2 |
| KV2.1 | voltage-gated potassium channel 2.1 |
| LC3-II | microtubule-associated protein 1A/1B-light chain 3 phos phatidylethanolamine conjugate |
| LPS | Lipopolysaccharide |
| MAPK | Mitogen Activated Protein Kinase |
| MHC-I | Major Histocompatibility Complex I |
| MNs | Motoneurons |
| Nox2 | NADPH Oxidase 2 |
| PAMPs | pathogen-associated molecular patterns |
| PLC | Phospholipase C |
| PNI | Peripheral Nerve Injury |
| P2XRs | ionotropic purinergic receptors |
| P2YRs | metabotropic purinergic receptors |
| RAG | recombination-activating genes |
| RFP | Red Fluorescent Protein |
| STAT-3 | Signal Transducer and Activator of Transcription 3 |
| SYK | Spleen Tyrosine Kinase |
| TLRs | Toll-Like Receptors |
| TNF-a | Tumor Necrosis Factor-Alpha |
| TGF-b1 | Transforming Growth Factor beta 1 |
| TREM2 | Triggering Receptor Expressed on Myeloid Cells-2 |
| TrkB | tropomyosin receptor kinase B |
| UTP | Uridine Triphosphate |
| VGLUT1 | Vesicular Glutamate Transporter 1 |
References
- Tsuda, M. P2 receptors, microglial cytokines and chemokines, and neuropathic pain. J. Neurosci. Res. 2017, 95, 1319–1329. [Google Scholar] [CrossRef]
- Eriksson, N.; Persson, J.; Svensson, M.; Arvidsson, J.; Molander, C.; Aldskogius, H. A quantitative analysis of the microglial cell reaction in central primary sensory projection territories following peripheral nerve injury in the adult rat. Exp. Brain Res. 1993, 96, 19–27. [Google Scholar] [CrossRef]
- Rotterman, T.M.; Akhter, E.T.; Lane, A.R.; MacPherson, K.P.; García, V.V.; Tansey, M.G.; Alvarez, F.J. Spinal Motor Circuit Synaptic Plasticity after Peripheral Nerve Injury Depends on Microglia Activation and a CCR2 Mechanism. J. Neurosci. 2019, 39, 3412–3433. [Google Scholar] [CrossRef] [Green Version]
- Aldskogius, H.; Liu, L.; Svensson, M. Glial responses to synaptic damage and plasticity. J. Neurosci. Res. 1999, 58, 33–41. [Google Scholar] [CrossRef]
- Aldskogius, H. Mechanisms and consequences of microglial responses to peripheral axotomy. Front. Biosci. 2011, 3, 857–868. [Google Scholar] [CrossRef] [Green Version]
- Beggs, S.; Trang, T.; Salter, M.W. P2X4R+ microglia drive neuropathic pain. Nat. Neurosci. 2012, 15, 1068–1073. [Google Scholar] [CrossRef]
- Echeverry, S.; Shi, X.Q.; Zhang, J. Characterization of cell proliferation in rat spinal cord following peripheral nerve injury and the relationship with neuropathic pain. Pain 2008, 135, 37–47. [Google Scholar] [CrossRef]
- Calvo, M.; Bennett, D.L. The mechanisms of microgliosis and pain following peripheral nerve injury. Exp. Neurol. 2012, 234, 271–282. [Google Scholar] [CrossRef]
- Inoue, K.; Tsuda, M. Microglia in neuropathic pain: Cellular and molecular mechanisms and therapeutic potential. Nat. Rev. Neurosci. 2018, 19, 138–152. [Google Scholar] [CrossRef]
- Trang, T.; Beggs, S.; Salter, M.W. ATP receptors gate microglia signaling in neuropathic pain. Exp. Neurol. 2012, 234, 354–361. [Google Scholar] [CrossRef] [Green Version]
- Sunderland, S. Nerve Injuries and Their Repair: A Critical Appraisal; Churchill Livingstone: Edinburgh, UK, 1991. [Google Scholar]
- Li, N.Y.; Onor, G.I.; Lemme, N.J.; Gil, J.A. Epidemiology of Peripheral Nerve Injuries in Sports, Exercise, and Recreation in the United States, 2009–2018. Phys. Sportsmed. 2021, 49, 355–362. [Google Scholar] [CrossRef] [PubMed]
- Taylor, C.A.; Braza, D.; Rice, J.B.; Dillingham, T. The Incidence of Peripheral Nerve Injury in Extremity Trauma. Am. J. Phys. Med. Rehabil. 2008, 87, 381–385. [Google Scholar] [CrossRef]
- Ciaramitaro, P.; Mondelli, M.; Logullo, F.; Grimaldi, S.; Battiston, B.; Sard, A.; Scarinzi, C.; Migliaretti, G.; Faccani, G.; Cocito, D. Traumatic peripheral nerve injuries: Epidemiological findings, neuropathic pain and quality of life in 158 patients. J. Peripher. Nerv. Syst. 2010, 15, 120–127. [Google Scholar] [CrossRef] [PubMed]
- Noble, J.; Munro, C.A.; Prasad, V.S.; Midha, R. Analysis of upper and lower extremity peripheral nerve injuries in a population of patients with multiple injuries. J. Trauma Acute Care Surg. 1998, 45, 116–122. [Google Scholar] [CrossRef]
- Menorca, R.M.; Fussell, T.S.; Elfar, J.C. Peripheral nerve trauma: Mechanisms of injury and recovery. Hand Clin. 2013, 29, 317. [Google Scholar] [CrossRef] [Green Version]
- Ashford, B.A.; Boche, D.; Cooper-Knock, J.; Heath, P.R.; Simpson, J.E.; Highley, J.R. Microglia in motor neuron disease. Neuropathol. Appl. Neurobiol. 2021, 47, 179–197. [Google Scholar] [CrossRef]
- Seddon, H. Three types of nerve injury. Brain A J. Neurol. 1943, 66, 237–288. [Google Scholar] [CrossRef]
- Brushart, T.M. Nerve Repair; Oxford University Press: Oxford, UK, 2011. [Google Scholar]
- Lundborg, G. Nerve injury and repair–a challenge to the plastic brain. J. Peripher. Nerv. Syst. 2003, 8, 209–226. [Google Scholar] [CrossRef] [Green Version]
- Lanier, S.T.; Hill, J.R.; Dy, C.J.; Brogan, D.M. Evolving Techniques in Peripheral Nerve Regeneration. J. Hand Surg. 2021, 46, 695–701. [Google Scholar] [CrossRef]
- Lundborg, G. A 25-year perspective of peripheral nerve surgery: Evolving neuroscientific concepts and clinical significance. J. Hand Surg. 2000, 25, 391–414. [Google Scholar] [CrossRef]
- Bullinger, K.L.; Nardelli, P.; Pinter, M.J.; Alvarez, F.J.; Cope, T.C. Permanent central synaptic disconnection of proprioceptors after nerve injury and regeneration. II. Loss of functional connectivity with motoneurons. J. Neurophysiol. 2011, 106, 2471–2485. [Google Scholar] [CrossRef] [Green Version]
- Yiu, G.; He, Z. Glial inhibition of CNS axon regeneration. Nat. Rev. Neurosci. 2006, 7, 617–627. [Google Scholar] [CrossRef] [Green Version]
- Richardson, P.M.; McGuinness, U.M.; Aguayo, A.J. Axons from CNS neurons regenerate into PNS grafts. Nature 1980, 284, 264–265. [Google Scholar] [CrossRef]
- Cote, M.P.; Amin, A.A.; Tom, V.J.; Houle, J.D. Peripheral nerve grafts support regeneration after spinal cord injury. Neurotherapeutics 2011, 8, 294–303. [Google Scholar] [CrossRef] [Green Version]
- Hanna, A.S.; Cote, M.P.; Houle, J.; Dempsey, R. Nerve grafting for spinal cord injury in cats: Are we close to translational research? Neurosurgery 2011, 68, N14–N15. [Google Scholar] [CrossRef]
- Danielsen, N. Nerve regeneration and repair. Diabet. Med. 1996, 13, 677–678. [Google Scholar] [CrossRef]
- Gordon, T.; Sulaiman, O. Nerve regeneration in the peripheral nervous system. In Neuroglia, 3rd ed.; Kettenmann, H., Ransom, B.F., Eds.; Oxford University Press: New York, NY, USA, 2013. [Google Scholar]
- Valls-Sole, J.; Castillo, C.D.; Casanova-Molla, J.; Costa, J. Clinical consequences of reinnervation disorders after focal peripheral nerve lesions. Clin. Neurophysiol. 2011, 122, 219–228. [Google Scholar] [CrossRef]
- Gordon, T.; Chan, K.M.; Sulaiman, O.A.; Udina, E.; Amirjani, N.; Brushart, T.M. Accelerating axon growth to overcome limitations in functional recovery after peripheral nerve injury. Neurosurgery 2009, 65, A132–A144. [Google Scholar] [CrossRef]
- Höke, A.; Brushart, T. Introduction to special issue: Challenges and opportunities for regeneration in the peripheral nervous system. Exp. Neurol. 2010, 223, 1–4. [Google Scholar] [CrossRef] [Green Version]
- Hart, A.M.; Terenghi, G.; Wiberg, M. Neuronal death after peripheral nerve injury and experimental strategies for neuroprotection. Neurol. Res. 2008, 30, 999–1011. [Google Scholar] [CrossRef]
- Ma, J.; Novikov, L.N.; Wiberg, M.; Kellerth, J.O. Delayed loss of spinal motoneurons after peripheral nerve injury in adult rats: A quantitative morphological study. Exp. Brain Res. 2001, 139, 216–223. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wang, H. Peripheral nerve injury induced changes in the spinal cord and strategies to counteract/enhance the changes to promote nerve regeneration. Neural Regen. Res. 2020, 15, 189–198. [Google Scholar] [CrossRef] [PubMed]
- Scholz, J.; Woolf, C.J. The neuropathic pain triad: Neurons, immune cells and glia. Nat. Neurosci. 2007, 10, 1361–1368. [Google Scholar] [CrossRef]
- Alvarez, F.J.; Bullinger, K.L.; Titus, H.E.; Nardelli, P.; Cope, T.C. Permanent reorganization of Ia afferent synapses on motoneurons after peripheral nerve injuries. Ann. N. Y. Acad. Sci. 2010, 1198, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Cope, T.C.; Bonasera, S.J.; Nichols, T.R. Reinnervated muscles fail to produce stretch reflexes. J. Neurophysiol. 1994, 71, 817–820. [Google Scholar] [CrossRef]
- Abelew, T.A.; Miller, M.D.; Cope, T.C.; Nichols, T.R. Local loss of proprioception results in disruption of interjoint coordination during locomotion in the cat. J. Neurophysiol 2000, 84, 2709–2714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabatier, M.J.; To, B.N.; Nicolini, J.; English, A.W. Effect of slope and sciatic nerve injury on ankle muscle recruitment and hindlimb kinematics during walking in the rat. J. Exp. Biol. 2011, 214, 1007–1016. [Google Scholar] [CrossRef] [Green Version]
- Chang, Y.H.; Housley, S.N.; Hart, K.S.; Nardelli, P.; Nichols, R.T.; Maas, H.; Cope, T.C. Progressive adaptation of whole-limb kinematics after peripheral nerve injury. Biol. Open 2018, 7, bio028852. [Google Scholar] [CrossRef] [Green Version]
- Alvarez, F.J.; Rotterman, T.M.; Akhter, E.T.; Lane, A.R.; English, A.W.; Cope, T.C. Synaptic Plasticity on Motoneurons After Axotomy: A Necessary Change in Paradigm. Front. Mol. Neurosci. 2020, 13, 68. [Google Scholar] [CrossRef]
- Blinzinger, K.; Kreutzberg, G. Displacement of synaptic terminals from regenerating motoneurons by microglial cells. Z. Zellforsch. Mikrosk. Anat. 1968, 85, 145–157. [Google Scholar] [CrossRef]
- Salvany, S.; Casanovas, A.; Piedrafita, L.; Tarabal, O.; Hernández, S.; Calderó, J.; Esquerda, J.E. Microglial recruitment and mechanisms involved in the disruption of afferent synaptic terminals on spinal cord motor neurons after acute peripheral nerve injury. Glia 2021, 69, 1216–1240. [Google Scholar] [CrossRef] [PubMed]
- Gilmore, S.A.; Skinner, R.D. Intraspinal non-neuronal cellular responses to peripheral nerve injury. Anat. Rec. 1979, 194, 369–387. [Google Scholar] [CrossRef] [PubMed]
- Gilmore, S.A. Proliferation of non-neuronal cells in spinal cords of irradiated, immature rats following transection of the sciatic nerve. Anat. Rec. 1975, 181, 799–811. [Google Scholar] [CrossRef] [PubMed]
- Jaggi, A.S.; Jain, V.; Singh, N. Animal models of neuropathic pain. Fundam. Clin. Pharmacol. 2011, 25, 1–28. [Google Scholar] [CrossRef]
- Colleoni, M.; Sacerdote, P. Murine models of human neuropathic pain. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2010, 1802, 924–933. [Google Scholar] [CrossRef] [Green Version]
- Sorkin, L.S.; Yaksh, T.L. Behavioral models of pain states evoked by physical injury to the peripheral nerve. Neurotherapeutics 2009, 6, 609–619. [Google Scholar] [CrossRef] [Green Version]
- Barrot, M. Tests and models of nociception and pain in rodents. Neuroscience 2012, 211, 39–50. [Google Scholar] [CrossRef]
- Nishihara, T.; Tanaka, J.; Sekiya, K.; Nishikawa, Y.; Abe, N.; Hamada, T.; Kitamura, S.; Ikemune, K.; Ochi, S.; Choudhury, M.E. Chronic constriction injury of the sciatic nerve in rats causes different activation modes of microglia between the anterior and posterior horns of the spinal cord. Neurochem. Int. 2020, 134, 104672. [Google Scholar] [CrossRef]
- Mogil, J.S. Qualitative sex differences in pain processing: Emerging evidence of a biased literature. Nat. Rev. Neurosci. 2020, 21, 353–365. [Google Scholar] [CrossRef]
- Kuhn, J.A.; Vainchtein, I.D.; Braz, J.; Hamel, K.; Bernstein, M.; Craik, V.; Dahlgren, M.W.; Ortiz-Carpena, J.; Molofsky, A.B.; Molofsky, A.V. Regulatory T-cells inhibit microglia-induced pain hypersensitivity in female mice. Elife 2021, 10, e69056. [Google Scholar] [CrossRef]
- Greenspan, J.D.; Craft, R.M.; LeResche, L.; Arendt-Nielsen, L.; Berkley, K.J.; Fillingim, R.B.; Gold, M.S.; Holdcroft, A.; Lautenbacher, S.; Mayer, E.A. Studying sex and gender differences in pain and analgesia: A consensus report. Pain 2007, 132, S26–S45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doyle, H.H.; Eidson, L.N.; Sinkiewicz, D.M.; Murphy, A.Z. Sex differences in microglia activity within the periaqueductal gray of the rat: A potential mechanism driving the dimorphic effects of morphine. J. Neurosci. 2017, 37, 3202–3214. [Google Scholar] [CrossRef] [PubMed]
- Fiore, N.T.; Yin, Z.; Guneykaya, D.; Gauthier, C.D.; Hayes, J.P.; D’Hary, A.; Butovsky, O.; Moalem-Taylor, G. Sex-specific transcriptome of spinal microglia in neuropathic pain due to peripheral nerve injury. Glia 2021, 70, 675–696. [Google Scholar] [CrossRef] [PubMed]
- Guan, Z.; Kuhn, J.A.; Wang, X.; Colquitt, B.; Solorzano, C.; Vaman, S.; Guan, A.K.; Evans-Reinsch, Z.; Braz, J.; Devor, M. Injured sensory neuron–derived CSF1 induces microglial proliferation and DAP12-dependent pain. Nat. Neurosci. 2016, 19, 94–101. [Google Scholar] [CrossRef]
- Sorge, R.E.; Mapplebeck, J.; Rosen, S.; Beggs, S.; Taves, S.; Alexander, J.K.; Martin, L.J.; Austin, J.-S.; Sotocinal, S.G.; Chen, D. Different immune cells mediate mechanical pain hypersensitivity in male and female mice. Nat. Neurosci. 2015, 18, 1081–1083. [Google Scholar] [CrossRef] [Green Version]
- Masuda, T.; Sankowski, R.; Staszewski, O.; Prinz, M. Microglia heterogeneity in the single-cell era. Cell Rep. 2020, 30, 1271–1281. [Google Scholar] [CrossRef]
- Andoh, M.; Koyama, R. Assessing Microglial Dynamics by Live Imaging. Front. Immunol. 2021, 12, 605. [Google Scholar] [CrossRef]
- Wake, H.; Moorhouse, A.; Jinno, S.K.; Nabekura, J. Resting Microglia Directly Monitor the Functional State of Synapses In Vivo and Determine the Fate of Ischemic Terminals. J. Neurosci. 2009, 29, 3974. [Google Scholar] [CrossRef] [Green Version]
- Kato, G.; Inada, H.; Wake, H.; Akiyoshi, R.; Miyamoto, A.; Eto, K.; Ishikawa, T.; Moorhouse, A.J.; Strassman, A.M.; Nabekura, J. Microglial contact prevents excess depolarization and rescues neurons from excitotoxicity. eNeuro 2016, 3, ENEURO.0004-16.2016. [Google Scholar] [CrossRef] [Green Version]
- Cserép, C.; Pósfai, B.; Lénárt, N.; Fekete, R.; László, Z.I.; Lele, Z.; Orsolits, B.; Molnár, G.; Heindl, S.; Schwarcz, A.D.; et al. Microglia monitor and protect neuronal function through specialized somatic purinergic junctions. Science 2020, 367, 528–537. [Google Scholar] [CrossRef]
- Šišková, Z.; Tremblay, M.-È. Microglia and synapse: Interactions in health and neurodegeneration. Neural Plast. 2013, 2013, 425845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolf, S.A.; Boddeke, H.W.G.M.; Kettenmann, H. Microglia in Physiology and Disease. Annu. Rev. Physiol. 2017, 79, 619–643. [Google Scholar] [CrossRef] [PubMed]
- Endo, Y.; Asanuma, D.; Namiki, S.; Sugihara, K.; Hirose, K.; Uemura, A.; Kubota, Y.; Miura, T. Quantitative modeling of regular retinal microglia distribution. Sci. Rep. 2021, 11, 22671. [Google Scholar] [CrossRef] [PubMed]
- Kohno, K.; Kitano, J.; Kohro, Y.; Tozaki-Saitoh, H.; Inoue, K.; Tsuda, M. Temporal kinetics of microgliosis in the spinal dorsal horn after peripheral nerve injury in rodents. Biol. Pharm. Bull. 2018, 41, 1096–1102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gehrmann, J.; Banati, R.B. Microglial turnover in the injured CNS: Activated microglia undergo delayed DNA fragmentation following peripheral nerve injury. J. Neuropathol. Exp. Neurol. 1995, 54, 680–688. [Google Scholar] [CrossRef]
- Rotterman, T.M.; Alvarez, F.J. Microglia dynamics and interactions with motoneurons axotomized after nerve injuries revealed by two-photon imaging. Sci. Rep. 2020, 10, 8648. [Google Scholar] [CrossRef]
- Maeda, M.; Tsuda, M.; Tozaki-Saitoh, H.; Inoue, K.; Kiyama, H. Nerve injury-activated microglia engulf myelinated axons in a P2Y12 signaling-dependent manner in the dorsal horn. Glia 2010, 58, 1838–1846. [Google Scholar] [CrossRef]
- Kalla, R.; Liu, Z.; Xu, S.; Koppius, A.; Imai, Y.; Kloss, C.U.; Kohsaka, S.; Gschwendtner, A.; Möller, J.C.; Werner, A. Microglia and the early phase of immune surveillance in the axotomized facial motor nucleus: Impaired microglial activation and lymphocyte recruitment but no effect on neuronal survival or axonal regeneration in macrophage-colony stimulating factor-deficient mice. J. Comp. Neurol. 2001, 436, 182–201. [Google Scholar]
- Sanagi, T.; Nakamura, Y.; Suzuki, E.; Uchino, S.; Aoki, M.; Warita, H.; Itoyama, Y.; Kohsaka, S.; Ohsawa, K. Involvement of activated microglia in increased vulnerability of motoneurons after facial nerve avulsion in presymptomatic amyotrophic lateral sclerosis model rats. Glia 2012, 60, 782–793. [Google Scholar] [CrossRef]
- Akhter, E.T.; Griffith, R.W.; English, A.W.; Alvarez, F.J. Removal of the potassium chloride co-transporter from the somatodendritic membrane of axotomized motoneurons is independent of BDNF/TrkB signaling but is controlled by neuromuscular innervation. eNeuro 2019, 6, ENEURO.0172-19.2019. [Google Scholar] [CrossRef] [Green Version]
- Chitu, V.; Gokhan, S.; Nandi, S.; Mehler, M.F.; Stanley, E.R. Emerging Roles for CSF-1 Receptor and its Ligands in the Nervous System. Trends Neurosci. 2016, 39, 378–393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raivich, G.; Moreno-Flores, M.T.; Möller, J.C.; Kreutzberg, G.W. Inhibition of posttraumatic microglial proliferation in a genetic model of macrophage colony-stimulating factor deficiency in the mouse. Eur. J. Neurosci. 1994, 6, 1615–1618. [Google Scholar] [CrossRef] [PubMed]
- Okubo, M.; Yamanaka, H.; Kobayashi, K.; Dai, Y.; Kanda, H.; Yagi, H.; Noguchi, K. Macrophage-colony stimulating factor derived from injured primary afferent induces proliferation of spinal microglia and neuropathic pain in rats. PLoS ONE 2016, 11, e0153375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, J.; Elwood, F.; Britschgi, M.; Villeda, S.; Zhang, H.; Ding, Z.; Zhu, L.; Alabsi, H.; Getachew, R.; Narasimhan, R.; et al. Colony-stimulating factor 1 receptor (CSF1R) signaling in injured neurons facilitates protection and survival. J. Exp. Med. 2013, 210, 157–172. [Google Scholar] [CrossRef] [Green Version]
- Hamilton, J.A. CSF-1 signal transduction. J. Leukoc. Biol. 1997, 62, 145–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pixley, F.J.; Stanley, E.R. CSF-1 regulation of the wandering macrophage: Complexity in action. Trends Cell Biol. 2004, 14, 628–638. [Google Scholar] [CrossRef]
- Mitrasinovic, O.M.; Vincent, V.A.; Simsek, D.; Murphy, G.M., Jr. Macrophage colony stimulating factor promotes phagocytosis by murine microglia. Neurosci. Lett. 2003, 344, 185–188. [Google Scholar] [CrossRef]
- Konishi, H.; Kiyama, H. Microglial TREM2/DAP12 Signaling: A Double-Edged Sword in Neural Diseases. Front. Cell. Neurosci. 2018, 12, 206. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.-W.; Lee, I.-H.; Iimura, T.; Kong, S.W. Two macrophages, osteoclasts and microglia: From development to pleiotropy. Bone Res. 2021, 9, 11. [Google Scholar] [CrossRef]
- Linnartz, B.; Bodea, L.-G.; Neumann, H. Microglial carbohydrate-binding receptors for neural repair. Cell Tissue Res. 2012, 349, 215–227. [Google Scholar] [CrossRef]
- Kobayashi, M.; Konishi, H.; Sayo, A.; Takai, T.; Kiyama, H. TREM2/DAP12 signal elicits proinflammatory response in microglia and exacerbates neuropathic pain. J. Neurosci. 2016, 36, 11138–11150. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Aldskogius, H.; Svensson, M. Ultrastructural localization of immunoglobulin G and complement C9 in the brain stem and spinal cord following peripheral nerve injury: An immunoelectron microscopic study. J. Neurocytol. 1998, 27, 737–748. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Zhang, Z.; Juan, Z.; Zhang, R.; Zhang, C. The high-affinity IgG receptor FcγRI modulates peripheral nerve injury-induced neuropathic pain in rats. Mol. Brain 2019, 12, 83. [Google Scholar] [CrossRef] [PubMed]
- Prinz, M.; Priller, J. Tickets to the brain: Role of CCR2 and CX3CR1 in myeloid cell entry in the CNS. J. Neuroimmunol. 2010, 224, 80–84. [Google Scholar] [CrossRef]
- Tanaka, T.; Minami, M.; Nakagawa, T.; Satoh, M. Enhanced production of monocyte chemoattractant protein-1 in the dorsal root ganglia in a rat model of neuropathic pain: Possible involvement in the development of neuropathic pain. Neurosci. Res. 2004, 48, 463–469. [Google Scholar] [CrossRef]
- Abbadie, C.; Lindia, J.A.; Cumiskey, A.M.; Peterson, L.B.; Mudgett, J.S.; Bayne, E.K.; DeMartino, J.A.; MacIntyre, D.E.; Forrest, M.J. Impaired neuropathic pain responses in mice lacking the chemokine receptor CCR2. Proc. Natl. Acad. Sci. USA 2003, 100, 7947–7952. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; De Koninck, Y. Spatial and temporal relationship between monocyte chemoattractant protein-1 expression and spinal glial activation following peripheral nerve injury. J. Neurochem. 2006, 97, 772–783. [Google Scholar] [CrossRef]
- Zhang, J.; Shi, X.Q.; Echeverry, S.; Mogil, J.S.; De Koninck, Y.; Rivest, S. Expression of CCR2 in Both resident and bone marrow-derived microglia plays a critical role in neuropathic pain. J. Neurosci. 2007, 27, 12396–12406. [Google Scholar] [CrossRef]
- Echeverry, S.; Shi, X.Q.; Rivest, S.; Zhang, J. Peripheral Nerve Injury Alters Blood–Spinal Cord Barrier Functional and Molecular Integrity through a Selective Inflammatory Pathway. J. Neurosci. 2011, 31, 10819–10828. [Google Scholar] [CrossRef]
- Zigmond, R.E.; Echevarria, F.D. Macrophage biology in the peripheral nervous system after injury. Prog. Neurobiol. 2019, 173, 102–121. [Google Scholar] [CrossRef]
- Gu, N.; Peng, J.; Murugan, M.; Wang, X.; Eyo, U.B.; Sun, D.; Ren, Y.; DiCicco-Bloom, E.; Young, W.; Dong, H. Spinal microgliosis due to resident microglial proliferation is required for pain hypersensitivity after peripheral nerve injury. Cell Rep. 2016, 16, 605–614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsou, C.-L.; Peters, W.; Si, Y.; Slaymaker, S.; Aslanian, A.M.; Weisberg, S.P.; Mack, M.; Charo, I.F. Critical roles for CCR2 and MCP-3 in monocyte mobilization from bone marrow and recruitment to inflammatory sites. J. Clin. Investig. 2007, 117, 902–909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rotterman, T.M.; MacPherson, K.P.; Chopra, T.; Fishers, S.; Tansey, M.G.; Alvarez, F.J. Comparison between sciatic nerve transection and sciatic nerve crush: Differences in regeneration outcomes peripherally, graded central neuroimmune responses and spinal circuit plasticity following injury. In Proceedings of the 47th Society for Neuroscience Meeting, Washington, DC, USA, 11–15 November 2017. [Google Scholar]
- Bazan, J.F.; Bacon, K.B.; Hardiman, G.; Wang, W.; Soo, K.; Rossi, D.; Greaves, D.R.; Zlotnik, A.; Schall, T.J. A new class of membrane-bound chemokine with a CX3C motif. Nature 1997, 385, 640–644. [Google Scholar] [CrossRef] [PubMed]
- Kierdorf, K.; Erny, D.; Goldmann, T.; Sander, V.; Schulz, C.; Perdiguero, E.G.; Wieghofer, P.; Heinrich, A.; Riemke, P.; Hölscher, C. Microglia emerge from erythromyeloid precursors via Pu. 1-and Irf8-dependent pathways. Nat. Neurosci. 2013, 16, 273–280. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.; Aliberti, J.; Graemmel, P.; Sunshine, M.J.; Kreutzberg, G.W.; Sher, A.; Littman, D.R. Analysis of fractalkine receptor CX3CR1 function by targeted deletion and green fluorescent protein reporter gene insertion. Mol. Cell. Biol. 2000, 20, 4106–4114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harrison, J.K.; Jiang, Y.; Chen, S.; Xia, Y.; Maciejewski, D.; McNamara, R.K.; Streit, W.J.; Salafranca, M.N.; Adhikari, S.; Thompson, D.A. Role for neuronally derived fractalkine in mediating interactions between neurons and CX3CR1-expressing microglia. Proc. Natl. Acad. Sci. USA 1998, 95, 10896–10901. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.-M.; Jeon, S.-M.; Cho, H.-J. Interleukin-6 induces microglial CX3CR1 expression in the spinal cord after peripheral nerve injury through the activation of p38 MAPK. Eur. J. Pain 2010, 14, 682.e1–682.e12. [Google Scholar] [CrossRef]
- Galiano, M.; Liu, Z.Q.; Kalla, R.; Bohatschek, M.; Koppius, A.; Gschwendtner, A.; Xu, S.; Werner, A.; Kloss, C.U.; Jones, L.L. Interleukin-6 (IL6) and cellular response to facial nerve injury: Effects on lymphocyte recruitment, early microglial activation and axonal outgrowth in IL6-deficient mice. Eur. J. Neurosci. 2001, 14, 327–341. [Google Scholar] [CrossRef]
- Silva, R.; Malcangio, M. Fractalkine/CX3CR1 Pathway in Neuropathic Pain: An Update. Front. Pain Res. 2021, 2, 684684. [Google Scholar] [CrossRef]
- Gao, Y.-J.; Ji, R.-R. Chemokines, neuronal–glial interactions, and central processing of neuropathic pain. Pharmacol. Ther. 2010, 126, 56–68. [Google Scholar] [CrossRef] [Green Version]
- Verge, G.M.; Milligan, E.D.; Maier, S.F.; Watkins, L.R.; Naeve, G.S.; Foster, A.C. Fractalkine (CX3CL1) and fractalkine receptor (CX3CR1) distribution in spinal cord and dorsal root ganglia under basal and neuropathic pain conditions. Eur. J. Neurosci. 2004, 20, 1150–1160. [Google Scholar] [CrossRef] [PubMed]
- Chapman, G.A.; Moores, K.; Harrison, D.; Campbell, C.A.; Stewart, B.R.; Strijbos, P.J. Fractalkine cleavage from neuronal membranes represents an acute event in the inflammatory response to excitotoxic brain damage. J. Neurosci. 2000, 20, Rc87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clark, A.K.; Wodarski, R.; Guida, F.; Sasso, O.; Malcangio, M. Cathepsin S release from primary cultured microglia is regulated by the P2X7 receptor. Glia 2010, 58, 1710–1726. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.-X.; Zhuang, Z.-Y.; Woolf, C.J.; Ji, R.-R. p38 mitogen-activated protein kinase is activated after a spinal nerve ligation in spinal cord microglia and dorsal root ganglion neurons and contributes to the generation of neuropathic pain. J. Neurosci. 2003, 23, 4017–4022. [Google Scholar] [CrossRef] [Green Version]
- Clark, A.K.; Yip, P.K.; Grist, J.; Gentry, C.; Staniland, A.A.; Marchand, F.; Dehvari, M.; Wotherspoon, G.; Winter, J.; Ullah, J. Inhibition of spinal microglial cathepsin S for the reversal of neuropathic pain. Proc. Natl. Acad. Sci. USA 2007, 104, 10655–10660. [Google Scholar] [CrossRef] [Green Version]
- Kawasaki, Y.; Zhang, L.; Cheng, J.-K.; Ji, R.-R. Cytokine mechanisms of central sensitization: Distinct and overlapping role of interleukin-1β, interleukin-6, and tumor necrosis factor-α in regulating synaptic and neuronal activity in the superficial spinal cord. J. Neurosci. 2008, 28, 5189–5194. [Google Scholar] [CrossRef] [Green Version]
- Aravalli, R.N.; Peterson, P.K.; Lokensgard, J.R. Toll-like receptors in defense and damage of the central nervous system. J. Neuroimmune Pharmacol. 2007, 2, 297–312. [Google Scholar] [CrossRef]
- Meller, S.T.; Dykstra, C.; Grzybycki, D.; Murphy, S.; Gebhart, G.F. The possible role of glia in nociceptive processing and hyperalgesia in the spinal cord of the rat. Neuropharmacology 1994, 33, 1471–1478. [Google Scholar] [CrossRef]
- Wu, F.-X.; Bian, J.-J.; Miao, X.-R.; Huang, S.-D.; Xu, X.-W.; Gong, D.-J.; Sun, Y.-M.; Lu, Z.-J.; Yu, W.-F. Intrathecal siRNA against Toll-like receptor 4 reduces nociception in a rat model of neuropathic pain. Int. J. Med. Sci. 2010, 7, 251. [Google Scholar] [CrossRef] [Green Version]
- Kuang, X.; Huang, Y.; Gu, H.-f.; Zu, X.-y.; Zou, W.-y.; Song, Z.-b.; Guo, Q.-l. Effects of intrathecal epigallocatechin gallate, an inhibitor of Toll-like receptor 4, on chronic neuropathic pain in rats. Eur. J. Pharmacol. 2012, 676, 51–56. [Google Scholar] [CrossRef]
- Tanga, F.Y.; Nutile-McMenemy, N.; DeLeo, J.A. The CNS role of Toll-like receptor 4 in innate neuroimmunity and painful neuropathy. Proc. Natl. Acad. Sci. USA 2005, 102, 5856–5861. [Google Scholar] [CrossRef] [Green Version]
- Woller, S.A.; Ravula, S.B.; Tucci, F.C.; Beaton, G.; Corr, M.; Isseroff, R.R.; Soulika, A.M.; Chigbrow, M.; Eddinger, K.A.; Yaksh, T.L. Systemic TAK-242 prevents intrathecal LPS evoked hyperalgesia in male, but not female mice and prevents delayed allodynia following intraplantar formalin in both male and female mice: The role of TLR4 in the evolution of a persistent pain state. Brain Behav. Immun. 2016, 56, 271–280. [Google Scholar] [CrossRef] [Green Version]
- Huck, N.A.; Siliezar-Doyle, J.; Haight, E.S.; Ishida, R.; Forman, T.E.; Wu, S.; Shen, H.; Takemura, Y.; Clark, J.D.; Tawfik, V.L. Temporal contribution of myeloid-lineage TLR4 to the transition to chronic pain: A focus on sex differences. J. Neurosci. 2021, 41, 4349–4365. [Google Scholar] [CrossRef]
- Szabo-Pardi, T.A.; Barron, L.R.; Lenert, M.E.; Burton, M.D. Sensory Neuron TLR4 mediates the development of nerve-injury induced mechanical hypersensitivity in female mice. Brain Behav. Immun. 2021, 97, 42–60. [Google Scholar] [CrossRef]
- Lim, H.; Kim, D.; Lee, S.J. Toll-like receptor 2 mediates peripheral nerve injury-induced NADPH oxidase 2 expression in spinal cord microglia. J. Biol. Chem. 2013, 288, 7572–7579. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Kim, M.A.; Cho, I.-H.; Kim, M.S.; Lee, S.; Jo, E.-K.; Choi, S.-Y.; Park, K.; Kim, J.S.; Akira, S. A critical role of toll-like receptor 2 in nerve injury-induced spinal cord glial cell activation and pain hypersensitivity. J. Biol. Chem. 2007, 282, 14975–14983. [Google Scholar] [CrossRef] [Green Version]
- Shi, X.Q.; Zekki, H.; Zhang, J. The role of TLR2 in nerve injury-induced neuropathic pain is essentially mediated through macrophages in peripheral inflammatory response. Glia 2011, 59, 231–241. [Google Scholar] [CrossRef]
- Lim, H.; Lee, J.; You, B.; Oh, J.H.; Mok, H.J.; Kim, Y.S.; Yoon, B.E.; Kim, B.G.; Back, S.K.; Park, J.S. GT 1b functions as a novel endogenous agonist of toll-like receptor 2 inducing neuropathic pain. EMBO J. 2020, 39, e102214. [Google Scholar] [CrossRef]
- Lee, J.; Hwang, H.; Lee, S.J. Distinct roles of GT1b and CSF-1 in microglia activation in nerve injury-induced neuropathic pain. Mol. Pain 2021, 17, 1–8. [Google Scholar] [CrossRef]
- Agalave, N.M.; Larsson, M.; Abdelmoaty, S.; Su, J.; Baharpoor, A.; Lundbäck, P.; Palmblad, K.; Andersson, U.; Harris, H.; Svensson, C.I. Spinal HMGB1 induces TLR4-mediated long-lasting hypersensitivity and glial activation and regulates pain-like behavior in experimental arthritis. Pain 2014, 155, 1802–1813. [Google Scholar] [CrossRef]
- Feldman, P.; Due, M.R.; Ripsch, M.S.; Khanna, R.; White, F.A. The persistent release of HMGB1 contributes to tactile hyperalgesia in a rodent model of neuropathic pain. J. Neuroinflamm. 2012, 9, 180. [Google Scholar] [CrossRef] [Green Version]
- Freria, C.M.; Velloso, L.A.; Oliveira, A.L. Opposing effects of Toll-like receptors 2 and 4 on synaptic stability in the spinal cord after peripheral nerve injury. J. Neuroinflamm. 2012, 9, 240. [Google Scholar] [CrossRef] [Green Version]
- Ribeiro, P.; Castro, M.V.; Perez, M.; Cartarozzi, L.P.; Spejo, A.B.; Chiarotto, G.B.; Augusto, T.M.; Oliveira, A.L. Toll-like receptor 4 (TLR4) influences the glial reaction in the spinal cord and the neural response to injury following peripheral nerve crush. Brain Res. Bull. 2020, 155, 67–80. [Google Scholar] [CrossRef]
- Freria, C.M.; Brennan, F.H.; Sweet, D.R.; Guan, Z.; Hall, J.C.; Kigerl, K.A.; Nemeth, D.P.; Liu, X.; Lacroix, S.; Quan, N. Serial Systemic Injections of Endotoxin (LPS) Elicit Neuroprotective Spinal Cord Microglia through IL-1-Dependent Cross Talk with Endothelial Cells. J. Neurosci. 2020, 40, 9103–9120. [Google Scholar] [CrossRef]
- Wainwright, D.A.; Xin, J.; Mesnard, N.A.; Sanders, V.M.; Jones, K.J. Toll-like receptor 2 and facial motoneuron survival after facial nerve axotomy. Neurosci. Lett. 2010, 471, 10–14. [Google Scholar] [CrossRef] [Green Version]
- Salter, M.W.; De Koninck, Y.; Henry, J.L. Physiological roles for adenosine and ATP in synaptic transmission in the spinal dorsal horn. Prog. Neurobiol. 1993, 41, 125–156. [Google Scholar] [CrossRef]
- Li, J.; Perl, E. ATP modulation of synaptic transmission in the spinal substantia gelatinosa. J. Neurosci. 1995, 15, 3357–3365. [Google Scholar] [CrossRef] [Green Version]
- Burnstock, G.; Wood, J.N. Purinergic receptors: Their role in nociception and primary afferent neurotransmission. Curr. Opin. Neurobiol. 1996, 6, 526–532. [Google Scholar] [CrossRef]
- Nakatsuka, T.; Gu, J.G. P2X purinoceptors and sensory transmission. Pflüg. Arch. 2006, 452, 598–607. [Google Scholar] [CrossRef]
- Garré, J.M.; Retamal, M.A.; Cassina, P.; Barbeito, L.; Bukauskas, F.F.; Sáez, J.C.; Bennett, M.V.; Abudara, V. FGF-1 induces ATP release from spinal astrocytes in culture and opens pannexin and connexin hemichannels. Proc. Natl. Acad. Sci. USA 2010, 107, 22659–22664. [Google Scholar] [CrossRef] [Green Version]
- Ohsawa, K.; Kohsaka, S. Dynamic motility of microglia: Purinergic modulation of microglial movement in the normal and pathological brain. Glia 2011, 59, 1793–1799. [Google Scholar] [CrossRef]
- Masuda, T.; Ozono, Y.; Mikuriya, S.; Kohro, Y.; Tozaki-Saitoh, H.; Iwatsuki, K.; Uneyama, H.; Ichikawa, R.; Salter, M.W.; Tsuda, M. Dorsal horn neurons release extracellular ATP in a VNUT-dependent manner that underlies neuropathic pain. Nat. Commun. 2016, 7, 12529. [Google Scholar] [CrossRef] [Green Version]
- Koizumi, S.; Ohsawa, K.; Inoue, K.; Kohsaka, S. Purinergic receptors in microglia: Functional modal shifts of microglia mediated by P2 and P1 receptors. Glia 2013, 61, 47–54. [Google Scholar] [CrossRef]
- Kobayashi, K.; Fukuoka, T.; Yamanaka, H.; Dai, Y.; Obata, K.; Tokunaga, A.; Noguchi, K. Neurons and glial cells differentially express P2Y receptor mRNAs in the rat dorsal root ganglion and spinal cord. J. Comp. Neurol. 2006, 498, 443–454. [Google Scholar] [CrossRef]
- Kobayashi, K.; Yamanaka, H.; Yanamoto, F.; Okubo, M.; Noguchi, K. Multiple P2Y subtypes in spinal microglia are involved in neuropathic pain after peripheral nerve injury. Glia 2012, 60, 1529–1539. [Google Scholar] [CrossRef]
- Kobayashi, K.; Yamanaka, H.; Fukuoka, T.; Dai, Y.; Obata, K.; Noguchi, K. P2Y12 receptor upregulation in activated microglia is a gateway of p38 signaling and neuropathic pain. J. Neurosci. 2008, 28, 2892–2902. [Google Scholar] [CrossRef]
- Tozaki-Saitoh, H.; Tsuda, M.; Miyata, H.; Ueda, K.; Kohsaka, S.; Inoue, K. P2Y12 receptors in spinal microglia are required for neuropathic pain after peripheral nerve injury. J. Neurosci. 2008, 28, 4949–4956. [Google Scholar] [CrossRef]
- Tsuda, M.; Shigemoto-Mogami, Y.; Koizumi, S.; Mizokoshi, A.; Kohsaka, S.; Salter, M.W.; Inoue, K. P2X4 receptors induced in spinal microglia gate tactile allodynia after nerve injury. Nature 2003, 424, 778–783. [Google Scholar] [CrossRef]
- Masuda, T.; Tsuda, M.; Yoshinaga, R.; Tozaki-Saitoh, H.; Ozato, K.; Tamura, T.; Inoue, K. IRF8 is a critical transcription factor for transforming microglia into a reactive phenotype. Cell Rep. 2012, 1, 334–340. [Google Scholar] [CrossRef] [Green Version]
- Horiuchi, M.; Wakayama, K.; Itoh, A.; Kawai, K.; Pleasure, D.; Ozato, K.; Itoh, T. Interferon regulatory factor 8/interferon consensus sequence binding protein is a critical transcription factor for the physiological phenotype of microglia. J. Neuroinflamm. 2012, 9, 227. [Google Scholar] [CrossRef] [Green Version]
- Gérard, C.; De Mot, L.; Cordi, S.; van Eyll, J.; Lemaigre, F.P. Temporal dynamics of a CSF1R signaling gene regulatory network involved in epilepsy. PLoS Comput. Biol. 2021, 17, e1008854. [Google Scholar] [CrossRef]
- Tsuda, M.; Inoue, K. Neuron-microglia interaction by purinergic signaling in neuropathic pain following neurodegeneration. Neuropharmacology 2016, 104, 76–81. [Google Scholar] [CrossRef] [Green Version]
- Tam, T.H.; Salter, M.W. Purinergic signalling in spinal pain processing. Purinergic Signal. 2021, 17, 49–54. [Google Scholar] [CrossRef]
- Castellano, B.; Bosch-Queralt, M.; Almolda, B.; Villacampa, N.; Gonzalez, B. Purine Signaling and Microglial Wrapping. Adv. Exp. Med. Biol. 2016, 949, 147–165. [Google Scholar] [CrossRef]
- Fields, R.D.; Burnstock, G. Purinergic signalling in neuron-glia interactions. Nat. Rev. Neurosci. 2006, 7, 423–436. [Google Scholar] [CrossRef]
- Cserép, C.; Pósfai, B.; Dénes, Á. Shaping neuronal fate: Functional heterogeneity of direct microglia-neuron interactions. Neuron 2020, 109, 222–240. [Google Scholar] [CrossRef]
- Kettenmann, H.; Hanisch, U.K.; Noda, M.; Verkhratsky, A. Physiology of microglia. Physiol. Rev. 2011, 91, 461–553. [Google Scholar] [CrossRef]
- Davalos, D.; Grutzendler, J.; Yang, G.; Kim, J.V.; Zuo, Y.; Jung, S.; Littman, D.R.; Dustin, M.L.; Gan, W.B. ATP mediates rapid microglial response to local brain injury in vivo. Nat. Neurosci. 2005, 8, 752–758. [Google Scholar] [CrossRef]
- Sipe, G.O.; Lowery, R.L.; Tremblay, M.E.; Kelly, E.A.; Lamantia, C.E.; Majewska, A.K. Microglial P2Y12 is necessary for synaptic plasticity in mouse visual cortex. Nat. Commun. 2016, 7, 10905. [Google Scholar] [CrossRef]
- Haynes, S.E.; Hollopeter, G.; Yang, G.; Kurpius, D.; Dailey, M.E.; Gan, W.B.; Julius, D. The P2Y12 receptor regulates microglial activation by extracellular nucleotides. Nat. Neurosci. 2006, 9, 1512–1519. [Google Scholar] [CrossRef]
- Ohsawa, K.; Irino, Y.; Sanagi, T.; Nakamura, Y.; Suzuki, E.; Inoue, K.; Kohsaka, S. P2Y12 receptor-mediated integrin-beta1 activation regulates microglial process extension induced by ATP. Glia 2010, 58, 790–801. [Google Scholar] [CrossRef]
- Ohsawa, K.; Sanagi, T.; Nakamura, Y.; Suzuki, E.; Inoue, K.; Kohsaka, S. Adenosine A3 receptor is involved in ADP-induced microglial process extension and migration. J. Neurochem. 2012, 121, 217–227. [Google Scholar] [CrossRef] [PubMed]
- Romer, S.H.; Dominguez, K.M.; Gelpi, M.W.; Deardorff, A.S.; Tracy, R.C.; Fyffe, R.E.W. Redistribution of Kv2.1 ion channels on spinal motoneurons following peripheral nerve injury. Brain Res. 2014, 1547, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernier, L.-P.; Bohlen, C.J.; York, E.M.; Choi, H.B.; Kamyabi, A.; Dissing-Olesen, L.; Hefendehl, J.K.; Collins, H.Y.; Stevens, B.; Barres, B.A. Nanoscale surveillance of the brain by microglia via cAMP-regulated filopodia. Cell Rep. 2019, 27, 2895–2908.e4. [Google Scholar] [CrossRef] [Green Version]
- Koizumi, S.; Shigemoto-Mogami, Y.; Nasu-Tada, K.; Shinozaki, Y.; Ohsawa, K.; Tsuda, M.; Joshi, B.V.; Jacobson, K.A.; Kohsaka, S.; Inoue, K. UDP acting at P2Y6 receptors is a mediator of microglial phagocytosis. Nature 2007, 446, 1091–1095. [Google Scholar] [CrossRef] [Green Version]
- Bura, S.A.; Nadal, X.; Ledent, C.; Maldonado, R.; Valverde, O. A2A adenosine receptor regulates glia proliferation and pain after peripheral nerve injury. Pain 2008, 140, 95–103. [Google Scholar] [CrossRef]
- Coull, J.A.; Beggs, S.; Boudreau, D.; Boivin, D.; Tsuda, M.; Inoue, K.; Gravel, C.; Salter, M.W.; De Koninck, Y. BDNF from microglia causes the shift in neuronal anion gradient underlying neuropathic pain. Nature 2005, 438, 1017–1021. [Google Scholar] [CrossRef]
- Rivera, C.; Li, H.; Thomas-Crusells, J.; Lahtinen, H.; Viitanen, T.; Nanobashvili, A.; Kokaia, Z.A.M.; Voipio, J.; Kaila, K.; Saarma, M. BDNF-induced TrkB activation down-regulates the K+-Cl− cotransporter KCC2 and impairs neuronal Cl− extrusion. J. Cell Biol. 2002, 159, 747–752. [Google Scholar] [CrossRef] [Green Version]
- Hildebrand, M.E.; Xu, J.; Dedek, A.; Li, Y.; Sengar, A.S.; Beggs, S.; Lombroso, P.J.; Salter, M.W. Potentiation of synaptic GluN2B NMDAR currents by Fyn kinase is gated through BDNF-mediated disinhibition in spinal pain processing. Cell Rep. 2016, 17, 2753–2765. [Google Scholar] [CrossRef] [Green Version]
- Ulmann, L.; Hatcher, J.P.; Hughes, J.P.; Chaumont, S.; Green, P.J.; Conquet, F.; Buell, G.N.; Reeve, A.J.; Chessell, I.P.; Rassendren, F. Up-regulation of P2X4 receptors in spinal microglia after peripheral nerve injury mediates BDNF release and neuropathic pain. J. Neurosci. 2008, 28, 11263–11268. [Google Scholar] [CrossRef]
- Chessell, I.P.; Hatcher, J.P.; Bountra, C.; Michel, A.D.; Hughes, J.P.; Green, P.; Egerton, J.; Murfin, M.; Richardson, J.; Peck, W.L.; et al. Disruption of the P2X7 purinoceptor gene abolishes chronic inflammatory and neuropathic pain. Pain 2005, 114, 386–396. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, K.; Takahashi, E.; Miyagawa, Y.; Yamanaka, H.; Noguchi, K. Induction of the P2X7 receptor in spinal microglia in a neuropathic pain model. Neurosci. Lett. 2011, 504, 57–61. [Google Scholar] [CrossRef] [PubMed]
- He, W.-J.; Cui, J.; Du, L.; Zhao, Y.-D.; Burnstock, G.; Zhou, H.-D.; Ruan, H.-Z. Spinal P2X7 receptor mediates microglia activation-induced neuropathic pain in the sciatic nerve injury rat model. Behav. Brain Res. 2012, 226, 163–170. [Google Scholar] [CrossRef] [PubMed]
- Pelegrin, P.; Surprenant, A. Pannexin-1 mediates large pore formation and interleukin-1β release by the ATP-gated P2X7 receptor. EMBO J. 2006, 25, 5071–5082. [Google Scholar] [CrossRef] [Green Version]
- Lowrie, M.; Vrbova, G. Dependence of postnatal motoneurones on their targets: Review and hypothesis. Trends Neurosci. 1992, 15, 80–84. [Google Scholar] [CrossRef]
- Angelov, D.; Krebs, C.; Walther, M.; Martinez-Portillo, F.; Gunkel, A.; Lay, C.; Streppel, M.; Guntinas-Lichius, O.; Stennert, E.; Neiss, W. Altered expression of immune-related antigens by neuronophages does not improve neuronal survival after severe lesion of the facial nerve in rats. Glia 1998, 24, 155–171. [Google Scholar] [CrossRef]
- Fukushima, N.; Kobayashi, T.; Kakegawa, A.; Sumitomo, N.; Nagira, A.; Moriizumi, T. Hypoglossal nerve injury with long nerve resection leading to slow motoneuron death. Neurosci. Lett. 2020, 715, 134668. [Google Scholar] [CrossRef]
- Kou, S.Y.; Chiu, A.Y.; Patterson, P.H. Differential regulation of motor neuron survival and choline acetyltransferase expression following axotomy. J. Neurobiol. 1995, 27, 561–572. [Google Scholar] [CrossRef]
- Serpe, C.J.; Kohm, A.P.; Huppenbauer, C.B.; Sanders, V.M.; Jones, K.J. Exacerbation of facial motoneuron loss after facial nerve transection in severe combined immunodeficient (scid) mice. J. Neurosci. 1999, 19, RC7. [Google Scholar] [CrossRef] [Green Version]
- Ferri, C.C.; Moore, F.A.; Bisby, M.A. Effects of facial nerve injury on mouse motoneurons lacking the p75 low-affinity neurotrophin receptor. J. Neurobiol. 1998, 34, 1–9. [Google Scholar] [CrossRef]
- Hottinger, A.F.; Azzouz, M.; Déglon, N.; Aebischer, P.; Zurn, A.D. Complete and long-term rescue of lesioned adult motoneurons by lentiviral-mediated expression of glial cell line-derived neurotrophic factor in the facial nucleus. J. Neurosci. 2000, 20, 5587–5593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiryu-Seo, S.; Hirayama, T.; Kato, R.; Kiyama, H. Noxa is a critical mediator of p53-dependent motor neuron death after nerve injury in adult mouse. J. Neurosci. 2005, 25, 1442–1447. [Google Scholar] [CrossRef] [PubMed]
- Snider, W.; Thanedar, S. Target dependence of hypoglossal motor neurons during development and in maturity. J. Comp. Neurol. 1989, 279, 489–498. [Google Scholar] [CrossRef] [PubMed]
- Yamada, J.; Jinno, S. Alterations in neuronal survival and glial reactions after axotomy by ceftriaxone and minocycline in the mouse hypoglossal nucleus. Neurosci. Lett. 2011, 504, 295–300. [Google Scholar] [CrossRef]
- Park, O.h.; Lee, K.J.; Rhyu, I.J.; Geum, D.; Kim, H.; Buss, R.; Oppenheim, R.W.; Sun, W. Bax-dependent and-independent death of motoneurons after facial nerve injury in adult mice. Eur. J. Neurosci. 2007, 26, 1421–1432. [Google Scholar] [CrossRef]
- Pollin, M.M.; McHanwell, S.; Slater, C.R. The effect of age on motor neurone death following axotomy in the mouse. Development 1991, 112, 83–89. [Google Scholar] [CrossRef]
- Tornqvist, E.; Aldskogius, H. Motoneuron survival is not affected by the proximo-distal level of axotomy but by the possibility of regenerating axons to gain access to the distal nerve stump. J. Neurosci. Res. 1994, 39, 159–165. [Google Scholar] [CrossRef]
- Koliatsos, V.E.; Price, W.L.; Pardo, C.A.; Price, D.L. Ventral root avulsion: An experimental model of death of adult motor neurons. J. Comp. Neurol. 1994, 342, 35–44. [Google Scholar] [CrossRef]
- Mattsson, P.; Meijer, B.; Svensson, M. Extensive neuronal cell death following intracranial transection of the facial nerve in the adult rat. Brain Res. Bull. 1999, 49, 333–341. [Google Scholar] [CrossRef]
- Yu, W.H.A. Spatial and temporal correlation of nitric oxide synthase expression with CuZn-superoxide dismutase reduction in motor neurons following axotomy. Ann. N. Y. Acad. Sci. 2002, 962, 111–121. [Google Scholar] [CrossRef]
- Parsadanian, A.S.; Cheng, Y.; Keller-Peck, C.R.; Holtzman, D.M.; Snider, W.D. Bcl-xL is an antiapoptotic regulator for postnatal CNS neurons. J. Neurosci. 1998, 18, 1009–1019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kinugasa, T.; Ozaki, S.; Hamanaka, S.; Kudo, N. The effects of sciatic nerve axotomy on spinal motoneurons in neonatal Bax-deficient mice. Neurosci. Res. 2002, 44, 439–446. [Google Scholar] [CrossRef]
- Farlie, P.G.; Dringen, R.; Rees, S.M.; Kannourakis, G.; Bernard, O. bcl-2 transgene expression can protect neurons against developmental and induced cell death. Proc. Natl. Acad. Sci. USA 1995, 92, 4397–4401. [Google Scholar] [CrossRef] [Green Version]
- Gillardon, F.; Klimaschewski, L.; Wickert, H.; Krajewski, S.; Reed, J.; Zimmermann, M. Expression pattern of candidate cell death effector proteins Bax, Bcl-2, Bcl-X, and c-Jun in sensory and motor neurons following sciatic nerve transection in the rat. Brain Res. 1996, 739, 244–250. [Google Scholar] [CrossRef]
- Perrelet, D.; Perrin, F.E.; Liston, P.; Korneluk, R.G.; MacKenzie, A.; Ferrer-Alcon, M.; Kato, A.C. Motoneuron resistance to apoptotic cell death in vivo correlates with the ratio between X-linked inhibitor of apoptosis proteins (XIAPs) and its inhibitor, XIAP-associated factor 1. J. Neurosci. 2004, 24, 3777–3785. [Google Scholar] [CrossRef] [Green Version]
- Chan, Y.-M.; Wu, W.; Yip, H.K.; So, K.-F.; Oppenheim, R.W. Caspase inhibitors promote the survival of avulsed spinal motoneurons in neonatal rats. Neuroreport 2001, 12, 541–545. [Google Scholar] [CrossRef]
- Harada, S.; Suzuki, S.O.; Seki, Y.; Nakamura, S.; Iwaki, T. Differential activation of proapoptotic molecules between mouse and rat models of distal motor trigeminal denervation. J. Oral Pathol. Med. 2012, 41, 354–360. [Google Scholar] [CrossRef]
- Li, L.; Houenou, L.J.; Wu, W.; Lei, M.; Prevette, D.M.; Oppenheim, R.W. Characterization of spinal motoneuron degeneration following different types of peripheral nerve injury in neonatal and adult mice. J. Comp. Neurol. 1998, 396, 158–168. [Google Scholar] [CrossRef]
- Baumgartner, B.J.; Shine, H.D. Neuroprotection of spinal motoneurons following targeted transduction with an adenoviral vector carrying the gene for glial cell line-derived neurotrophic factor. Exp. Neurol. 1998, 153, 102–112. [Google Scholar] [CrossRef]
- Cuevas, P.; Carceller, F.; Giménez-Gallego, G. Acidic fibroblast growth factor prevents death of spinal cord motoneurons in newborn rats after nerve section. Neurol. Res. 1995, 17, 396–399. [Google Scholar] [CrossRef]
- Ikeda, K.; Iwasaki, Y.; Shiojima, T.; Kinoshita, M. Neuroprotective effect of various cytokines on developing spinal motoneurons following axotomy. J. Neurol. Sci. 1996, 135, 109–113. [Google Scholar] [CrossRef]
- Li, L.; Oppenheim, R.W.; Lei, M.; Houenou, L.J. Neurotrophic agents prevent motoneuron death following sciatic nerve section in the neonatal mouse. J. Neurobiol. 1994, 25, 759–766. [Google Scholar] [CrossRef] [PubMed]
- Zurn, A.D.; Baetge, E.E.; Hammang, J.P.; Tan, S.A.; Aebischer, P. Glial cell line-derived neurotrophic factor (GDNF), a new neurotrophic factor for motoneurones. Neuroreport 1994, 6, 113–118. [Google Scholar] [CrossRef]
- Pu, S.F.; Zhuang, H.X.; Marsh, D.J.; Ishii, D.N. Insulin-like growth factor-II increases and IGF is required for postnatal rat spinal motoneuron survival following sciatic nerve axotomy. J. Neurosci. Res. 1999, 55, 9–16. [Google Scholar] [CrossRef]
- Gravel, C.; Götz, R.; Lorrain, A.; Sendtner, M. Adenoviral gene transfer of ciliary neurotrophic factor and brain-derived neurotrophic factor leads to long-term survival of axotomized motor neurons. Nat. Med. 1997, 3, 765–770. [Google Scholar] [CrossRef]
- Koliatsos, V.E.; Clatterbuck, R.E.; Winslow, J.W.; Cayouette, M.H.; Prices, D.L. Evidence that brain-derived neurotrophic factor is a trophic factor for motor neurons in vivo. Neuron 1993, 10, 359–367. [Google Scholar] [CrossRef]
- Koliatsos, V.E.; Cayouette, M.H.; Berkemeier, L.R.; Clatterbuck, R.E.; Price, D.L.; Rosenthal, A. Neurotrophin 4/5 is a trophic factor for mammalian facial motor neurons. Proc. Natl. Acad. Sci. USA 1994, 91, 3304–3308. [Google Scholar] [CrossRef] [Green Version]
- Matheson, C.R.; Wang, J.; Collins, F.D.; Yan, Q. Long-term survival effects of GDNF on neonatal rat facial motoneurons after axotomy. Neuroreport 1997, 8, 1739–1742. [Google Scholar] [CrossRef]
- Sendtner, M.; Götz, R.; Holtmann, B.; Thoenen, H. Endogenous ciliary neurotrophic factor is a lesion factor for axotomized motoneurons in adult mice. J. Neurosci. 1997, 17, 6999–7006. [Google Scholar] [CrossRef]
- Yan, Q.; Matheson, C.; Lopez, O.T. In vivo neurotrophic effects of GDNF on neonatal and adult facial motor neurons. Nature 1995, 373, 341–344. [Google Scholar] [CrossRef]
- Yan, Q.; Matheson, C.; Lopez, O.T.; Miller, J.A. The biological responses of axotomized adult motoneurons to brain-derived neurotrophic factor. J. Neurosci. 1994, 14, 5281–5291. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Wu, W.; Lin, L.F.; Lei, M.; Oppenheim, R.W.; Houenou, L.J. Rescue of adult mouse motoneurons from injury-induced cell death by glial cell line-derived neurotrophic factor. Proc. Natl. Acad. Sci. USA 1995, 92, 9771–9775. [Google Scholar] [CrossRef] [Green Version]
- Hammarberg, H.; Piehl, F.; Risling, M.; Cullheim, S. Differential regulation of trophic factor receptor mRNAs in spinal motoneurons after sciatic nerve transection and ventral root avulsion in the rat. J. Comput. Neurol. 2000, 426, 587–601. [Google Scholar] [CrossRef]
- Yamada, J.; Nakanishi, H.; Jinno, S. Differential involvement of perineuronal astrocytes and microglia in synaptic stripping after hypoglossal axotomy. Neuroscience 2011, 182, 1–10. [Google Scholar] [CrossRef]
- Raivich, G.; Jones, L.L.; Kloss, C.U.; Werner, A.; Neumann, H.; Kreutzberg, G.W. Immune surveillance in the injured nervous system: T-lymphocytes invade the axotomized mouse facial motor nucleus and aggregate around sites of neuronal degeneration. J. Neurosci. 1998, 18, 5804–5816. [Google Scholar] [CrossRef] [Green Version]
- Setter, D.O.; Haulcomb, M.M.; Beahrs, T.; Meadows, R.M.; Schartz, N.D.; Custer, S.K.; Sanders, V.M.; Jones, K.J. Identification of a resilient mouse facial motoneuron population following target disconnection by injury or disease. Restor. Neurol. Neurosci. 2018, 36, 417–422. [Google Scholar] [CrossRef]
- Udina, E.; Putman, C.T.; Harris, L.R.; Tyreman, N.; Cook, V.E.; Gordon, T. Compensatory axon sprouting for very slow axonal die-back in a transgenic model of spinal muscular atrophy type III. J. Physiol. 2017, 595, 1815–1829. [Google Scholar] [CrossRef]
- Svensson, M.; Aldskogius, H. Infusion of cytosine-arabinoside into the cerebrospinal fluid of the rat brain inhibits the microglial cell proliferation after hypoglossal nerve injury. Glia 1993, 7, 286–298. [Google Scholar] [CrossRef]
- Spittau, B.; Dokalis, N.; Prinz, M. The role of TGFβ signaling in microglia maturation and activation. Trends Immunol. 2020, 41, 836–848. [Google Scholar] [CrossRef]
- Makwana, M.; Jones, L.L.; Cuthill, D.; Heuer, H.; Bohatschek, M.; Hristova, M.; Friedrichsen, S.; Ormsby, I.; Bueringer, D.; Koppius, A. Endogenous transforming growth factor β1 suppresses inflammation and promotes survival in adult CNS. J. Neurosci. 2007, 27, 11201–11213. [Google Scholar] [CrossRef]
- Hao, H.P.; Doh-ura, K.; Nakanishi, H. Impairment of microglial responses to facial nerve axotomy in cathepsin S–deficient mice. J. Neurosci. Res. 2007, 85, 2196–2206. [Google Scholar] [CrossRef]
- Kobayashi, M.; Konishi, H.; Takai, T.; Kiyama, H. A DAP12-dependent signal promotes pro-inflammatory polarization in microglia following nerve injury and exacerbates degeneration of injured neurons. Glia 2015, 63, 1073–1082. [Google Scholar] [CrossRef]
- Sun, L.; Wu, Z.; Baba, M.; Peters, C.; Uchiyama, Y.; Nakanishi, H. Cathepsin B-dependent motor neuron death after nerve injury in the adult mouse. Biochem. Biophys. Res. Commun. 2010, 399, 391–395. [Google Scholar] [CrossRef]
- Möller, T.; Bard, F.; Bhattacharya, A.; Biber, K.; Campbell, B.; Dale, E.; Eder, C.; Gan, L.; Garden, G.A.; Hughes, Z.A. Critical data-based re-evaluation of minocycline as a putative specific microglia inhibitor. Glia 2016, 64, 1788–1794. [Google Scholar] [CrossRef]
- Tanaka, T.; Murakami, K.; Bando, Y.; Nomura, T.; Isonishi, A.; Morita-Takemura, S.; Tatsumi, K.; Wanaka, A.; Yoshida, S. Microglia support ATF3-positive neurons following hypoglossal nerve axotomy. Neurochem. Int. 2017, 108, 332–342. [Google Scholar] [CrossRef]
- Sumner, B. A quantitative analysis of the response of presynaptic boutons to postsynaptic motor neuron axotomy. Exp. Neurol. 1975, 46, 605–615. [Google Scholar] [CrossRef]
- Sunico, C.R.; Gonzalez-Forero, D.; Dominguez, G.; Garcia-Verdugo, J.M.; Moreno-Lopez, B. Nitric oxide induces pathological synapse loss by a protein kinase G-, Rho kinase-dependent mechanism preceded by myosin light chain phosphorylation. J. Neurosci. 2010, 30, 973–984. [Google Scholar] [CrossRef]
- Lindå, H.; Shupliakov, O.; Örnung, G.; Ottersen, O.P.; Storm-Mathisen, J.; Risling, M.; Cullheim, S. Ultrastructural evidence for a preferential elimination of glutamate-immunoreactive synaptic terminals from spinal motoneurons after intramedullary axotomy. J. Comp. Neurol. 2000, 425, 10–23. [Google Scholar] [CrossRef]
- Alvarez, F.J.; Titus-Mitchell, H.E.; Bullinger, K.L.; Kraszpulski, M.; Nardelli, P.; Cope, T.C. Permanent central synaptic disconnection of proprioceptors after nerve injury and regeneration. I. Loss of VGLUT1/IA synapses on motoneurons. J. Neurophysiol. 2011, 106, 2450–2470. [Google Scholar] [CrossRef]
- Barron, K.D. Neuronal Responses to Axotomy: Consequences and Possibilities for Rescue from Permanent Atrophy or Cell Death; Seil, F.J., Ed.; Liss: New York, NY, USA, 1989. [Google Scholar]
- Cullheim, S.; Thams, S. The microglial networks of the brain and their role in neuronal network plasticity after lesion. Brain Res. Rev. 2007, 55, 89–96. [Google Scholar] [CrossRef]
- Navarro, X.; Vivó, M.; Valero-Cabré, A. Neural plasticity after peripheral nerve injury and regeneration. Prog. Neurobiol. 2007, 82, 163–201. [Google Scholar] [CrossRef]
- Mentis, G.; Greensmith, L.; Vrbova, G. Motoneurons destined to die are rescued by blocking N-methyl-D-aspartate receptors by MK-801. Neuroscience 1993, 54, 283–285. [Google Scholar] [CrossRef]
- Greensmith, L.; Mentis, G.; Vrbová, G. Blockade of N-methyl-D-aspartate receptors by MK-801 (dizocilpine maleate) rescues motoneurones in developing rats. Dev. Brain Res. 1994, 81, 162–170. [Google Scholar] [CrossRef]
- Iwasaki, Y.; Ikeda, K.; Shiojima, T.; Kinoshita, M. CNQX prevents spinal motor neuron death following sciatic nerve transection in newborn rats. J. Neurol. Sci. 1995, 134, 21–25. [Google Scholar] [CrossRef]
- Spejo, A.; Carvalho, J.; Goes, A.; Oliveira, A. Neuroprotective effects of mesenchymal stem cells on spinal motoneurons following ventral root axotomy: Synapse stability and axonal regeneration. Neuroscience 2013, 250, 715–732. [Google Scholar] [CrossRef]
- Dekkers, J.; Bayley, P.; Dick, J.; Schwaller, B.; Berchtold, M.; Greensmith, L. Over-expression of parvalbumin in transgenic mice rescues motoneurons from injury-induced cell death. Neuroscience 2004, 123, 459–466. [Google Scholar] [CrossRef] [Green Version]
- Toyoda, H.; Ohno, K.; Yamada, J.; Ikeda, M.; Okabe, A.; Sato, K.; Hashimoto, K.; Fukuda, A. Induction of NMDA and GABAA receptor-mediated Ca2+ oscillations with KCC2 mRNA downregulation in injured facial motoneurons. J. Neurophysiol. 2003, 89, 1353–1362. [Google Scholar] [CrossRef] [Green Version]
- Tatetsu, M.; Kim, J.; Kina, S.; Sunakawa, H.; Takayama, C. GABA/glycine signaling during degeneration and regeneration of mouse hypoglossal nerves. Brain Res. 2012, 1446, 22–33. [Google Scholar] [CrossRef]
- Gordon, T.; English, A.W. Strategies to promote peripheral nerve regeneration: Electrical stimulation and/or exercise. Eur. J. Neurosci. 2016, 43, 336–350. [Google Scholar] [CrossRef] [Green Version]
- Zuo, K.J.; Gordon, T.; Chan, K.M.; Borschel, G.H. Electrical stimulation to enhance peripheral nerve regeneration: Update in molecular investigations and clinical translation. Exp. Neurol. 2020, 332, 113397. [Google Scholar] [CrossRef]
- Svensson, M.; Aldskogius, H. Synaptic density of axotomized hypoglossal motorneurons following pharmacological blockade of the microglial cell proliferation. Exp. Neurol. 1993, 120, 123–131. [Google Scholar] [CrossRef]
- Berg, A.; Zelano, J.; Thams, S.; Cullheim, S. The extent of synaptic stripping of motoneurons after axotomy is not correlated to activation of surrounding glia or downregulation of postsynaptic adhesion molecules. PLoS ONE 2013, 8, e59647. [Google Scholar] [CrossRef] [Green Version]
- Raivich, G.; Bohatschek, M.; Kloss, C.U.; Werner, A.; Jones, L.L.; Kreutzberg, G.W. Neuroglial activation repertoire in the injured brain: Graded response, molecular mechanisms and cues to physiological function. Brain Res. Rev. 1999, 30, 77–105. [Google Scholar] [CrossRef]
- Berg, A.; Zelano, J.; Stephan, A.; Thams, S.; Barres, B.A.; Pekny, M.; Pekna, M.; Cullheim, S. Reduced removal of synaptic terminals from axotomized spinal motoneurons in the absence of complement C3. Exp. Neurol. 2012, 237, 8–17. [Google Scholar] [CrossRef] [Green Version]
- Novikov, L.; Novikova, L.; Kellerth, J.O. Brain-derived neurotrophic factor promotes axonal regeneration and long-term survival of adult rat spinal motoneurons in vivo. Neuroscience 1997, 79, 765–774. [Google Scholar] [CrossRef]
- Oliveira, A.L.; Thams, S.; Lidman, O.; Piehl, F.; Hökfelt, T.; Kärre, K.; Lindå, H.; Cullheim, S. A role for MHC class I molecules in synaptic plasticity and regeneration of neurons after axotomy. Proc. Natl. Acad. Sci. USA 2004, 101, 17843–17848. [Google Scholar] [CrossRef] [Green Version]
- Sabha Jr, M.; Emirandetti, A.; Cullheim, S.; De Oliveira, A.L.R. MHC I expression and synaptic plasticity in different mice strains after axotomy. Synapse 2008, 62, 137–148. [Google Scholar] [CrossRef]
- Kiefer, R.; Lindholm, D.; Kreutzberg, G.W. Interleukin-6 and transforming growth factor-β1 mRNAs are induced in rat facial nucleus following motoneuron axotomy. Eur. J. Neurosci. 1993, 5, 775–781. [Google Scholar] [CrossRef]
- Allan, S.M.; Rothwell, N.J. Cytokines and acute neurodegeneration. Nat. Rev. Neurosci. 2001, 2, 734–744. [Google Scholar] [CrossRef]
- De Bilbao, F.; Giannakopoulos, P.; Srinivasan, A.; Dubois-Dauphin, M. In vivo study of motoneuron death induced by nerve injury in mice deficient in the caspase 1/interleukin-1β-converting enzyme. Neuroscience 2000, 98, 573–583. [Google Scholar] [CrossRef]
- de Bilbao, F.; Dubois-Dauphin, M. Acute application of an interleukin-1 beta-converting enzyme-specific inhibitor delays axotomy-induced motoneurone death. Neuroreport 1996, 7, 3051–3054. [Google Scholar] [CrossRef]
- Sasaki, M.; Seo-Kiryu, S.; Kato, R.; Kita, S.-I.; Kiyama, H. A disintegrin and metalloprotease with thrombospondin type1 motifs (ADAMTS-1) and IL-1 receptor type 1 mRNAs are simultaneously induced in nerve injured motor neurons. Mol. Brain Res. 2001, 89, 158–163. [Google Scholar] [CrossRef]
- Raivich, G.; Liu, Z.; Kloss, C.; Labow, M.; Bluethmann, H.; Bohatschek, M. Cytotoxic potential of proinflammatory cytokines: Combined deletion of TNF receptors TNFR1 and TNFR2 prevents motoneuron cell death after facial axotomy in adult mouse. Exp. Neurol. 2002, 178, 186–193. [Google Scholar] [CrossRef]
- Terrado, J.; Monnier, D.; Perrelet, D.; Vesin, D.; Jemelin, S.; Buurman, W.A.; Mattenberger, L.; King, B.; Kato, A.C.; Garcia, I. Soluble TNF receptors partially protect injured motoneurons in the postnatal CNS. Eur. J. Neurosci. 2000, 12, 3443–3447. [Google Scholar] [CrossRef]
- Olsson, T.; Kristensson, K.; Ljungdahl, A.; Maehlen, J.; Holmdahl, R.; Klareskog, L. Gamma-interferon-like immunoreactivity in axotomized rat motor neurons. J. Neurosci. 1989, 9, 3870–3875. [Google Scholar] [CrossRef]
- Kristensson, K.; Aldskogius, M.; Peng, Z.-C.; Olsson, T.; Aldskogius, H.; Bentivoglio, M. Co-induction of neuronal interferon-gamma and nitric oxide synthase in rat motor neurons after axotomy: A role in nerve repair or death? J. Neurocytol. 1994, 23, 453–459. [Google Scholar] [CrossRef]
- Watabe, K.; Ohashi, T.; Sakamoto, T.; Kawazoe, Y.; Takeshima, T.; Oyanagi, K.; Inoue, K.; Eto, Y.; Kim, S. Rescue of lesioned adult rat spinal motoneurons by adenoviral gene transfer of glial cell line-derived neurotrophic factor. J. Neurosci. Res. 2000, 60, 511–519. [Google Scholar] [CrossRef]
- Xin, J.; Wainwright, D.A.; Serpe, C.J.; Sanders, V.M.; Jones, K.J. Phenotype of CD4+ T cell subsets that develop following mouse facial nerve axotomy. Brain Behav. Immun. 2008, 22, 528–537. [Google Scholar] [CrossRef] [Green Version]
- Kawamura, M.F.; Yamasaki, R.; Kawamura, N.; Tateishi, T.; Nagara, Y.; Matsushita, T.; Ohyagi, Y.; Kira, J.-i. Impaired recruitment of neuroprotective microglia and T cells during acute neuronal injury coincides with increased neuronal vulnerability in an amyotrophic lateral sclerosis model. Exp. Neurol. 2012, 234, 437–445. [Google Scholar]
- Jones, K.J.; Lovett-Racke, A.E.; Walker, C.L.; Sanders, V.M. CD4 + T Cells and Neuroprotection: Relevance to Motoneuron Injury and Disease. J. Neuroimmune Pharmacol. 2015, 10, 587–594. [Google Scholar] [CrossRef] [Green Version]
- Byram, S.C.; Carson, M.J.; DeBoy, C.A.; Serpe, C.J.; Sanders, V.M.; Jones, K.J. CD4-positive T cell-mediated neuroprotection requires dual compartment antigen presentation. J. Neurosci. 2004, 24, 4333–4339. [Google Scholar] [CrossRef]
- Wainwright, D.A.; Xin, J.; Mesnard, N.A.; Beahrs, T.R.; Politis, C.M.; Sanders, V.M.; Jones, K.J. Exacerbation of facial motoneuron loss after facial nerve axotomy in CCR3-deficient mice. ASN Neuro. 2009, 1, e00024. [Google Scholar]
- Raivich, G.; Bohatschek, M.; Werner, A.; Jones, L.L.; Galiano, M.; Kloss, C.U.; Zhu, X.Z.; Pfeffer, K.; Liu, Z.Q. Lymphocyte infiltration in the injured brain: Role of proinflammatory cytokines. J. Neurosci. Res. 2003, 72, 726–733. [Google Scholar] [CrossRef]
- Wainwright, D.A.; Xin, J.; Mesnard, N.A.; Politis, C.M.; Sanders, V.M.; Jones, K.J. Effects of facial nerve axotomy on Th2- and Th1-associated chemokine expression in the facial motor nucleus of wild-type and presymptomatic mSOD1 mice. J. Neuroimmunol. 2009, 216, 66–75. [Google Scholar] [CrossRef] [Green Version]
- Deboy, C.A.; Xin, J.; Byram, S.C.; Serpe, C.J.; Sanders, V.M.; Jones, K.J. Immune-mediated neuroprotection of axotomized mouse facial motoneurons is dependent on the IL-4/STAT6 signaling pathway in CD4(+) T cells. Exp. Neurol. 2006, 201, 212–224. [Google Scholar] [CrossRef]
- Serpe, C.J.; Sanders, V.M.; Jones, K.J. Kinetics of facial motoneuron loss following facial nerve transection in severe combined immunodeficient mice. J. Neurosci. Res. 2000, 62, 273–278. [Google Scholar] [CrossRef]
- Xin, J.; Wainwright, D.A.; Mesnard, N.A.; Serpe, C.J.; Sanders, V.M.; Jones, K.J. IL-10 within the CNS is necessary for CD4+ T cells to mediate neuroprotection. Brain Behav. Immun. 2011, 25, 820–829. [Google Scholar] [CrossRef] [Green Version]
- Strle, K.; Zhou, J.H.; Shen, W.H.; Broussard, S.R.; Johnson, R.W.; Freund, G.G.; Dantzer, R.; Kelley, K.W. Interleukin-10 in the brain. Crit. Rev. Immunol. 2001, 21, 427–449. [Google Scholar] [CrossRef]
- Boyd, Z.S.; Kriatchko, A.; Yang, J.; Agarwal, N.; Wax, M.B.; Patil, R.V. Interleukin-10 receptor signaling through STAT-3 regulates the apoptosis of retinal ganglion cells in response to stress. Investig. Ophthalmol. Vis. Sci. 2003, 44, 5206–5211. [Google Scholar] [CrossRef] [Green Version]
- Villacampa, N.; Almolda, B.; Vilella, A.; Campbell, I.L.; Gonzalez, B.; Castellano, B. Astrocyte-targeted production of IL-10 induces changes in microglial reactivity and reduces motor neuron death after facial nerve axotomy. Glia 2015, 63, 1166–1184. [Google Scholar] [CrossRef]
- Kloss, C.U.; Werner, A.; Klein, M.A.; Shen, J.; Menuz, K.; Probst, J.C.; Kreutzberg, G.W.; Raivich, G. Integrin family of cell adhesion molecules in the injured brain: Regulation and cellular localization in the normal and regenerating mouse facial motor nucleus. J. Comp. Neurol. 1999, 411, 162–178. [Google Scholar] [CrossRef]
- Almolda, B.; Villacampa, N.; Manders, P.; Hidalgo, J.; Campbell, I.L.; Gonzalez, B.; Castellano, B. Effects of astrocyte-targeted production of interleukin-6 in the mouse on the host response to nerve injury. Glia 2014, 62, 1142–1161. [Google Scholar] [CrossRef]
- Tröscher, A.R.; Wimmer, I.; Quemada-Garrido, L.; Köck, U.; Gessl, D.; Verberk, S.G.; Martin, B.; Lassmann, H.; Bien, C.G.; Bauer, J. Microglial nodules provide the environment for pathogenic T cells in human encephalitis. Acta Neuropathol. 2019, 137, 619–635. [Google Scholar] [CrossRef] [Green Version]
- Butovsky, O.; Jedrychowski, M.P.; Moore, C.S.; Cialic, R.; Lanser, A.J.; Gabriely, G.; Koeglsperger, T.; Dake, B.; Wu, P.M.; Doykan, C.E. Identification of a unique TGF-β–dependent molecular and functional signature in microglia. Nat. Neurosci. 2014, 17, 131–143. [Google Scholar] [CrossRef] [Green Version]
- Vega-Avelaira, D.; Moss, A.; Fitzgerald, M. Age-related changes in the spinal cord microglial and astrocytic response profile to nerve injury. Brain Behav. Immun. 2007, 21, 617–623. [Google Scholar] [CrossRef]
- Sulaiman, W.; Gordon, T. Neurobiology of peripheral nerve injury, regeneration, and functional recovery: From bench top research to bedside application. Ochsner J. 2013, 13, 100–108. [Google Scholar]
- Gehrmann, J.; Monaco, S.; Kreutzberg, G.W. Spinal cord microglial cells and DRG satellite cells rapidly respond to transection of the rat sciatic nerve. Restor. Neurol. Neurosci. 1991, 2, 181–198. [Google Scholar] [CrossRef]
- Lu, X.; Richardson, P.M. Inflammation near the nerve cell body enhances axonal regeneration. J. Neurosci. 1991, 11, 972–978. [Google Scholar] [CrossRef] [Green Version]
- McQuarrie, I.G.; Grafstein, B. Axon outgrowth enhanced by a previous nerve injury. Arch. Neurol. 1973, 29, 53–55. [Google Scholar] [CrossRef]
- Svensson, M.; Aldskogius, H. Regeneration of hypoglossal nerve axons following blockade of the axotomy-induced microglial cell reaction in the rat. Eur. J. Neurosci. 1993, 5, 85–94. [Google Scholar] [CrossRef]
- Horvat, A.; Schwaiger, F.-W.; Hager, G.; Bröcker, F.; Streif, R.; Knyazev, P.G.; Ullrich, A.; Kreutzberg, G.W. A novel role for protein tyrosine phosphatase shp1 in controlling glial activation in the normal and injured nervous system. J. Neurosci. 2001, 21, 865–874. [Google Scholar] [CrossRef]
- Armstrong, B.; Abad, C.; Chhith, S.; Cheung-Lau, G.; Hajji, O.; Nobuta, H.; Waschek, J. Impaired nerve regeneration and enhanced neuroinflammatory response in mice lacking pituitary adenylyl cyclase activating peptide. Neuroscience 2008, 151, 63–73. [Google Scholar] [CrossRef] [Green Version]
- Molnár, K.; Nógrádi, B.; Kristóf, R.; Mészáros, Á.; Pajer, K.; Siklós, L.; Nógrádi, A.; Wilhelm, I.; Krizbai, I.A. Motoneuronal inflammasome activation triggers excessive neuroinflammation and impedes regeneration after sciatic nerve injury. J. Neuroinflamm. 2022, 19, 68. [Google Scholar] [CrossRef]
- Shokouhi, B.N.; Wong, B.Z.; Siddiqui, S.; Lieberman, A.R.; Campbell, G.; Tohyama, K.; Anderson, P.N. Microglial responses around intrinsic CNS neurons are correlated with axonal regeneration. BMC Neurosci. 2010, 11, 13. [Google Scholar] [CrossRef] [Green Version]
- Gordon, T.; Tyreman, N.; Raji, M.A. The basis for diminished functional recovery after delayed peripheral nerve repair. J. Neurosci. 2011, 31, 5325–5334. [Google Scholar] [CrossRef] [Green Version]
- Guo, S.; Moore, R.M.; Charlesworth, M.C.; Johnson, K.L.; Spinner, R.J.; Windebank, A.J.; Wang, H. The proteome of distal nerves: Implication in delayed repair and poor functional recovery. Neural Regen. Res. 2022, 17, 1998. [Google Scholar]
- Tozaki-Saitoh, H.; Takeda, H.; Inoue, K. The Role of Microglial Purinergic Receptors in Pain Signaling. Molecules 2022, 27, 1919. [Google Scholar] [CrossRef]
- Boakye, P.A.; Tang, S.-J.; Smith, P.A. Mediators of Neuropathic Pain; Focus on Spinal Microglia, CSF-1, BDNF, CCL21, TNF-α, Wnt Ligands, and Interleukin 1β. Front. Pain Res. 2021, 2, 698157. [Google Scholar] [CrossRef]
- Sideris-Lampretsas, G.; Malcangio, M. Microglial heterogeneity in chronic pain. Brain Behav. Immun. 2021, 96, 279–289. [Google Scholar] [CrossRef]
- Yu, X.; Basbaum, A.; Guan, Z. Contribution of colony-stimulating factor 1 to neuropathic pain. Pain Rep. 2021, 6, e883. [Google Scholar] [CrossRef]
- Kohno, K.; Tsuda, M. Role of microglia and P2X4 receptors in chronic pain. Pain Rep. 2021, 6, e864. [Google Scholar] [CrossRef]
- Donnelly, C.R.; Andriessen, A.S.; Chen, G.; Wang, K.; Jiang, C.; Maixner, W.; Ji, R.-R. Central nervous system targets: Glial cell mechanisms in chronic pain. Neurotherapeutics 2020, 17, 846–860. [Google Scholar] [CrossRef]
- Salter, M.W.; Beggs, S. Sublime microglia: Expanding roles for the guardians of the CNS. Cell 2014, 158, 15–24. [Google Scholar] [CrossRef] [Green Version]
- Ferrini, F.; De Koninck, Y. Microglia control neuronal network excitability via BDNF signalling. Neural Plast. 2013, 2013, 429815. [Google Scholar] [CrossRef]
- Persson, J.K.; Aldskogius, H.; Arvidsson, J.; Holmberg, A. Ultrastructural changes in the gracile nucleus of the rat after sciatic nerve transection. Anat. Embryol. 1991, 184, 591–604. [Google Scholar] [CrossRef]
- Arvidsson, J. An ultrastructural study of transganglionic degeneration in the main sensory trigeminal nucleus of the rat. J. Neurocytol. 1979, 8, 31–45. [Google Scholar] [CrossRef]
- Aldskogius, H.; Arvidsson, J.; Grant, G. The reaction of primary sensory neurons to peripheral nerve injury with particular emphasis on transganglionic changes. Brain Res. Rev. 1985, 10, 27–46. [Google Scholar] [CrossRef]
- Arvidsson, J. Transganglionic degeneration in vibrissae innervating primary sensory neurons of the rat: A light and electron microscopic study. J. Comp. Neurol. 1986, 249, 392–403. [Google Scholar] [CrossRef]
- Bjelke, K.; Aldskogius, H.; Arvidsson, J. Short-and long-term transganglionic changes in the central terminations of transected vibrissal afferents in the rat. Exp. Brain Res. 1996, 112, 268–276. [Google Scholar] [CrossRef]
- Svensson, M.; Eriksson, P.; Persson, J.; Molander, C.; Arvidsson, J.; Aldskogius, H. The response of central glia to peripheral nerve injury. Brain Res. Bull. 1993, 30, 499–506. [Google Scholar] [CrossRef]
- Ueta, Y.; Miyata, M. Brainstem local microglia induce whisker map plasticity in the thalamus after peripheral nerve injury. Cell Rep. 2021, 34, 108823. [Google Scholar] [CrossRef]
- Imai, F.; Yoshida, Y. Molecular mechanisms underlying monosynaptic sensory-motor circuit development in the spinal cord. Dev. Dyn. 2018, 247, 581–587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rotterman, T.M.; Nardelli, P.; Cope, T.C.; Alvarez, F.J. Normal Distribution of VGLUT1 Synapses on Spinal Motoneuron Dendrites and Their Reorganization after Nerve Injury. J. Neurosci. 2014, 34, 3475–3492. [Google Scholar] [CrossRef] [Green Version]
- Schultz, A.J.; Rotterman, T.M.; Dwarakanath, A.; Alvarez, F.J. VGLUT1 synapses and P-boutons on regenerating motoneurons after nerve crush. J. Comp. Neurol. 2017, 525, 2876–2889. [Google Scholar] [CrossRef]
- Brushart, T.; Henry, E.; Mesulam, M.-M. Reorganization of muscle afferent projections accompanies peripheral nerve regeneration. Neuroscience 1981, 6, 2053–2061. [Google Scholar] [CrossRef]
- Brushart, T.M.; Mesulam, M.-M. Alteration in connections between muscle and anterior horn motoneurons after peripheral nerve repair. Science 1980, 208, 603–605. [Google Scholar] [CrossRef]
- Bosco, G.; Poppele, R. Proprioception from a spinocerebellar perspective. Physiol. Rev. 2001, 81, 539–568. [Google Scholar] [CrossRef]
- Tenner, A.J.; Stevens, B.; Woodruff, T.M. New tricks for an ancient system: Physiological and pathological roles of complement in the CNS. Mol. Immunol. 2018, 102, 3–13. [Google Scholar] [CrossRef]
- Stephan, A.H.; Barres, B.A.; Stevens, B. The complement system: An unexpected role in synaptic pruning during development and disease. Annu. Rev. Neurosci. 2012, 35, 369–389. [Google Scholar] [CrossRef] [Green Version]
- Stevens, B.; Allen, N.J.; Vazquez, L.E.; Howell, G.R.; Christopherson, K.S.; Nouri, N.; Micheva, K.D.; Mehalow, A.K.; Huberman, A.D.; Stafford, B. The classical complement cascade mediates CNS synapse elimination. Cell 2007, 131, 1164–1178. [Google Scholar] [CrossRef] [Green Version]
- Schafer, D.P.; Lehrman, E.K.; Kautzman, A.G.; Koyama, R.; Mardinly, A.R.; Yamasaki, R.; Ransohoff, R.M.; Greenberg, M.E.; Barres, B.A.; Stevens, B. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron 2012, 74, 691–705. [Google Scholar] [CrossRef] [Green Version]
- Esser, A.F. Big MAC attack: Complement proteins cause leaky patches. Immunol. Today 1991, 12, 316–318. [Google Scholar] [CrossRef]
- Kinoshita, T. Biology of complement: The overture. Immunol. Today 1991, 12, 291–295. [Google Scholar] [CrossRef]
- Perry, V.H.; O’Connor, V. C1q: The perfect complement for a synaptic feast? Nat. Rev. Neurosci. 2008, 9, 807–811. [Google Scholar] [CrossRef]
- Ramaglia, V.; Daha, M.R.; Baas, F. The complement system in the peripheral nerve: Friend or foe? Mol. Immunol. 2008, 45, 3865–3877. [Google Scholar] [CrossRef]
- Svensson, M.; Aldskogius, H. Evidence for activation of the complement cascade in the hypoglossal nucleus following peripheral nerve injury. J. Neuroimmunol. 1992, 40, 99–109. [Google Scholar] [CrossRef]
- Liu, L.; Törnqvist, E.; Mattsson, P.; Eriksson, N.P.; Persson, J.K.E.; Morgan, B.P.; Aldskogius, H.; Svensson, M. Complement and clusterin in the spinal cord dorsal horn and gracile nucleus following sciatic nerve injury in the adult rat. Neuroscience 1995, 68, 167–179. [Google Scholar] [CrossRef]
- Alvarez, F.J.; Villalba, R.M.; Zerda, R.; Schneider, S.P. Vesicular glutamate transporters in the spinal cord, with special reference to sensory primary afferent synapses. J. Comp. Neurol. 2004, 472, 257–280. [Google Scholar] [CrossRef]
- Vukojicic, A.; Delestrée, N.; Fletcher, E.V.; Pagiazitis, J.G.; Sankaranarayanan, S.; Yednock, T.A.; Barres, B.A.; Mentis, G.Z. The classical complement pathway mediates microglia-dependent remodeling of spinal motor circuits during development and in SMA. Cell Rep. 2019, 29, 3087–3100.e7. [Google Scholar] [CrossRef] [Green Version]
- Vasek, M.J.; Garber, C.; Dorsey, D.; Durrant, D.M.; Bollman, B.; Soung, A.; Yu, J.; Perez-Torres, C.; Frouin, A.; Wilton, D.K.; et al. A complement-microglial axis drives synapse loss during virus-induced memory impairment. Nature 2016, 534, 538–543. [Google Scholar] [CrossRef] [Green Version]
- Ye, Y.; Salvo, E.; Romero-Reyes, M.; Akerman, S.; Shimizu, E.; Kobayashi, Y.; Michot, B.; Gibbs, J. Glia and orofacial pain: Progress and future directions. Int. J. Mol. Sci. 2021, 22, 5345. [Google Scholar] [CrossRef]
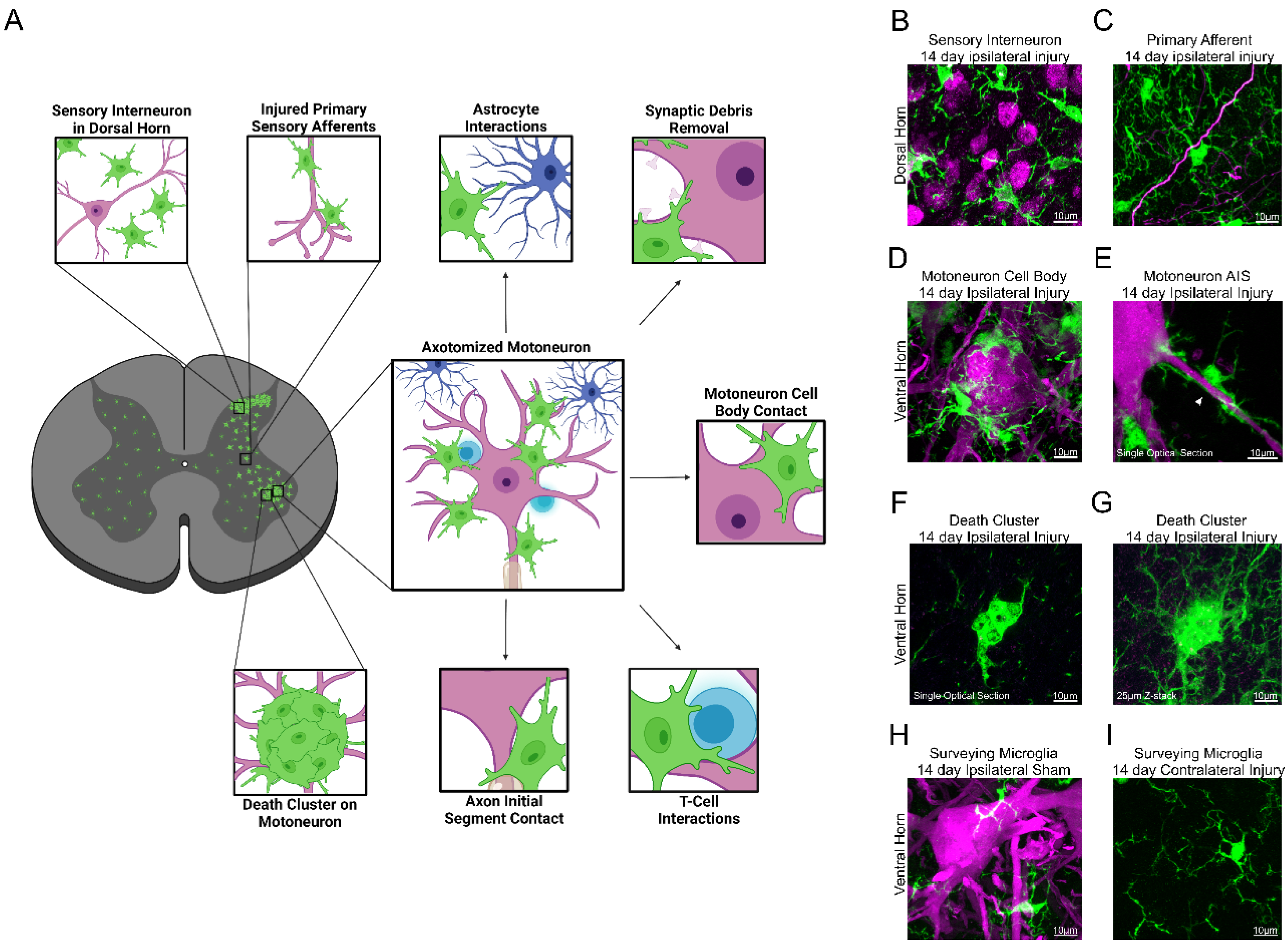
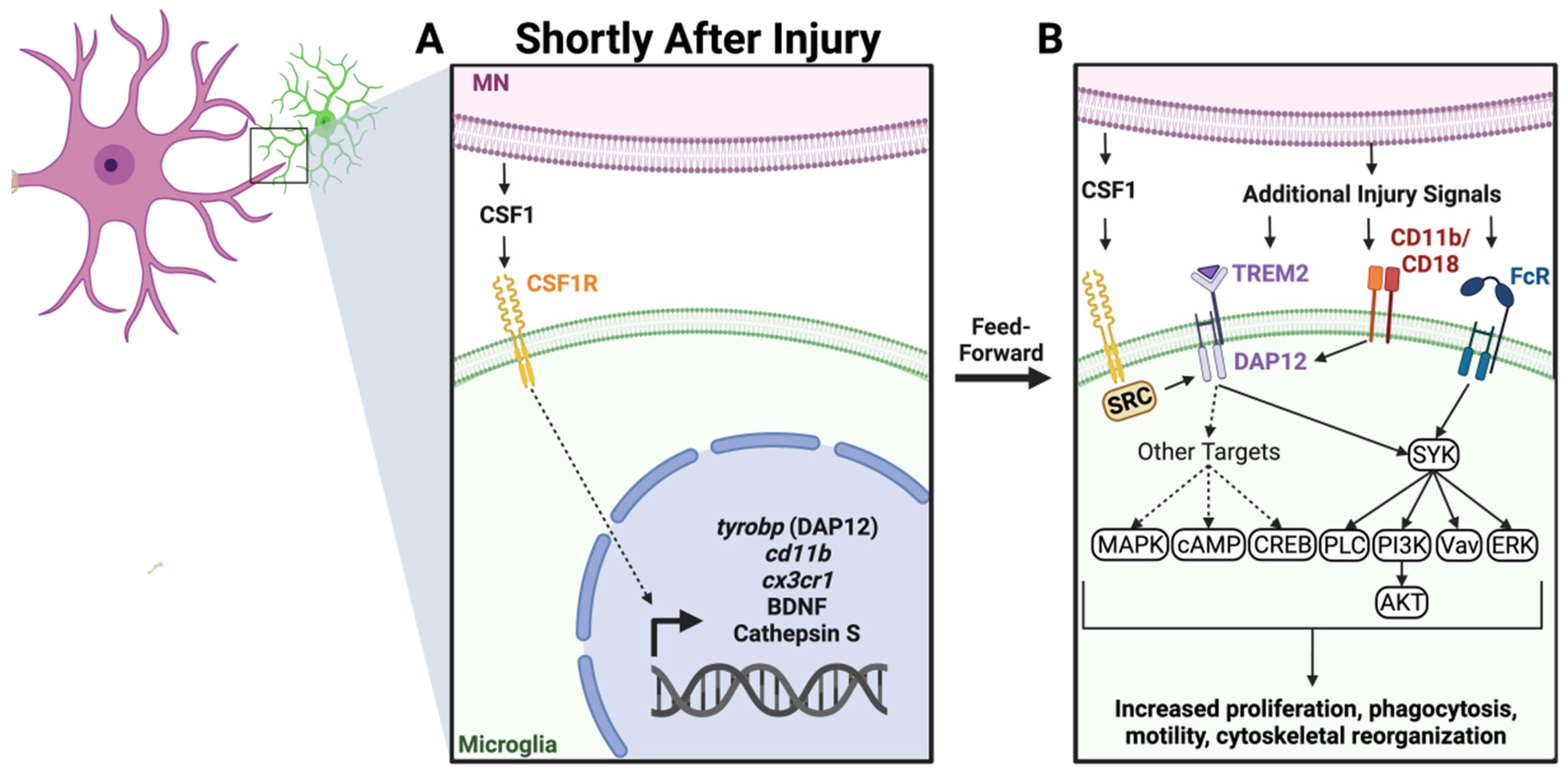
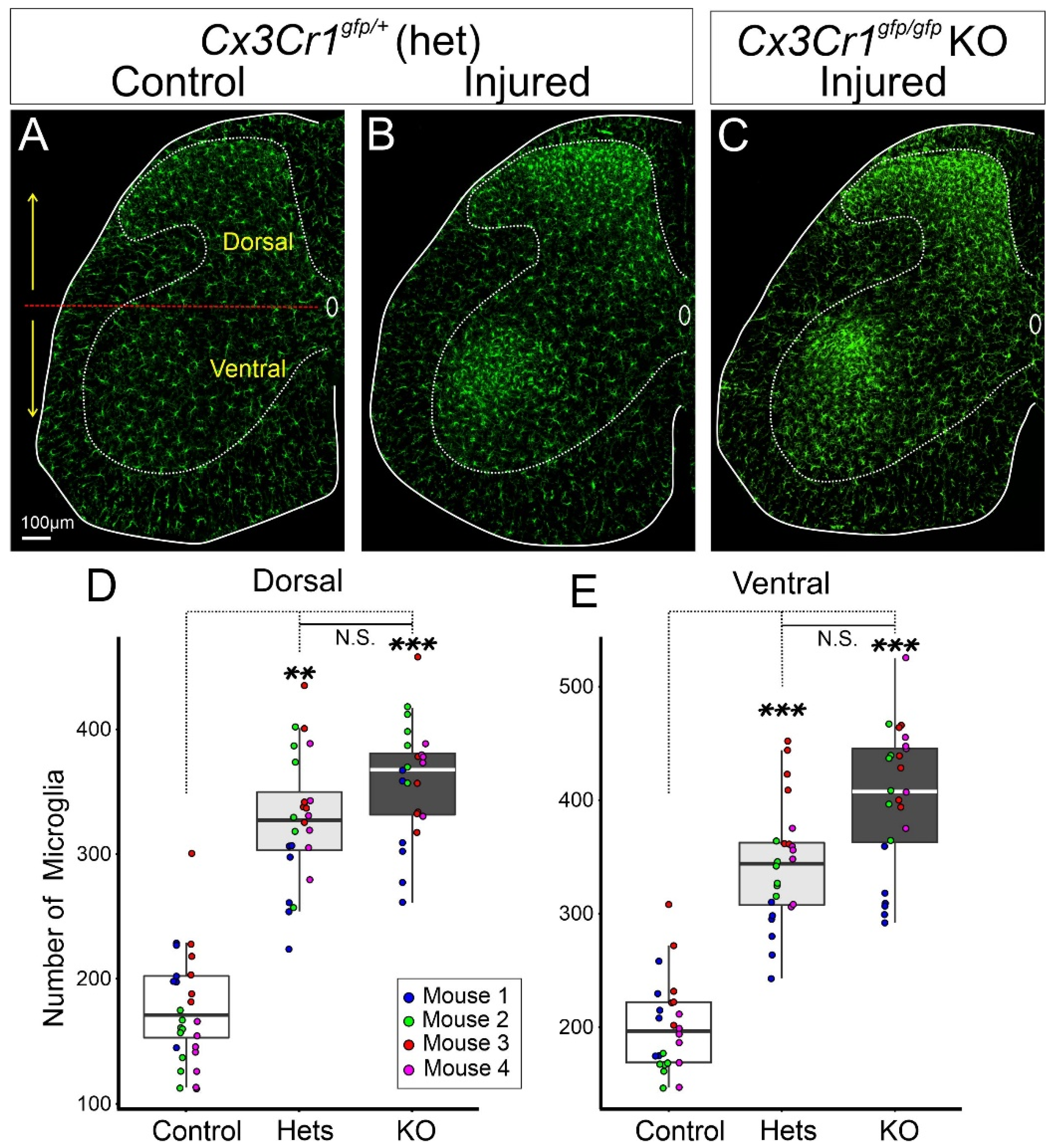

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pottorf, T.S.; Rotterman, T.M.; McCallum, W.M.; Haley-Johnson, Z.A.; Alvarez, F.J. The Role of Microglia in Neuroinflammation of the Spinal Cord after Peripheral Nerve Injury. Cells 2022, 11, 2083. https://doi.org/10.3390/cells11132083
Pottorf TS, Rotterman TM, McCallum WM, Haley-Johnson ZA, Alvarez FJ. The Role of Microglia in Neuroinflammation of the Spinal Cord after Peripheral Nerve Injury. Cells. 2022; 11(13):2083. https://doi.org/10.3390/cells11132083
Chicago/Turabian StylePottorf, Tana S., Travis M. Rotterman, William M. McCallum, Zoë A. Haley-Johnson, and Francisco J. Alvarez. 2022. "The Role of Microglia in Neuroinflammation of the Spinal Cord after Peripheral Nerve Injury" Cells 11, no. 13: 2083. https://doi.org/10.3390/cells11132083
APA StylePottorf, T. S., Rotterman, T. M., McCallum, W. M., Haley-Johnson, Z. A., & Alvarez, F. J. (2022). The Role of Microglia in Neuroinflammation of the Spinal Cord after Peripheral Nerve Injury. Cells, 11(13), 2083. https://doi.org/10.3390/cells11132083