1. Introduction
Parkinson’s disease (PD) is the second most common neurodegenerative disease after Alzheimer’s disease. The principal hallmarks of PD are the death of the dopaminergic neurons, specifically in the Substantia nigra pars compacta (SNpc), and the presence of gliosis (pathological proliferation of reactive glial cells) [
1,
2]. However, the pathogeny of PD is still unknown, despite its impact on health, society, and the economy [
2,
3]. Several cellular alterations play important roles in PD development, for example, oxidative stress, mitochondrial dysfunction, loss of proteostasis, impaired autophagic flux, and endoplasmic reticulum stress [
3]. Oxidative stress is provoked by the high energetic demand of dopaminergic neurons, dopamine metabolism itself, as well as mitochondrial dysfunction [
4,
5]. Moreover, autophagic flux alterations, a loss of proteostasis, and the relationship between both alterations play principal roles in PD development [
3]. In recent years, lipid metabolism alterations have also become relevant in PD pathology, including the alteration of lipid droplet dynamics. The modification of lipid metabolism has emerged as a possible therapeutic approach for this disease [
6,
7].
Lipid droplets (LDs) are cellular organelles formed by a core of neutral lipids surrounded by a phospholipid monolayer with different proteins on their surface, like those of the perilipin family [
8]. The main components of these LDs are triacylglycerides which are synthesized in the endoplasmic reticulum by several enzymes, such as the diacylglyceroltransferases (DGAT), [
9]. The membrane of the endoplasmic reticulum accumulates these triacylglycerides until reaching a determined concentration. At that time, the membrane of the reticulum expands and curls to give rise to the nascent LDs. Then, the LDs grow up further and ends up separating from the endoplasmic reticulum to be released into the cytoplasm [
10]. Later, these LDs can be degraded in two ways: lipolysis and lipophagy. Lipolysis is a type of neutral degradation carried out by various enzymes in the cytoplasm. Lipophagy is an acidic degradation of the LDs accomplished through autophagy, either micro- or macro-autophagy. The latter process starts with the generation of a phagophore that includes all LDs or a portion of them to give rise to a lipoautophagosome. This lipoautophagosome fuses with a lysosome to generate the autolysosome, from which the LDs are degraded [
11].
The relationship between PD and lipid metabolism (LD biogenesis and lipophagy) has not been studied in depth. Shimabukuro et al. suggested that neural cells could accumulate LDs together with autophagy markers during the aging process [
12]. Other authors have demonstrated that neurons increase lipid production and astrocytes enhance LD accumulation under stressful conditions, possibly due to a neuron–astrocyte coupling involved in lipid metabolism [
13,
14]. Interestingly, an increasing body of evidence is indicating that LDs could be a sink for free radicals [
15,
16,
17] and, consequently, could play an antioxidant and protective role in the pathogeny of PD. On the other hand, several fatty acids (e.g., linoleic or oleic acid) modify LD levels by regulating the autophagic flux and LD biogenesis [
18,
19]. Each of these pieces of evidence suggests the importance of LDs in the pathogeny of PD. However, it is necessary to further study the connections between these processes.
Given this background, this research aims to demonstrate the neuroprotective and anti-inflammatory effects of linoleic acid (LA) using in vivo and in vitro models of PD. We also aim to confirm that these effects are due to the stimulation of LD biogenesis and lipophagic flux in our in vitro model of PD. We hypothesize that the increased number of LDs act as scavengers of free radicals. Then, their clearance by enhanced lipophagy could act as an antioxidant mechanism. We name this mechanism the “LD recycler mechanism”.
Our results partially demonstrate this hypothesis. We confirm the neuroprotective and anti-inflammatory effect of LA using in vitro and in vivo models of PD. LA appears to act as an antioxidant, LD biogenesis stimulant, and an inducer of lipophagy. This new mechanism of action for LA opens new avenues for the development of novel treatments against PD.
2. Materials and Methods
2.1. SH-SY5Y Cell Cultures
SH-SY5Y, a human-neuroblastoma-derived cell line (ATCC (Ref.CRL-2266)), was cultured and propagated with RPMI medium (Sigma-Aldrich, Madrid, Spain, R8758) supplemented with 2 mM of glutamine, 10% fetal bovine serum, and 100 μg/mL of gentamicin.
2.2. Cell Culture Treatments
In order to attain 70% confluence, cells were seeded onto 96-well plates (3 × 104 cells/well) for the cell viability evaluation and nitrite measurement, onto 24-well plates (1.0 × 105 cells per well) for the immunocytochemical analysis, and onto 100 mm culture plates (1.0 × 106 cells per well) for the flow cytometer and Western blot assays.
To evaluate the cytotoxicity of LA, cell cultures were treated with LA at different concentrations and incubated in the presence or not of 6-OHDA (35 µM). Stemming from these results, an LA concentration of 25 μM and incubation time of 24 h were chosen as the optimal conditions for the experiments. Some other cultures were treated with PF-06424439 (PF, 40 nM, Sigma) or chloroquine (CQ, 40 μM, Sigma, St. Louis, MI, USA) prior to 6-OHDA exposure. Finally, after 18 h of treatment with 6-OHDA, different analyses were performed.
2.3. Cell Viability Assay (MTT)
Cell viability was determined by a colorimetric assay with tetrazolium salt (3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazoliumbromide; MTT; Roche Diagnostic GmbH, Manmheim, Germany, 11465007001), a method that is based on the ability of viable cells to reduce the MTT to formazan. After exposure to 6-OHDA, cells were incubated with MTT (0.5 mg/mL) diluted in cell medium for 1 h. Then, the medium was removed, and the resulting formazan crystals were dissolved with dimethylsulfoxide (DMSO, Sigma). Finally, the reduced MTT was quantified by 595/650 nm absorbance in a spectrophotometer.
2.4. Griess Assay
The nitrite concentration in the cell culture medium was measured by Griess assay. For this purpose, 100 μL of media was mixed with 100 μL of Griess reagent (Sigma, final concentration of 40 mg/mL) and incubated for 15 min at room temperature while protected from light. Finally, the absorbance at 540 nm was measured in a spectrophotometer, and the nitrite concentration was calculated by absorbance data extrapolation using a standard curve with sodium nitrite.
2.5. Immunocytochemistry Assay
Cells were seeded on glass coverslips in 24-well cell culture plates precoated with poly-D-lysine (20 μg/mL) and treated as mentioned above. After treatment, coverslips were washed in PBS 1X and fixed in paraformaldehyde (4%) for 15 min. After several rinses in PBS, cells were blocked in PBS containing 0.01% Triton X-100 (Sigma) and 10% goat serum for 30 min at 37 °C and incubated with primary antibodies diluted in PBS with 0.01% Triton X-100 and 5% goat serum for 1 h at 37 °C. The following antibodies were used: α-cleaved caspase 3 (1:500, R&D Bio-Tech., Abingdon, UK, AF835), α-COX2 (1:200, Santa Cruz Biotechnology, Heidelberg, Germany, sc-376861), α-SQSTM1 (p62, 1:300, Cusabio, Houston, TX, USA, CSB-PA615696LA01HU), and/or α-ADRP (1:200, Santa Cruz, sc-377429). Then, after several rinses in PBS, cells were incubated with secondary antibodies diluted in PBS with 0.01% Triton X-100 and 5% goat serum for 1 h at 37 °C. The following antibodies were used: Alexa Fluor 488 (ThermoFisher Scientific, Madrid, Spain; Ex 490 nm/Em 525 nm), Alexa Fluor 568 (ThermoFisher; Ex 578 nm/Em 603 nm), and/or Alexa Fluor 647 (ThermoFisher; Ex 650 nm/Em 665 nm). DAPI (4’,6-diamino-2-phenylindole) was used as a nuclear marker. Finally, glasses were mounted with Vectashield (Vector Labs, Newark, CA, USA, H-1000-10), and images were acquired with a confocal microscope LSM710 (Carl Zeiss Inc, Carl Zeiss Iberia, S.L-Division Microscopy, Tres Cantos, Madrid, Spain) or an epifluorescence microscope (Nikon 90i). Fiji Software 1.53c (plus) [
20] and JaCop Plugin [
21] were used to carry out the different analyses of the obtained images.
2.6. BODIPY 493/503 and BODIPY 581/591 C11 Staining
Staining with BODIPY 493/503 (Sigma, 1 μM) and BODIPY 581/591 C11 (Sigma, 2 μM) was performed prior to fixation or flow cytometry analysis. BODIPY 581/591 C11 was added to the 6-OHDA treatment. BODIPY 493/503 was added to cultures 15 min before finishing the incubation with 6-OHDA. Then, immunocytochemistry or flow cytometry analysis was performed.
2.7. Flow Cytometry
After treatments, 1.0 × 10
6 cells per experimental group were stained with BODIPY 493/503 (Sigma, 1 μM) or BODIPY 581/591 C11 (Sigma, 2 μM) [
22]. BODIPY 581/591 C11 was added to the 6-OHDA treatment. BODIPY 493/503 was added to cultures 15 min before finishing the incubation with 6-OHDA. Then, cells were washed with PBS 1X, collected by trypsinization, and diluted in 0.5 mL PBS 1X in flow cytometry tubes. Finally, samples were analyzed in a FACS Canto II flow cytometer, and light emissions at different wavelengths of stained cells were measured (530/30 and 582/15 filters for BODIPY 581/591 C11 and 530/30 filter for BODIPY 493/503). Data from cell aggregates, dead cells, and cell debris signals were removed.
2.8. Immunoblot Analysis
After 6-OHDA exposure, cells were washed with PBS 1X, and their proteins were isolated using ice-cold RIPA buffer with protease and phosphatase inhibitors (10 mM sodium pyrophosphate, 1 mM phenylmethylsulphonyl fluoride (PMSF), and 1 mM sodium orthovanadate). Then, a total protein amount of 35 µg was subjected to 12% SDS-PAGE and transferred into PVDF membranes (Protran, Whatman). After blocking with 5% fat-free milk in T-TBS (Tween-20 0.05% and 20 mM Tris-HCl pH 7.5 in distilled water) for 1 h at room temperature, the membranes were incubated with different primary antibodies diluted in the same blocking medium for 18 h at 4 °C. A α-LC3β antibody (1:500, Santa Cruz, sc-376404) was used. As a control, an anti-tubulin (1:2000, Sigma, T5168) antibody was used. Then, the membranes were washed with T-TBS and incubated at room temperature with the corresponding secondary antibody diluted in blocking medium for 1 h at room temperature. Proteins were finally detected by the ECL chemiluminescence system (Amersham) in accordance with the manufacturer’s instructions. Fiji software was used for quantification [
20].
2.9. Animals
All animal experiments were specifically approved by the “Ethics Committee for Animal Experimentation” of the Instituto de Investigaciones Biomedicas (CSIC- UAM) and carried out in accordance with the European Communities Council Directive (2010/63/EEC) and National regulations (RD1386/2018). The experimental design was planned to minimize the number of animals used and to reduce damage to animals. Adult male C57BL/6 mice (3 months) were housed under standard temperature and humidity conditions with 12-h light–dark cycles and ad libitum access to food and water.
2.10. Parkinson’s Disease Animal Model
The PD experimental model is based on the intracerebral injection of 6-OHDA into the Substantia nigra pars compacta (SNpc) of mice. 6-OHDA is the most frequently used neurotoxin to model PD since, as a selective catecholaminergic neurotoxin, it produces specific lesions in the nigrostriatal system with progressive retrograde neuronal degeneration that follows a very similar pattern to that described in PD patients [
23].
2.11. Treatment of Animals
Treatments were intracerebrally injected in mice using a stereotaxic apparatus (Kopf Instruments, Tujunga, CA, USA, Model 900LS). After anesthesia with a mixture of ketamine (60 mg/kg) and medetomidine (0.5 mg/kg), an intracerebral injection was made unilaterally in the SNpc (according to the coordinates of Paxinos and Franklin, from Bregma: 3.2 mm posterior, 2.0 mm lateral, and 4.7 mm depth) with a micropump. Three animals per experimental group were injected with the vehicle (PBS 1X), LA (16 μg) and/or 6-OHDA (1 μg). After injection, animals were individually housed to recover, and they were sacrificed 7 days after lesioning.
2.12. Tissue Processing
After animal perfusion with 4% paraformaldehyde, brains were removed, postfixed overnight in the same solution at 4 °C, cryoprotected in 30% sucrose, frozen, and finally, coronal sections of 30 μm thickness were obtained in a cryostat (Cryocut 1900, Leica Biosystems, Barcelona, Spain, CM1900).
2.13. Immunohistochemical Assays
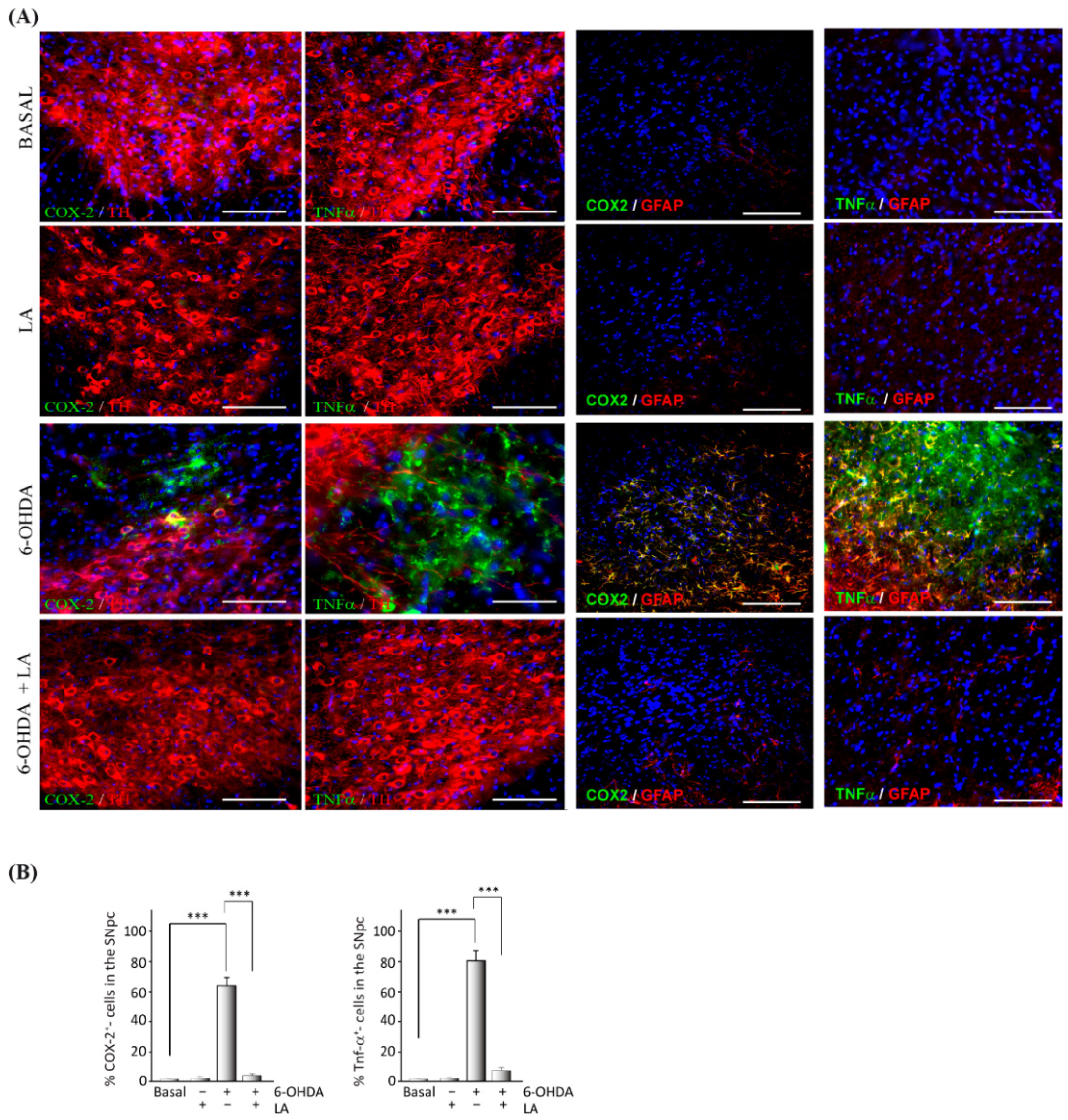
Brain sections containing the SNpc were used to perform the immunohistochemical assays. These free-floating sections were first blocked at room temperature for 1 h (PBS 1X, 0.1% of Triton X-100 and 3% of goat serum) and then were incubated with the primary antibodies (α-TH, 1:200, Millipore, Merck Life Science, Madrid, Spain, AB152; α-TH, 1:300, Sigma-Aldrich, Merck Life Science, Madrid, Spain, T2928; α-GFAP, 1:250, Dako, Agilent Technologies, Madrid, Spain, Z0334; α-GFAP, 1:250, Sigma, G3893; α-COX2, 1:200, Cayman Chemical, Ann Arbor, MI, USA, 160106, 160,106; α-TNFα, 1:200, Abcam plc, Cambridge, UK, ab1793) diluted in blocking solution at 4 °C for 24 h. After this, they were incubated for 1 h with the corresponding secondary antibody and 4′, 6-diamidino-2- phenylindole (DAPI; Calbiochem, Merck Life Science, Madrid, Spain 268298) for nuclear staining and diluted in blocking solution at room temperature. Some sections were stained with the fluorescent marker Tomato Lectin (1:150, Vector Labs, Newark, CA, USA, TL1176). Finally, sections were mounted with wet mounting medium (Vectashield; Vector Labs) and observed under an epifluorescence microscope (Nikon 90i).
2.14. Fluoro-Jade B Staining
This type of histochemical staining reveals degenerating neurons [
24]. Brain sections, previously mounted on gelatinized slides, were immersed in absolute ethanol, followed by immersion in 70% ethanol and distilled water. Then, slides were incubated in 0.06% potassium permanganate for 15 min at room temperature. After this, sections were incubated in the staining solution (0.001% Fluoro-Jade B (Chemicon, Madrid, Spain, AG310) in acetic acid) for 30 min at room temperature and finally rinsed in distilled water, dried at room temperature, and mounted with DePeX (Serva). Images were acquired by confocal microscopy. To compare fluorescence signals from different preparations, confocal microscope settings were fixed for all samples within the same analysis group and adjusted to produce the optimum signal-to-noise ratio.
2.15. TUNEL (Free DNA End Marking by Biotinylated dUTPs)
DNA fragmentation in apoptotic cells was detected by marking the free DNA end with biotinylated dUTPs (TUNEL) using the in situ Cell Death Detection kit (Ref.11684817910; Sigma-Aldrich) and following the manufacturer’s instructions. Briefly, coronal brain sections containing SNpc were fixed with 4% paraformaldehyde for 20 min, and 3% H2O2 was used for endogenous peroxidase inactivation. Tissue was later permeabilized with 0.1% triton and 0.1% sodium citrate for 2 min at 4 °C, and free DNA ends were marked with biotinylated dUTPs in TdT buffer for 60 min. After that, sections were incubated with horseradish peroxidase (HRP)-conjugated streptavidin for 30 min at 37 °C. DNA breaks were observed using a 3-3′-diaminobendicina (DAB) chromogen at a final concentration of 50 μg/mL and H2O2. Brain sections treated with 2U of DNAse (Promega, Promega Biotech Ibérica, Alcobendas, Madrid, Spain, Ref.M610A) were used as positive controls for DNA fragmentation. Pictures were taken with a Nikon Eclipse 80i microscope coupled with a Nikon DS-Fi camera.
2.16. Cell Count Analysis
The numbers of dopaminergic neurons (TH-immunoreactive cells), astroglia (GFAP-positive cells), microglial cells (tomato-lectin-stained cells), Fluoro-Jade B and active caspase 3 marked cells as well as COX-2 and TNFα expressing cells in the SNpc were estimated. To that end, a modified stereological approach was used, as previously described [
25]. Confocal images of serial coronal sections (30 μm) containing the entire SNpc (rostrocaudal extent) were acquired under an objective (×63) to avoid oversampling errors. The number of positive cells was counted in a 1:5 series of sections. The boundaries of the SNpc were determined with reference to internal anatomic landmarks [
26]. Images were analyzed using computer-assisted image analysis software (Soft Imaging System Corporation, Lakewood, CO, USA). Four animals were used per group. The total number of positive cells for a particular marker in the SNpc was determined by multiplying the average number of labeled cells/section by the total number of 30-μm-thick sections containing the SNpc. Data are expressed as percentages to allow better understanding.
2.17. Statistical Analysis and Figures
Data were statistically analyzed using SPSS statistical software (IBM Corp., Armonk, NY, USA, Published in 2016. IBM SPSS Statistics for Windows, version 24.0. Armonk, NY, USA) through one-way analysis of variance (ANOVA), followed by a multiple comparison test with Bonferroni (if the data showed normality and homoscedasticity) or Games–Howell correction (if the data showed normality but not homoscedasticity) or the Kruskal–Wallis test, followed by Dunn’s multiple comparisons test (if data showed neither normality nor homoscedasticity). p-value < 0.05 was considered statistically significant. Figures were made with the Adobe Illustrator software (CS6, Adobe System Incorporated, San Jose, CA, USA).
4. Discussion
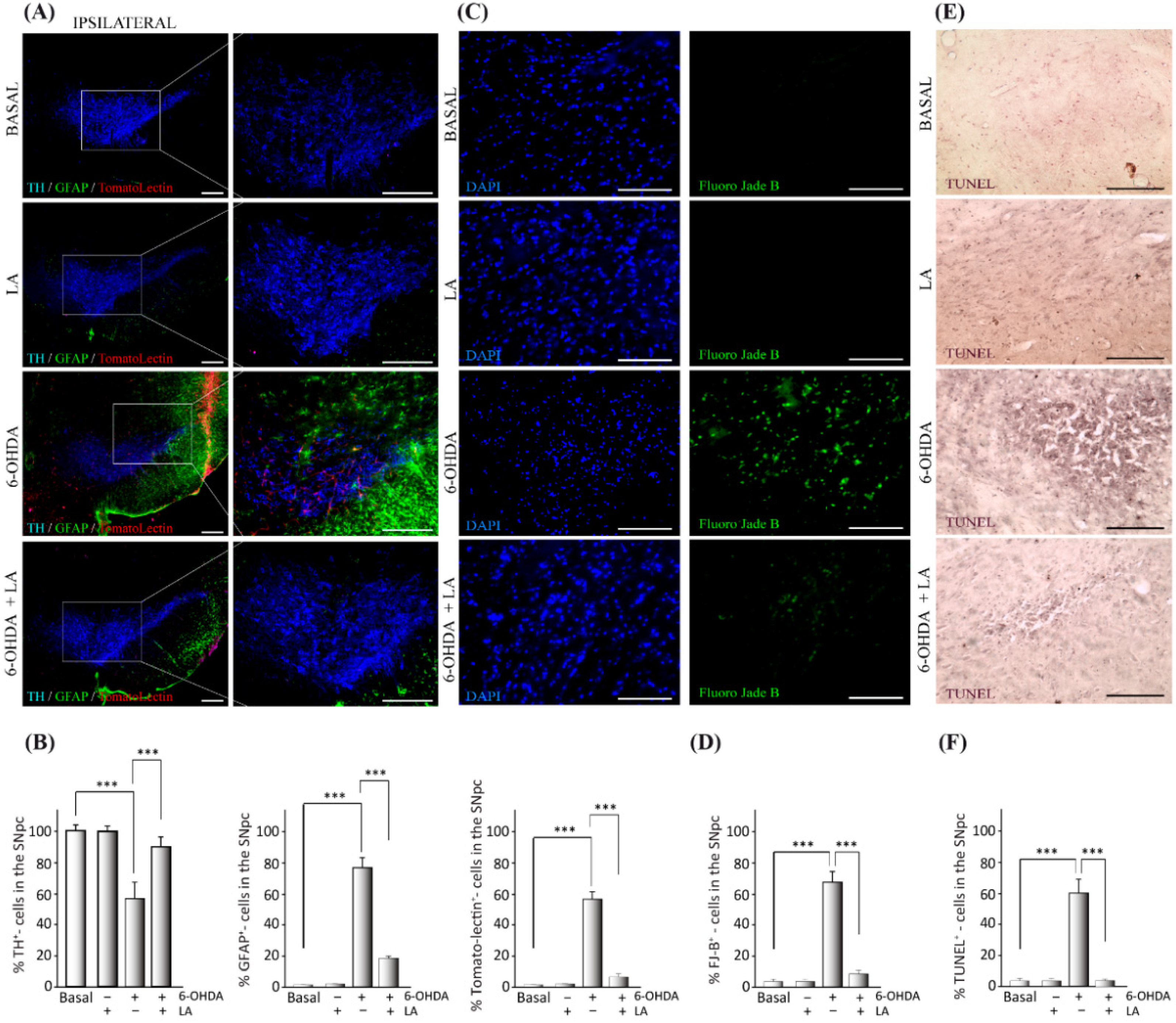
Our study demonstrates LA’s neuroprotective and anti-inflammatory effects using in vitro and in vivo models of PD. LA prevented the neurodegeneration of dopaminergic neurons in the SNpc of 6-OHDA damaged mice (
Figure 1 and
Figure 2) and the death of the SH-SY5Y cell line after damage with 6-OHDA (
Figure 3). Additionally, LA prevented the neuroinflammation presented in both in vitro and in vivo models (
Figure 1,
Figure 2 and
Figure 3). Furthermore, our study shows, for the first time, that the possible mechanism of action for LA in an in vitro PD model is through increasing the genesis of LDs and lipophagic flux to achieve an antioxidant effect. Further experiments are warranted to demonstrate the participation of lipids, lipid droplets, and autophagy/lipophagy in the pathogenesis of neurodegenerative diseases as well as the possible use of their regulation to treat these pathologies [
7,
31,
32].
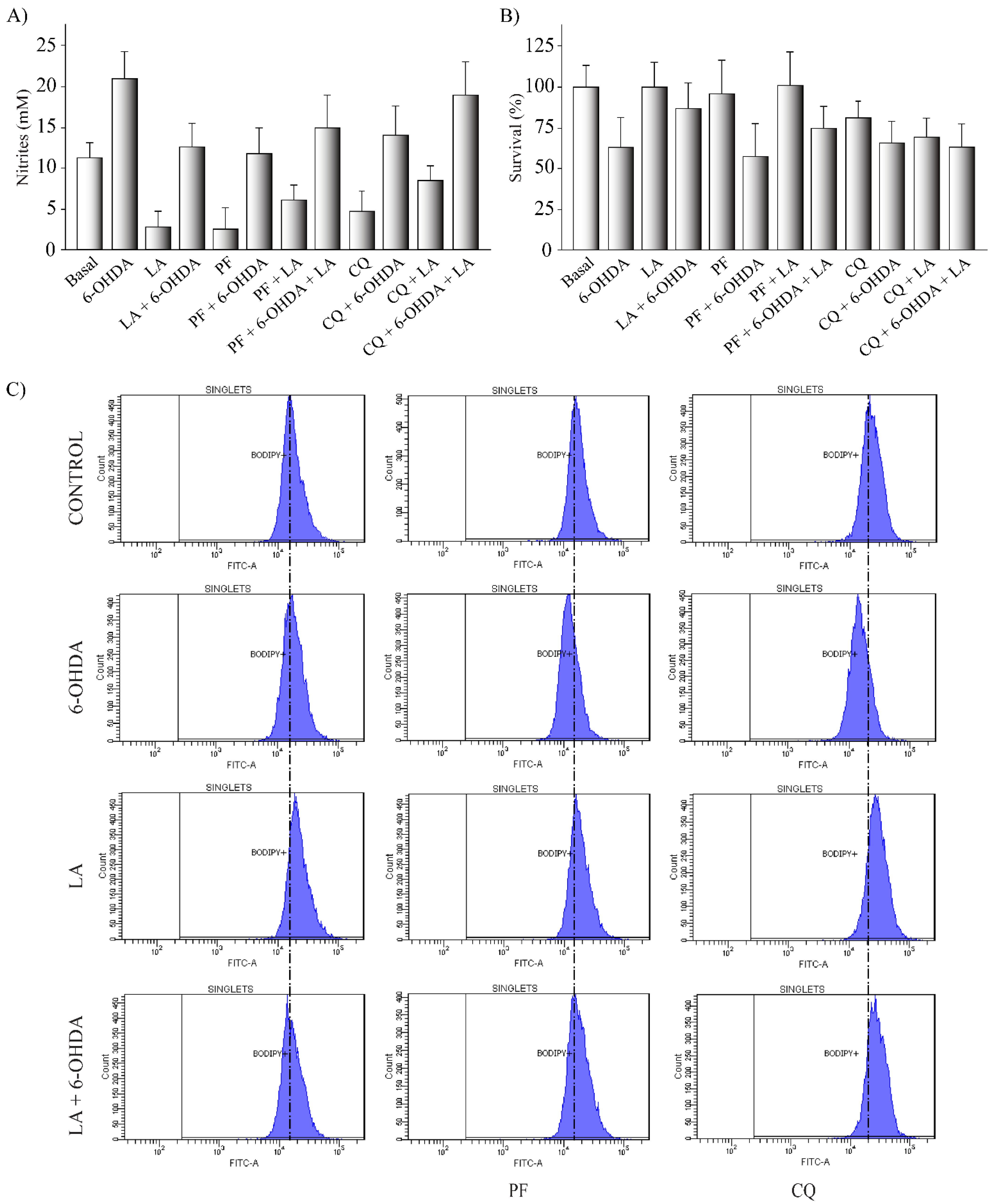
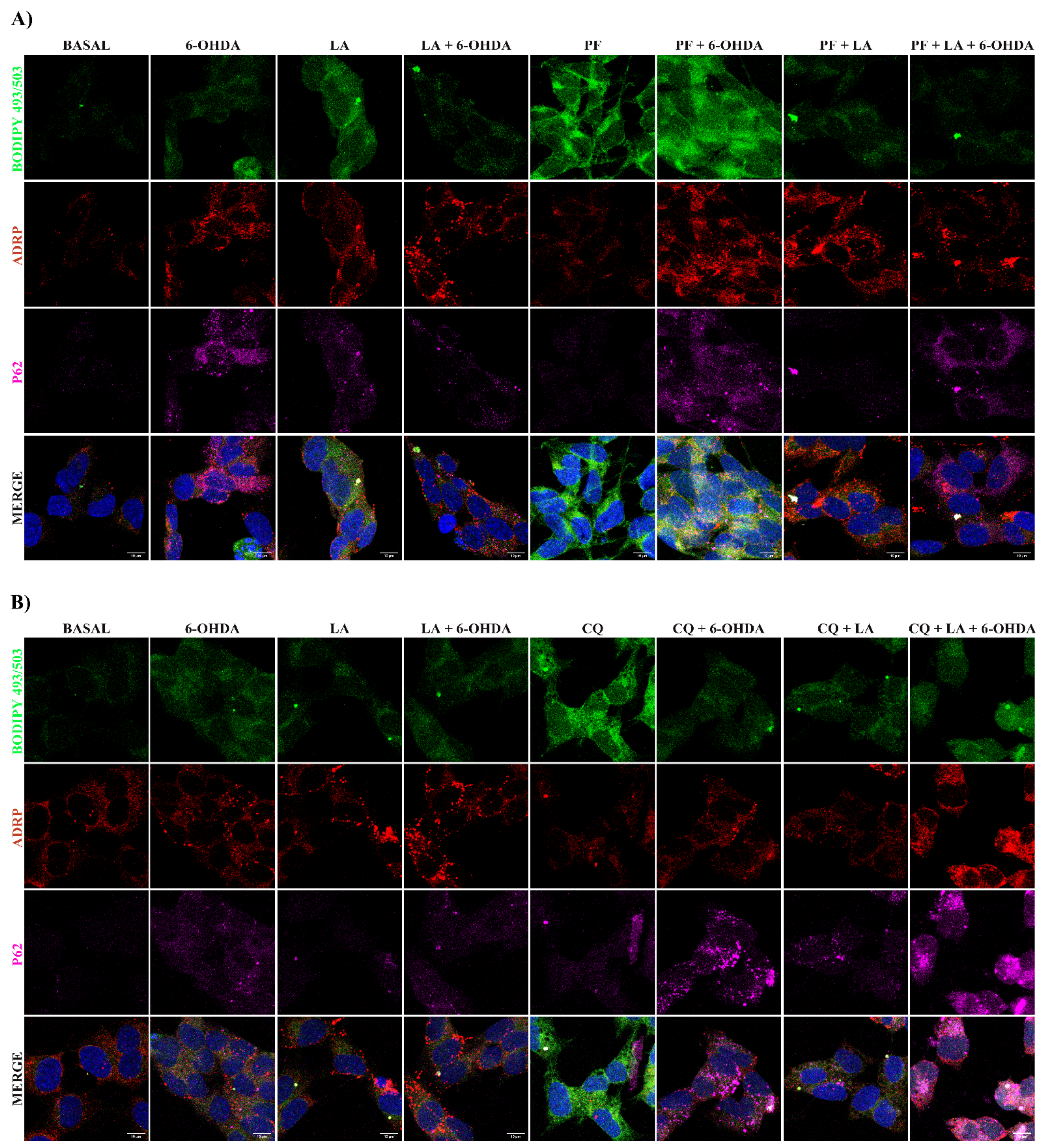
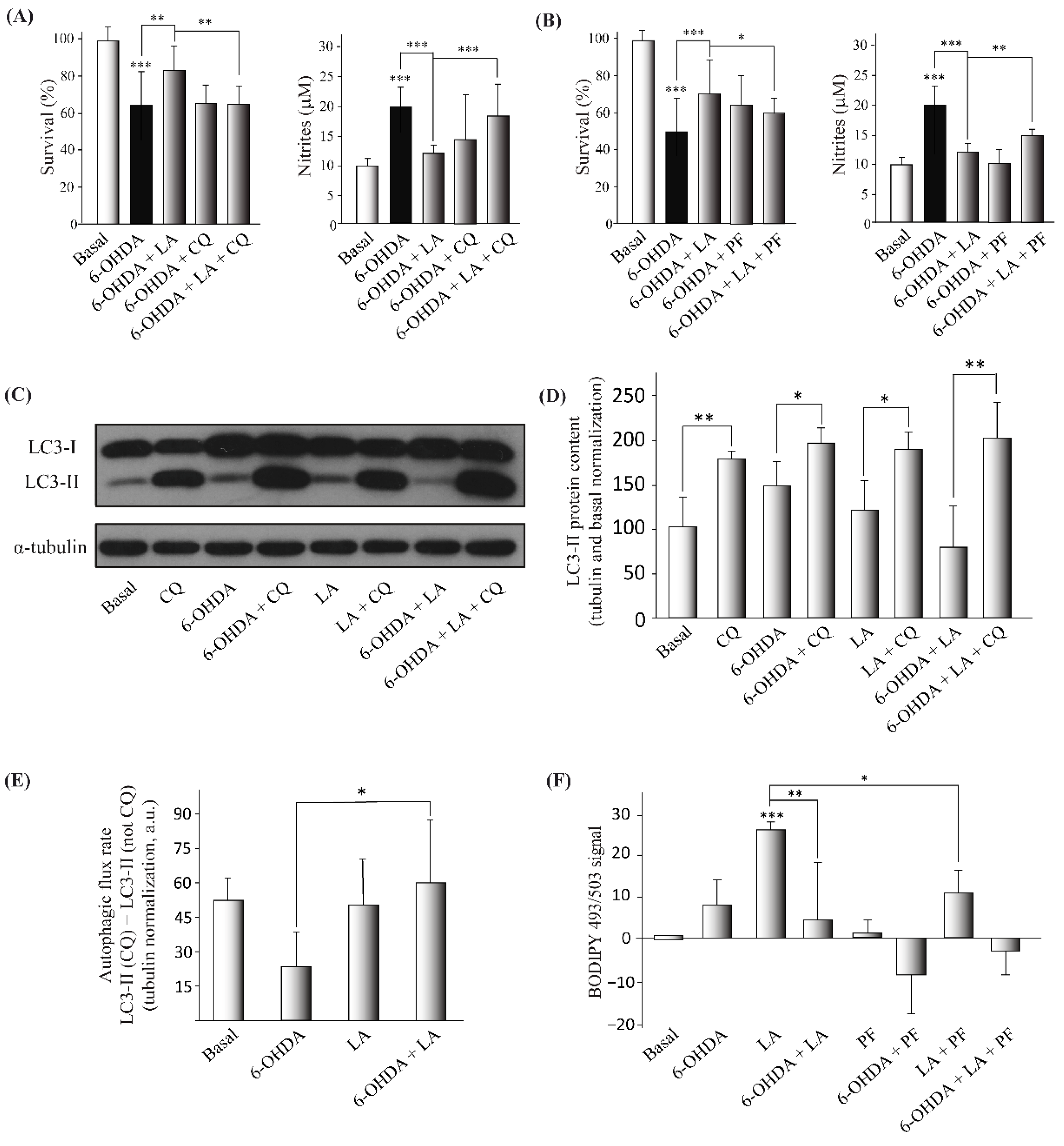
Regarding the possible mechanism of action of LA, we demonstrated that this fatty acid can induce the genesis of LDs under basal conditions (
Figure 4 and
Figure 5). This stimulation of the genesis of LDs by LA is dependent (at least in part) on the DGAT-2 enzyme (
Figure 4 and
Figure 5). However, the specific involvement of DGAT-2, as well as that of the DGAT-1 enzyme, needs to be verified in future studies. In this regard, the possible action mechanism of other fatty acids through lipid droplets biogenesis has been studied. Nakajima et al. [
19] showed that oleic acid induces the genesis of lipid droplets in astrocytes, achieving significant stimulation of their biogenesis after 24 h of treatment. This is the same amount of time that it takes for LA to achieve its best neuroprotective effect. Additionally, they verified that this lipid droplet genesis is dependent on DGAT-2 (although it is more dependent on DGAT-1). Thus, they found that oleic acid esterifies in triacylglycerols, so it is not unreasonable to think that the same could happen with LA in the SH-SY5Y cell line.
However, the PD model treated with LA did not show an increase in the level of LDs. This difference was probably due to the degradation of LDs generated by LA. Indeed, we observed a possible induction of lipophagy (triggered by LA), which could underlie the decrease in the LD levels in SH-SY5Y cells treated with LA and 6-OHDA (
Figure 4 and
Figure 5). Although we also observed a tendency of LA to induce lipophagy in a basal situation (
Figure 4 and
Figure 5), its induction by LA mainly occurred in the PD model (cellular stress). However, more studies are needed to analyze other possible mechanisms for LD degradation (such as lipolysis) and to prevent possible side effects of the treatment. Yang et al. [
33] proved that LA stimulates autophagic flux and an antioxidant response in hepatocytes and demonstrated the presence of a positive feedback loop between autophagy induction and antioxidant response by LA, which may coincide with our proposed “LD’ recycling mechanism”.
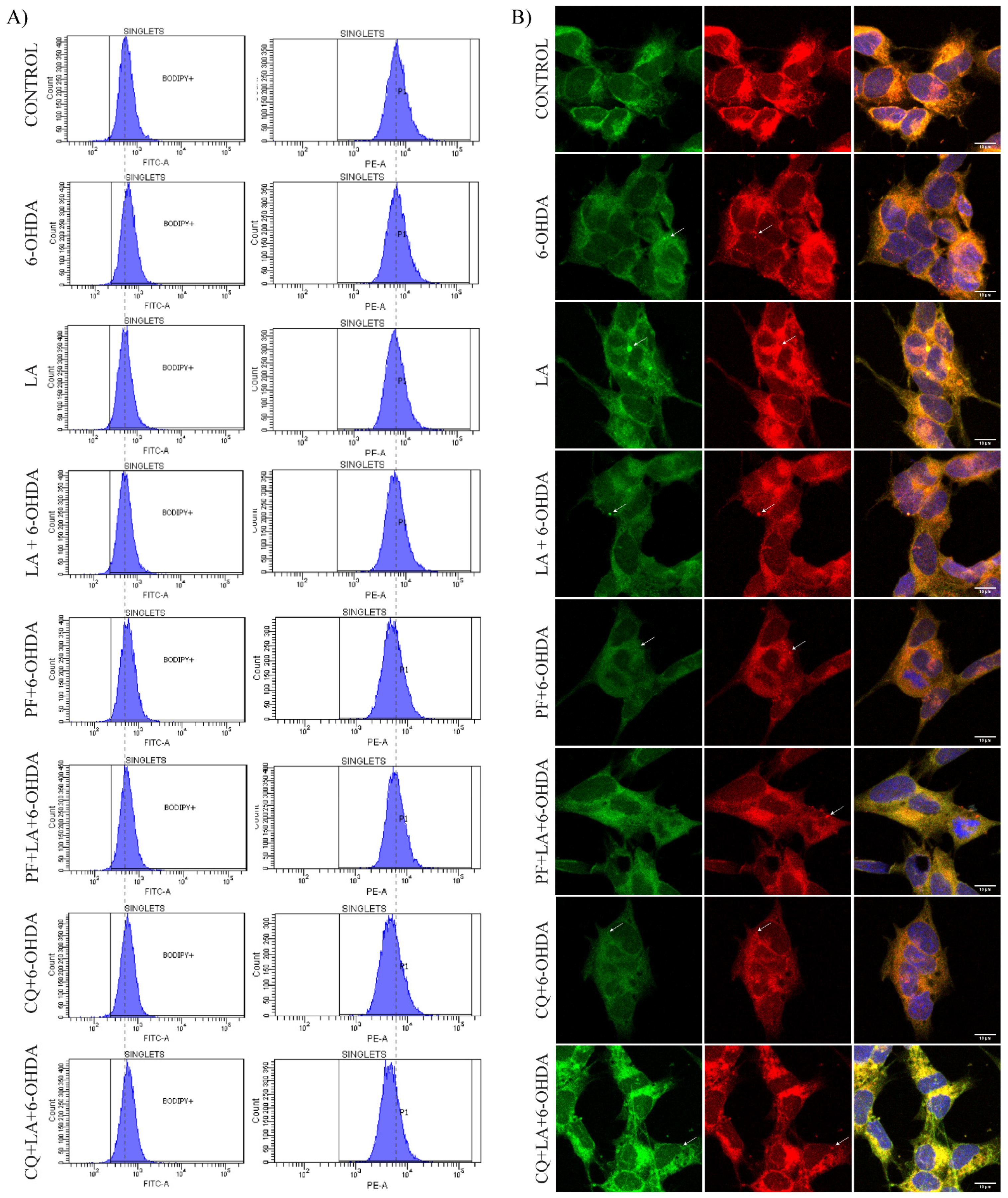
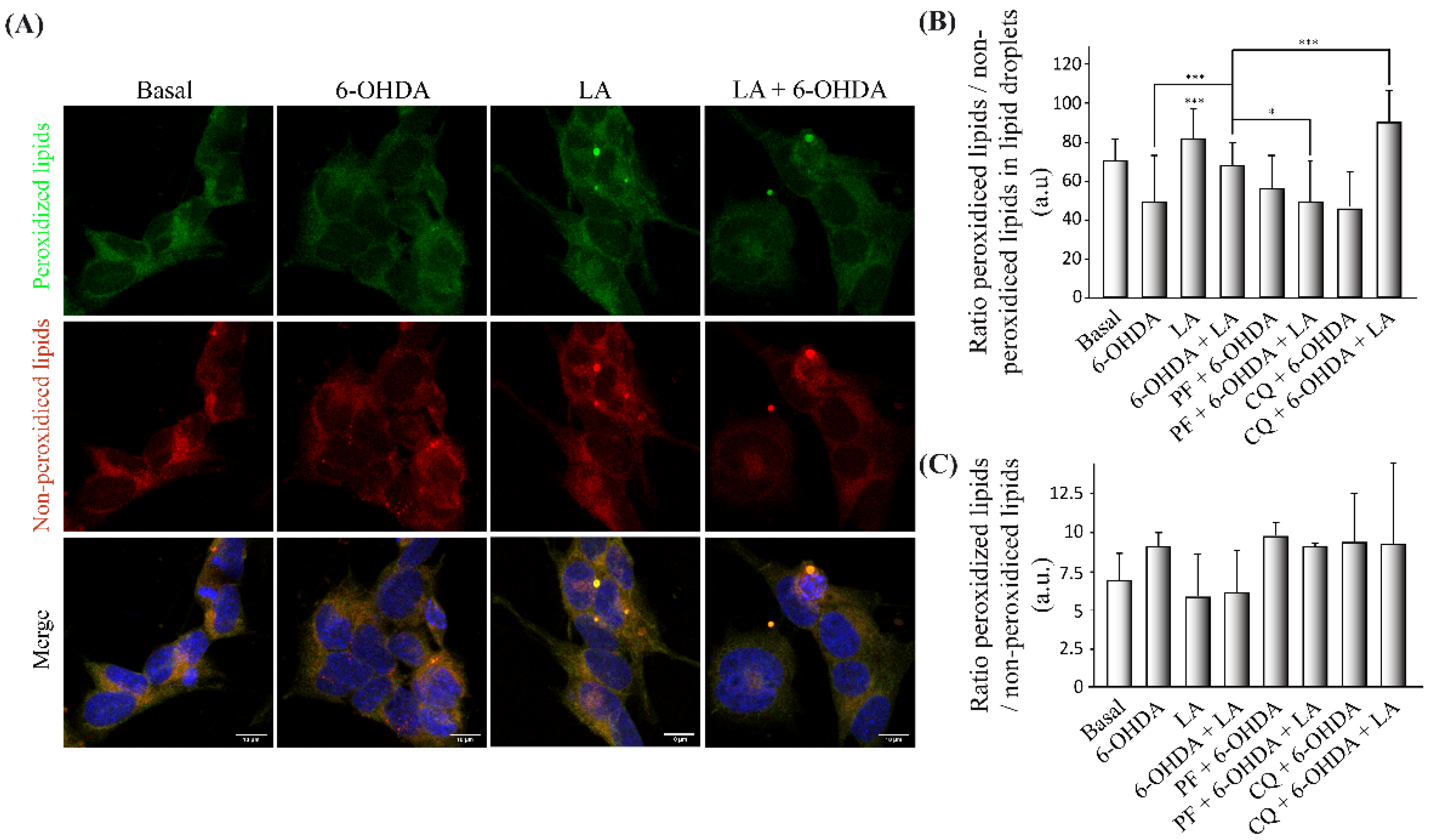
Regarding this antioxidant response of LA, we observed that LA caused an increase in the oxidative state of LDs and a possible decrease in the lipid peroxidation state of the cells (
Figure 6) under SH-SY5Y basal conditions and in the PD model. Thus, LDs may sequester the oxidative stress present in the PD model. In this way, the inhibition of DGAT-2 with PF prevented the peroxidation of LD and produced a lipid peroxidation state improvement for LA in the SH-SY5Y PD model. Moreover, inhibition of the autophagic flux with CQ increased the peroxidation of LD and improved the lipid peroxidation state in the SH-SY5Y PD model treated with LA. These results show the importance of the stimulation of LD biogenesis by LA for its oxidant scavenger effect and to stimulate autophagic/lipophagic flux by LA to remove the accumulation of peroxidized LD. Given these data, it is necessary to verify the exact mechanism by which LDs sequester free radicals, given that we already have previous indications of the possible existence of this mechanism [
34,
35]. Additionally, other studies have observed the ability of LDs to protect different nerve cell types from oxidative stress [
13,
15,
16].
Thus, our proposed mechanism of action, “LD recycling” (increasing genesis as well as degradation), may be a protective mechanism against lipocytotoxity (which causes a high level of oxidative stress) that gets rid of excess lipids. The accumulation of altered LDs in neural cell types can lead to pathological situations [
12,
31,
36,
37]. However, this mechanism could reduce the oxidative stress present in neurodegenerative diseases with specific concentrations of fatty acids (as has been shown in this study). This therapeutic strategy may have positive effects on the cellular oxidative state [
13,
15,
16], proteostasis [
6,
32], the stability of the membranes [
34], etc. Thus, our study increases the body of knowledge regarding the genesis of LDs, their antioxidant capacity, and the participation of lipophagy in a neuroprotective physiological process using in vitro and in vivo PD models. On the other hand, Cui et al. [
38] recently demonstrated an extracellular flux of fatty acids due to lipophagy through exocytosis that could explain the future of LDs degraded by lipophagy found in this study. Furthermore, this evidence may relate to recent research showing a lipid metabolic coupling between neurons and astrocytes [
13,
14], whose failure is related to the presence of Apolipoprotein E4 and its alterations (the principal risk factor for Alzheimer’s disease) [
14,
31]. Consequently, these cellular effects of LA may be part of a broader physiological mechanism of action against lipocytoxicity. This mechanism could participate in the pathogenesis of neurodegenerative diseases and could be used as a novel treatment for these disorders. A limitation of the in vitro model used in this study is the partially oncogenic phenotype of the SH-SY5Y cells. Hence, carrying out future experiments in cultures that include all neural cell lines, such as mesencephalic organoids [
39], is needed to allow the analysis of the relationship between neurons and glial cells on primary cultures and to verify our results.
In summary, the results presented here corroborate the neuroprotective and anti-inflammatory effects of LA against PD and suggest, for the first time, a new mechanism of action of LA for the treatment of neurodegenerative disorders, which underlies LDs, lipophagy, and oxidative stress processes. Thus, our study opens the door for further research on the use of coordinated regulation of the genesis of lipid droplets and lipophagy to regulate oxidative stress as a new therapeutic strategy against these diseases.


 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}