
Neuroprotective and Anti-Inflammatory Effects of Linoleic Acid in Models of Parkinson’s Disease: The Implication of Lipid Droplets and Lipophagy

 ,
,  , ,
, ,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. SH-SY5Y Cell Cultures
2.2. Cell Culture Treatments
2.3. Cell Viability Assay (MTT)
2.4. Griess Assay
2.5. Immunocytochemistry Assay
2.6. BODIPY 493/503 and BODIPY 581/591 C11 Staining
2.7. Flow Cytometry
2.8. Immunoblot Analysis
2.9. Animals
2.10. Parkinson’s Disease Animal Model
2.11. Treatment of Animals
2.12. Tissue Processing
2.13. Immunohistochemical Assays
2.14. Fluoro-Jade B Staining
2.15. TUNEL (Free DNA End Marking by Biotinylated dUTPs)
2.16. Cell Count Analysis
2.17. Statistical Analysis and Figures
3. Results
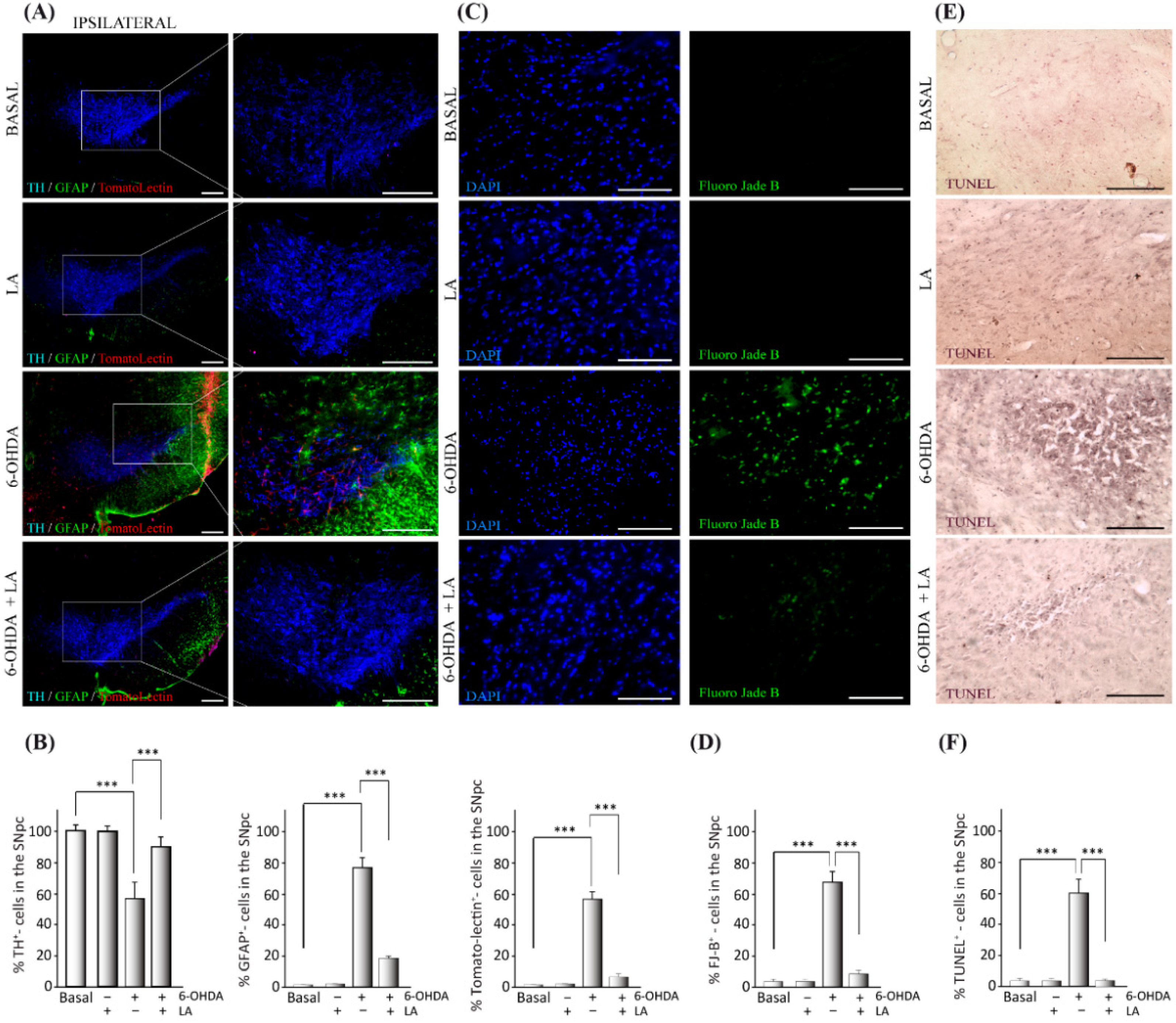
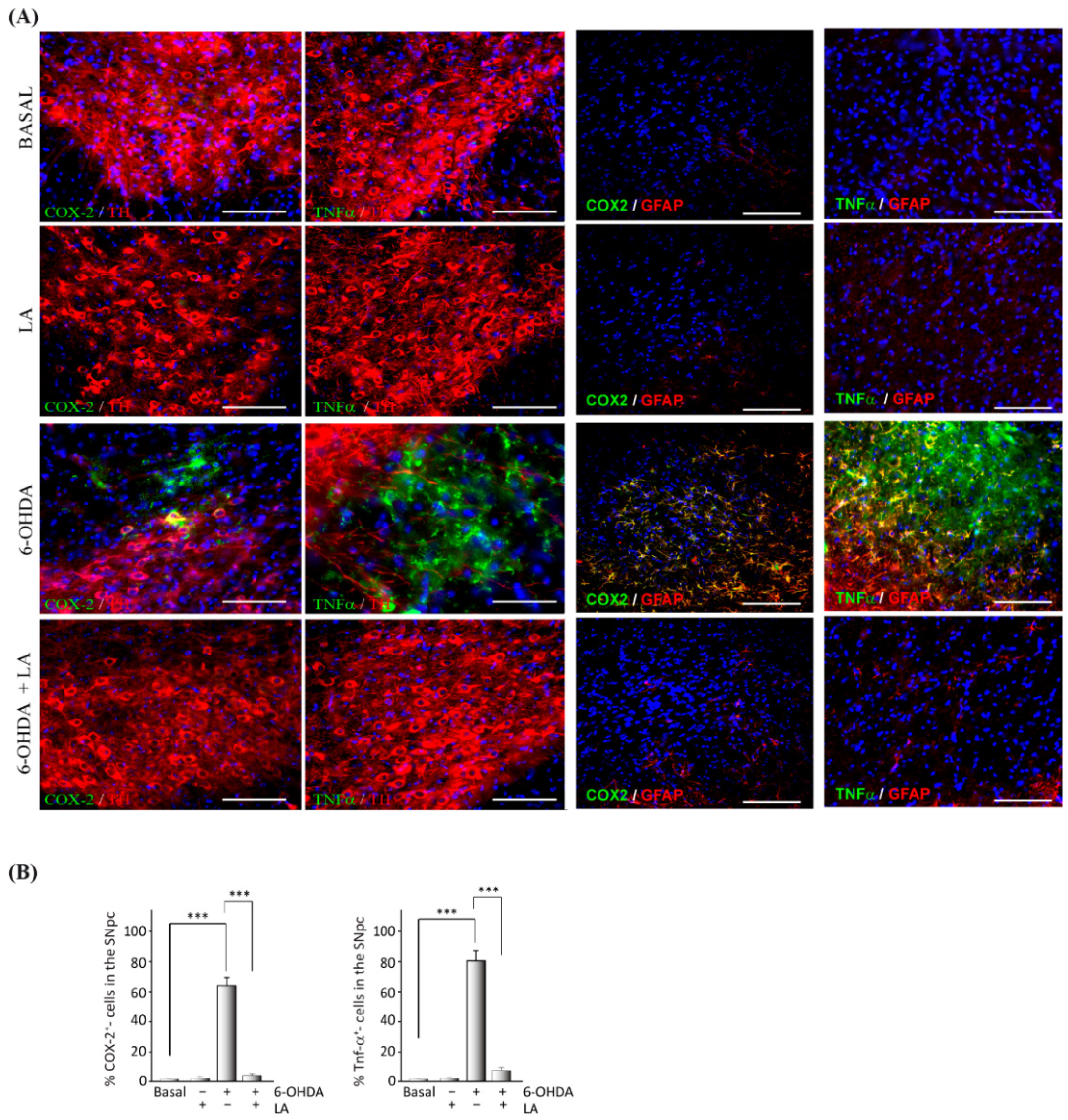
3.1. In Vivo Effect of LA
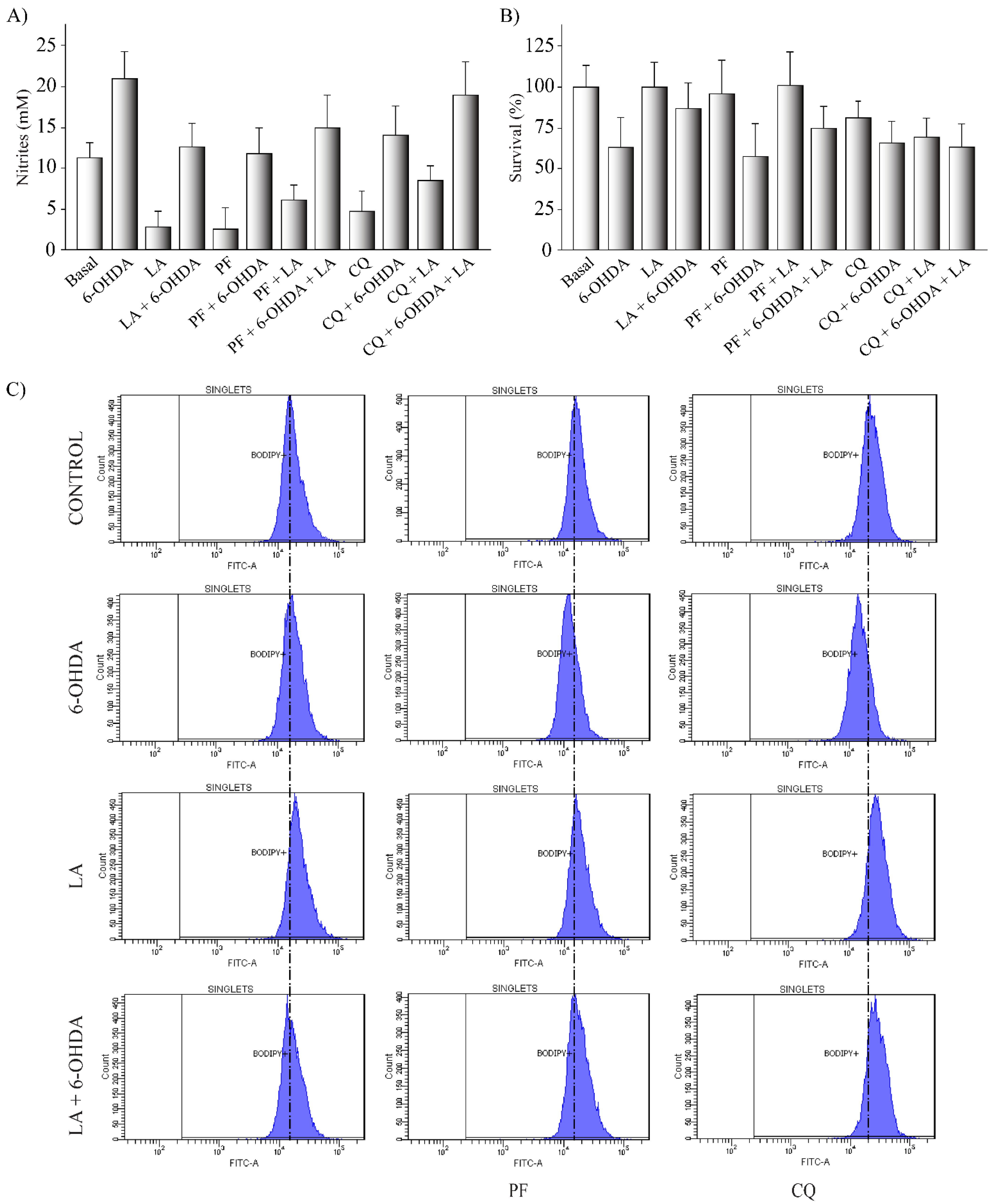
3.2. In Vitro Effect of LA
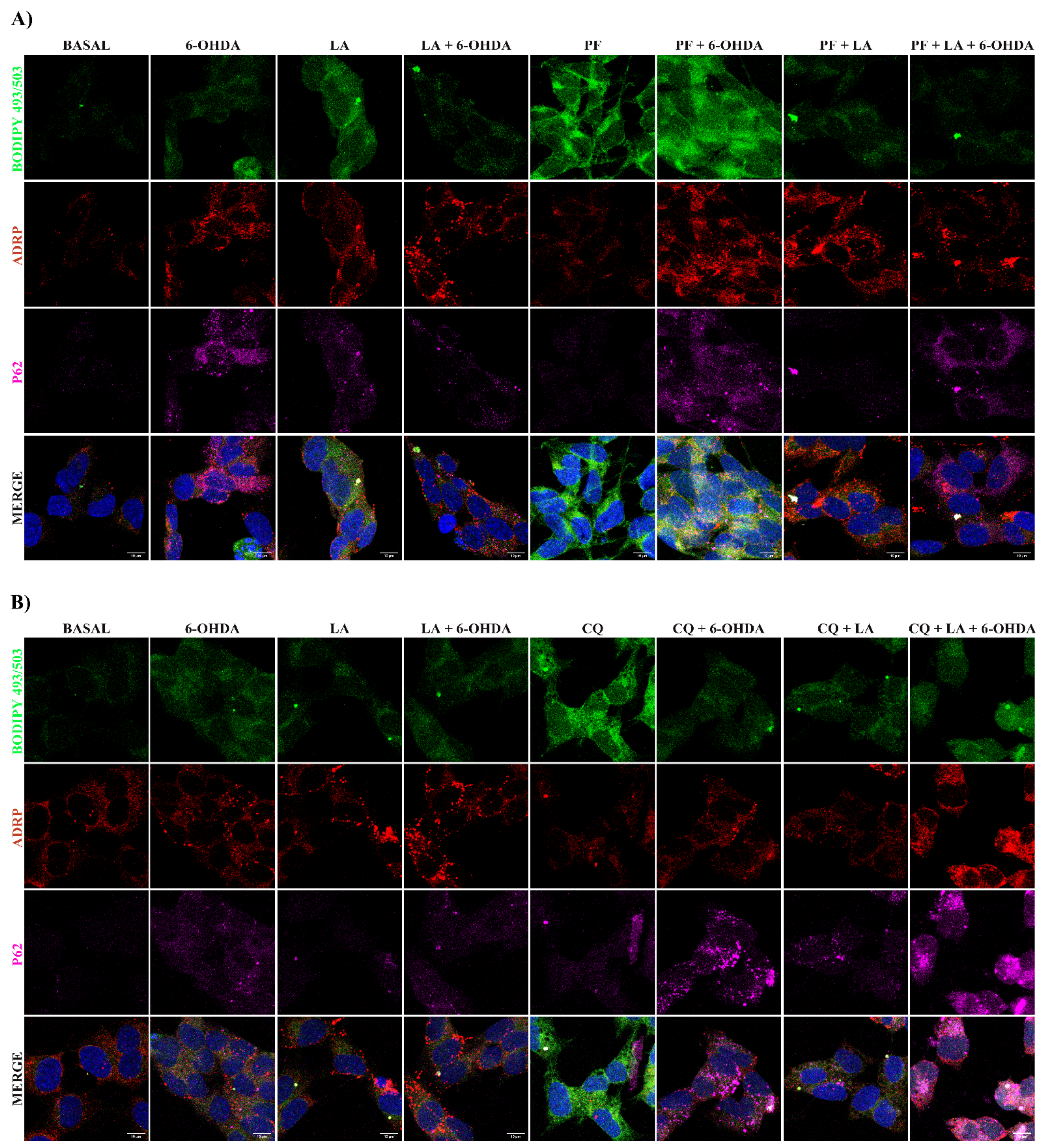
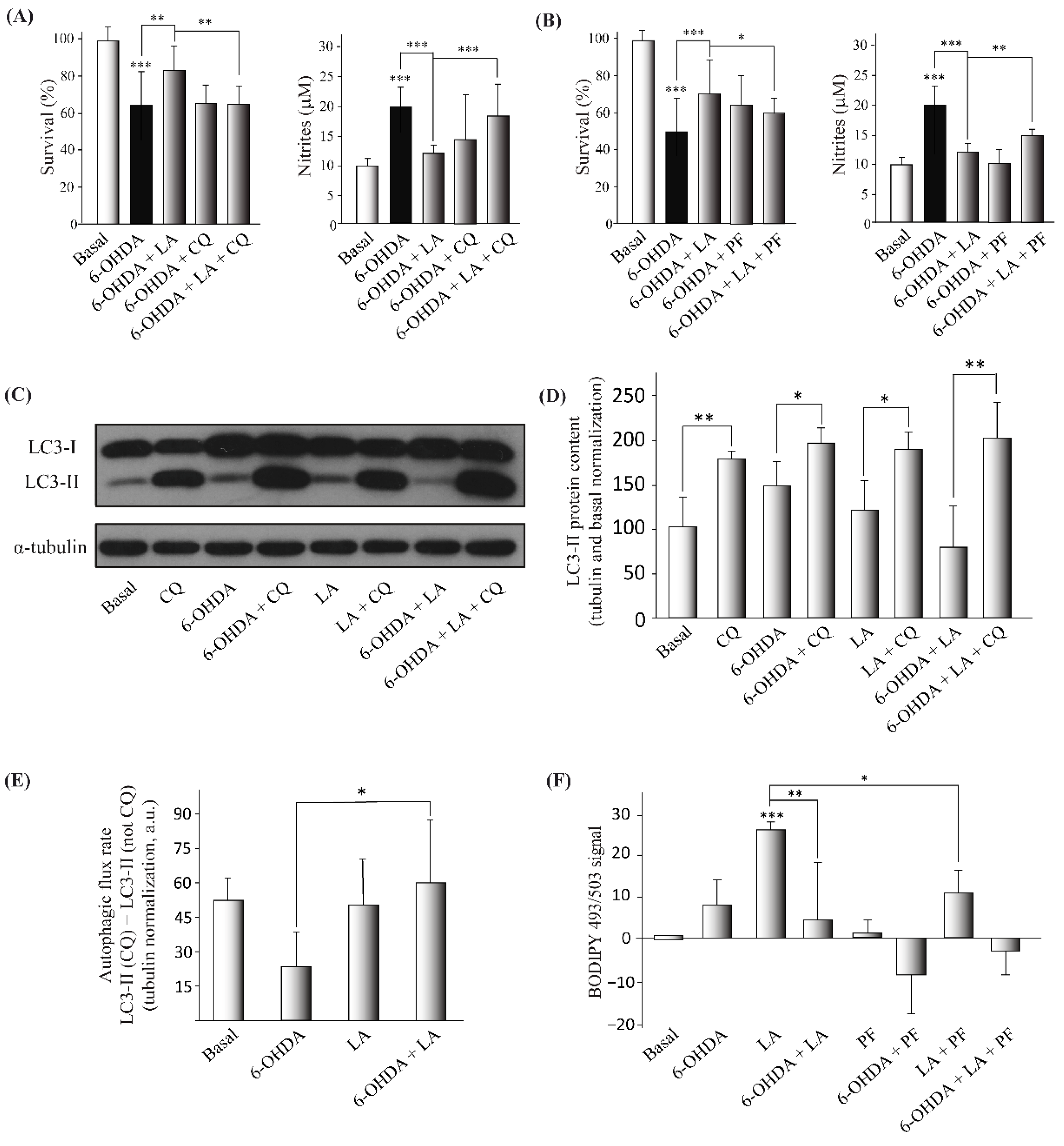
3.3. Mechanism of Action of LA Regulation of Autophagic Flux and Lipid Droplet Levels
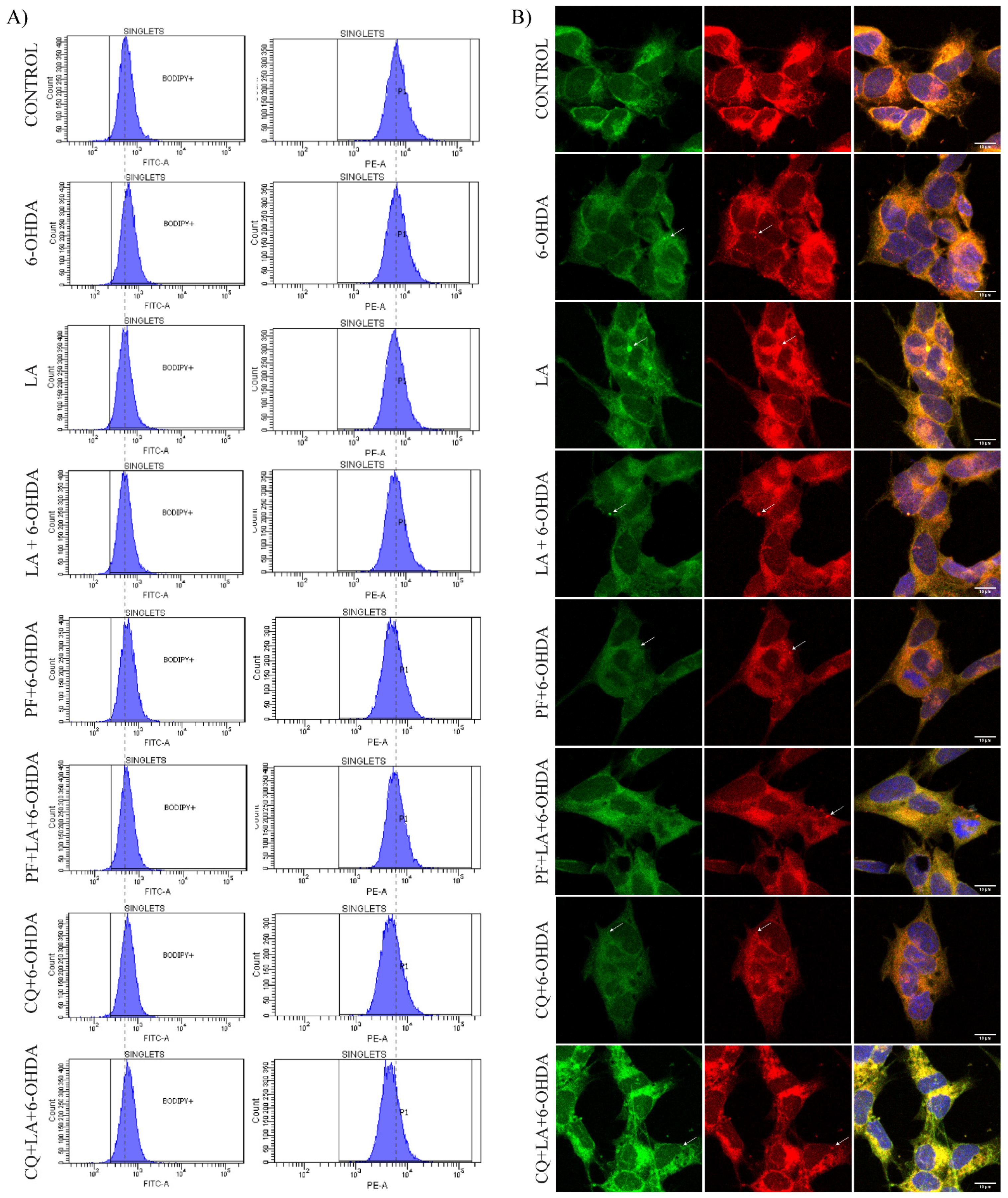
3.4. Lipid Droplet Recycling Mechanism
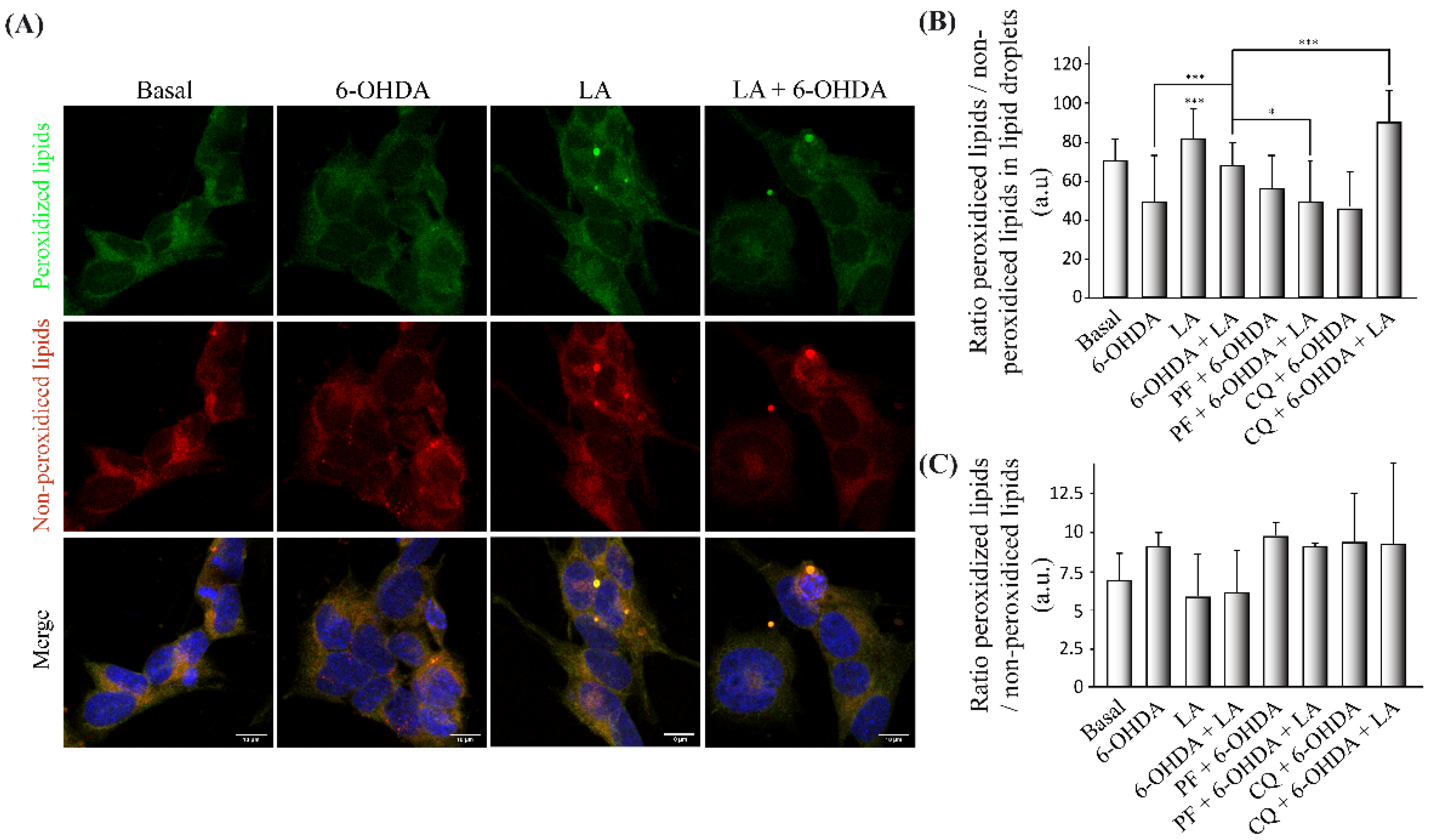
3.5. Regulation of Lipid Peroxidation by LA
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A


References
- Reich, S.G.; Savitt, J.M. Parkinson’s Disease. Med. Clin. N. Am. 2019, 103, 337–350. [Google Scholar] [CrossRef] [PubMed]
- Dorsey, E.R.; Sherer, T.; Okun, M.S.; Bloem, B.R. The Emerging Evidence of the Parkinson Pandemic. J. Parkinsons Dis. 2018, 8, S3–S8. [Google Scholar] [CrossRef] [Green Version]
- Van Bulck, M.; Sierra-Magro, A.; Alarcon-Gil, J.; Perez-Castillo, A.; Morales-Garcia, J.A. Novel Approaches for the Treatment of Alzheimer’s and Parkinson’s Disease. Int. J. Mol. Sci. 2019, 20, 719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diederich, N.J.; James Surmeier, D.; Uchihara, T.; Grillner, S.; Goetz, C.G. Parkinson’s disease: Is it a consequence of human brain evolution? Mov. Disord. 2019, 34, 453–459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trist, B.G.; Hare, D.J.; Double, K.L. Oxidative stress in the aging substantia nigra and the etiology of Parkinson’s disease. Aging Cell 2019, 18, e13031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cole, N.B.; Murphy, D.D.; Grider, T.; Rueter, S.; Brasaemle, D.; Nussbaum, R.L. Lipid droplet binding and oligomerization properties of the Parkinson’s disease protein alpha-synuclein. J. Biol. Chem. 2002, 277, 6344–6352. [Google Scholar] [CrossRef] [Green Version]
- Fanning, S.; Selkoe, D.; Dettmer, U. Parkinson’s disease: Proteinopathy or lipidopathy? NPJ Parkinsons Dis. 2020, 6, 3. [Google Scholar] [CrossRef] [Green Version]
- Sztalryd, C.; Brasaemle, D.L. The perilipin family of lipid droplet proteins: Gatekeepers of intracellular lipolysis. Biochim. Biophys. Acta. Mol. Cell. Biol. Lipids 2017, 1862, 1221–1232. [Google Scholar] [CrossRef]
- Bhatt-Wessel, B.; Jordan, T.W.; Miller, J.H.; Peng, L. Role of DGAT enzymes in triacylglycerol metabolism. Arch. Biochem. Biophys. 2018, 655, 1–11. [Google Scholar] [CrossRef]
- Olzmann, J.A.; Carvalho, P. Dynamics and functions of lipid droplets. Nat. Rev. Mol. Cell. Biol. 2019, 20, 137–155. [Google Scholar] [CrossRef]
- Zechner, R.; Madeo, F.; Kratky, D. Cytosolic lipolysis and lipophagy: Two sides of the same coin. Nat. Rev. Mol. Cell. Biol. 2017, 18, 671–684. [Google Scholar] [CrossRef] [PubMed]
- Shimabukuro, M.K.; Langhi, L.G.; Cordeiro, I.; Brito, J.M.; Batista, C.M.; Mattson, M.P.; Mello Coelho, V. Lipid-laden cells differentially distributed in the aging brain are functionally active and correspond to distinct phenotypes. Sci. Rep. 2016, 6, 23795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, L.; MacKenzie, K.R.; Putluri, N.; Maletic-Savatic, M.; Bellen, H.J. The Glia-Neuron Lactate Shuttle and Elevated ROS Promote Lipid Synthesis in Neurons and Lipid Droplet Accumulation in Glia via APOE/D. Cell. Metab. 2017, 26, 719–737.e6. [Google Scholar] [CrossRef] [Green Version]
- Ioannou, M.S.; Jackson, J.; Sheu, S.H.; Chang, C.L.; Weigel, A.V.; Liu, H.; Pasolli, H.A.; Xu, C.S.; Pang, S.; Matthies, D.; et al. Neuron-Astrocyte Metabolic Coupling Protects against Activity-Induced Fatty Acid Toxicity. Cell 2019, 177, 1522–1535.e14. [Google Scholar] [CrossRef]
- Bailey, A.P.; Koster, G.; Guillermier, C.; Hirst, E.M.; MacRae, J.I.; Lechene, C.P.; Postle, A.D.; Gould, A.P. Antioxidant Role for Lipid Droplets in a Stem Cell Niche of Drosophila. Cell 2015, 163, 340–353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Islam, A.; Kagawa, Y.; Miyazaki, H.; Shil, S.K.; Umaru, B.A.; Yasumoto, Y.; Yamamoto, Y.; Owada, Y. FABP7 Protects Astrocytes Against ROS Toxicity via Lipid Droplet Formation. Mol. Neurobiol. 2019, 56, 5763–5779. [Google Scholar] [CrossRef]
- Li, L.; Zhou, F.; Gao, Q.; Lu, Y.; Xu, X.; Hu, R.; Wang, Z.; Peng, M.; Yang, Z.; Tang, B.Z. Visualizing Dynamic Performance of Lipid Droplets in a Parkinson’s Disease Model via a Smart Photostable Aggregation-Induced Emission Probe. iScience 2019, 21, 261–272. [Google Scholar] [CrossRef] [Green Version]
- Niso-Santano, M.; Malik, S.A.; Pietrocola, F.; Bravo-San Pedro, J.M.; Marino, G.; Cianfanelli, V.; Ben-Younes, A.; Troncoso, R.; Markaki, M.; Sica, V.; et al. Unsaturated fatty acids induce non-canonical autophagy. EMBO J. 2015, 34, 1025–1041. [Google Scholar] [CrossRef]
- Nakajima, S.; Gotoh, M.; Fukasawa, K.; Murakami-Murofushi, K.; Kunugi, H. Oleic acid is a potent inducer for lipid droplet accumulation through its esterification to glycerol by diacylglycerol acyltransferase in primary cortical astrocytes. Brain. Res. 2019, 1725, 146484. [Google Scholar] [CrossRef]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]
- Bolte, S.; Cordelieres, F.P. A guided tour into subcellular colocalization analysis in light microscopy. J. Microsc. 2006, 224, 213–232. [Google Scholar] [CrossRef] [PubMed]
- Qiu, B.; Simon, M.C. BODIPY 493/503 Staining of Neutral Lipid Droplets for Microscopy and Quantification by Flow Cytometry. Bio. Protoc. 2016, 6, e1912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bove, J.; Perier, C. Neurotoxin-based models of Parkinson’s disease. Neuroscience 2012, 211, 51–76. [Google Scholar] [CrossRef] [PubMed]
- Schmued, L.C.; Hopkins, K.J. Fluoro-Jade B: A high affinity fluorescent marker for the localization of neuronal degeneration. Brain Res 2000, 874, 123–130. [Google Scholar] [CrossRef] [Green Version]
- Morales-Garcia, J.A.; Alonso-Gil, S.; Santos, A.; Perez-Castillo, A. Phosphodiesterase 7 Regulation in Cellular and Rodent Models of Parkinson’s Disease. Mol. Neurobiol. 2020, 57, 806–822. [Google Scholar] [CrossRef] [PubMed]
- Paxinos, G.; Watson, C.; Petrides, M.; Rosa, M.; Tokuno, H. The Marmoset Brain in Stereotaxic Coordinates; Elsevier Academic Press: Amsterdam, The Netherlands, 2012. [Google Scholar]
- Krishna, A.; Biryukov, M.; Trefois, C.; Antony, P.M.; Hussong, R.; Lin, J.; Heinaniemi, M.; Glusman, G.; Koglsberger, S.; Boyd, O.; et al. Systems genomics evaluation of the SH-SY5Y neuroblastoma cell line as a model for Parkinson’s disease. BMC Genom. 2014, 15, 1154. [Google Scholar] [CrossRef]
- Wu, F.; Xu, H.D.; Guan, J.J.; Hou, Y.S.; Gu, J.H.; Zhen, X.C.; Qin, Z.H. Rotenone impairs autophagic flux and lysosomal functions in Parkinson’s disease. Neuroscience 2015, 284, 900–911. [Google Scholar] [CrossRef]
- Yan, Y.; Wang, H.; Wei, C.; Xiang, Y.; Liang, X.; Phang, C.W.; Jiao, R. HDAC6 regulates lipid droplet turnover in response to nutrient deprivation via p62-mediated selective autophagy. J. Genet. Genom. 2019, 46, 221–229. [Google Scholar] [CrossRef]
- Kaushik, S.; Cuervo, A.M. AMPK-dependent phosphorylation of lipid droplet protein PLIN2 triggers its degradation by CMA. Autophagy 2016, 12, 432–438. [Google Scholar] [CrossRef] [Green Version]
- Farmer, B.C.; Kluemper, J.; Johnson, L.A. Apolipoprotein E4 Alters Astrocyte Fatty Acid Metabolism and Lipid Droplet Formation. Cells 2019, 8, 182. [Google Scholar] [CrossRef] [Green Version]
- Palhegyi, A.M.; Seranova, E.; Dimova, S.; Hoque, S.; Sarkar, S. Biomedical Implications of Autophagy in Macromolecule Storage Disorders. Front. Cell. Dev. Biol. 2019, 7, 179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, B.; Zhou, Y.; Wu, M.; Li, X.; Mai, K.; Ai, Q. omega-6 Polyunsaturated fatty acids (linoleic acid) activate both autophagy and antioxidation in a synergistic feedback loop via TOR-dependent and TOR-independent signaling pathways. Cell. Death Dis. 2020, 11, 607. [Google Scholar] [CrossRef] [PubMed]
- Mohammadyani, D.; Tyurin, V.A.; O’Brien, M.; Sadovsky, Y.; Gabrilovich, D.I.; Klein-Seetharaman, J.; Kagan, V.E. Molecular speciation and dynamics of oxidized triacylglycerols in lipid droplets: Mass spectrometry and coarse-grained simulations. Free Radic. Biol. Med. 2014, 76, 53–60. [Google Scholar] [CrossRef] [Green Version]
- Thiam, A.R.; Antonny, B.; Wang, J.; Delacotte, J.; Wilfling, F.; Walther, T.C.; Beck, R.; Rothman, J.E.; Pincet, F. COPI buds 60-nm lipid droplets from reconstituted water-phospholipid-triacylglyceride interfaces, suggesting a tension clamp function. Proc. Natl. Acad. Sci. USA 2013, 110, 13244–13249. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Zhang, K.; Sandoval, H.; Yamamoto, S.; Jaiswal, M.; Sanz, E.; Li, Z.; Hui, J.; Graham, B.H.; Quintana, A.; et al. Glial lipid droplets and ROS induced by mitochondrial defects promote neurodegeneration. Cell 2015, 160, 177–190. [Google Scholar] [CrossRef] [Green Version]
- Marschallinger, J.; Iram, T.; Zardeneta, M.; Lee, S.E.; Lehallier, B.; Haney, M.S.; Pluvinage, J.V.; Mathur, V.; Hahn, O.; Morgens, D.W.; et al. Lipid-droplet-accumulating microglia represent a dysfunctional and proinflammatory state in the aging brain. Nat. Neurosci. 2020, 23, 194–208. [Google Scholar] [CrossRef] [PubMed]
- Cui, W.; Sathyanarayan, A.; Lopresti, M.; Aghajan, M.; Chen, C.; Mashek, D.G. Lipophagy-derived fatty acids undergo extracellular efflux via lysosomal exocytosis. Autophagy 2020, 17, 690–705. [Google Scholar] [CrossRef]
- Monzel, A.S.; Smits, L.M.; Hemmer, K.; Hachi, S.; Moreno, E.L.; van Wuellen, T.; Jarazo, J.; Walter, J.; Bruggemann, I.; Boussaad, I.; et al. Derivation of Human Midbrain-Specific Organoids from Neuroepithelial Stem Cells. Stem. Cell. Rep. 2017, 8, 1144–1154. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alarcon-Gil, J.; Sierra-Magro, A.; Morales-Garcia, J.A.; Sanz-SanCristobal, M.; Alonso-Gil, S.; Cortes-Canteli, M.; Niso-Santano, M.; Martínez-Chacón, G.; Fuentes, J.M.; Santos, A.; et al. Neuroprotective and Anti-Inflammatory Effects of Linoleic Acid in Models of Parkinson’s Disease: The Implication of Lipid Droplets and Lipophagy. Cells 2022, 11, 2297. https://doi.org/10.3390/cells11152297
Alarcon-Gil J, Sierra-Magro A, Morales-Garcia JA, Sanz-SanCristobal M, Alonso-Gil S, Cortes-Canteli M, Niso-Santano M, Martínez-Chacón G, Fuentes JM, Santos A, et al. Neuroprotective and Anti-Inflammatory Effects of Linoleic Acid in Models of Parkinson’s Disease: The Implication of Lipid Droplets and Lipophagy. Cells. 2022; 11(15):2297. https://doi.org/10.3390/cells11152297
Chicago/Turabian StyleAlarcon-Gil, Jesus, Ana Sierra-Magro, Jose A. Morales-Garcia, Marina Sanz-SanCristobal, Sandra Alonso-Gil, Marta Cortes-Canteli, Mireia Niso-Santano, Guadalupe Martínez-Chacón, Jose M. Fuentes, Angel Santos, and et al. 2022. "Neuroprotective and Anti-Inflammatory Effects of Linoleic Acid in Models of Parkinson’s Disease: The Implication of Lipid Droplets and Lipophagy" Cells 11, no. 15: 2297. https://doi.org/10.3390/cells11152297
APA StyleAlarcon-Gil, J., Sierra-Magro, A., Morales-Garcia, J. A., Sanz-SanCristobal, M., Alonso-Gil, S., Cortes-Canteli, M., Niso-Santano, M., Martínez-Chacón, G., Fuentes, J. M., Santos, A., & Perez-Castillo, A. (2022). Neuroprotective and Anti-Inflammatory Effects of Linoleic Acid in Models of Parkinson’s Disease: The Implication of Lipid Droplets and Lipophagy. Cells, 11(15), 2297. https://doi.org/10.3390/cells11152297