
AKT1 Transcriptomic Landscape in Breast Cancer Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
3. Results
3.1. Expression AKTs in Breast Cancer Cells
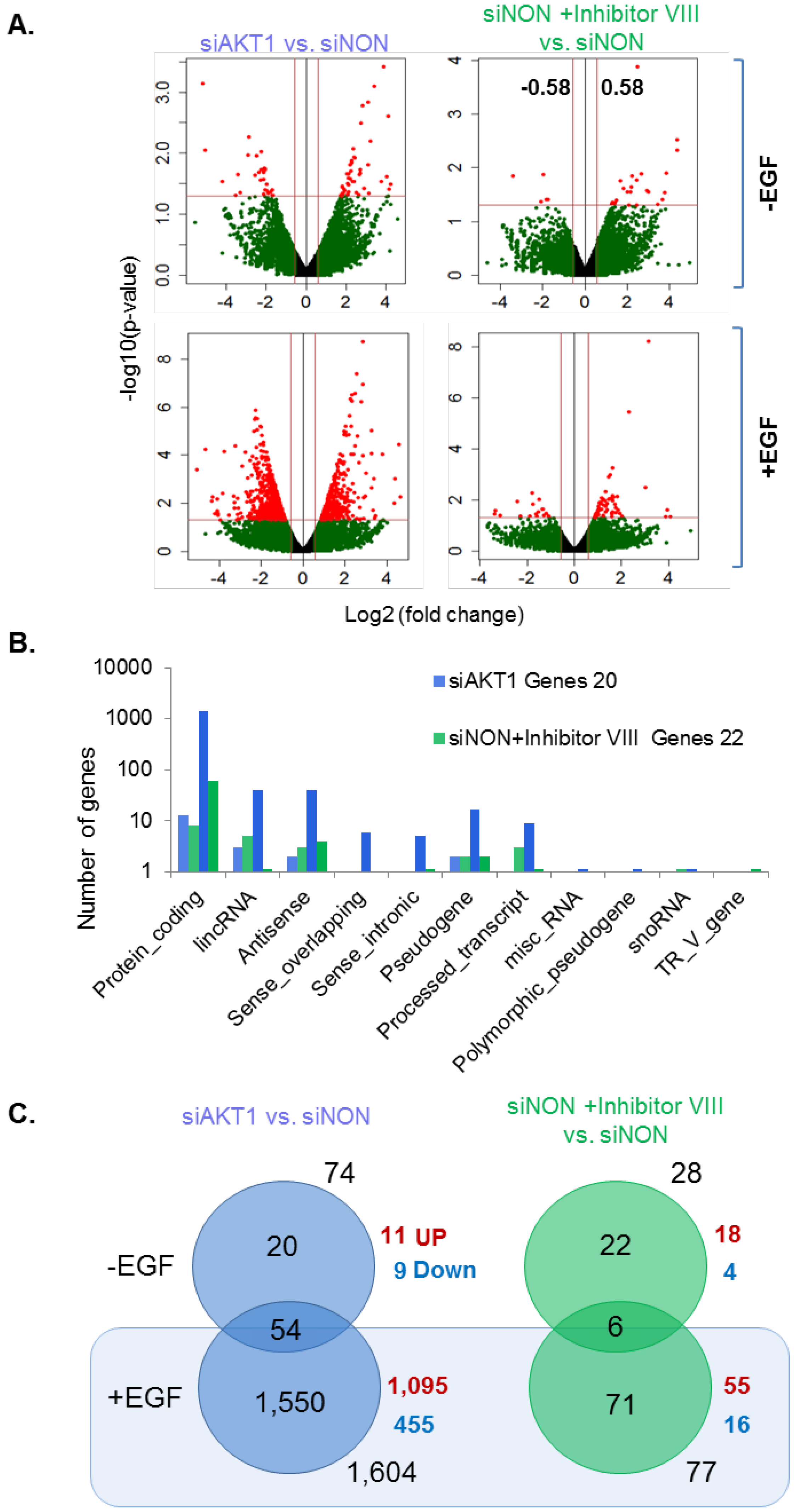
3.2. Analysis of AKT1 Transcriptome in Breast Cancer Cells
3.3. Influence of AKT1 on the Status of Growth Factor Induced Genes
3.4. EGF Modulation of AKT-Dependent Transcriptome
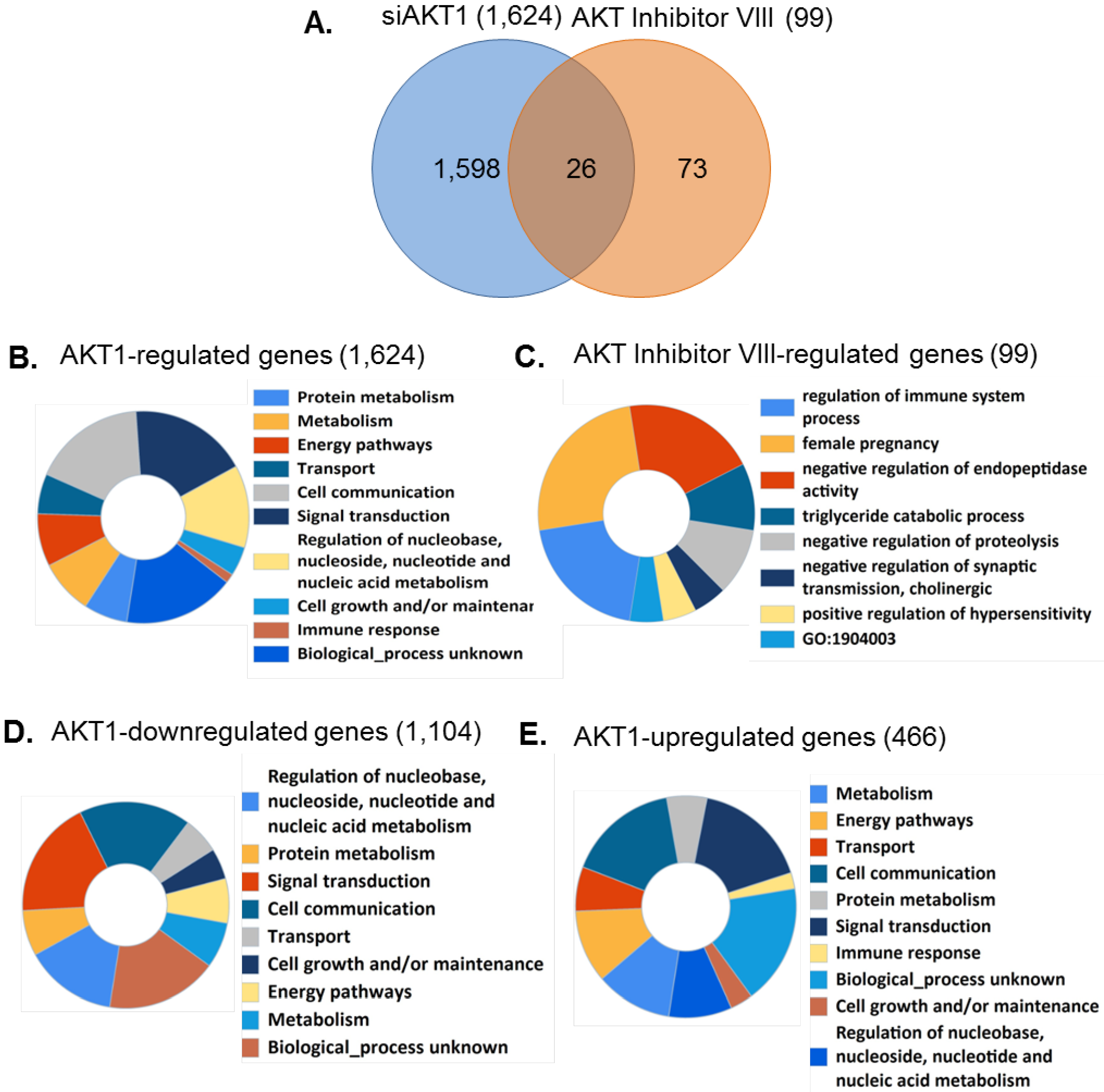
3.5. Identification of AKT1 Specific Regulatory Pathways
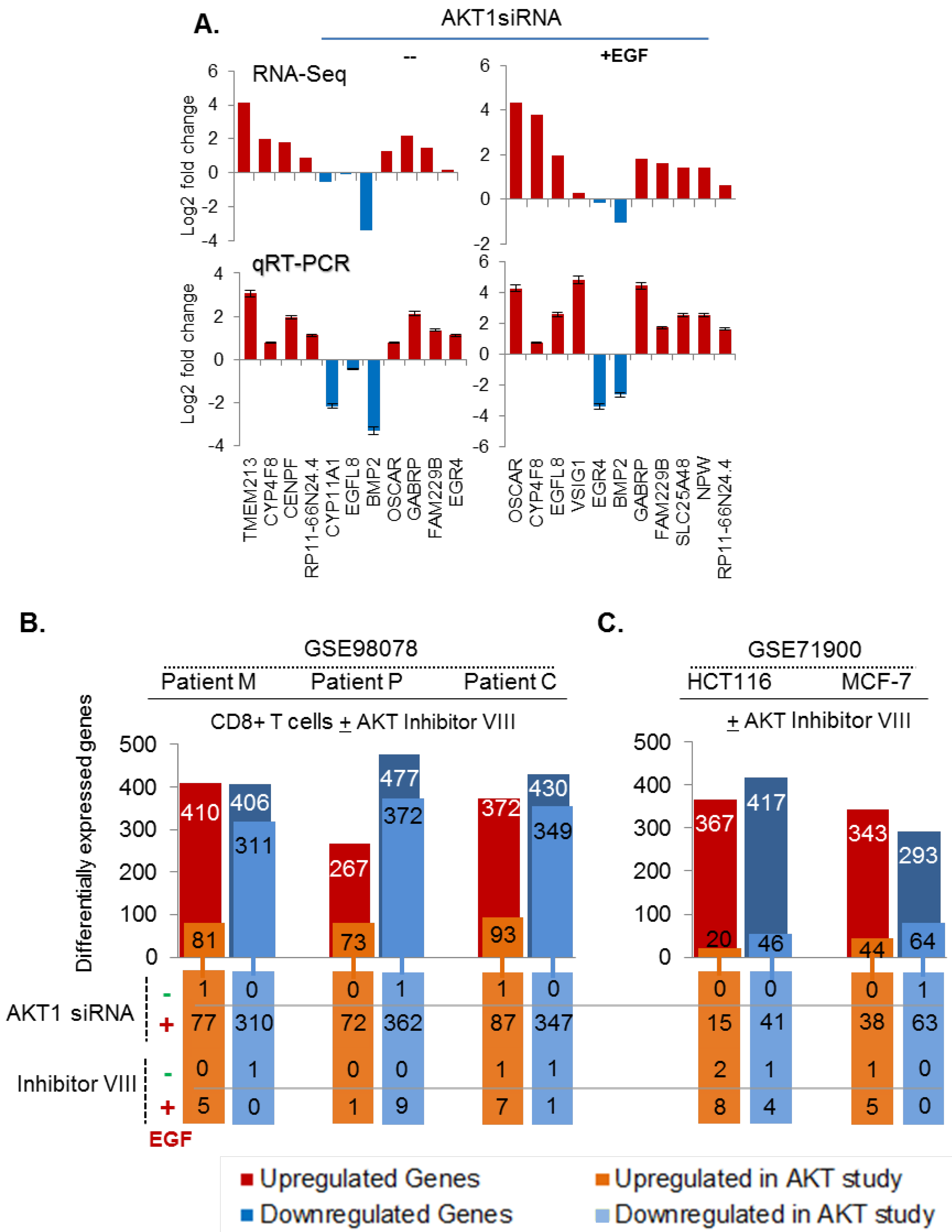
3.6. Validation of AKT1 Regulated Significant Genes
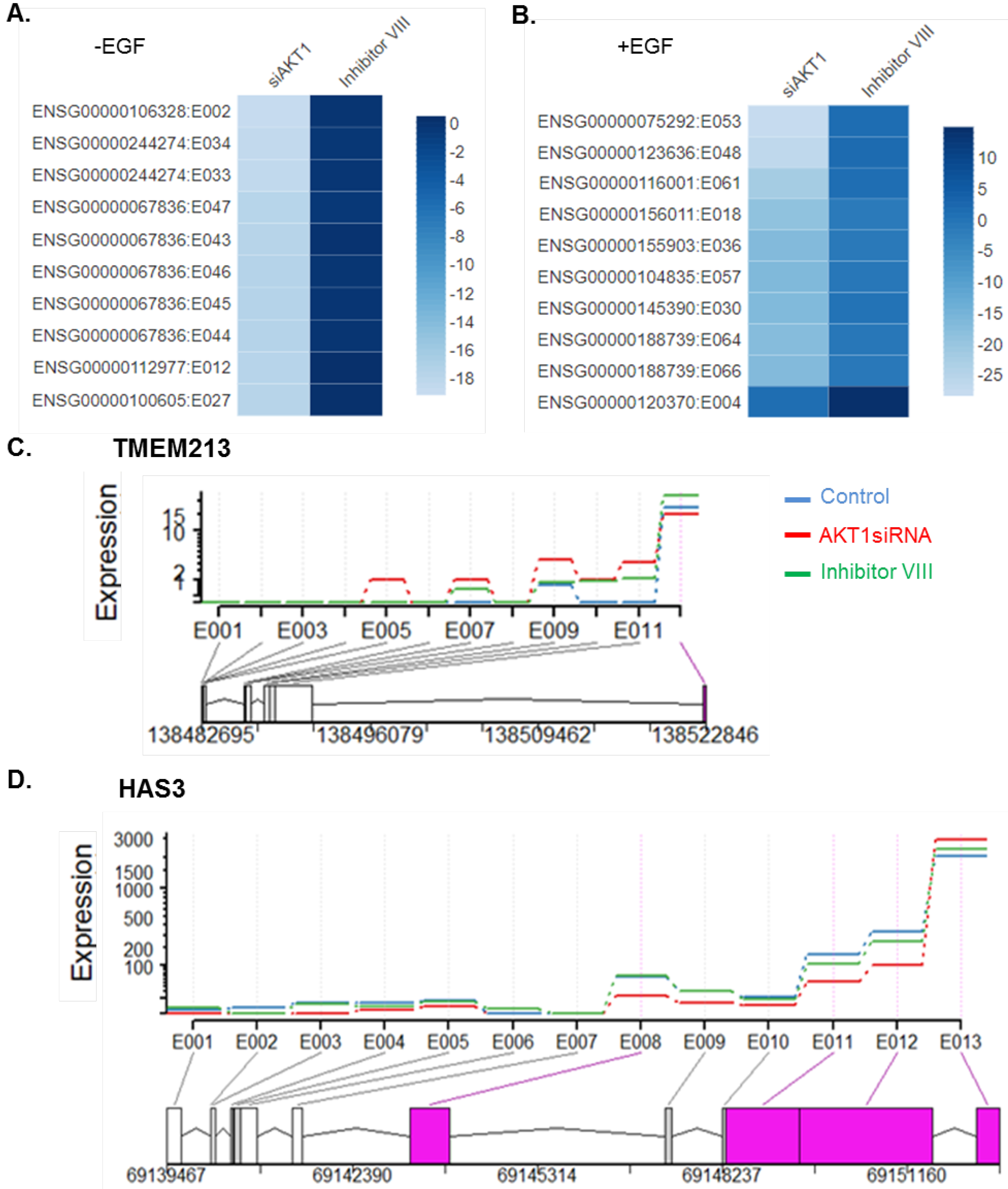
3.7. Influence of the Endogenous Status of AKT1 on Splice Variation
3.8. Top 10 Highly Dispersed Splicing Events between the AKT1 Knockdown
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- International Agency for Research on Cancer. Cancer Today. Available online: https://gco.iarc.fr/today/online-analysis-pie?v=2020&mode=cancer&mode_population=continents&population=900&populations=900&key=total&sex=2&cancer=39&type=0&statistic=5&prevalence=0&population_group=0&ages_group%5B%5D=0&ages_group%5B%5D=17&nb_items=7&group (accessed on 15 June 2021).
- Eroles, P.; Bosch, A.; Pérez-Fidalgo, J.A.; Lluch, A. Molecular Biology in Breast Cancer: Intrinsic Subtypes and Signaling Pathways. Cancer Treat. Rev. 2012, 38, 698–707. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. The Hallmarks of Cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [Green Version]
- Vadlamudi, R.K.; Kumar, R. P21-Activated Kinases in Human Cancer. Cancer Metastasis Rev. 2003, 22, 385–393. [Google Scholar] [CrossRef] [PubMed]
- Manavathi, B.; Acconcia, F.; Rayala, S.K.; Kumar, R. An inherent role of microtubule network in the action of nuclear receptor. Proc. Natl. Acad. Sci. USA 2006, 103, 15981–15986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nair, S.S.; Mishra, S.K.; Yang, Z.; Balasenthil, S.; Kumar, R.; Vadlamudi, R.K. Potential Role of a Novel Transcriptional Coactivator PELP1 in Histone H1 Displacement in Cancer Cells. Cancer Res. 2004, 64, 6416–6423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nair, S.S.; Kumar, R. Chronatin remodeling in cancer: A gateway to regulate gene transcription. Mol. Oncol. 2012, 6, 611–619. [Google Scholar] [CrossRef]
- Salony; Solé, X.; Alves, C.P.; Dey-Guha, I.; Ritsma, L.; Boukhali, M.; Lee, J.H.; Chowdhury, J.; Ross, K.N.; Haas, W.; et al. AKT Inhibition Promotes Nonautonomous Cancer Cell Survival. Mol. Cancer Ther. 2016, 15, 142–153. [Google Scholar] [CrossRef] [Green Version]
- Santi, S.A.; Douglas, A.C.; Lee, H. The Akt Isoforms, Their Unique Functions and Potential as Anticancer Therapeutic Targets. Biomol. Concepts 2010, 1, 389–401. [Google Scholar] [CrossRef]
- Burgering, B.M.; Coffer, P.J. Protein Kinase B (c-Akt) in Phosphatidylinositol-3-OH Kinase Signal Transduction. Nature 1995, 376, 599–602. [Google Scholar] [CrossRef]
- Okano, J.; Gaslightwala, I.; Birnbaum, M.J.; Rustgi, A.K.; Nakagawa, H. Akt/Protein Kinase B Isoforms Are Differentially Regulated by Epidermal Growth Factor Stimulation. J. Biol. Chem. 2000, 275, 30934–30942. [Google Scholar] [CrossRef] [Green Version]
- Hinz, N.; Jücker, M. Distinct Functions of AKT Isoforms in Breast Cancer: A Comprehensive Review. Cell Commun. Signal. 2019, 17, 154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carpten, J.D.; Faber, A.L.; Horn, C.; Donoho, G.P.; Briggs, S.L.; Robbins, C.M.; Hostetter, G.; Boguslawski, S.; Moses, T.Y.; Savage, S.; et al. A Transforming Mutation in the Pleckstrin Homology Domain of AKT1 in Cancer. Nature 2007, 448, 439–444. [Google Scholar] [CrossRef]
- Landgraf, K.E.; Pilling, C.; Falke, J.J. Molecular Mechanism of an Oncogenic Mutation That Alters Membrane Targeting: Glu17Lys Modifies the PIP Lipid Specificity of the AKT1 PH Domain. Biochemistry 2008, 47, 12260–12269. [Google Scholar] [CrossRef] [Green Version]
- Brown, J.S.; Banerji, U. Maximising the Potential of AKT Inhibitors as Anti-Cancer Treatments. Pharmacol. Ther. 2017, 172, 101–115. [Google Scholar] [CrossRef] [PubMed]
- Polytarchou, C.; Iliopoulos, D.; Hatziapostolou, M.; Kottakis, F.; Maroulakou, I.; Struhl, K.; Tsichlis, P.N. Akt2 Regulates All Akt Isoforms and Promotes Resistance to Hypoxia through Induction of MiR-21 upon Oxygen Deprivation. Cancer Res. 2011, 71, 4720–4731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watson, K.L.; Moorehead, R.A. Loss of Akt1 or Akt2 Delays Mammary Tumor Onset and Suppresses Tumor Growth Rate in MTB-IGFIR Transgenic Mice. BMC Cancer 2013, 13, 375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gargini, R.; Cerliani, J.P.; Escoll, M.; Antón, I.M.; Wandosell, F. Cancer Stem Cell-like Phenotype and Survival Are Coordinately Regulated by Akt/FoxO/Bim Pathway. Stem Cells 2015, 33, 646–660. [Google Scholar] [CrossRef]
- Hutchinson, J.; Jin, J.; Cardiff, R.D.; Woodgett, J.R.; Muller, W.J. Activation of Akt (Protein Kinase B) in Mammary Epithelium Provides a Critical Cell Survival Signal Required for Tumor Progression. Mol. Cell. Biol. 2001, 21, 2203–2212. [Google Scholar] [CrossRef] [Green Version]
- Dillon, R.L.; Marcotte, R.; Hennessy, B.T.; Woodgett, J.R.; Mills, G.B.; Muller, W.J. Akt1 and Akt2 Play Distinct Roles in the Initiation and Metastatic Phases of Mammary Tumor Progression. Cancer Res. 2009, 69, 5057–5064. [Google Scholar] [CrossRef] [Green Version]
- Riggio, M.; Polo, M.L.; Blaustein, M.; Colman-Lerner, A.; Lüthy, I.; Lanari, C.; Novaro, V. PI3K/AKT Pathway Regulates Phosphorylation of Steroid Receptors, Hormone Independence and Tumor Differentiation in Breast Cancer. Carcinogenesis 2012, 33, 509–518. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Radisky, D.C.; Nelson, C.M.; Zhang, H.; Fata, J.E.; Roth, R.A.; Bissell, M.J. Mechanism of Akt1 Inhibition of Breast Cancer Cell Invasion Reveals a Protumorigenic Role for TSC2. Proc. Natl. Acad. Sci. USA 2006, 103, 4134–4139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.S.; Xu, P.Z.; Gottlob, K.; Chen, M.L.; Sokol, K.; Shiyanova, T.; Roninson, I.; Weng, W.; Suzuki, R.; Tobe, K.; et al. Growth Retardation and Increased Apoptosis in Mice with Homozygous Disruption of the Akt1 Gene. Genes Dev. 2001, 15, 2203–2208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garofalo, R.S.; Orena, S.J.; Rafidi, K.; Torchia, A.J.; Stock, J.L.; Hildebrandt, A.L.; Coskran, T.; Black, S.C.; Brees, D.J.; Wicks, J.R.; et al. Severe Diabetes, Age-Dependent Loss of Adipose Tissue, and Mild Growth Deficiency in Mice Lacking Akt2/PKB Beta. J. Clin. Investig. 2003, 112, 197–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, X.-D.; Xu, P.-Z.; Chen, M.-L.; Hahn-Windgassen, A.; Skeen, J.; Jacobs, J.; Sundararajan, D.; Chen, W.S.; Crawford, S.E.; Coleman, K.G.; et al. Dwarfism, Impaired Skin Development, Skeletal Muscle Atrophy, Delayed Bone Development, and Impeded Adipogenesis in Mice Lacking Akt1 and Akt2. Genes Dev. 2003, 17, 1352–1365. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.-Z.; Tschopp, O.; Di-Poï, N.; Bruder, E.; Baudry, A.; Dümmler, B.; Wahli, W.; Hemmings, B.A. Dosage-Dependent Effects of Akt1/Protein Kinase Balpha (PKBalpha) and Akt3/PKBgamma on Thymus, Skin, and Cardiovascular and Nervous System Development in Mice. Mol. Cell. Biol. 2005, 25, 10407–10418. [Google Scholar] [CrossRef] [Green Version]
- Dummler, B.; Tschopp, O.; Hynx, D.; Yang, Z.-Z.; Dirnhofer, S.; Hemmings, B.A. Life with a Single Isoform of Akt: Mice Lacking Akt2 and Akt3 Are Viable but Display Impaired Glucose Homeostasis and Growth Deficiencies. Mol. Cell. Biol. 2006, 26, 8042–8051. [Google Scholar] [CrossRef] [Green Version]
- Ju, X.; Katiyar, S.; Wang, C.; Liu, M.; Jiao, X.; Li, S.; Zhou, J.; Turner, J.; Lisanti, M.P.; Russell, R.G.; et al. Akt1 Governs Breast Cancer Progression in Vivo. Proc. Natl. Acad. Sci. USA 2007, 104, 7438–7443. [Google Scholar] [CrossRef] [Green Version]
- Maroulakou, I.G.; Oemler, W.; Naber, S.P.; Tsichlis, P.N. Akt1 Ablation Inhibits, Whereas Akt2 Ablation Accelerates, the Development of Mammary Adenocarcinomas in Mouse Mammary Tumor Virus (MMTV)-ErbB2/Neu and MMTV-Polyoma Middle T Transgenic Mice. Cancer Res. 2007, 67, 167–177. [Google Scholar] [CrossRef] [Green Version]
- Bilodeau, M.T.; Balitza, A.E.; Hoffman, J.M.; Manley, P.J.; Barnett, S.F.; Defeo-Jones, D.; Haskell, K.; Jones, R.E.; Leander, K.; Robinson, R.G.; et al. Allosteric Inhibitors of Akt1 and Akt2: A Naphthyridinone with Efficacy in an A2780 Tumor Xenograft Model. Bioorg. Med. Chem. Lett. 2008, 18, 3178–3182. [Google Scholar] [CrossRef]
- Wang, J.; Wan, W.; Sun, R.; Liu, Y.; Sun, X.; Ma, D.; Zhang, N. Reduction of Akt2 Expression Inhibits Chemotaxis Signal Transduction in Human Breast Cancer Cells. Cell. Signal. 2008, 20, 1025–1034. [Google Scholar] [CrossRef]
- Grabinski, N.; Möllmann, K.; Milde-Langosch, K.; Müller, V.; Schumacher, U.; Brandt, B.; Pantel, K.; Jücker, M. AKT3 Regulates ErbB2, ErbB3 and Estrogen Receptor α Expression and Contributes to Endocrine Therapy Resistance of ErbB2+ Breast Tumor Cells from Balb-NeuT Mice. Cell. Signal. 2014, 26, 1021–1029. [Google Scholar] [CrossRef]
- Santi, S.A.; Lee, H. Ablation of Akt2 Induces Autophagy through Cell Cycle Arrest, the Downregulation of P70S6K, and the Deregulation of Mitochondria in MDA-MB231 Cells. PLoS ONE 2011, 6, e14614. [Google Scholar]
- Xia, X.; Li, X.; Li, F.; Wu, X.; Zhang, M.; Zhou, H.; Huang, N.; Yang, X.; Xiao, F.; Liu, D.; et al. A novel tumor suppressor protein encoded by circular AKT3 RNA inhibits glioblastoma tumorigenicity by competing with active phosphoinositide-dependent Kinase-1. Mol. Cancer 2019, 18, 131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barnett, S.F.; Defeo-Jones, D.; Fu, S.; Hancock, P.J.; Haskell, K.M.; Jones, R.E.; Kahana, J.A.; Kral, A.M.; Leander, K.; Lee, L.L.; et al. Identification and Characterization of Pleckstrin-Homology-Domain-Dependent and Isoenzyme-Specific Akt Inhibitors. Biochem. J. 2005, 385, 399–408. [Google Scholar] [CrossRef] [PubMed]
- Gills, J.J.; Dennis, P.A. Perifosine: Update on a Novel Akt Inhibitor. Curr. Oncol. Rep. 2009, 11, 102–110. [Google Scholar] [CrossRef] [PubMed]
- Nitulescu, G.M.; Margina, D.; Juzenas, P.; Peng, Q.; Olaru, O.T.; Saloustros, E.; Fenga, C.; Spandidos, D.A.; Libra, M.; Tsatsakis, A.M. Akt Inhibitors in Cancer Treatment: The Long Journey from Drug Discovery to Clinical Use (Review). Int. J. Oncol. 2016, 48, 869–885. [Google Scholar] [CrossRef] [Green Version]
- Bhat-Nakshatri, P.; Gao, H.; Sheng, L.; McGuire, P.C.; Xuei, X.; Wan, J.; Liu, Y.; Althouse, S.K.; Colter, A.; Sandusky, G.; et al. A Single-Cell Atlas of the Healthy Breast Tissues Reveals Clinically Relevant Clusters of Breast Epithelial Cells. Cell Rep. Med. 2021, 2, 100219. [Google Scholar] [CrossRef]
- Kim, D.; Pertea, G.; Trapnell, C.; Pimentel, H.; Kelley, R.; Salzberg, S.L. TopHat2: Accurate Alignment of Transcriptomes in the Presence of Insertions, Deletions and Gene Fusions. Genome Biol. 2013, 14, R36. [Google Scholar] [CrossRef] [Green Version]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and Memory-Efficient Alignment of Short DNA Sequences to the Human Genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef] [Green Version]
- Dodt, M.; Roehr, J.T.; Ahmed, R.; Dieterich, C. FLEXBAR-Flexible Barcode and Adapter Processing for Next-Generation Sequencing Platforms. Biology 2012, 1, 895–905. [Google Scholar] [CrossRef] [Green Version]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python Framework to Work with High-Throughput Sequencing Data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Huber, W. Differential Expression Analysis for Sequence Count Data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eswaran, J.; Cyanam, D.; Mudvari, P.; Reddy, S.D.N.; Pakala, S.B.; Nair, S.S.; Florea, L.; Fuqua, S.A.W.; Godbole, S.; Kumar, R. Transcriptomic Landscape of Breast Cancers through MRNA Sequencing. Sci. Rep. 2012, 2, 264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schafer, S.; Miao, K.; Benson, C.C.; Heinig, M.; Cook, S.A.; Hubner, N. Alternative Splicing Signatures in RNA-Seq Data: Percent Spliced in (PSI). Curr. Protoc. Hum. Genet. 2015, 87, 11.16.1–11.16.14. [Google Scholar] [CrossRef] [PubMed]
- Eswaran, J.; Horvath, A.; Godbole, S.; Reddy, S.D.; Mudvari, P.; Ohshiro, K.; Cyanam, D.; Nair, S.; Fuqua, S.A.W.; Polyak, K.; et al. RNA Sequencing of Cancer Reveals Novel Splicing Alterations. Sci. Rep. 2013, 3, 1689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anders, S.; Reyes, A.; Huber, W. Detecting Differential Usage of Exons from RNA-Seq Data. Genome Res. 2012, 22, 2008–2017. [Google Scholar] [CrossRef]
- Klebanoff, C.A.; Crompton, J.G.; Leonardi, A.J.; Yamamoto, T.N.; Chandran, S.S.; Eil, R.L.; Sukumar, M.; Vodnala, S.K.; Hu, J.; Ji, Y.; et al. Inhibition of AKT Signaling Uncouples T Cell Differentiation from Expansion for Receptor-Engineered Adoptive Immunotherapy. JCI Insight 2017, 2, e95103. [Google Scholar] [CrossRef] [Green Version]
- Pereira, B.; Chin, S.F.; Rueda, O.M.; Vollan, H.K.; Provenzano, E.; Bardwell, H.A.; Pugh, M.; Jones, L.; Russell, R.; Sammut, S.J.; et al. The Somatic Mutation Profiles of 2,433 Breast Cancers Refines Their Genomic and Transcriptomic Landscapes. Nat. Commun. 2016, 7, 11479. [Google Scholar] [CrossRef] [Green Version]
- Rueda, O.M.; Sammut, S.-J.; Seoane, J.A.; Chin, S.-F.; Caswell-Jin, J.L.; Callari, M.; Batra, R.; Pereira, B.; Bruna, A.; Ali, H.R.; et al. Dynamics of Breast-Cancer Relapse Reveal Late-Recurring ER-Positive Genomic Subgroups. Nature 2019, 567, 399–404. [Google Scholar] [CrossRef]
- Curtis, C.; Shah, S.P.; Chin, S.-F.; Turashvili, G.; Rueda, O.M.; Dunning, M.J.; Speed, D.; Lynch, A.G.; Samarajiwa, S.; Yuan, Y.; et al. The Genomic and Transcriptomic Architecture of 2,000 Breast Tumours Reveals Novel Subgroups. Nature 2012, 486, 346–352. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The CBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the CBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fonseka, P.; Pathan, M.; Chitti, S.V.; Kang, T.; Mathivanan, S. FunRich Enables Enrichment Analysis of OMICs Datasets. J. Mol. Biol. 2021, 433, 166747. [Google Scholar] [CrossRef] [PubMed]
- Franceschini, A.; Meier, R.; Casanova, A.; Kreibich, S.; Daga, N.; Andritschke, D.; Dilling, S.; Rämö, P.; Emmenlauer, M.; Kaufmann, A.; et al. Specific Inhibition of Diverse Pathogens in Human Cells by Synthetic MicroRNA-like Oligonucleotides Inferred from RNAi Screens. Proc. Natl. Acad. Sci. USA 2014, 111, 4548–4553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turner, K.M.; Sun, Y.; Ji, P.; Granberg, K.J.; Bernard, B.; Hu, L.; Cogdell, D.E.; Zhou, X.; Yli-Harja, O.; Nykter, M.; et al. Genomically Amplified Akt3 Activates DNA Repair Pathway and Promotes Glioma Progression. Proc. Natl. Acad. Sci. USA 2015, 112, 3421–3426. [Google Scholar] [CrossRef] [Green Version]
- Halacli, S.O.; Dogan, A.L. FOXP1 Regulation via the PI3K/Akt/P70S6K Signaling Pathway in Breast Cancer Cells. Oncol. Lett. 2015, 9, 1482–1488. [Google Scholar] [CrossRef] [Green Version]
- Zhu, L.; Derijard, B.; Chakrabandhu, K.; Wang, B.-S.; Chen, H.-Z.; Hueber, A.-O. Synergism of PI3K/Akt Inhibition and Fas Activation on Colon Cancer Cell Death. Cancer Lett. 2014, 354, 355–364. [Google Scholar] [CrossRef]
- Mi, H.; Muruganujan, A.; Ebert, D.; Huang, X.; Thomas, P.D. PANTHER Version 14: More Genomes, a New PANTHER GO-Slim and Improvements in Enrichment Analysis Tools. Nucleic Acids Res. 2018, 47, D419–D426. [Google Scholar] [CrossRef]
- The Gene Ontology Consortium. The Gene Ontology Resource: Enriching a GOld Mine. Nucleic Acids Res. 2021, 49, D325–D334. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene Ontology: Tool for the Unification of Biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [Green Version]
- Bhat-Nakshatri, P.; Wang, G.; Appaiah, H.; Luktuke, N.; Carroll, J.S.; Geistlinger, T.R.; Brown, M.; Badve, S.; Liu, Y.; Nakshatri, H. AKT Alters Genome-Wide Estrogen Receptor Alpha Binding and Impacts Estrogen Signaling in Breast Cancer. Mol. Cell. Biol. 2008, 28, 7487–7503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Privat, M.; Rudewicz, J.; Sonnier, N.; Tamisier, C.; Ponelle-Chachuat, F.; Bignon, Y.-J. Antioxydation and Cell Migration Genes Are Identified as Potential Therapeutic Targets in Basal-Like and BRCA1 Mutated Breast Cancer Cell Lines. Int. J. Med. Sci. 2018, 15, 46–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhat-Nakshatri, P.; Song, E.-K.; Collins, N.R.; Uversky, V.N.; Dunker, A.K.; O’Malley, B.W.; Geistlinger, T.R.; Carroll, J.S.; Brown, M.; Nakshatri, H. Interplay between Estrogen Receptor and AKT in Estradiol-Induced Alternative Splicing. BMC Med. Genom. 2013, 6, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horvath, A.; Pakala, S.B.; Mudvari, P.; Reddy, S.D.N.; Ohshiro, K.; Casimiro, S.; Pires, R.; Fuqua, S.A.W.; Toi, M.; Costa, L.; et al. Novel Insights into Breast Cancer Genetic Variance through RNA Sequencing. Sci. Rep. 2013, 3, 2256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, K.; Patel, N.A.; Watson, J.E.; Apostolatos, H.; Kleiman, E.; Hanson, O.; Hagiwara, M.; Cooper, D.R. Akt2 Regulation of Cdc2-like Kinases (Clk/Sty), Serine/Arginine-Rich (SR) Protein Phosphorylation, and Insulin-Induced Alternative Splicing of PKCbetaII Messenger Ribonucleic Acid. Endocrinology 2009, 150, 2087–2097. [Google Scholar] [CrossRef]
- Yea, S.; Narla, G.; Zhao, X.; Garg, R.; Tal-Kremer, S.; Hod, E.; Villanueva, A.; Loke, J.; Tarocchi, M.; Akita, K.; et al. Ras Promotes Growth by Alternative Splicing-Mediated Inactivation of the KLF6 Tumor Suppressor in Hepatocellular Carcinoma. Gastroenterology 2008, 134, 1521–1531. [Google Scholar] [CrossRef] [Green Version]
- Whitsett, T.G.; Inge, L.J.; Dhruv, H.D.; Cheung, P.Y.; Weiss, G.J.; Bremner, R.M.; Winkles, J.A.; Tran, N.L. Molecular Determinants of Lung Cancer Metastasis to the Central Nervous System. Transl. Lung Cancer Res. 2013, 2, 273–283. [Google Scholar]
- Speers, C.; Tsimelzon, A.; Sexton, K.; Herrick, A.M.; Gutierrez, C.; Culhane, A.; Quackenbush, J.; Hilsenbeck, S.; Chang, J.; Brown, P. Identification of Novel Kinase Targets for the Treatment of Estrogen Receptor-Negative Breast Cancer. Clin. Cancer Res. 2009, 15, 6327–6340. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.-F.; Cho, J.J.; Huang, T.-H.; Tseng, C.-N.; Huang, E.-Y.; Cho, C.-L. Downregulation of a Novel Human Gene, ROGDI, Increases Radiosensitivity in Cervical Cancer Cells. Cancer Biol. Ther. 2016, 17, 1070–1078. [Google Scholar] [CrossRef] [Green Version]
- Hong, S. RNA Binding Protein as an Emerging Therapeutic Target for Cancer Prevention and Treatment. J. Cancer Prev. 2017, 22, 203–210. [Google Scholar] [CrossRef] [Green Version]
- Thomassen, M.; Tan, Q.; Kruse, T.A. Gene Expression Meta-Analysis Identifies Chromosomal Regions and Candidate Genes Involved in Breast Cancer Metastasis. Breast Cancer Res. Treat. 2009, 113, 239–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, Q.; Rayala, S.K.; Gururaj, A.E.; Talukder, A.H.; O’Malley, B.W.; Kumar, R. Signaling-Dependent and Coordinated Regulation of Transcription, Splicing, and Translation Resides in a Single Coregulator, PCBP1. Proc. Natl. Acad. Sci. USA 2007, 104, 5866–5871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghosh, D.K.; Roy, A.; Ranjan, A. Aggregation-Prone Regions in HYPK Help It to Form Sequestration Complex for Toxic Protein Aggregates. J. Mol. Biol. 2018, 430, 963–986. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Huang, J.; Sun, J.; Xiang, S.; Yang, D.; Ying, X.; Lu, M.; Li, H.; Ren, G. The Transcription Levels and Prognostic Values of Seven Proteasome Alpha Subunits in Human Cancers. Oncotarget 2017, 8, 4501–4519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takei, N.; Yoneda, A.; Sakai-Sawada, K.; Kosaka, M.; Minomi, K.; Tamura, Y. Hypoxia-Inducible ERO1α Promotes Cancer Progression through Modulation of Integrin-Β1 Modification and Signalling in HCT116 Colorectal Cancer Cells. Sci. Rep. 2017, 7, 9389. [Google Scholar] [CrossRef]
- Sarajlić, A.; Filipović, A.; Janjić, V.; Coombes, R.C.; Pržulj, N. The Role of Genes Co-Amplified with Nicastrin in Breast Invasive Carcinoma. Breast Cancer Res. Treat. 2014, 143, 393–401. [Google Scholar] [CrossRef]
- Dolezal, J.M.; Dash, A.P.; Prochownik, E.V. Diagnostic and Prognostic Implications of Ribosomal Protein Transcript Expression Patterns in Human Cancers. BMC Cancer 2018, 18, 275. [Google Scholar] [CrossRef] [Green Version]
- Kikuchi, M.; Katoh, H.; Waraya, M.; Tanaka, Y.; Ishii, S.; Tanaka, T.; Nishizawa, N.; Yokoi, K.; Minatani, N.; Ema, A.; et al. Epigenetic Silencing of HOPX Contributes to Cancer Aggressiveness in Breast Cancer. Cancer Lett. 2017, 384, 70–78. [Google Scholar] [CrossRef]
- Drew, B.G.; Hamidi, H.; Zhou, Z.; Villanueva, C.J.; Krum, S.A.; Calkin, A.C.; Parks, B.W.; Ribas, V.; Kalajian, N.Y.; Phun, J.; et al. Estrogen Receptor (ER)α-Regulated Lipocalin 2 Expression in Adipose Tissue Links Obesity with Breast Cancer Progression. J. Biol. Chem. 2015, 290, 5566–5581. [Google Scholar] [CrossRef] [Green Version]
- Satih, S.; Chalabi, N.; Rabiau, N.; Bosviel, R.; Fontana, L.; Bignon, Y.-J.; Bernard-Gallon, D.J. Gene Expression Profiling of Breast Cancer Cell Lines in Response to Soy Isoflavones Using a Pangenomic Microarray Approach. OMICS 2010, 14, 231–238. [Google Scholar] [CrossRef] [Green Version]
- Wrzesiński, T.; Szelag, M.; Cieślikowski, W.A.; Ida, A.; Giles, R.; Zodro, E.; Szumska, J.; Poźniak, J.; Kwias, Z.; Bluyssen, H.A.R.; et al. Expression of Pre-Selected TMEMs with Predicted ER Localization as Potential Classifiers of CcRCC Tumors. BMC Cancer 2015, 15, 518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zou, J.; Li, Z.; Deng, H.; Hao, J.; Ding, R.; Zhao, M. TMEM213 as a Novel Prognostic and Predictive Biomarker for Patients with Lung Adenocarcinoma after Curative Resection: A Study Based on Bioinformatics Analysis. J. Thorac. Dis. 2019, 11, 3399–3410. [Google Scholar] [CrossRef]
- Chen, Y.; Pan, K.; Li, S.; Xia, J.; Wang, W.; Chen, J.; Zhao, J.; Lü, L.; Wang, D.; Pan, Q.; et al. Decreased Expression of V-Set and Immunoglobulin Domain Containing 1 (VSIG1) Is Associated with Poor Prognosis in Primary Gastric Cancer. J. Surg. Oncol. 2012, 106, 286–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oidovsambuu, O.; Nyamsuren, G.; Liu, S.; Göring, W.; Engel, W.; Adham, I.M. Adhesion Protein VSIG1 Is Required for the Proper Differentiation of Glandular Gastric Epithelia. PLoS ONE 2011, 6, e25908. [Google Scholar] [CrossRef] [Green Version]
- COSMIC Catalogue of Somatic Mutations in Cancer. Available online: https://cancer.sanger.ac.uk/cosmic/mutation/overview?id=95736066#references (accessed on 15 February 2019).
- Lehmann, U.; Celikkaya, G.; Hasemeier, B.; Länger, F.; Kreipe, H. Promoter Hypermethylation of the Death-Associated Protein Kinase Gene in Breast Cancer Is Associated with the Invasive Lobular Subtype. Cancer Res. 2002, 62, 6634–6638. [Google Scholar] [PubMed]
- O’Reilly, J.-A.; Fitzgerald, J.; Fitzgerald, S.; Kenny, D.; Kay, E.W.; O’Kennedy, R.; Kijanka, G.S. Diagnostic Potential of Zinc Finger Protein-Specific Autoantibodies and Associated Linear B-Cell Epitopes in Colorectal Cancer. PLoS ONE 2015, 10, e0123469. [Google Scholar] [CrossRef]
- Zhu, Z.; Teng, Z.; van Duijnhoven, F.J.B.; Dong, M.; Qian, Y.; Yu, H.; Yang, J.; Han, R.; Su, J.; Du, W.; et al. Interactions between RASA2, CADM1, HIF1AN Gene Polymorphisms and Body Fatness with Breast Cancer: A Population-Based Case-Control Study in China. Oncotarget 2017, 8, 98258–98269. [Google Scholar] [CrossRef] [Green Version]
- Suber, T.L.; Nikolli, I.; O’Brien, M.E.; Londino, J.; Zhao, J.; Chen, K.; Mallampalli, R.K.; Zhao, Y. FBXO17 Promotes Cell Proliferation through Activation of Akt in Lung Adenocarcinoma Cells. Respir. Res. 2018, 19, 206. [Google Scholar] [CrossRef] [Green Version]
- Cancer Dependency Map. Available online: https://score.depmap.sanger.ac.uk (accessed on 13 September 2019).
- Zhao, X.; Wu, X.; Wang, H.; Yu, H.; Wang, J. USP53 Promotes Apoptosis and Inhibits Glycolysis in Lung Adenocarcinoma through FKBP51-AKT1 Signaling. Mol. Carcinog. 2020, 59, 1000–1011. [Google Scholar] [CrossRef]
- Winter, S.F.; Lukes, L.; Hunter, K.W. Arid4b Is a Potential Breast Cancer Progression Modifier Gene. Cancer Res. 2010, 70, 2371. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
George, B.; Gui, B.; Raguraman, R.; Paul, A.M.; Nakshatri, H.; Pillai, M.R.; Kumar, R. AKT1 Transcriptomic Landscape in Breast Cancer Cells. Cells 2022, 11, 2290. https://doi.org/10.3390/cells11152290
George B, Gui B, Raguraman R, Paul AM, Nakshatri H, Pillai MR, Kumar R. AKT1 Transcriptomic Landscape in Breast Cancer Cells. Cells. 2022; 11(15):2290. https://doi.org/10.3390/cells11152290
Chicago/Turabian StyleGeorge, Bijesh, Bin Gui, Rajeswari Raguraman, Aswathy Mary Paul, Harikrishna Nakshatri, Madhavan Radhakrishna Pillai, and Rakesh Kumar. 2022. "AKT1 Transcriptomic Landscape in Breast Cancer Cells" Cells 11, no. 15: 2290. https://doi.org/10.3390/cells11152290
APA StyleGeorge, B., Gui, B., Raguraman, R., Paul, A. M., Nakshatri, H., Pillai, M. R., & Kumar, R. (2022). AKT1 Transcriptomic Landscape in Breast Cancer Cells. Cells, 11(15), 2290. https://doi.org/10.3390/cells11152290