
Inhibition of DNA Repair by Inappropriate Activation of ATM, PARP, and DNA-PK with the Drug Agonist AsiDNA

 , and
, and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines and Treatments
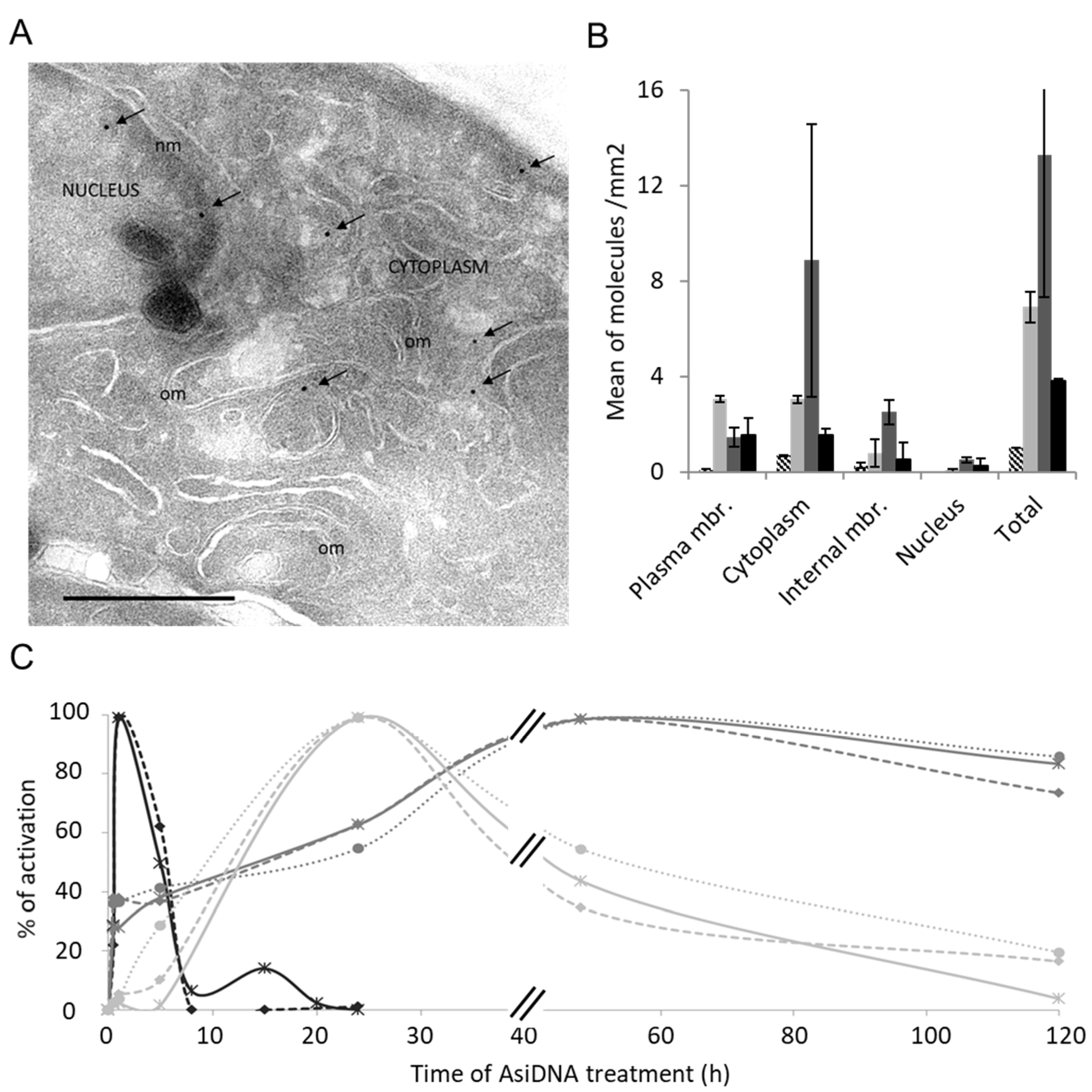
2.2. Electronic Microscopy
2.3. Antibodies, Irradiation-Induced DNA Damage, and Immunostaining
2.4. Inducing Photodamage with Laser
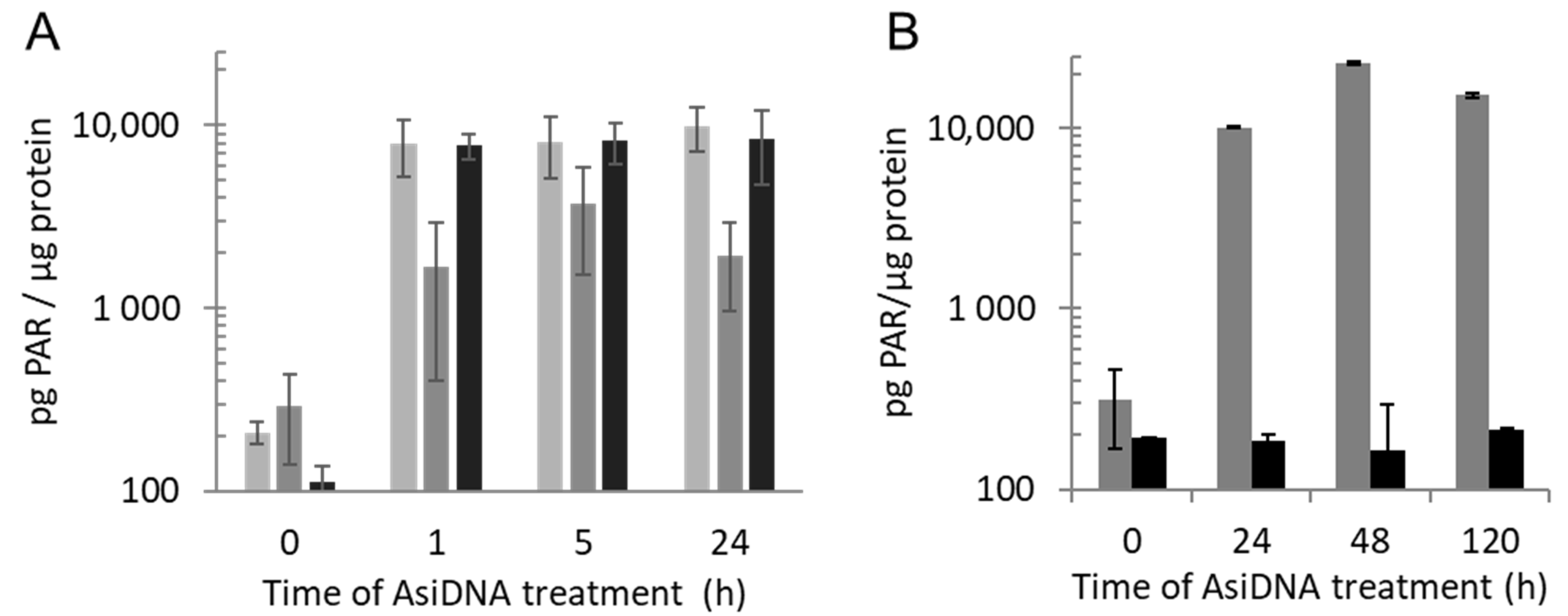
2.5. ELISA Assay for PARylation
3. Results and Discussion
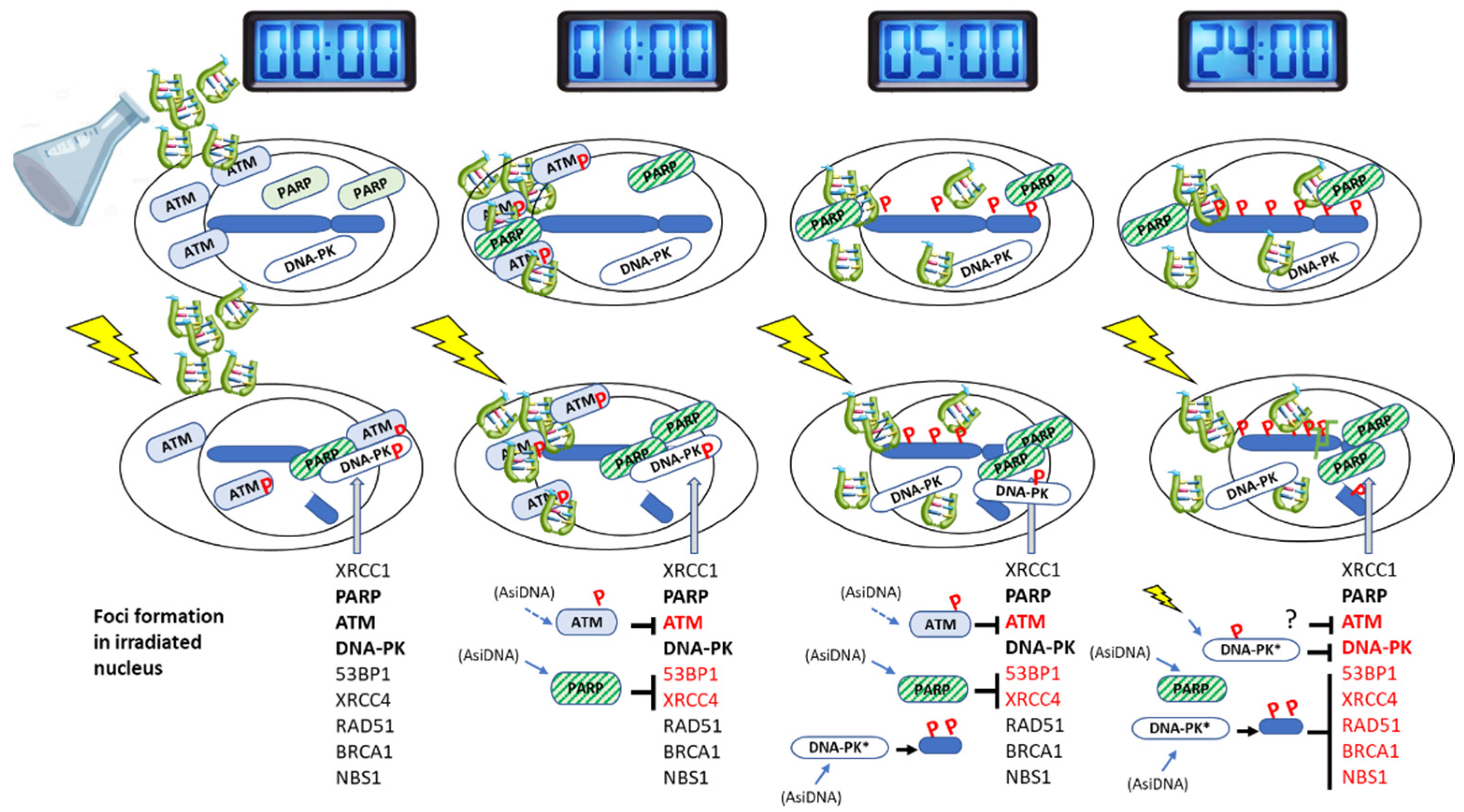
3.1. Small DNA Fragments AsiDNA Activate DNA-PK, PARP, and ATM with Different Kinetics
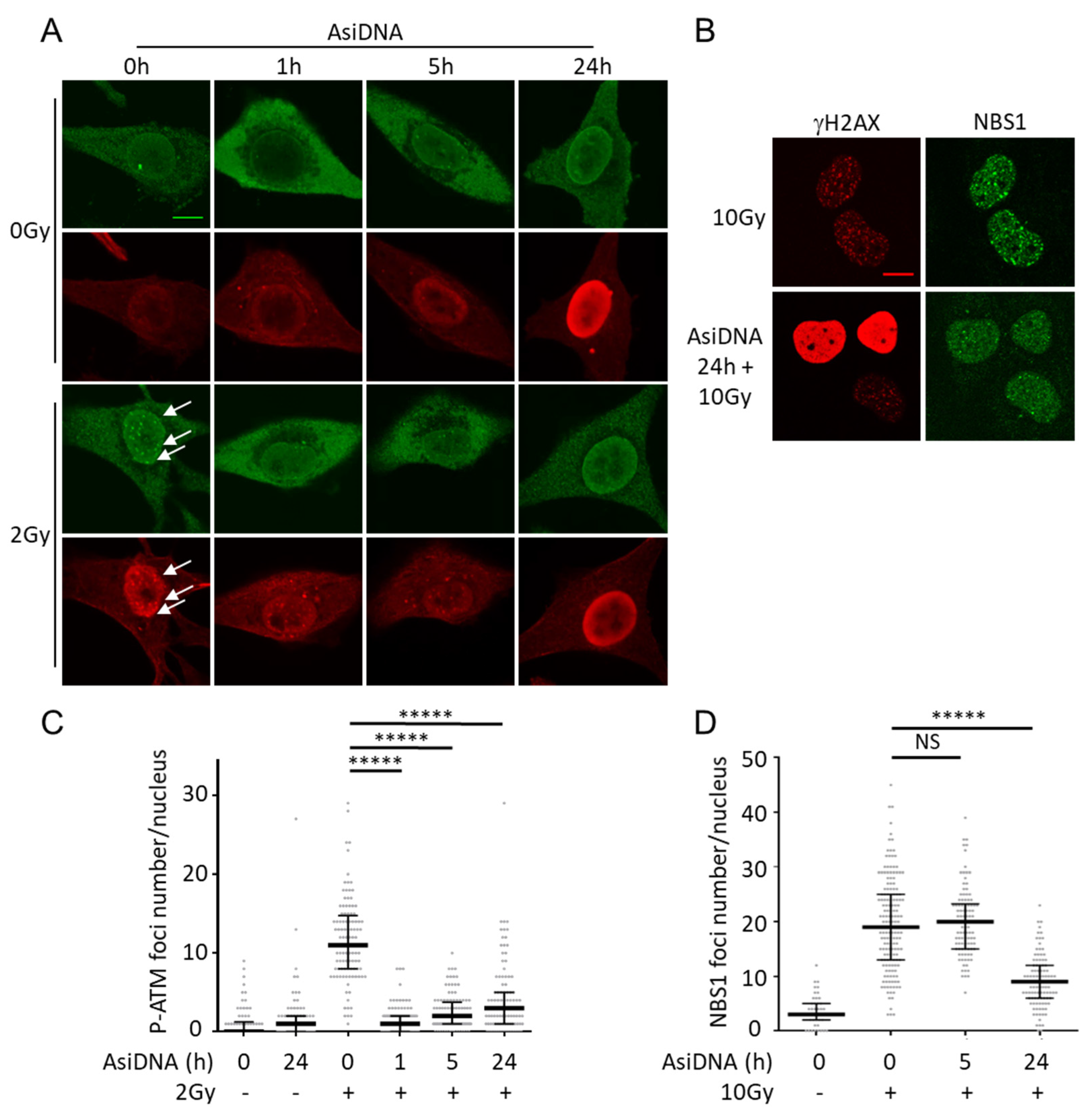
3.2. ATM Activation and Trapping in the Cytoplasm
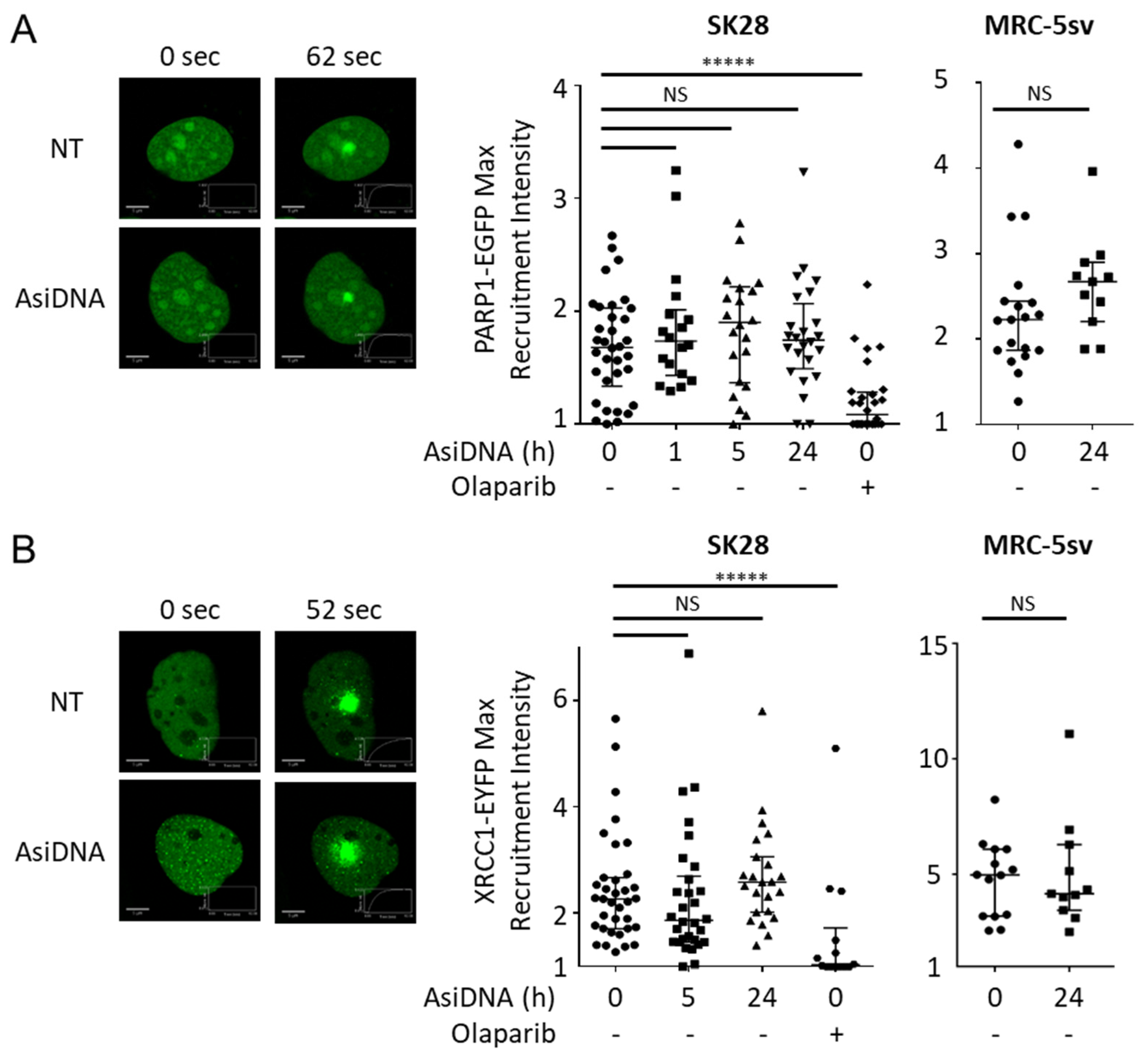
3.3. PARP Activation Properties
3.4. DNA-PK Activation Properties
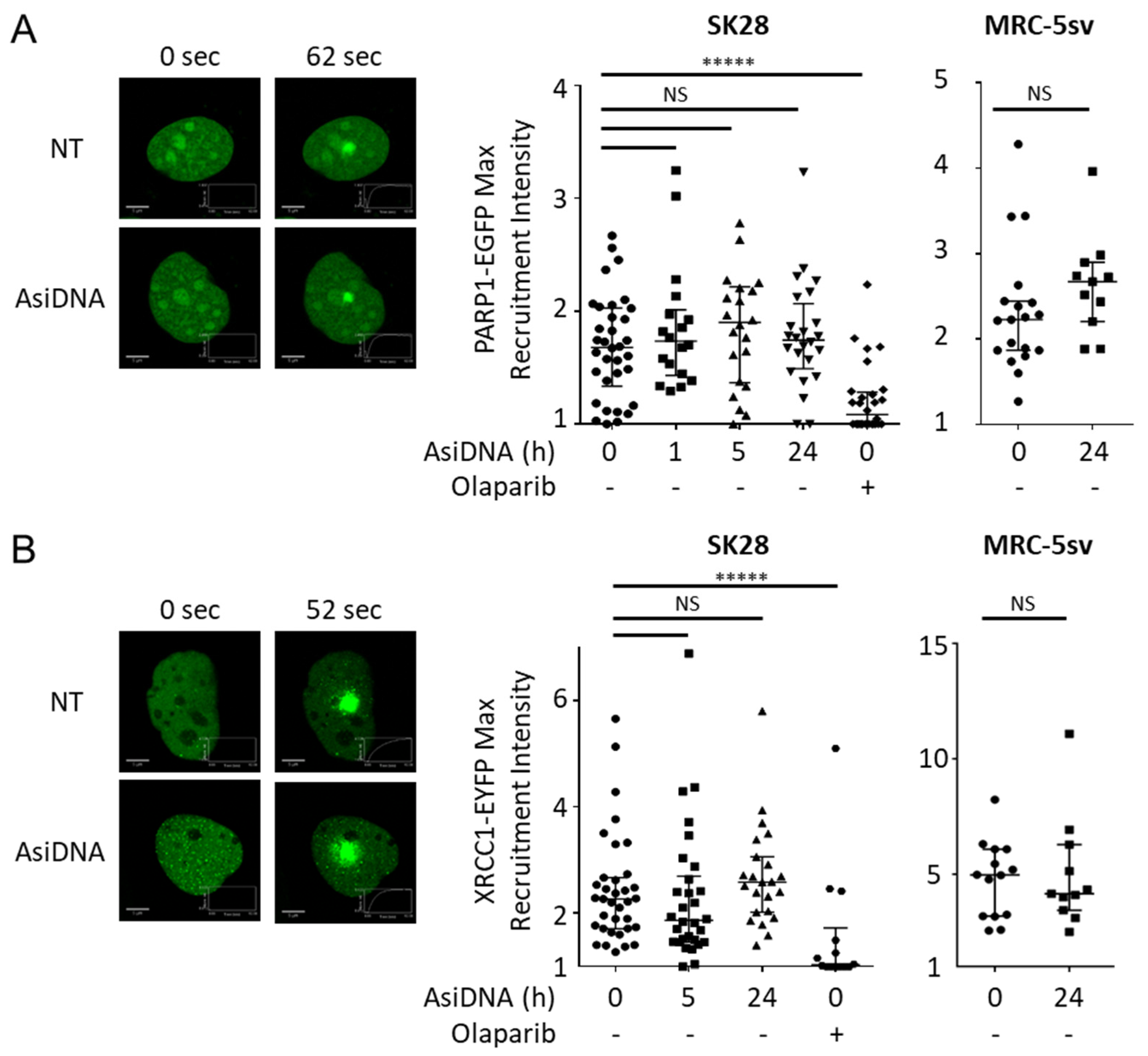
3.5. Consequences of the Activation of AsiDNA-Dependent Signaling on the Recruitment of Repair Proteins
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hoeijmakers, J.H. Genome maintenance mechanisms for preventing cancer. Nature 2001, 411, 366–374. [Google Scholar] [CrossRef]
- Ciccia, A.; Elledge, S.J. The DNA damage response: Making it safe to play with knives. Mol. Cell 2010, 40, 179–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shrivastav, M.; De Haro, L.P.; Nickoloff, J.A. Regulation of DNA double-strand break repair pathway choice. Cell Res. 2008, 18, 134–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saha, J.; Wang, S.Y.; Davis, A.J. Examining DNA Double-Strand Break Repair in a Cell Cycle-Dependent Manner. Methods Enzymol. 2017, 591, 97–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brandsma, I.; Gent, D.C. Pathway choice in DNA double strand break repair: Observations of a balancing act. Genome Integr. 2012, 3, 9. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Q.; Barboule, N.; Frit, P.; Gomez, D.; Bombarde, O.; Couderc, B.; Ren, G.S.; Salles, B.; Calsou, P. Ku counteracts mobilization of PARP1 and MRN in chromatin damaged with DNA double-strand breaks. Nucleic Acids Res. 2011, 39, 9605–9619. [Google Scholar] [CrossRef] [Green Version]
- Shibata, A.; Moiani, D.; Arvai, A.S.; Perry, J.; Harding, S.M.; Genois, M.M.; Maity, R.; van Rossum-Fikkert, S.; Kertokalio, A.; Romoli, F.; et al. DNA double-strand break repair pathway choice is directed by distinct MRE11 nuclease activities. Mol. Cell 2014, 53, 7–18. [Google Scholar] [CrossRef] [Green Version]
- Haince, J.F.; McDonald, D.; Rodrigue, A.; Dery, U.; Masson, J.Y.; Hendzel, M.J.; Poirier, G.G. PARP1-dependent kinetics of recruitment of MRE11 and NBS1 proteins to multiple DNA damage sites. J. Biol. Chem. 2008, 283, 1197–1208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shiloh, Y. ATM and related protein kinases: Safeguarding genome integrity. Nat. Rev. Cancer 2003, 3, 155–168. [Google Scholar] [CrossRef]
- Wang, M.; Wu, W.; Wu, W.; Rosidi, B.; Zhang, L.; Wang, H.; Iliakis, G. PARP-1 and Ku compete for repair of DNA double strand breaks by distinct NHEJ pathways. Nucleic Acids Res. 2006, 34, 6170–6182. [Google Scholar] [CrossRef]
- Yang, G.; Liu, C.; Chen, S.H.; Kassab, M.A.; Hoff, J.D.; Walter, N.G.; Yu, X. Super-resolution imaging identifies PARP1 and the Ku complex acting as DNA double-strand break sensors. Nucleic Acids Res. 2018, 46, 3446–3457. [Google Scholar] [CrossRef] [PubMed]
- Stiff, T.; O’Driscoll, M.; Rief, N.; Iwabuchi, K.; Lobrich, M.; Jeggo, P.A. ATM and DNA-PK function redundantly to phosphorylate H2AX after exposure to ionizing radiation. Cancer Res. 2004, 64, 2390–2396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Wang, M.; Wang, H.; Bocker, W.; Iliakis, G. Complex H2AX phosphorylation patterns by multiple kinases including ATM and DNA-PK in human cells exposed to ionizing radiation and treated with kinase inhibitors. J. Cell Physiol. 2005, 202, 492–502. [Google Scholar] [CrossRef] [PubMed]
- Quanz, M.; Berthault, N.; Roulin, C.; Roy, M.; Herbette, A.; Agrario, C.; Alberti, C.; Josserand, V.; Coll, J.L.; Sastre-Garau, X.; et al. Small-molecule drugs mimicking DNA damage: A new strategy for sensitizing tumors to radiotherapy. Clin. Cancer Res. 2009, 15, 1308–1316. [Google Scholar] [CrossRef] [Green Version]
- Croset, A.; Cordelieres, F.P.; Berthault, N.; Buhler, C.; Sun, J.S.; Quanz, M.; Dutreix, M. Inhibition of DNA damage repair by artificial activation of PARP with siDNA. Nucleic Acids Res. 2013, 41, 7344–7355. [Google Scholar] [CrossRef] [Green Version]
- Quanz, M.; Chassoux, D.; Berthault, N.; Agrario, C.; Sun, J.S.; Dutreix, M. Hyperactivation of DNA-PK by double-strand break mimicking molecules disorganizes DNA damage response. PLoS ONE 2009, 4, e6298. [Google Scholar] [CrossRef] [Green Version]
- Girard, P.M.; Pozzebon, M.; Delacote, F.; Douki, T.; Smirnova, V.; Sage, E. Inhibition of S-phase progression triggered by UVA-induced ROS does not require a functional DNA damage checkpoint response in mammalian cells. DNA Repair 2008, 7, 1500–1516. [Google Scholar] [CrossRef]
- Godon, C.; Cordelieres, F.P.; Biard, D.; Giocanti, N.; Megnin-Chanet, F.; Hall, J.; Favaudon, V. PARP inhibition versus PARP-1 silencing: Different outcomes in terms of single-strand break repair and radiation susceptibility. Nucleic Acids Res. 2008, 36, 4454–4464. [Google Scholar] [CrossRef] [Green Version]
- Fan, J.; Otterlei, M.; Wong, H.K.; Tomkinson, A.E.; Wilson, D.M., 3rd. XRCC1 co-localizes and physically interacts with PCNA. Nucleic Acids Res. 2004, 32, 2193–2201. [Google Scholar] [CrossRef]
- Dantzer, F.; Ame, J.C.; Schreiber, V.; Nakamura, J.; Menissier-de Murcia, J.; de Murcia, G. Poly(ADP-ribose) polymerase-1 activation during DNA damage and repair. Methods Enzymol. 2006, 409, 493–510. [Google Scholar] [CrossRef]
- Britton, S.; Coates, J.; Jackson, S.P. A new method for high-resolution imaging of Ku foci to decipher mechanisms of DNA double-strand break repair. J. Cell Biol. 2013, 202, 579–595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, K.; Bose, P.; Leong-Quong, R.Y.; Fujita, D.J.; Riabowol, K. REAP: A two minute cell fractionation method. BMC Res. Notes 2010, 3, 294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quanz, M.; Herbette, A.; Sayarath, M.; de Koning, L.; Dubois, T.; Sun, J.S.; Dutreix, M. Heat shock protein 90alpha (Hsp90alpha) is phosphorylated in response to DNA damage and accumulates in repair foci. J. Biol. Chem. 2012, 287, 8803–8815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- So, S.; Davis, A.J.; Chen, D.J. Autophosphorylation at serine 1981 stabilizes ATM at DNA damage sites. J. Cell Biol. 2009, 187, 977–990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindahl, T.; Satoh, M.S.; Poirier, G.G.; Klungland, A. Post-translational modification of poly(ADP-ribose) polymerase induced by DNA strand breaks. Trends Biochem. Sci. 1995, 20, 405–411. [Google Scholar] [CrossRef]
- Berthel, E.; Foray, N.; Ferlazzo, M.L. The Nucleoshuttling of the ATM Protein: A Unified Model to Describe the Individual Response to High- and Low-Dose of Radiation? Cancers 2019, 11, 905. [Google Scholar] [CrossRef] [Green Version]
- Uziel, T.; Lerenthal, Y.; Moyal, L.; Andegeko, Y.; Mittelman, L.; Shiloh, Y. Requirement of the MRN complex for ATM activation by DNA damage. EMBO J. 2003, 22, 5612–5621. [Google Scholar] [CrossRef] [Green Version]
- Jang, E.R.; Choi, J.D.; Jeong, G.; Lee, J.S. Phosphorylation of p300 by ATM controls the stability of NBS1. Biochem. Biophys. Res. Commun. 2010, 397, 637–643. [Google Scholar] [CrossRef]
- Masson, M.; Niedergang, C.; Schreiber, V.; Muller, S.; Menissier-de Murcia, J.; de Murcia, G. XRCC1 is specifically associated with poly(ADP-ribose) polymerase and negatively regulates its activity following DNA damage. Mol. Cell Biol. 1998, 18, 3563–3571. [Google Scholar] [CrossRef] [Green Version]
- El-Khamisy, S.F.; Masutani, M.; Suzuki, H.; Caldecott, K.W. A requirement for PARP-1 for the assembly or stability of XRCC1 nuclear foci at sites of oxidative DNA damage. Nucleic Acids Res. 2003, 31, 5526–5533. [Google Scholar] [CrossRef] [Green Version]
- Fell, V.L.; Schild-Poulter, C. The Ku heterodimer: Function in DNA repair and beyond. Mutat. Res. Rev. Mutat. Res. 2015, 763, 15–29. [Google Scholar] [CrossRef] [PubMed]
- Yue, X.; Bai, C.; Xie, D.; Ma, T.; Zhou, P.K. DNA-PKcs: A Multi-Faceted Player in DNA Damage Response. Front. Genet. 2020, 11, 607428. [Google Scholar] [CrossRef] [PubMed]
- Kurosawa, A. Autophosphorylation and Self-Activation of DNA-Dependent Protein Kinase. Genes 2021, 12, 1091. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Lees-Miller, J.P.; He, Y.; Lees-Miller, S.P. Structural insights into the role of DNA-PK as a master regulator in NHEJ. Genome Instab. Dis. 2021, 2, 195–210. [Google Scholar] [CrossRef] [PubMed]
- Berthault, N.; Maury, B.; Agrario, C.; Herbette, A.; Sun, J.S.; Peyrieras, N.; Dutreix, M. Comparison of distribution and activity of nanoparticles with short interfering DNA (Dbait) in various living systems. Cancer Gene Ther. 2011, 18, 695–706. [Google Scholar] [CrossRef] [Green Version]
- Subecz, C.; Sun, J.S.; Roger, L. Effect of DNA repair inhibitor AsiDNA on the incidence of telomere fusion in crisis. Hum. Mol. Genet. 2021, 30, 172–181. [Google Scholar] [CrossRef]
- Luijsterburg, M.S.; de Krijger, I.; Wiegant, W.W.; Shah, R.G.; Smeenk, G.; de Groot, A.J.L.; Pines, A.; Vertegaal, A.C.O.; Jacobs, J.J.L.; Shah, G.M.; et al. PARP1 Links CHD2-Mediated Chromatin Expansion and H3.3 Deposition to DNA Repair by Non-homologous End-Joining. Mol. Cell 2016, 61, 547–562. [Google Scholar] [CrossRef] [Green Version]
- Devun, F.; Bousquet, G.; Biau, J.; Herbette, A.; Roulin, C.; Berger, F.; Sun, J.S.; Robine, S.; Dutreix, M. Preclinical study of the DNA repair inhibitor Dbait in combination with chemotherapy in colorectal cancer. J. Gastroenterol. 2012, 47, 266–275. [Google Scholar] [CrossRef]
- Biau, J.; Devun, F.; Jdey, W.; Kotula, E.; Quanz, M.; Chautard, E.; Sayarath, M.; Sun, J.S.; Verrelle, P.; Dutreix, M. A preclinical study combining the DNA repair inhibitor Dbait with radiotherapy for the treatment of melanoma. Neoplasia 2014, 16, 835–844. [Google Scholar] [CrossRef] [Green Version]
- Herath, N.I.; Devun, F.; Lienafa, M.C.; Herbette, A.; Denys, A.; Sun, J.S.; Dutreix, M. The DNA Repair Inhibitor DT01 as a Novel Therapeutic Strategy for Chemosensitization of Colorectal Liver Metastasis. Mol. Cancer Ther. 2016, 15, 15–22. [Google Scholar] [CrossRef] [Green Version]
- Herath, N.I.; Berthault, N.; Thierry, S.; Jdey, W.; Lienafa, M.C.; Bono, F.; Noguiez-Hellin, P.; Sun, J.S.; Dutreix, M. Preclinical Studies Comparing Efficacy and Toxicity of DNA Repair Inhibitors, Olaparib, and AsiDNA, in the Treatment of Carboplatin-Resistant Tumors. Front. Oncol. 2019, 9, 1097. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| AsiDNA treatment 1 h | AsiDNA treatment 5 h | AsiDNA treatment 24 h | ||||||||||||
| Enzyme response | in DDR proficient cells | in cells inactivated for | in DDR proficient cells | in cells inactivated for | in DDR proficient cells | in cells inactivated for | ||||||||
| ATM | DNA-PK | PARP1 | ATM | DNA-PK | PARP1 | ATM | DNA-PK | PARP1 | ||||||
| No irradiation | ATM | Autophosphorylation | Yes | Irr | Yes | Yes | Decrease | Irr | Yes | Yes | No | Irr | Irr | Irr |
| DNA-PK | Pan nuclear phosphorylation of H2AX and HSP90 | No | Irr | Irr | Irr | No | Irr | Irr | Irr | Yes | Yes | No | Yes | |
| PARP1 | PARylation activity | Yes | Yes | Yes | No | Yes | Yes | Yes | No | Yes | Yes | Yes | No | |
| Gamma irradiation | P-ATM | Inhibition of nuclear foci | Yes | Irr | - (1) | Yes | Yes | Irr | Yes | Yes | Yes | Irr | Yes | - (1) |
| NBS1 | Inhibition of nuclear foci | - (1) | Irr | Irr | Irr | No | Irr | Irr | Irr | Yes | - (1) | - (1) | - (1) | |
| BRCA1 | Inhibition of nuclear foci (3) | - (1) | Irr | Irr | Irr | No | Irr | Irr | Irr | Yes | Yes | No | Yes | |
| Rad51 | Inhibition of nuclear foci (3) | - (1) | Irr | Irr | Irr | No | Irr | Irr | Irr | Yes | Yes | No | - (1) | |
| P-DNA-PK | Inhibition of nuclear foci | - (1) | Irr | Irr | Irr | No | Irr | Irr | Irr | Yes | - (1) | Irr | - (1) | |
| γ-H2AX | Pan nuclear phosphorylation | No | Irr | Irr | Irr | No | Irr | Irr | Irr | Yes | Yes | No | Yes | |
| Inhibition of nuclear foci? (4) | No | Irr | Irr | Irr | No | Irr | Irr | Irr | No | Irr | Irr | Irr | ||
| 53BP1 | Inhibition of nuclear foci | Yes | Yes | Yes | No | Yes | Yes | Yes | No | Yes | Yes | No | Yes | |
| Laser induced damage | XRCC4 | Inhibition of recruitment at damage | Yes | Yes | - (2) | No | Yes | Yes | - (2) | No | Yes | Yes | - (2) | - (1) |
| PARP1 | Inhibition of recruitment at damage | No | Irr | Irr | Irr | No | Irr | Irr | Irr | No | Irr | Irr | Irr | |
| XRCC1 | Inhibition of recruitment at damage | No | Irr | Irr | Irr | No | Irr | Irr | Irr | No | Irr | Irr | Irr | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Berthault, N.; Bergam, P.; Pereira, F.; Girard, P.-M.; Dutreix, M. Inhibition of DNA Repair by Inappropriate Activation of ATM, PARP, and DNA-PK with the Drug Agonist AsiDNA. Cells 2022, 11, 2149. https://doi.org/10.3390/cells11142149
Berthault N, Bergam P, Pereira F, Girard P-M, Dutreix M. Inhibition of DNA Repair by Inappropriate Activation of ATM, PARP, and DNA-PK with the Drug Agonist AsiDNA. Cells. 2022; 11(14):2149. https://doi.org/10.3390/cells11142149
Chicago/Turabian StyleBerthault, Nathalie, Ptissam Bergam, Floriane Pereira, Pierre-Marie Girard, and Marie Dutreix. 2022. "Inhibition of DNA Repair by Inappropriate Activation of ATM, PARP, and DNA-PK with the Drug Agonist AsiDNA" Cells 11, no. 14: 2149. https://doi.org/10.3390/cells11142149
APA StyleBerthault, N., Bergam, P., Pereira, F., Girard, P. -M., & Dutreix, M. (2022). Inhibition of DNA Repair by Inappropriate Activation of ATM, PARP, and DNA-PK with the Drug Agonist AsiDNA. Cells, 11(14), 2149. https://doi.org/10.3390/cells11142149