
Peroxisome Proliferator-Activated Receptors and the Hallmarks of Cancer



Abstract
:1. Introduction
2. PPARs and Cell Proliferation
2.1. PPARα
2.2. PPARβ/δ
2.3. PPARγ
3. PPARs and Cell Death
3.1. PPARα
3.2. PPARβ/δ
3.3. PPARγ
4. PPARs and Angiogenesis
4.1. PPARα
4.2. PPARβ/δ
4.3. PPARγ
5. PPARs and Tumor Suppressors
5.1. PPARα
5.2. PPARβ/δ
5.3. PPARγ
6. PPARs in Invasion and Metastasis
6.1. PPARα
6.2. PPARβ/δ
6.3. PPARγ
7. PPARs and Replicative Immortality
7.1. PPARα
7.2. PPARβ/δ
7.3. PPARγ
8. PPARs and Tumor Metabolism
8.1. PPARα
8.2. PPARβ/δ
8.3. PPARγ
9. PPARs and Cancer Immunity
9.1. PPARα
9.2. PPARβ/δ
9.3. PPARγ
10. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Wagner, K.D.; Wagner, N. Peroxisome proliferator-activated receptor beta/delta (PPARβ/δ) acts as regulator of metabolism linked to multiple cellular functions. Pharmacol. Ther. 2010, 125, 423–435. [Google Scholar] [CrossRef]
- Miyachi, H. Structural Biology-Based Exploration of Subtype-Selective Agonists for Peroxisome Proliferator-Activated Receptors. Int. J. Mol. Sci. 2021, 22, 9223. [Google Scholar] [CrossRef] [PubMed]
- Moody, D.E.; Reddy, J.K. Increase in hepatic carnitine acetyltransferase activity associated with peroxisomal (microbody) proliferation induced by the hypolipidemic drugs clofibrate, nafenopin, and methyl clofenapate. Res. Commun. Chem. Pathol. Pharmacol. 1974, 9, 501–510. [Google Scholar] [PubMed]
- De Duve, C. Evolution of the peroxisome. Ann. N. Y. Acad. Sci. 1969, 168, 369–381. [Google Scholar] [CrossRef] [PubMed]
- Issemann, I.; Green, S. Activation of a member of the steroid hormone receptor superfamily by peroxisome proliferators. Nature 1990, 347, 645–650. [Google Scholar] [CrossRef] [PubMed]
- Göttlicher, M.; Widmark, E.; Li, Q.; Gustafsson, J.A. Fatty acids activate a chimera of the clofibric acid-activated receptor and the glucocorticoid receptor. Proc. Natl. Acad. Sci. USA 1992, 89, 4653–4657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dreyer, C.; Krey, G.; Keller, H.; Givel, F.; Helftenbein, G.; Wahli, W. Control of the peroxisomal β-oxidation pathway by a novel family of nuclear hormone receptors. Cell 1992, 68, 879–887. [Google Scholar] [CrossRef]
- Plutzky, J. The PPAR-RXR transcriptional complex in the vasculature: Energy in the balance. Circ. Res. 2011, 108, 1002–1016. [Google Scholar] [CrossRef]
- De Bosscher, K.; Desmet, S.J.; Clarisse, D.; Estébanez-Perpiña, E.; Brunsveld, L. Nuclear receptor crosstalk—Defining the mechanisms for therapeutic innovation. Nat. Rev. Endocrinol. 2020, 16, 363–377. [Google Scholar] [CrossRef] [PubMed]
- Palmer, C.N.; Hsu, M.H.; Griffin, H.J.; Johnson, E.F. Novel sequence determinants in peroxisome proliferator signaling. J. Biol. Chem. 1995, 270, 16114–16121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagner, K.D.; Du, S.; Martin, L.; Leccia, N.; Michiels, J.F.; Wagner, N. Vascular PPARβ/δ Promotes Tumor Angiogenesis and Progression. Cells 2019, 8, 1623. [Google Scholar] [CrossRef] [Green Version]
- Fougerat, A.; Montagner, A.; Loiseau, N.; Guillou, H.; Wahli, W. Peroxisome Proliferator-Activated Receptors and Their Novel Ligands as Candidates for the Treatment of Non-Alcoholic Fatty Liver Disease. Cells 2020, 9, 1638. [Google Scholar] [CrossRef] [PubMed]
- Feige, J.N.; Auwerx, J. Transcriptional coregulators in the control of energy homeostasis. Trends Cell Biol. 2007, 17, 292–301. [Google Scholar] [CrossRef] [PubMed]
- Grygiel-Górniak, B. Peroxisome proliferator-activated receptors and their ligands: Nutritional and clinical implications—A review. Nutr. J. 2014, 13, 17. [Google Scholar] [CrossRef] [Green Version]
- Montaigne, D.; Butruille, L.; Staels, B. PPAR control of metabolism and cardiovascular functions. Nat. Rev. Cardiol. 2021, 18, 809–823. [Google Scholar] [CrossRef]
- Wagner, K.D.; Wagner, N. PPARs and Myocardial Infarction. Int. J. Mol. Sci. 2020, 21, 9436. [Google Scholar] [CrossRef] [PubMed]
- Marx, N.; Davies, M.J.; Grant, P.J.; Mathieu, C.; Petrie, J.R.; Cosentino, F.; Buse, J.B. Guideline recommendations and the positioning of newer drugs in type 2 diabetes care. Lancet Diabetes Endocrinol. 2021, 9, 46–52. [Google Scholar] [CrossRef]
- Wagner, N.; Wagner, K.D. The Role of PPARs in Disease. Cells 2020, 9, 2367. [Google Scholar] [CrossRef] [PubMed]
- Sáez-Orellana, F.; Octave, J.N.; Pierrot, N. Alzheimer’s Disease, a Lipid Story: Involvement of Peroxisome Proliferator-Activated Receptor α. Cells 2020, 9, 1215. [Google Scholar] [CrossRef] [PubMed]
- Matheson, J.; Le Foll, B. Therapeutic Potential of Peroxisome Proliferator-Activated Receptor (PPAR) Agonists in Substance Use Disorders: A Synthesis of Preclinical and Human Evidence. Cells 2020, 9, 1996. [Google Scholar] [CrossRef]
- Quiroga, C.; Barberena, J.J.; Alcaraz-Silva, J.; Machado, S.; Imperatori, C.; Yadollahpour, A.; Budde, H.; Yamamoto, T.; Telles-Correia, D.; Murillo-Rodríguez, E. The Role of Peroxisome Proliferator-Activated Receptor in Addiction: A Novel Drug Target. Curr. Top. Med. Chem. 2021, 21, 964–975. [Google Scholar] [CrossRef] [PubMed]
- Elias, E.; Zhang, A.Y.; Manners, M.T. Novel Pharmacological Approaches to the Treatment of Depression. Life 2022, 12, 196. [Google Scholar] [CrossRef] [PubMed]
- Luan, Z.L.; Zhang, C.; Ming, W.H.; Huang, Y.Z.; Guan, Y.F.; Zhang, X.Y. Nuclear receptors in renal health and disease. EBioMedicine 2022, 76, 103855. [Google Scholar] [CrossRef]
- Mantovani, A.; Byrne, C.D.; Targher, G. Efficacy of peroxisome proliferator-activated receptor agonists, glucagon-like peptide-1 receptor agonists, or sodium-glucose cotransporter-2 inhibitors for treatment of non-alcoholic fatty liver disease: A systematic review. Lancet Gastroenterol. Hepatol. 2022, 7, 367–378. [Google Scholar] [CrossRef]
- Kökény, G.; Calvier, L.; Hansmann, G. PPARγ and TGFβ-Major Regulators of Metabolism, Inflammation, and Fibrosis in the Lungs and Kidneys. Int. J. Mol. Sci. 2021, 22, 10431. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, J.; Luo, S.; Zhan, Y.; Lu, Q. The roles of PPARγ and its agonists in autoimmune diseases: A comprehensive review. J. Autoimmun. 2020, 113, 102510. [Google Scholar] [CrossRef] [PubMed]
- Toobian, D.; Ghosh, P.; Katkar, G.D. Parsing the Role of PPARs in Macrophage Processes. Front. Immunol. 2021, 12, 783780. [Google Scholar] [CrossRef] [PubMed]
- Gerussi, A.; Lucà, M.; Cristoferi, L.; Ronca, V.; Mancuso, C.; Milani, C.; D’Amato, D.; O’Donnell, S.E.; Carbone, M.; Invernizzi, P. New Therapeutic Targets in Autoimmune Cholangiopathies. Front. Med. 2020, 7, 117. [Google Scholar] [CrossRef] [PubMed]
- Rzemieniec, J.; Castiglioni, L.; Gelosa, P.; Muluhie, M.; Mercuriali, B.; Sironi, L. Nuclear Receptors in Myocardial and Cerebral Ischemia-Mechanisms of Action and Therapeutic Strategies. Int. J. Mol. Sci. 2021, 22, 12326. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.S.; Yip, Y.S.; Lim, E.K.Y.; Wahli, W.; Tan, N.S. PPARs and Tumor Microenvironment: The Emerging Roles of the Metabolic Master Regulators in Tumor Stromal-Epithelial Crosstalk and Carcinogenesis. Cancers 2021, 13, 2153. [Google Scholar] [CrossRef] [PubMed]
- Wagner, K.D.; Cherfils-Vicini, J.; Hosen, N.; Hohenstein, P.; Gilson, E.; Hastie, N.D.; Michiels, J.F.; Wagner, N. The Wilms’ tumour suppressor Wt1 is a major regulator of tumour angiogenesis and progression. Nat. Commun. 2014, 5, 5852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Erez, N.; Truitt, M.; Olson, P.; Arron, S.T.; Hanahan, D. Cancer-Associated Fibroblasts Are Activated in Incipient Neoplasia to Orchestrate Tumor-Promoting Inflammation in an NF-κB-Dependent Manner. Cancer Cell 2010, 17, 135–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nozawa, H.; Chiu, C.; Hanahan, D. Infiltrating neutrophils mediate the initial angiogenic switch in a mouse model of multistage carcinogenesis. Proc. Natl. Acad. Sci. USA 2006, 103, 12493–12498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergers, G.; Brekken, R.; McMahon, G.; Vu, T.H.; Itoh, T.; Tamaki, K.; Tanzawa, K.; Thorpe, P.; Itohara, S.; Werb, Z.; et al. Matrix metalloproteinase-9 triggers the angiogenic switch during carcinogenesis. Nat. Cell Biol. 2000, 2, 737–744. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Folkman, J. Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell 1996, 86, 353–364. [Google Scholar] [CrossRef] [Green Version]
- Wagner, N.; Wagner, K.D. PPAR Beta/Delta and the Hallmarks of Cancer. Cells 2020, 9, 1133. [Google Scholar] [CrossRef] [PubMed]
- Suchanek, K.M.; May, F.J.; Robinson, J.A.; Lee, W.J.; Holman, N.A.; Monteith, G.R.; Roberts-Thomson, S.J. Peroxisome proliferator-activated receptor α in the human breast cancer cell lines MCF-7 and MDA-MB-231. Mol. Carcinog. 2002, 34, 165–171. [Google Scholar] [CrossRef] [PubMed]
- Okumura, M.; Yamamoto, M.; Sakuma, H.; Kojima, T.; Maruyama, T.; Jamali, M.; Cooper, D.R.; Yasuda, K. Leptin and high glucose stimulate cell proliferation in MCF-7 human breast cancer cells: Reciprocal involvement of PKC-α and PPAR expression. Biochim. Biophys. Acta 2002, 1592, 107–116. [Google Scholar] [CrossRef] [Green Version]
- Chang, N.W.; Wu, C.T.; Chen, D.R.; Yeh, C.Y.; Lin, C. High levels of arachidonic acid and peroxisome proliferator-activated receptor-alpha in breast cancer tissues are associated with promoting cancer cell proliferation. J. Nutr. Biochem. 2013, 24, 274–281. [Google Scholar] [CrossRef]
- Bocca, C.; Bozzo, F.; Martinasso, G.; Canuto, R.A.; Miglietta, A. Involvement of PPARα in the growth inhibitory effect of arachidonic acid on breast cancer cells. Br. J. Nutr. 2008, 100, 739–750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, T.; Zhang, Q.; Zhang, J.; Yang, G.; Shao, Z.; Luo, J.; Fan, M.; Ni, C.; Wu, Z.; Hu, X. Fenofibrate induces apoptosis of triple-negative breast cancer cells via activation of NF-κB pathway. BMC Cancer 2014, 14, 96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandran, K.; Goswami, S.; Sharma-Walia, N. Implications of a peroxisome proliferator-activated receptor alpha (PPARα) ligand clofibrate in breast cancer. Oncotarget 2016, 7, 15577–15599. [Google Scholar] [CrossRef] [PubMed]
- Tauber, Z.; Koleckova, M.; Cizkova, K. Peroxisome proliferator-activated receptor ɑ (PPARɑ)-cytochrome P450 epoxygenases-soluble epoxide hydrolase axis in ER + PR + HER2− breast cancer. Med. Mol. Morphol. 2020, 53, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Jiang, H.Y.; Wang, Z.H.; Ma, Y.C.; Bao, Y.N.; Jin, Y. Effect of fenofibrate on proliferation of SMMC-7721 cells via regulating cell cycle. Hum. Exp. Toxicol. 2021, 40, 1208–1221. [Google Scholar] [CrossRef]
- Morimura, K.; Cheung, C.; Ward, J.M.; Reddy, J.K.; Gonzalez, F.J. Differential susceptibility of mice humanized for peroxisome proliferator-activated receptor α to Wy-14,643-induced liver tumorigenesis. Carcinogenesis 2006, 27, 1074–1080. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, N.; Moriya, K.; Kiyosawa, K.; Koike, K.; Aoyama, T. Hepatitis C virus core protein induces spontaneous and persistent activation of peroxisome proliferator-activated receptor α in transgenic mice: Implications for HCV-associated hepatocarcinogenesis. Int. J. Cancer 2008, 122, 124–131. [Google Scholar] [CrossRef] [Green Version]
- Yang, Q.; Ito, S.; Gonzalez, F.J. Hepatocyte-restricted constitutive activation of PPARα induces hepatoproliferation but not hepatocarcinogenesis. Carcinogenesis 2007, 28, 1171–1177. [Google Scholar] [CrossRef]
- Gervois, P.; Torra, I.P.; Chinetti, G.; Grötzinger, T.; Dubois, G.; Fruchart, J.C.; Fruchart-Najib, J.; Leitersdorf, E.; Staels, B. A truncated human peroxisome proliferator-activated receptor α splice variant with dominant negative activity. Mol. Endocrinol. 1999, 13, 1535–1549. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.; Bayha, C.; Klein, K.; Müller, S.; Weiss, T.S.; Schwab, M.; Zanger, U.M. The truncated splice variant of peroxisome proliferator-activated receptor alpha, PPARα-tr, autonomously regulates proliferative and pro-inflammatory genes. BMC Cancer 2015, 15, 488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, N.; Chu, E.S.; Zhang, J.; Li, X.; Liang, Q.; Chen, J.; Chen, M.; Teoh, N.; Farrell, G.; Sung, J.J.; et al. Peroxisome proliferator activated receptor alpha inhibits hepatocarcinogenesis through mediating NF-κB signaling pathway. Oncotarget 2014, 5, 8330–8340. [Google Scholar] [CrossRef] [Green Version]
- Kaipainen, A.; Kieran, M.W.; Huang, S.; Butterfield, C.; Bielenberg, D.; Mostoslavsky, G.; Mulligan, R.; Folkman, J.; Panigrahy, D. PPARα deficiency in inflammatory cells suppresses tumor growth. PLoS ONE 2007, 2, e260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hichami, A.; Yessoufou, A.; Ghiringhelli, F.; Salvadori, F.; Moutairou, K.; Zwetyenga, N.; Khan, N.A. Peroxisome proliferator-activated receptor alpha deficiency impairs regulatory T cell functions: Possible application in the inhibition of melanoma tumor growth in mice. Biochimie 2016, 131, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Saidi, S.A.; Holland, C.M.; Charnock-Jones, D.S.; Smith, S.K. In vitro and in vivo effects of the PPAR-alpha agonists fenofibrate and retinoic acid in endometrial cancer. Mol. Cancer 2006, 5, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yokoyama, Y.; Xin, B.; Shigeto, T.; Umemoto, M.; Kasai-Sakamoto, A.; Futagami, M.; Tsuchida, S.; Al-Mulla, F.; Mizunuma, H. Clofibric acid, a peroxisome proliferator-activated receptor α ligand, inhibits growth of human ovarian cancer. Mol. Cancer Ther. 2007, 6, 1379–1386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urbanska, K.; Pannizzo, P.; Grabacka, M.; Croul, S.; Del Valle, L.; Khalili, K.; Reiss, K. Activation of PPARα inhibits IGF-I-mediated growth and survival responses in medulloblastoma cell lines. Int. J. Cancer 2008, 123, 1015–1024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, D.F.; Zhang, J.X.; Wei, W.J.; Tao, T.; Hu, Q.; Wang, Y.Y.; Wang, X.F.; Liu, N.; You, Y.P. Fenofibrate induces G0/G1 phase arrest by modulating the PPARα/FoxO1/p27 kip pathway in human glioblastoma cells. Tumour Biol. 2015, 36, 3823–3829. [Google Scholar] [CrossRef]
- Su, C.; Shi, A.; Cao, G.; Tao, T.; Chen, R.; Hu, Z.; Shen, Z.; Tao, H.; Cao, B.; Hu, D.; et al. Fenofibrate suppressed proliferation and migration of human neuroblastoma cells via oxidative stress dependent of TXNIP upregulation. Biochem. Biophys. Res. Commun. 2015, 460, 983–988. [Google Scholar] [CrossRef] [PubMed]
- Haynes, H.R.; Scott, H.L.; Killick-Cole, C.L.; Shaw, G.; Brend, T.; Hares, K.M.; Redondo, J.; Kemp, K.C.; Ballesteros, L.S.; Herman, A.; et al. shRNA-mediated PPARα knockdown in human glioma stem cells reduces in vitro proliferation and inhibits orthotopic xenograft tumour growth. J. Pathol. 2019, 247, 422–434. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wang, Y.; Huang, Y.; Zeng, H.; Hu, B.; Guan, L.; Zhang, H.; Yu, A.M.; Johnson, C.H.; Gonzalez, F.J.; et al. PPARα regulates tumor cell proliferation and senescence via a novel target gene carnitine palmitoyltransferase 1C. Carcinogenesis 2017, 38, 474–483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stebbins, K.J.; Broadhead, A.R.; Cabrera, G.; Correa, L.D.; Messmer, D.; Bundey, R.; Baccei, C.; Bravo, Y.; Chen, A.; Stock, N.S.; et al. In vitro and in vivo pharmacology of NXT629, a novel and selective PPARα antagonist. Eur. J. Pharmacol. 2017, 809, 130–140. [Google Scholar] [CrossRef] [PubMed]
- Li, M.Y.; Yuan, H.; Ma, L.T.; Kong, A.W.; Hsin, M.K.; Yip, J.H.; Underwood, M.J.; Chen, G.G. Roles of peroxisome proliferator-activated receptor-α and -γ in the development of non-small cell lung cancer. Am. J. Respir. Cell Mol. Biol. 2010, 43, 674–683. [Google Scholar] [CrossRef]
- Liang, H.; Kowalczyk, P.; Junco, J.J.; Klug-De Santiago, H.L.; Malik, G.; Wei, S.J.; Slaga, T.J. Differential effects on lung cancer cell proliferation by agonists of glucocorticoid and PPARα receptors. Mol. Carcinog. 2014, 53, 753–763. [Google Scholar] [CrossRef] [PubMed]
- Skrypnyk, N.; Chen, X.; Hu, W.; Su, Y.; Mont, S.; Yang, S.; Gangadhariah, M.; Wei, S.; Falck, J.R.; Jat, J.L.; et al. PPARα activation can help prevent and treat non-small cell lung cancer. Cancer Res. 2014, 74, 621–631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pozzi, A.; Ibanez, M.R.; Gatica, A.E.; Yang, S.; Wei, S.; Mei, S.; Falck, J.R.; Capdevila, J.H. Peroxisomal proliferator-activated receptor-α-dependent inhibition of endothelial cell proliferation and tumorigenesis. J. Biol. Chem. 2007, 282, 17685–17695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, J.; Das, S.K.; Jha, P.; Al Zoughbi, W.; Schauer, S.; Claudel, T.; Sexl, V.; Vesely, P.; Birner-Gruenberger, R.; Kratky, D.; et al. The PPARα agonist fenofibrate suppresses B-cell lymphoma in mice by modulating lipid metabolism. Biochim. Biophys. Acta 2013, 1831, 1555–1565. [Google Scholar] [CrossRef] [Green Version]
- Kong, R.; Wang, N.; Han, W.; Bao, W.; Lu, J. Fenofibrate Exerts Antitumor Effects in Colon Cancer via Regulation of DNMT1 and CDKN2A. PPAR Res. 2021, 2021, 6663782. [Google Scholar] [CrossRef] [PubMed]
- Berger, H.; Végran, F.; Chikh, M.; Gilardi, F.; Ladoire, S.; Bugaut, H.; Mignot, G.; Chalmin, F.; Bruchard, M.; Derangère, V.; et al. SOCS3 transactivation by PPARγ prevents IL-17-driven cancer growth. Cancer Res. 2013, 73, 3578–3590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takayama, O.; Yamamoto, H.; Damdinsuren, B.; Sugita, Y.; Ngan, C.Y.; Xu, X.; Tsujino, T.; Takemasa, I.; Ikeda, M.; Sekimoto, M.; et al. Expression of PPARδ in multistage carcinogenesis of the colorectum: Implications of malignant cancer morphology. Br. J. Cancer 2006, 95, 889–895. [Google Scholar] [CrossRef]
- Yoshinaga, M.; Taki, K.; Somada, S.; Sakiyama, Y.; Kubo, N.; Kaku, T.; Tsuruta, S.; Kusumoto, T.; Sakai, H.; Nakamura, K.; et al. The expression of both peroxisome proliferator-activated receptor delta and cyclooxygenase-2 in tissues is associated with poor prognosis in colorectal cancer patients. Dig. Dis. Sci. 2011, 56, 1194–1200. [Google Scholar] [CrossRef] [PubMed]
- Foreman, J.E.; Chang, W.C.; Palkar, P.S.; Zhu, B.; Borland, M.G.; Williams, J.L.; Kramer, L.R.; Clapper, M.L.; Gonzalez, F.J.; Peters, J.M. Functional characterization of peroxisome proliferator-activated receptor-β/δ expression in colon cancer. Mol. Carcinog. 2011, 50, 884–900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, D.; Wang, H.; Guo, Y.; Ning, W.; Katkuri, S.; Wahli, W.; Desvergne, B.; Dey, S.K.; DuBois, R.N. Crosstalk between peroxisome proliferator-activated receptor δ and VEGF stimulates cancer progression. Proc. Natl. Acad. Sci. USA 2006, 103, 19069–19074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Röhrl, C.; Kaindl, U.; Koneczny, I.; Hudec, X.; Baron, D.M.; König, J.S.; Marian, B. Peroxisome-proliferator-activated receptors γ and β/δ mediate vascular endothelial growth factor production in colorectal tumor cells. J. Cancer Res. Clin. Oncol. 2011, 137, 29–39. [Google Scholar] [CrossRef]
- Zuo, X.; Peng, Z.; Moussalli, M.J.; Morris, J.S.; Broaddus, R.R.; Fischer, S.M.; Shureiqi, I. Targeted genetic disruption of peroxisome proliferator-activated receptor-δ and colonic tumorigenesis. J. Natl. Cancer Inst. 2009, 101, 762–767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Zhou, J.; Ma, Q.; Wang, C.; Chen, K.; Meng, W.; Yu, Y.; Zhou, Z.; Sun, X. Knockdown of PPAR δ gene promotes the growth of colon cancer and reduces the sensitivity to bevacizumab in nude mice model. PLoS ONE 2013, 8, e60715. [Google Scholar] [CrossRef] [Green Version]
- Gupta, R.A.; Wang, D.; Katkuri, S.; Wang, H.; Dey, S.K.; DuBois, R.N. Activation of nuclear hormone receptor peroxisome proliferator-activated receptor-δ accelerates intestinal adenoma growth. Nat. Med. 2004, 10, 245–247. [Google Scholar] [CrossRef]
- Ding, J.; Gou, Q.; Jin, J.; Shi, J.; Liu, Q.; Hou, Y. Metformin inhibits PPARδ agonist-mediated tumor growth by reducing Glut1 and SLC1A5 expressions of cancer cells. Eur. J. Pharmacol. 2019, 857, 172425. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Deguchi, Y.; Tian, R.; Wei, D.; Wu, L.; Chen, W.; Xu, W.; Xu, M.; Liu, F.; Gao, S.; et al. Pleiotropic Effects of PPARD Accelerate Colorectal Tumorigenesis, Progression, and Invasion. Cancer Res. 2019, 79, 954–969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuo, X.; Deguchi, Y.; Xu, W.; Liu, Y.; Li, H.S.; Wei, D.; Tian, R.; Chen, W.; Xu, M.; Yang, Y.; et al. PPARD and Interferon Gamma Promote Transformation of Gastric Progenitor Cells and Tumorigenesis in Mice. Gastroenterology 2019, 157, 163–178. [Google Scholar] [CrossRef]
- Zhou, D.; Jin, J.; Liu, Q.; Shi, J.; Hou, Y. PPARδ agonist enhances colitis-associated colorectal cancer. Eur. J. Pharmacol. 2019, 842, 248–254. [Google Scholar] [CrossRef] [PubMed]
- Zuo, X.; Xu, M.; Yu, J.; Wu, Y.; Moussalli, M.J.; Manyam, G.C.; Lee, S.I.; Liang, S.; Gagea, M.; Morris, J.S.; et al. Potentiation of colon cancer susceptibility in mice by colonic epithelial PPAR-δ/β overexpression. J. Natl. Cancer Inst. 2014, 106, dju052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marin, H.E.; Peraza, M.A.; Billin, A.N.; Willson, T.M.; Ward, J.M.; Kennett, M.J.; Gonzalez, F.J.; Peters, J.M. Ligand activation of peroxisome proliferator-activated receptor β inhibits colon carcinogenesis. Cancer Res. 2006, 66, 4394–4401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harman, F.S.; Nicol, C.J.; Marin, H.E.; Ward, J.M.; Gonzalez, F.J.; Peters, J.M. Peroxisome proliferator-activated receptor-δ attenuates colon carcinogenesis. Nat. Med. 2004, 10, 481–483. [Google Scholar] [CrossRef] [PubMed]
- Beyaz, S.; Mana, M.D.; Roper, J.; Kedrin, D.; Saadatpour, A.; Hong, S.J.; Bauer-Rowe, K.E.; Xifaras, M.E.; Akkad, A.; Arias, E.; et al. High-fat diet enhances stemness and tumorigenicity of intestinal progenitors. Nature 2016, 531, 53–58. [Google Scholar] [CrossRef]
- Nagy, T.A.; Wroblewski, L.E.; Wang, D.; Piazuelo, M.B.; Delgado, A.; Romero-Gallo, J.; Noto, J.; Israel, D.A.; Ogden, S.R.; Correa, P.; et al. β-Catenin and p120 mediate PPARδ-dependent proliferation induced by Helicobacter pylori in human and rodent epithelia. Gastroenterology 2011, 141, 553–564. [Google Scholar] [CrossRef] [Green Version]
- Girroir, E.E.; Hollingshead, H.E.; Billin, A.N.; Willson, T.M.; Robertson, G.P.; Sharma, A.K.; Amin, S.; Gonzalez, F.J.; Peters, J.M. Peroxisome proliferator-activated receptor-β/δ (PPARβ/δ) ligands inhibit growth of UACC903 and MCF7 human cancer cell lines. Toxicology 2008, 243, 236–243. [Google Scholar] [CrossRef] [Green Version]
- Yao, P.L.; Morales, J.L.; Zhu, B.; Kang, B.H.; Gonzalez, F.J.; Peters, J.M. Activation of peroxisome proliferator-activated receptor-β/δ (PPAR-β/δ) inhibits human breast cancer cell line tumorigenicity. Mol. Cancer Ther. 2014, 13, 1008–1017. [Google Scholar] [CrossRef] [Green Version]
- Yao, P.L.; Chen, L.; Dobrzański, T.P.; Zhu, B.; Kang, B.H.; Müller, R.; Gonzalez, F.J.; Peters, J.M. Peroxisome proliferator-activated receptor-β/δ inhibits human neuroblastoma cell tumorigenesis by inducing p53- and SOX2-mediated cell differentiation. Mol. Carcinog. 2017, 56, 1472–1483. [Google Scholar] [CrossRef]
- Yao, P.L.; Chen, L.P.; Dobrzański, T.P.; Phillips, D.A.; Zhu, B.; Kang, B.H.; Gonzalez, F.J.; Peters, J.M. Inhibition of testicular embryonal carcinoma cell tumorigenicity by peroxisome proliferator-activated receptor-β/δ- and retinoic acid receptor-dependent mechanisms. Oncotarget 2015, 6, 36319–36337. [Google Scholar] [CrossRef] [Green Version]
- Yuan, H.; Lu, J.; Xiao, J.; Upadhyay, G.; Umans, R.; Kallakury, B.; Yin, Y.; Fant, M.E.; Kopelovich, L.; Glazer, R.I. PPARδ induces estrogen receptor-positive mammary neoplasia through an inflammatory and metabolic phenotype linked to mTOR activation. Cancer Res. 2013, 73, 4349–4361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pollock, C.B.; Yin, Y.; Yuan, H.; Zeng, X.; King, S.; Li, X.; Kopelovich, L.; Albanese, C.; Glazer, R.I. PPARδ activation acts cooperatively with 3-phosphoinositide-dependent protein kinase-1 to enhance mammary tumorigenesis. PLoS ONE 2011, 6, e16215. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Russell, R.G.; Dettin, L.E.; Bai, R.; Wei, Z.L.; Kozikowski, A.P.; Kopelovich, L.; Kopleovich, L.; Glazer, R.I. Peroxisome proliferator-activated receptor δ and γ agonists differentially alter tumor differentiation and progression during mammary carcinogenesis. Cancer Res. 2005, 65, 3950–3957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghosh, M.; Ai, Y.; Narko, K.; Wang, Z.; Peters, J.M.; Hla, T. PPARδ is pro-tumorigenic in a mouse model of COX-2-induced mammary cancer. Prostaglandins Other Lipid Mediat. 2009, 88, 97–100. [Google Scholar] [CrossRef] [Green Version]
- Levi, L.; Lobo, G.; Doud, M.K.; von Lintig, J.; Seachrist, D.; Tochtrop, G.P.; Noy, N. Genetic ablation of the fatty acid-binding protein FABP5 suppresses HER2-induced mammary tumorigenesis. Cancer Res. 2013, 73, 4770–4780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schug, T.T.; Berry, D.C.; Toshkov, I.A.; Cheng, L.; Nikitin, A.Y.; Noy, N. Overcoming retinoic acid-resistance of mammary carcinomas by diverting retinoic acid from PPARβ/δ to RAR. Proc. Natl. Acad. Sci. USA 2008, 105, 7546–7551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morgan, E.; Kannan-Thulasiraman, P.; Noy, N. Involvement of Fatty Acid Binding Protein 5 and PPARβ/δ in Prostate Cancer Cell Growth. PPAR Res. 2010, 2010, 234629. [Google Scholar] [CrossRef] [Green Version]
- Her, N.G.; Jeong, S.I.; Cho, K.; Ha, T.K.; Han, J.; Ko, K.P.; Park, S.K.; Lee, J.H.; Lee, M.G.; Ryu, B.K.; et al. PPARδ promotes oncogenic redirection of TGF-β1 signaling through the activation of the ABCA1-Cav1 pathway. Cell Cycle 2013, 12, 1521–1535. [Google Scholar] [CrossRef] [Green Version]
- Martín-Martín, N.; Zabala-Letona, A.; Fernández-Ruiz, S.; Arreal, L.; Camacho, L.; Castillo-Martin, M.; Cortazar, A.R.; Torrano, V.; Astobiza, I.; Zúñiga-García, P.; et al. PPARδ Elicits Ligand-Independent Repression of Trefoil Factor Family to Limit Prostate Cancer Growth. Cancer Res. 2018, 78, 399–409. [Google Scholar] [CrossRef] [Green Version]
- Balandaram, G.; Kramer, L.R.; Kang, B.H.; Murray, I.A.; Perdew, G.H.; Gonzalez, F.J.; Peters, J.M. Ligand activation of peroxisome proliferator-activated receptor-β/δ suppresses liver tumorigenesis in hepatitis B transgenic mice. Toxicology 2016, 363–364, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Xu, L.; Han, C.; Lim, K.; Wu, T. Cross-talk between peroxisome proliferator-activated receptor δ and cytosolic phospholipase A(2)α/cyclooxygenase-2/prostaglandin E(2) signaling pathways in human hepatocellular carcinoma cells. Cancer Res. 2006, 66, 11859–11868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michiels, J.F.; Perrin, C.; Leccia, N.; Massi, D.; Grimaldi, P.; Wagner, N. PPARβ activation inhibits melanoma cell proliferation involving repression of the Wilms’ tumour suppressor WT1. Pflugers Arch. 2010, 459, 689–703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagner, N.; Panelos, J.; Massi, D.; Wagner, K.D. The Wilms’ tumor suppressor WT1 is associated with melanoma proliferation. Pflugers Arch. 2008, 455, 839–847. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.J.; Bility, M.T.; Billin, A.N.; Willson, T.M.; Gonzalez, F.J.; Peters, J.M. PPARβ/δ selectively induces differentiation and inhibits cell proliferation. Cell Death Differ. 2006, 13, 53–60. [Google Scholar] [CrossRef]
- Borland, M.G.; Kehres, E.M.; Lee, C.; Wagner, A.L.; Shannon, B.E.; Albrecht, P.P.; Zhu, B.; Gonzalez, F.J.; Peters, J.M. Inhibition of tumorigenesis by peroxisome proliferator-activated receptor (PPAR)-dependent cell cycle blocks in human skin carcinoma cells. Toxicology 2018, 404–405, 25–32. [Google Scholar] [CrossRef]
- Zhu, B.; Bai, R.; Kennett, M.J.; Kang, B.H.; Gonzalez, F.J.; Peters, J.M. Chemoprevention of chemically induced skin tumorigenesis by ligand activation of peroxisome proliferator-activated receptor-β/δ and inhibition of cyclooxygenase 2. Mol. Cancer Ther. 2010, 9, 3267–3277. [Google Scholar] [CrossRef] [Green Version]
- Bility, M.T.; Zhu, B.; Kang, B.H.; Gonzalez, F.J.; Peters, J.M. Ligand activation of peroxisome proliferator-activated receptor-β/δ and inhibition of cyclooxygenase-2 enhances inhibition of skin tumorigenesis. Toxicol. Sci. 2010, 113, 27–36. [Google Scholar] [CrossRef]
- Kim, D.J.; Prabhu, K.S.; Gonzalez, F.J.; Peters, J.M. Inhibition of chemically induced skin carcinogenesis by sulindac is independent of peroxisome proliferator-activated receptor-β/δ (PPARβ/δ). Carcinogenesis 2006, 27, 1105–1112. [Google Scholar] [CrossRef] [Green Version]
- Montagner, A.; Delgado, M.B.; Tallichet-Blanc, C.; Chan, J.S.; Sng, M.K.; Mottaz, H.; Degueurce, G.; Lippi, Y.; Moret, C.; Baruchet, M.; et al. Src is activated by the nuclear receptor peroxisome proliferator-activated receptor β/δ in ultraviolet radiation-induced skin cancer. EMBO Mol. Med. 2014, 6, 80–98. [Google Scholar] [CrossRef]
- Tan, M.W.Y.; Sng, M.K.; Cheng, H.S.; Low, Z.S.; Leong, B.J.J.; Chua, D.; Tan, E.H.P.; Chan, J.S.K.; Yip, Y.S.; Lee, Y.H.; et al. Deficiency in fibroblast PPARβ/δ reduces nonmelanoma skin cancers in mice. Cell Death Differ. 2020, 27, 2668–2680. [Google Scholar] [CrossRef]
- Pedchenko, T.V.; Gonzalez, A.L.; Wang, D.; DuBois, R.N.; Massion, P.P. Peroxisome proliferator—Activated receptor β/δ expression and activation in lung cancer. Am. J. Respir. Cell Mol. Biol. 2008, 39, 689–696. [Google Scholar] [CrossRef] [Green Version]
- Genini, D.; Garcia-Escudero, R.; Carbone, G.M.; Catapano, C.V. Transcriptional and Non-Transcriptional Functions of PPARβ/δ in Non-Small Cell Lung Cancer. PLoS ONE 2012, 7, e46009. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Ritzenthaler, J.D.; Zhong, X.; Zheng, Y.; Roman, J.; Han, S. Nicotine stimulates PPARβ/δ expression in human lung carcinoma cells through activation of PI3K/mTOR and suppression of AP-2α. Cancer Res. 2009, 69, 6445–6453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, P.; Borland, M.G.; Zhu, B.; Sharma, A.K.; Amin, S.; El-Bayoumy, K.; Gonzalez, F.J.; Peters, J.M. Effect of ligand activation of peroxisome proliferator-activated receptor-β/δ (PPARβ/δ) in human lung cancer cell lines. Toxicology 2008, 254, 112–117. [Google Scholar] [CrossRef] [Green Version]
- Müller-Brüsselbach, S.; Ebrahimsade, S.; Jäkel, J.; Eckhardt, J.; Rapp, U.R.; Peters, J.M.; Moll, R.; Müller, R. Growth of transgenic RAF-induced lung adenomas is increased in mice with a disrupted PPARβ/δ gene. Int. J. Oncol. 2007, 31, 607–611. [Google Scholar] [CrossRef]
- Gu, L.; Shi, Y.; Xu, W.; Ji, Y. PPARβ/δ Agonist GW501516 Inhibits Tumorigenesis and Promotes Apoptosis of the Undifferentiated Nasopharyngeal Carcinoma C666-1 Cells by Regulating miR-206. Oncol. Res. 2019, 27, 923–933. [Google Scholar] [CrossRef]
- Wagner, K.D.; Benchetrit, M.; Bianchini, L.; Michiels, J.F.; Wagner, N. Peroxisome proliferator-activated receptor β/δ (PPARβ/δ) is highly expressed in liposarcoma and promotes migration and proliferation. J. Pathol. 2011, 224, 575–588. [Google Scholar] [CrossRef] [PubMed]
- Zeng, L.; Geng, Y.; Tretiakova, M.; Yu, X.; Sicinski, P.; Kroll, T.G. Peroxisome proliferator-activated receptor-δ induces cell proliferation by a cyclin E1-dependent mechanism and is up-regulated in thyroid tumors. Cancer Res. 2008, 68, 6578–6586. [Google Scholar] [CrossRef] [Green Version]
- Daikoku, T.; Tranguch, S.; Chakrabarty, A.; Wang, D.; Khabele, D.; Orsulic, S.; Morrow, J.D.; Dubois, R.N.; Dey, S.K. Extracellular signal-regulated kinase is a target of cyclooxygenase-1-peroxisome proliferator-activated receptor-δ signaling in epithelial ovarian cancer. Cancer Res. 2007, 67, 5285–5292. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.J.; Sun, L.; Shi, Y.; Wang, G.; Wang, X.; Dunn, S.E.; Iorio, C.; Screaton, R.A.; Spaner, D.E. PPAR-delta promotes survival of chronic lymphocytic leukemia cells in energetically unfavorable conditions. Leukemia 2017, 31, 1905–1914. [Google Scholar] [CrossRef]
- Sun, L.; Shi, Y.; Wang, G.; Wang, X.; Zeng, S.; Dunn, S.E.; Fairn, G.D.; Li, Y.J.; Spaner, D.E. PPAR-delta modulates membrane cholesterol and cytokine signaling in malignant B cells. Leukemia 2018, 32, 184–193. [Google Scholar] [CrossRef] [PubMed]
- Lefebvre, A.M.; Chen, I.; Desreumaux, P.; Najib, J.; Fruchart, J.C.; Geboes, K.; Briggs, M.; Heyman, R.; Auwerx, J. Activation of the peroxisome proliferator-activated receptor γ promotes the development of colon tumors in C57BL/6J-APCMin/+ mice. Nat. Med. 1998, 4, 1053–1057. [Google Scholar] [CrossRef]
- Saez, E.; Tontonoz, P.; Nelson, M.C.; Alvarez, J.G.; Ming, U.T.; Baird, S.M.; Thomazy, V.A.; Evans, R.M. Activators of the nuclear receptor PPARγ enhance colon polyp formation. Nat. Med. 1998, 4, 1058–1061. [Google Scholar] [CrossRef] [PubMed]
- Brockman, J.A.; Gupta, R.A.; Dubois, R.N. Activation of PPARγ leads to inhibition of anchorage-independent growth of human colorectal cancer cells. Gastroenterology 1998, 115, 1049–1055. [Google Scholar] [CrossRef]
- Sarraf, P.; Mueller, E.; Jones, D.; King, F.J.; DeAngelo, D.J.; Partridge, J.B.; Holden, S.A.; Chen, L.B.; Singer, S.; Fletcher, C.; et al. Differentiation and reversal of malignant changes in colon cancer through PPARγ. Nat. Med. 1998, 4, 1046–1052. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, S.; Miyazaki, Y.; Shinomura, Y.; Kondo, S.; Kanayama, S.; Matsuzawa, Y. Peroxisome proliferator-activated receptor γ induces growth arrest and differentiation markers of human colon cancer cells. Jpn. J. Cancer Res. 1999, 90, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Chintharlapalli, S.; Papineni, S.; Safe, S. 1,1-Bis(3′-indolyl)-1-(p-substituted phenyl)methanes inhibit colon cancer cell and tumor growth through PPARγ-dependent and PPARγ-independent pathways. Mol. Cancer Ther. 2006, 5, 1362–1370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kubota, T.; Koshizuka, K.; Williamson, E.A.; Asou, H.; Said, J.W.; Holden, S.; Miyoshi, I.; Koeffler, H.P. Ligand for peroxisome proliferator-activated receptor γ (troglitazone) has potent antitumor effect against human prostate cancer both in vitro and in vivo. Cancer Res. 1998, 58, 3344–3352. [Google Scholar]
- Shappell, S.B.; Gupta, R.A.; Manning, S.; Whitehead, R.; Boeglin, W.E.; Schneider, C.; Case, T.; Price, J.; Jack, G.S.; Wheeler, T.M.; et al. 15S-Hydroxyeicosatetraenoic acid activates peroxisome proliferator-activated receptor γ and inhibits proliferation in PC3 prostate carcinoma cells. Cancer Res. 2001, 61, 497–503. [Google Scholar] [PubMed]
- Yoshimura, R.; Matsuyama, M.; Hase, T.; Tsuchida, K.; Kuratsukuri, K.; Kawahito, Y.; Sano, H.; Segawa, Y.; Nakatani, T. The effect of peroxisome proliferator-activated receptor-γ ligand on urological cancer cells. Int. J. Mol. Med. 2003, 12, 861–865. [Google Scholar] [CrossRef] [PubMed]
- Tontonoz, P.; Singer, S.; Forman, B.M.; Sarraf, P.; Fletcher, J.A.; Fletcher, C.D.; Brun, R.P.; Mueller, E.; Altiok, S.; Oppenheim, H.; et al. Terminal differentiation of human liposarcoma cells induced by ligands for peroxisome proliferator-activated receptor γ and the retinoid X receptor. Proc. Natl. Acad. Sci. USA 1997, 94, 237–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, N.; Okumura, T.; Motomura, W.; Fujimoto, Y.; Kawabata, I.; Kohgo, Y. Activation of PPARγ inhibits cell growth and induces apoptosis in human gastric cancer cells. FEBS Lett. 1999, 455, 135–139. [Google Scholar] [CrossRef] [Green Version]
- Sato, H.; Ishihara, S.; Kawashima, K.; Moriyama, N.; Suetsugu, H.; Kazumori, H.; Okuyama, T.; Rumi, M.A.; Fukuda, R.; Nagasue, N.; et al. Expression of peroxisome proliferator-activated receptor (PPAR)γ in gastric cancer and inhibitory effects of PPARγ agonists. Br. J. Cancer 2000, 83, 1394–1400. [Google Scholar] [CrossRef] [Green Version]
- Kassouf, W.; Chintharlapalli, S.; Abdelrahim, M.; Nelkin, G.; Safe, S.; Kamat, A.M. Inhibition of bladder tumor growth by 1,1-bis(3′-indolyl)-1-(p-substitutedphenyl)methanes: A new class of peroxisome proliferator-activated receptor γ agonists. Cancer Res. 2006, 66, 412–418. [Google Scholar] [CrossRef] [Green Version]
- Han, S.W.; Greene, M.E.; Pitts, J.; Wada, R.K.; Sidell, N. Novel expression and function of peroxisome proliferator-activated receptor γ (PPARγ) in human neuroblastoma cells. Clin. Cancer Res. 2001, 7, 98–104. [Google Scholar] [PubMed]
- Cellai, I.; Benvenuti, S.; Luciani, P.; Galli, A.; Ceni, E.; Simi, L.; Baglioni, S.; Muratori, M.; Ottanelli, B.; Serio, M.; et al. Antineoplastic effects of rosiglitazone and PPARγ transactivation in neuroblastoma cells. Br. J. Cancer 2006, 95, 879–888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chearwae, W.; Bright, J.J. PPARγ agonists inhibit growth and expansion of CD133+ brain tumour stem cells. Br. J. Cancer 2008, 99, 2044–2053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, P.; Yu, J.; Yin, Q.; Li, W.; Ren, X.; Hao, X. Rosiglitazone suppresses glioma cell growth and cell cycle by blocking the transforming growth factor-delta mediated pathway. Neurochem. Res. 2012, 37, 2076–2084. [Google Scholar] [CrossRef] [PubMed]
- Jozkowicz, A.; Dulak, J.; Piatkowska, E.; Placha, W.; Dembinska-Kiec, A. Ligands of peroxisome proliferator-activated receptor-γ increase the generation of vascular endothelial growth factor in vascular smooth muscle cells and in macrophages. Acta Biochim. Pol. 2000, 47, 1147–1157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freudlsperger, C.; Moll, I.; Schumacher, U.; Thies, A. Anti-proliferative effect of peroxisome proliferator-activated receptor γ agonists on human malignant melanoma cells in vitro. Anticancer Drugs 2006, 17, 325–332. [Google Scholar] [CrossRef]
- Botton, T.; Puissant, A.; Bahadoran, P.; Annicotte, J.S.; Fajas, L.; Ortonne, J.P.; Gozzerino, G.; Zamoum, T.; Tartare-Deckert, S.; Bertolotto, C.; et al. In vitro and in vivo anti-melanoma effects of ciglitazone. J. Investig. Dermatol. 2009, 129, 1208–1218. [Google Scholar] [CrossRef] [Green Version]
- Placha, W.; Gil, D.; Dembińska-Kieć, A.; Laidler, P. The effect of PPARγ ligands on the proliferation and apoptosis of human melanoma cells. Melanoma Res. 2003, 13, 447–456. [Google Scholar] [CrossRef] [PubMed]
- Keshamouni, V.G.; Reddy, R.C.; Arenberg, D.A.; Joel, B.; Thannickal, V.J.; Kalemkerian, G.P.; Standiford, T.J. Peroxisome proliferator-activated receptor-γ activation inhibits tumor progression in non-small-cell lung cancer. Oncogene 2004, 23, 100–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, S.; Roman, J. Rosiglitazone suppresses human lung carcinoma cell growth through PPARγ-dependent and PPARγ-independent signal pathways. Mol. Cancer Ther. 2006, 5, 430–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferruzzi, P.; Ceni, E.; Tarocchi, M.; Grappone, C.; Milani, S.; Galli, A.; Fiorelli, G.; Serio, M.; Mannelli, M. Thiazolidinediones inhibit growth and invasiveness of the human adrenocortical cancer cell line H295R. J. Clin. Endocrinol. Metab. 2005, 90, 1332–1339. [Google Scholar] [CrossRef] [Green Version]
- Betz, M.J.; Shapiro, I.; Fassnacht, M.; Hahner, S.; Reincke, M.; Beuschlein, F.; Network, G.a.A.A. Peroxisome proliferator-activated receptor-γ agonists suppress adrenocortical tumor cell proliferation and induce differentiation. J. Clin. Endocrinol. Metab. 2005, 90, 3886–3896. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.; Qiao, L.; Zimmermann, L.; Ebert, M.P.; Zhang, H.; Lin, W.; Röcken, C.; Malfertheiner, P.; Farrell, G.C. Troglitazone inhibits tumor growth in hepatocellular carcinoma in vitro and in vivo. Hepatology 2006, 43, 134–143. [Google Scholar] [CrossRef] [PubMed]
- Ota, K.; Ito, K.; Suzuki, T.; Saito, S.; Tamura, M.; Hayashi, S.; Okamura, K.; Sasano, H.; Yaegashi, N. Peroxisome proliferator-activated receptor γ and growth inhibition by its ligands in uterine endometrial carcinoma. Clin. Cancer Res. 2006, 12, 4200–4208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lei, P.; Abdelrahim, M.; Safe, S. 1,1-Bis(3′-indolyl)-1-(p-substituted phenyl)methanes inhibit ovarian cancer cell growth through peroxisome proliferator-activated receptor-dependent and independent pathways. Mol. Cancer Ther. 2006, 5, 2324–2336. [Google Scholar] [CrossRef] [Green Version]
- Vignati, S.; Albertini, V.; Rinaldi, A.; Kwee, I.; Riva, C.; Oldrini, R.; Capella, C.; Bertoni, F.; Carbone, G.M.; Catapano, C.V. Cellular and molecular consequences of peroxisome proliferator-activated receptor-γ activation in ovarian cancer cells. Neoplasia 2006, 8, 851–861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Bates, T.M.; Bernstein, S.H.; Phipps, R.P. Peroxisome proliferator-activated receptor γ overexpression suppresses growth and induces apoptosis in human multiple myeloma cells. Clin. Cancer Res. 2008, 14, 6414–6425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Bates, T.M.; Peslak, S.A.; Baglole, C.J.; Maggirwar, S.B.; Bernstein, S.H.; Phipps, R.P. Peroxisome proliferator-activated receptor gamma overexpression and knockdown: Impact on human B cell lymphoma proliferation and survival. Cancer Immunol. Immunother. 2009, 58, 1071–1083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamaguchi, N.; Hamada, H.; Miyoshi, S.; Irifune, K.; Ito, R.; Miyazaki, T.; Higaki, J. In vitro and in vivo therapeutic efficacy of the PPAR-γ agonist troglitazone in combination with cisplatin against malignant pleural mesothelioma cell growth. Cancer Sci. 2010, 101, 1955–1964. [Google Scholar] [CrossRef] [PubMed]
- Sawayama, H.; Ishimoto, T.; Watanabe, M.; Yoshida, N.; Sugihara, H.; Kurashige, J.; Hirashima, K.; Iwatsuki, M.; Baba, Y.; Oki, E.; et al. Small molecule agonists of PPAR-γ exert therapeutic effects in esophageal cancer. Cancer Res. 2014, 74, 575–585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prost, S.; Relouzat, F.; Spentchian, M.; Ouzegdouh, Y.; Saliba, J.; Massonnet, G.; Beressi, J.P.; Verhoeyen, E.; Raggueneau, V.; Maneglier, B.; et al. Erosion of the chronic myeloid leukaemia stem cell pool by PPARγ agonists. Nature 2015, 525, 380–383. [Google Scholar] [CrossRef] [PubMed]
- Rousselot, P.; Prost, S.; Guilhot, J.; Roy, L.; Etienne, G.; Legros, L.; Charbonnier, A.; Coiteux, V.; Cony-Makhoul, P.; Huguet, F.; et al. Pioglitazone together with imatinib in chronic myeloid leukemia: A proof of concept study. Cancer 2017, 123, 1791–1799. [Google Scholar] [CrossRef] [PubMed]
- Kato, Y.; Ying, H.; Zhao, L.; Furuya, F.; Araki, O.; Willingham, M.C.; Cheng, S.Y. PPARγ insufficiency promotes follicular thyroid carcinogenesis via activation of the nuclear factor-κB signaling pathway. Oncogene 2006, 25, 2736–2747. [Google Scholar] [CrossRef] [Green Version]
- Wu, L.; Yan, C.; Czader, M.; Foreman, O.; Blum, J.S.; Kapur, R.; Du, H. Inhibition of PPARγ in myeloid-lineage cells induces systemic inflammation, immunosuppression, and tumorigenesis. Blood 2012, 119, 115–126. [Google Scholar] [CrossRef] [Green Version]
- Pignatelli, M.; Cortés-Canteli, M.; Lai, C.; Santos, A.; Perez-Castillo, A. The peroxisome proliferator-activated receptor γ is an inhibitor of ErbBs activity in human breast cancer cells. J. Cell Sci. 2001, 114, 4117–4126. [Google Scholar] [CrossRef]
- Qin, C.; Burghardt, R.; Smith, R.; Wormke, M.; Stewart, J.; Safe, S. Peroxisome proliferator-activated receptor γ agonists induce proteasome-dependent degradation of cyclin D1 and estrogen receptor α in MCF-7 breast cancer cells. Cancer Res. 2003, 63, 958–964. [Google Scholar]
- Houston, K.D.; Copland, J.A.; Broaddus, R.R.; Gottardis, M.M.; Fischer, S.M.; Walker, C.L. Inhibition of proliferation and estrogen receptor signaling by peroxisome proliferator-activated receptor γ ligands in uterine leiomyoma. Cancer Res. 2003, 63, 1221–1227. [Google Scholar] [PubMed]
- Catalano, S.; Mauro, L.; Bonofiglio, D.; Pellegrino, M.; Qi, H.; Rizza, P.; Vizza, D.; Bossi, G.; Andò, S. In vivo and in vitro evidence that PPARγ ligands are antagonists of leptin signaling in breast cancer. Am. J. Pathol. 2011, 179, 1030–1040. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Kim, J.Y.; Meng, Z.; Wang, L.H.; Liu, F.; Conrads, T.P.; Burke, T.R.; Veenstra, T.D.; Farrar, W.L. 15-deoxy-Δ12,14-prostaglandin J2 inhibits transcriptional activity of estrogen receptor-α via covalent modification of DNA-binding domain. Cancer Res. 2007, 67, 2595–2602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanderlaag, K.; Su, Y.; Frankel, A.E.; Grage, H.; Smith, R.; Khan, S.; Safe, S. 1,1-Bis(3′-indolyl)-1-(p-substituted phenyl)methanes inhibit proliferation of estrogen receptor-negative breast cancer cells by activation of multiple pathways. Breast Cancer Res. Treat. 2008, 109, 273–283. [Google Scholar] [CrossRef] [PubMed]
- Saez, E.; Rosenfeld, J.; Livolsi, A.; Olson, P.; Lombardo, E.; Nelson, M.; Banayo, E.; Cardiff, R.D.; Izpisua-Belmonte, J.C.; Evans, R.M. PPAR γ signaling exacerbates mammary gland tumor development. Genes Dev. 2004, 18, 528–540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avena, P.; Anselmo, W.; Whitaker-Menezes, D.; Wang, C.; Pestell, R.G.; Lamb, R.S.; Hulit, J.; Casaburi, I.; Andò, S.; Martinez-Outschoorn, U.E.; et al. Compartment-specific activation of PPARγ governs breast cancer tumor growth, via metabolic reprogramming and symbiosis. Cell Cycle 2013, 12, 1360–1370. [Google Scholar] [CrossRef] [Green Version]
- Apostoli, A.J.; Skelhorne-Gross, G.E.; Rubino, R.E.; Peterson, N.T.; Di Lena, M.A.; Schneider, M.M.; SenGupta, S.K.; Nicol, C.J. Loss of PPARγ expression in mammary secretory epithelial cells creates a pro-breast tumorigenic environment. Int. J. Cancer 2014, 134, 1055–1066. [Google Scholar] [CrossRef] [Green Version]
- Yee, L.D.; Williams, N.; Wen, P.; Young, D.C.; Lester, J.; Johnson, M.V.; Farrar, W.B.; Walker, M.J.; Povoski, S.P.; Suster, S.; et al. Pilot study of rosiglitazone therapy in women with breast cancer: Effects of short-term therapy on tumor tissue and serum markers. Clin. Cancer Res. 2007, 13, 246–252. [Google Scholar] [CrossRef] [Green Version]
- He, G.; Muga, S.; Thuillier, P.; Lubet, R.A.; Fischer, S.M. The effect of PPARγ ligands on UV- or chemically-induced carcinogenesis in mouse skin. Mol. Carcinog. 2005, 43, 198–206. [Google Scholar] [CrossRef]
- Palakurthi, S.S.; Aktas, H.; Grubissich, L.M.; Mortensen, R.M.; Halperin, J.A. Anticancer effects of thiazolidinediones are independent of peroxisome proliferator-activated receptor γ and mediated by inhibition of translation initiation. Cancer Res. 2001, 61, 6213–6218. [Google Scholar]
- Lucarelli, E.; Sangiorgi, L.; Maini, V.; Lattanzi, G.; Marmiroli, S.; Reggiani, M.; Mordenti, M.; Alessandra Gobbi, G.; Scrimieri, F.; Zambon Bertoja, A.; et al. Troglitazione affects survival of human osteosarcoma cells. Int. J. Cancer 2002, 98, 344–351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haydon, R.C.; Zhou, L.; Feng, T.; Breyer, B.; Cheng, H.; Jiang, W.; Ishikawa, A.; Peabody, T.; Montag, A.; Simon, M.A.; et al. Nuclear receptor agonists as potential differentiation therapy agents for human osteosarcoma. Clin. Cancer Res. 2002, 8, 1288–1294. [Google Scholar] [PubMed]
- Srivastava, N.; Kollipara, R.K.; Singh, D.K.; Sudderth, J.; Hu, Z.; Nguyen, H.; Wang, S.; Humphries, C.G.; Carstens, R.; Huffman, K.E.; et al. Inhibition of cancer cell proliferation by PPARγ is mediated by a metabolic switch that increases reactive oxygen species levels. Cell Metab. 2014, 20, 650–661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Musicant, A.M.; Parag-Sharma, K.; Gong, W.; Sengupta, M.; Chatterjee, A.; Henry, E.C.; Tsai, Y.H.; Hayward, M.C.; Sheth, S.; Betancourt, R.; et al. CRTC1/MAML2 directs a PGC-1α-IGF-1 circuit that confers vulnerability to PPARγ inhibition. Cell Rep. 2021, 34, 108768. [Google Scholar] [CrossRef]
- Pishvaian, M.J.; Marshall, J.L.; Wagner, A.J.; Hwang, J.J.; Malik, S.; Cotarla, I.; Deeken, J.F.; He, A.R.; Daniel, H.; Halim, A.B.; et al. A phase 1 study of efatutazone, an oral peroxisome proliferator-activated receptor gamma agonist, administered to patients with advanced malignancies. Cancer 2012, 118, 5403–5413. [Google Scholar] [CrossRef]
- Demetri, G.D.; Fletcher, C.D.; Mueller, E.; Sarraf, P.; Naujoks, R.; Campbell, N.; Spiegelman, B.M.; Singer, S. Induction of solid tumor differentiation by the peroxisome proliferator-activated receptor-γ ligand troglitazone in patients with liposarcoma. Proc. Natl. Acad. Sci. USA 1999, 96, 3951–3956. [Google Scholar] [CrossRef] [Green Version]
- Wagner, N.; Wagner, K.D. PPARs and Angiogenesis-Implications in Pathology. Int. J. Mol. Sci. 2020, 21, 5723. [Google Scholar] [CrossRef]
- Jiao, H.L.; Zhao, B.L. Cytotoxic effect of peroxisome proliferator fenofibrate on human HepG2 hepatoma cell line and relevant mechanisms. Toxicol. Appl. Pharmacol. 2002, 185, 172–179. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Liu, Q.; Xu, Y.; Gong, X.; Zhang, R.; Zhou, C.; Su, Z.; Jin, J.; Shi, H.; Shi, J.; et al. PPARα induces cell apoptosis by destructing Bcl2. Oncotarget 2015, 6, 44635–44642. [Google Scholar] [CrossRef] [Green Version]
- Holland, C.M.; Saidi, S.A.; Evans, A.L.; Sharkey, A.M.; Latimer, J.A.; Crawford, R.A.; Charnock-Jones, D.S.; Print, C.G.; Smith, S.K. Transcriptome analysis of endometrial cancer identifies peroxisome proliferator-activated receptors as potential therapeutic targets. Mol. Cancer Ther. 2004, 3, 993–1001. [Google Scholar] [CrossRef]
- Crowe, D.L.; Chandraratna, R.A. A retinoid X receptor (RXR)-selective retinoid reveals that RXR-α is potentially a therapeutic target in breast cancer cell lines, and that it potentiates antiproliferative and apoptotic responses to peroxisome proliferator-activated receptor ligands. Breast Cancer Res. 2004, 6, R546–R555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strakova, N.; Ehrmann, J.; Bartos, J.; Malikova, J.; Dolezel, J.; Kolar, Z. Peroxisome proliferator-activated receptors (PPAR) agonists affect cell viability, apoptosis and expression of cell cycle related proteins in cell lines of glial brain tumors. Neoplasma 2005, 52, 126–136. [Google Scholar] [PubMed]
- Martinasso, G.; Oraldi, M.; Trombetta, A.; Maggiora, M.; Bertetto, O.; Canuto, R.A.; Muzio, G. Involvement of PPARs in Cell Proliferation and Apoptosis in Human Colon Cancer Specimens and in Normal and Cancer Cell Lines. PPAR Res. 2007, 2007, 93416. [Google Scholar] [CrossRef] [Green Version]
- Xue, J.; Zhu, W.; Song, J.; Jiao, Y.; Luo, J.; Yu, C.; Zhou, J.; Wu, J.; Chen, M.; Ding, W.Q.; et al. Activation of PPARα by clofibrate sensitizes pancreatic cancer cells to radiation through the Wnt/β-catenin pathway. Oncogene 2018, 37, 953–962. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.S.; Han, Q.S.; Jia, Z.R.; Chen, C.S.; Qiao, C.; Liu, Q.Q.; Zhang, Y.M.; Wang, K.W.; Wang, J.; Xiao, K.; et al. PPARα agonist fenofibrate relieves acquired resistance to gefitinib in non-small cell lung cancer by promoting apoptosis via PPARα/AMPK/AKT/FoxO1 pathway. Acta Pharmacol. Sin. 2022, 43, 167–176. [Google Scholar] [CrossRef]
- Maggiora, M.; Bologna, M.; Cerù, M.P.; Possati, L.; Angelucci, A.; Cimini, A.; Miglietta, A.; Bozzo, F.; Margiotta, C.; Muzio, G.; et al. An overview of the effect of linoleic and conjugated-linoleic acids on the growth of several human tumor cell lines. Int. J. Cancer 2004, 112, 909–919. [Google Scholar] [CrossRef]
- Tuller, E.R.; Brock, A.L.; Yu, H.; Lou, J.R.; Benbrook, D.M.; Ding, W.Q. PPARα signaling mediates the synergistic cytotoxicity of clioquinol and docosahexaenoic acid in human cancer cells. Biochem. Pharmacol. 2009, 77, 1480–1486. [Google Scholar] [CrossRef]
- Zang, C.; Liu, H.; Bertz, J.; Possinger, K.; Koeffler, H.P.; Elstner, E.; Eucker, J. Induction of endoplasmic reticulum stress response by TZD18, a novel dual ligand for peroxisome proliferator-activated receptor α/γ, in human breast cancer cells. Mol. Cancer Ther. 2009, 8, 2296–2307. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.; Wang, B.; Li, L.; Wang, F.; Xia, X. The administration of peroxisome proliferator-activated receptors α/γ agonist TZD18 inhibits cell growth and induces apoptosis in human gastric cancer cell lines. J. Cancer Res. Ther. 2019, 15, 120–125. [Google Scholar] [CrossRef]
- Zak, Z.; Gelebart, P.; Lai, R. Fenofibrate induces effective apoptosis in mantle cell lymphoma by inhibiting the TNFα/NF-κB signaling axis. Leukemia 2010, 24, 1476–1486. [Google Scholar] [CrossRef] [Green Version]
- Deepa, P.R.; Vandhana, S.; Krishnakumar, S. Fatty acid synthase inhibition induces differential expression of genes involved in apoptosis and cell proliferation in ocular cancer cells. Nutr. Cancer 2013, 65, 311–316. [Google Scholar] [CrossRef] [PubMed]
- Casella, M.L.; Parody, J.P.; Ceballos, M.P.; Quiroga, A.D.; Ronco, M.T.; Francés, D.E.; Monti, J.A.; Pisani, G.B.; Carnovale, C.E.; Carrillo, M.C.; et al. Quercetin prevents liver carcinogenesis by inducing cell cycle arrest, decreasing cell proliferation and enhancing apoptosis. Mol. Nutr. Food Res. 2014, 58, 289–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abu Aboud, O.; Wettersten, H.I.; Weiss, R.H. Inhibition of PPARα induces cell cycle arrest and apoptosis, and synergizes with glycolysis inhibition in kidney cancer cells. PLoS ONE 2013, 8, e71115. [Google Scholar] [CrossRef]
- Abu Aboud, O.; Donohoe, D.; Bultman, S.; Fitch, M.; Riiff, T.; Hellerstein, M.; Weiss, R.H. PPARα inhibition modulates multiple reprogrammed metabolic pathways in kidney cancer and attenuates tumor growth. Am. J. Physiol. Cell Physiol. 2015, 308, C890–C898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Florio, R.; De Lellis, L.; di Giacomo, V.; Di Marcantonio, M.C.; Cristiano, L.; Basile, M.; Verginelli, F.; Verzilli, D.; Ammazzalorso, A.; Prasad, S.C.; et al. Effects of PPARα inhibition in head and neck paraganglioma cells. PLoS ONE 2017, 12, e0178995. [Google Scholar] [CrossRef] [Green Version]
- He, T.C.; Chan, T.A.; Vogelstein, B.; Kinzler, K.W. PPARδ is an APC-regulated target of nonsteroidal anti-inflammatory drugs. Cell 1999, 99, 335–345. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Wang, H.; Shi, Q.; Katkuri, S.; Walhi, W.; Desvergne, B.; Das, S.K.; Dey, S.K.; DuBois, R.N. Prostaglandin E(2) promotes colorectal adenoma growth via transactivation of the nuclear peroxisome proliferator-activated receptor δ. Cancer Cell 2004, 6, 285–295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cutler, N.S.; Graves-Deal, R.; LaFleur, B.J.; Gao, Z.; Boman, B.M.; Whitehead, R.H.; Terry, E.; Morrow, J.D.; Coffey, R.J. Stromal production of prostacyclin confers an antiapoptotic effect to colonic epithelial cells. Cancer Res. 2003, 63, 1748–1751. [Google Scholar] [PubMed]
- Liou, J.Y.; Lee, S.; Ghelani, D.; Matijevic-Aleksic, N.; Wu, K.K. Protection of endothelial survival by peroxisome proliferator-activated receptor-δ mediated 14-3-3 upregulation. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 1481–1487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, D.; Ning, W.; Xie, D.; Guo, L.; DuBois, R.N. Peroxisome proliferator-activated receptor δ confers resistance to peroxisome proliferator-activated receptor γ-induced apoptosis in colorectal cancer cells. Oncogene 2012, 31, 1013–1023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bell, E.; Ponthan, F.; Whitworth, C.; Westermann, F.; Thomas, H.; Redfern, C.P. Cell survival signalling through PPARδ and arachidonic acid metabolites in neuroblastoma. PLoS ONE 2013, 8, e68859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tong-Lin Wu, T.; Tong, Y.C.; Chen, I.H.; Niu, H.S.; Li, Y.; Cheng, J.T. Induction of apoptosis in prostate cancer by ginsenoside Rh2. Oncotarget 2018, 9, 11109–11118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, T.T.; Niu, H.S.; Chen, L.J.; Cheng, J.T.; Tong, Y.C. Increase of human prostate cancer cell (DU145) apoptosis by telmisartan through PPAR-delta pathway. Eur. J. Pharmacol. 2016, 775, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Péchery, A.; Fauconnet, S.; Bittard, H.; Lascombe, I. Apoptotic effect of the selective PPARβ/δ agonist GW501516 in invasive bladder cancer cells. Tumour Biol. 2016, 37, 14789–14802. [Google Scholar] [CrossRef] [PubMed]
- Shen, B.; Li, A.; Wan, Y.Y.; Shen, G.; Zhu, J.; Nie, Y. Lack of PPAR. Biomed Res. Int. 2020, 2020, 9563851. [Google Scholar] [CrossRef] [PubMed]
- Padilla, J.; Kaur, K.; Harris, S.G.; Phipps, R.P. PPAR-γ-mediated regulation of normal and malignant B lineage cells. Ann. N. Y. Acad. Sci. 2000, 905, 97–109. [Google Scholar] [CrossRef] [PubMed]
- Padilla, J.; Kaur, K.; Cao, H.J.; Smith, T.J.; Phipps, R.P. Peroxisome proliferator activator receptor-γ agonists and 15-deoxy-Δ12,1412,14-PGJ2 induce apoptosis in normal and malignant B-lineage cells. J. Immunol. 2000, 165, 6941–6948. [Google Scholar] [CrossRef] [Green Version]
- Piva, R.; Gianferretti, P.; Ciucci, A.; Taulli, R.; Belardo, G.; Santoro, M.G. 15-Deoxy-Δ12,14-prostaglandin J2 induces apoptosis in human malignant B cells: An effect associated with inhibition of NF-κB activity and down-regulation of antiapoptotic proteins. Blood 2005, 105, 1750–1758. [Google Scholar] [CrossRef] [Green Version]
- Tsao, T.; Kornblau, S.; Safe, S.; Watt, J.C.; Ruvolo, V.; Chen, W.; Qiu, Y.; Coombes, K.R.; Ju, Z.; Abdelrahim, M.; et al. Role of peroxisome proliferator-activated receptor-γ and its coactivator DRIP205 in cellular responses to CDDO (RTA-401) in acute myelogenous leukemia. Cancer Res. 2010, 70, 4949–4960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clay, C.E.; Monjazeb, A.; Thorburn, J.; Chilton, F.H.; High, K.P. 15-Deoxy-Δ12,14-prostaglandin J2-induced apoptosis does not require PPARγ in breast cancer cells. J. Lipid Res. 2002, 43, 1818–1828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaffer, C.L.; Thomas, D.M.; Thompson, E.W.; Williams, E.D. PPARγ-independent induction of growth arrest and apoptosis in prostate and bladder carcinoma. BMC Cancer 2006, 6, 53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kondoh, K.; Tsuji, N.; Asanuma, K.; Kobayashi, D.; Watanabe, N. Inhibition of estrogen receptor β-mediated human telomerase reverse transcriptase gene transcription via the suppression of mitogen-activated protein kinase signaling plays an important role in 15-deoxy-Δ12,14-prostaglandin J2-induced apoptosis in cancer cells. Exp. Cell Res. 2007, 313, 3486–3496. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Kwan, T.; Yu, C.; Chen, F.; Freedman, B.; Schafer, J.M.; Lee, E.J.; Jameson, J.L.; Jordan, V.C.; Cryns, V.L. Peroxisome proliferator-activated receptor γ agonists promote TRAIL-induced apoptosis by reducing survivin levels via cyclin D3 repression and cell cycle arrest. J. Biol. Chem. 2005, 280, 6742–6751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bräutigam, K.; Biernath-Wüpping, J.; Bauerschlag, D.O.; von Kaisenberg, C.S.; Jonat, W.; Maass, N.; Arnold, N.; Meinhold-Heerlein, I. Combined treatment with TRAIL and PPARγ ligands overcomes chemoresistance of ovarian cancer cell lines. J. Cancer Res. Clin. Oncol. 2011, 137, 875–886. [Google Scholar] [CrossRef] [PubMed]
- Bonofiglio, D.; Cione, E.; Qi, H.; Pingitore, A.; Perri, M.; Catalano, S.; Vizza, D.; Panno, M.L.; Genchi, G.; Fuqua, S.A.; et al. Combined low doses of PPARγ and RXR ligands trigger an intrinsic apoptotic pathway in human breast cancer cells. Am. J. Pathol. 2009, 175, 1270–1280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kazberuk, A.; Chalecka, M.; Palka, J.; Surazynski, A. Nonsteroidal Anti-Inflammatory Drugs as PPARγ Agonists Can Induce PRODH/POX-Dependent Apoptosis in Breast Cancer Cells: New Alternative Pathway in NSAID-Induced Apoptosis. Int. J. Mol. Sci. 2022, 23, 1510. [Google Scholar] [CrossRef]
- Guan, Y.F.; Zhang, Y.H.; Breyer, R.M.; Davis, L.; Breyer, M.D. Expression of peroxisome proliferator-activated receptor γ (PPARγ) in human transitional bladder cancer and its role in inducing cell death. Neoplasia 1999, 1, 330–339. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.; Imamura, K.; Nomura, S.; Mafune, K.; Nakajima, A.; Kadowaki, T.; Kubota, N.; Terauchi, Y.; Ishii, G.; Ochiai, A.; et al. Chemopreventive effect of peroxisome proliferator-activated receptor γ on gastric carcinogenesis in mice. Cancer Res. 2005, 65, 4769–4774. [Google Scholar] [CrossRef] [Green Version]
- Tsubouchi, Y.; Sano, H.; Kawahito, Y.; Mukai, S.; Yamada, R.; Kohno, M.; Inoue, K.; Hla, T.; Kondo, M. Inhibition of human lung cancer cell growth by the peroxisome proliferator-activated receptor-γ agonists through induction of apoptosis. Biochem. Biophys. Res. Commun. 2000, 270, 400–405. [Google Scholar] [CrossRef]
- Takashima, T.; Fujiwara, Y.; Higuchi, K.; Arakawa, T.; Yano, Y.; Hasuma, T.; Otani, S. PPAR-γ ligands inhibit growth of human esophageal adenocarcinoma cells through induction of apoptosis, cell cycle arrest and reduction of ornithine decarboxylase activity. Int. J. Oncol. 2001, 19, 465–471. [Google Scholar] [CrossRef]
- Eibl, G.; Wente, M.N.; Reber, H.A.; Hines, O.J. Peroxisome proliferator-activated receptor γ induces pancreatic cancer cell apoptosis. Biochem. Biophys. Res. Commun. 2001, 287, 522–529. [Google Scholar] [CrossRef] [PubMed]
- Li, M.Y.; Deng, H.; Zhao, J.M.; Dai, D.; Tan, X.Y. PPARγ pathway activation results in apoptosis and COX-2 inhibition in HepG2 cells. World J. Gastroenterol. 2003, 9, 1220–1226. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.J.; Park, K.S.; Chung, S.Y.; Sheen, Y.Y.; Moon, D.C.; Song, Y.S.; Kim, K.S.; Song, S.; Yun, Y.P.; Lee, M.K.; et al. Peroxisome proliferator-activated receptor-γ activator 15-deoxy-Δ12,14-prostaglandin J2 inhibits neuroblastoma cell growth through induction of apoptosis: Association with extracellular signal-regulated kinase signal pathway. J. Pharmacol. Exp. Ther. 2003, 307, 505–517. [Google Scholar] [CrossRef] [PubMed]
- Strakova, N.; Ehrmann, J.; Dzubak, P.; Bouchal, J.; Kolar, Z. The synthetic ligand of peroxisome proliferator-activated receptor-γ ciglitazone affects human glioblastoma cell lines. J. Pharmacol. Exp. Ther. 2004, 309, 1239–1247. [Google Scholar] [CrossRef] [Green Version]
- Konopleva, M.; Elstner, E.; McQueen, T.J.; Tsao, T.; Sudarikov, A.; Hu, W.; Schober, W.D.; Wang, R.Y.; Chism, D.; Kornblau, S.M.; et al. Peroxisome proliferator-activated receptor γ and retinoid X receptor ligands are potent inducers of differentiation and apoptosis in leukemias. Mol. Cancer Ther. 2004, 3, 1249–1262. [Google Scholar] [CrossRef]
- Nam, D.H.; Ramachandran, S.; Song, D.K.; Kwon, K.Y.; Jeon, D.S.; Shin, S.J.; Kwon, S.H.; Cha, S.D.; Bae, I.; Cho, C.H. Growth inhibition and apoptosis induced in human leiomyoma cells by treatment with the PPAR gamma ligand ciglitizone. Mol. Hum. Reprod. 2007, 13, 829–836. [Google Scholar] [CrossRef] [Green Version]
- Shimada, T.; Kojima, K.; Yoshiura, K.; Hiraishi, H.; Terano, A. Characteristics of the peroxisome proliferator activated receptor γ (PPARγ) ligand induced apoptosis in colon cancer cells. Gut 2002, 50, 658–664. [Google Scholar] [CrossRef] [Green Version]
- Hernandez-Quiles, M.; Broekema, M.F.; Kalkhoven, E. PPARgamma in Metabolism, Immunity, and Cancer: Unified and Diverse Mechanisms of Action. Front. Endocrinol. 2021, 12, 624112. [Google Scholar] [CrossRef]
- Kim, H.J.; Hwang, J.Y.; Choi, W.S.; Lee, J.H.; Chang, K.C.; Nishinaka, T.; Yabe-Nishimura, C.; Seo, H.G. Expression of a peroxisome proliferator-activated receptor γ 1 splice variant that was identified in human lung cancers suppresses cell death induced by cisplatin and oxidative stress. Clin. Cancer Res. 2007, 13, 2577–2583. [Google Scholar] [CrossRef] [Green Version]
- Sun, H.; Berquin, I.M.; Owens, R.T.; O’Flaherty, J.T.; Edwards, I.J. Peroxisome proliferator-activated receptor γ-mediated up-regulation of syndecan-1 by n-3 fatty acids promotes apoptosis of human breast cancer cells. Cancer Res. 2008, 68, 2912–2919. [Google Scholar] [CrossRef] [Green Version]
- Baek, S.J.; Wilson, L.C.; Hsi, L.C.; Eling, T.E. Troglitazone, a peroxisome proliferator-activated receptor γ (PPAR γ) ligand, selectively induces the early growth response-1 gene independently of PPAR γ. A novel mechanism for its anti-tumorigenic activity. J. Biol. Chem. 2003, 278, 5845–5853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Funao, K.; Matsuyama, M.; Kawahito, Y.; Sano, H.; Chargui, J.; Touraine, J.L.; Nakatani, T.; Yoshimura, R. Telmisartan is a potent target for prevention and treatment in human prostate cancer. Oncol. Rep. 2008, 20, 295–300. [Google Scholar] [PubMed] [Green Version]
- Funao, K.; Matsuyama, M.; Kawahito, Y.; Sano, H.; Chargui, J.; Touraine, J.L.; Nakatani, T.; Yoshimura, R. Telmisartan as a peroxisome proliferator-activated receptor-γ ligand is a new target in the treatment of human renal cell carcinoma. Mol. Med. Rep. 2009, 2, 193–198. [Google Scholar] [CrossRef] [Green Version]
- Matsuyama, M.; Funao, K.; Kuratsukuri, K.; Tanaka, T.; Kawahito, Y.; Sano, H.; Chargui, J.; Touraine, J.L.; Yoshimura, N.; Yoshimura, R. Telmisartan inhibits human urological cancer cell growth through early apoptosis. Exp. Ther. Med. 2010, 1, 301–306. [Google Scholar] [CrossRef]
- Zaytseva, Y.Y.; Wang, X.; Southard, R.C.; Wallis, N.K.; Kilgore, M.W. Down-regulation of PPARgamma1 suppresses cell growth and induces apoptosis in MCF-7 breast cancer cells. Mol. Cancer 2008, 7, 90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khandekar, M.J.; Banks, A.S.; Laznik-Bogoslavski, D.; White, J.P.; Choi, J.H.; Kazak, L.; Lo, J.C.; Cohen, P.; Wong, K.K.; Kamenecka, T.M.; et al. Noncanonical agonist PPARγ ligands modulate the response to DNA damage and sensitize cancer cells to cytotoxic chemotherapy. Proc. Natl. Acad. Sci. USA 2018, 115, 561–566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaefer, K.L.; Wada, K.; Takahashi, H.; Matsuhashi, N.; Ohnishi, S.; Wolfe, M.M.; Turner, J.R.; Nakajima, A.; Borkan, S.C.; Saubermann, L.J. Peroxisome proliferator-activated receptor γ inhibition prevents adhesion to the extracellular matrix and induces anoikis in hepatocellular carcinoma cells. Cancer Res. 2005, 65, 2251–2259. [Google Scholar] [CrossRef] [Green Version]
- Masuda, T.; Wada, K.; Nakajima, A.; Okura, M.; Kudo, C.; Kadowaki, T.; Kogo, M.; Kamisaki, Y. Critical role of peroxisome proliferator-activated receptor γ on anoikis and invasion of squamous cell carcinoma. Clin. Cancer Res. 2005, 11, 4012–4021. [Google Scholar] [CrossRef] [Green Version]
- Cerquetti, L.; Sampaoli, C.; Amendola, D.; Bucci, B.; Masuelli, L.; Marchese, R.; Misiti, S.; De Venanzi, A.; Poggi, M.; Toscano, V.; et al. Rosiglitazone induces autophagy in H295R and cell cycle deregulation in SW13 adrenocortical cancer cells. Exp. Cell Res. 2011, 317, 1397–1410. [Google Scholar] [CrossRef]
- Rovito, D.; Giordano, C.; Vizza, D.; Plastina, P.; Barone, I.; Casaburi, I.; Lanzino, M.; De Amicis, F.; Sisci, D.; Mauro, L.; et al. Omega-3 PUFA ethanolamides DHEA and EPEA induce autophagy through PPARγ activation in MCF-7 breast cancer cells. J. Cell. Physiol. 2013, 228, 1314–1322. [Google Scholar] [CrossRef]
- To, K.K.W.; Wu, W.K.K.; Loong, H.H.F. PPARgamma agonists sensitize PTEN-deficient resistant lung cancer cells to EGFR tyrosine kinase inhibitors by inducing autophagy. Eur. J. Pharmacol. 2018, 823, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Yun, C.W.; Lee, S.H. The Roles of Autophagy in Cancer. Int. J. Mol. Sci. 2018, 19, 3466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baron, D.M.; Kaindl, U.; Haudek-Prinz, V.J.; Bayer, E.; Röhrl, C.; Gerner, C.; Marian, B. Autonomous inhibition of apoptosis correlates with responsiveness of colon carcinoma cell lines to ciglitazone. PLoS ONE 2014, 9, e114158. [Google Scholar] [CrossRef] [PubMed]
- Nijsten, T.; Geluyckens, E.; Colpaert, C.; Lambert, J. Peroxisome proliferator-activated receptors in squamous cell carcinoma and its precursors. J. Cutan. Pathol. 2005, 32, 340–347. [Google Scholar] [CrossRef] [PubMed]
- Panigrahy, D.; Kaipainen, A.; Huang, S.; Butterfield, C.E.; Barnés, C.M.; Fannon, M.; Laforme, A.M.; Chaponis, D.M.; Folkman, J.; Kieran, M.W. PPARα agonist fenofibrate suppresses tumor growth through direct and indirect angiogenesis inhibition. Proc. Natl. Acad. Sci. USA 2008, 105, 985–990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pozzi, A.; Popescu, V.; Yang, S.; Mei, S.; Shi, M.; Puolitaival, S.M.; Caprioli, R.M.; Capdevila, J.H. The anti-tumorigenic properties of peroxisomal proliferator-activated receptor α are arachidonic acid epoxygenase-mediated. J. Biol. Chem. 2010, 285, 12840–12850. [Google Scholar] [CrossRef] [Green Version]
- Wu, L.; Wang, W.; Dai, M.; Li, H.; Chen, C.; Wang, D. PPARα ligand, AVE8134, and cyclooxygenase inhibitor therapy synergistically suppress lung cancer growth and metastasis. BMC Cancer 2019, 19, 1166. [Google Scholar] [CrossRef]
- Garrido-Urbani, S.; Jemelin, S.; Deffert, C.; Carnesecchi, S.; Basset, O.; Szyndralewiez, C.; Heitz, F.; Page, P.; Montet, X.; Michalik, L.; et al. Targeting vascular NADPH oxidase 1 blocks tumor angiogenesis through a PPARα mediated mechanism. PLoS ONE 2011, 6, e14665. [Google Scholar] [CrossRef]
- Abdollahi, A.; Schwager, C.; Kleeff, J.; Esposito, I.; Domhan, S.; Peschke, P.; Hauser, K.; Hahnfeldt, P.; Hlatky, L.; Debus, J.; et al. Transcriptional network governing the angiogenic switch in human pancreatic cancer. Proc. Natl. Acad. Sci. USA 2007, 104, 12890–12895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshinaga, M.; Kitamura, Y.; Chaen, T.; Yamashita, S.; Tsuruta, S.; Hisano, T.; Ikeda, Y.; Sakai, H.; Nakamura, K.; Takayanagi, R.; et al. The simultaneous expression of peroxisome proliferator-activated receptor Delta and cyclooxygenase-2 may enhance angiogenesis and tumor venous invasion in tissues of colorectal cancers. Dig. Dis. Sci. 2009, 54, 1108–1114. [Google Scholar] [CrossRef]
- Müller-Brüsselbach, S.; Kömhoff, M.; Rieck, M.; Meissner, W.; Kaddatz, K.; Adamkiewicz, J.; Keil, B.; Klose, K.J.; Moll, R.; Burdick, A.D.; et al. Deregulation of tumor angiogenesis and blockade of tumor growth in PPARβ-deficient mice. EMBO J. 2007, 26, 3686–3698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuo, X.; Xu, W.; Xu, M.; Tian, R.; Moussalli, M.J.; Mao, F.; Zheng, X.; Wang, J.; Morris, J.S.; Gagea, M.; et al. Metastasis regulation by PPARD expression in cancer cells. JCI Insight 2017, 2, e91419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meissner, M.; Hrgovic, I.; Doll, M.; Naidenow, J.; Reichenbach, G.; Hailemariam-Jahn, T.; Michailidou, D.; Gille, J.; Kaufmann, R. Peroxisome proliferator-activated receptor δ activators induce IL-8 expression in nonstimulated endothelial cells in a transcriptional and posttranscriptional manner. J. Biol. Chem. 2010, 285, 33797–33804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piqueras, L.; Reynolds, A.R.; Hodivala-Dilke, K.M.; Alfranca, A.; Redondo, J.M.; Hatae, T.; Tanabe, T.; Warner, T.D.; Bishop-Bailey, D. Activation of PPARβ/δ induces endothelial cell proliferation and angiogenesis. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 63–69. [Google Scholar] [CrossRef] [Green Version]
- Wagner, K.D.; Vukolic, A.; Baudouy, D.; Michiels, J.F.; Wagner, N. Inducible Conditional Vascular-Specific Overexpression of Peroxisome Proliferator-Activated Receptor Beta/Delta Leads to Rapid Cardiac Hypertrophy. PPAR Res. 2016, 2016, 7631085. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Yang, W.; Zhao, D.; Han, Y.; Liu, B.; Zhao, H.; Wang, H.; Zhang, Q.; Xu, G. Correlation between TSP-1, TGF-β and PPAR-γ expression levels and glioma microvascular density. Oncol. Lett. 2014, 7, 95–100. [Google Scholar] [CrossRef] [Green Version]
- Panigrahy, D.; Singer, S.; Shen, L.Q.; Butterfield, C.E.; Freedman, D.A.; Chen, E.J.; Moses, M.A.; Kilroy, S.; Duensing, S.; Fletcher, C.; et al. PPARγ ligands inhibit primary tumor growth and metastasis by inhibiting angiogenesis. J. Clin. Investig. 2002, 110, 923–932. [Google Scholar] [CrossRef]
- Huang, H.; Campbell, S.C.; Bedford, D.F.; Nelius, T.; Veliceasa, D.; Shroff, E.H.; Henkin, J.; Schneider, A.; Bouck, N.; Volpert, O.V. Peroxisome proliferator-activated receptor γ ligands improve the antitumor efficacy of thrombospondin peptide ABT510. Mol. Cancer Res. 2004, 2, 541–550. [Google Scholar] [CrossRef]
- Keshamouni, V.G.; Arenberg, D.A.; Reddy, R.C.; Newstead, M.J.; Anthwal, S.; Standiford, T.J. PPAR-γ activation inhibits angiogenesis by blocking ELR+CXC chemokine production in non-small cell lung cancer. Neoplasia 2005, 7, 294–301. [Google Scholar] [CrossRef] [Green Version]
- Copland, J.A.; Marlow, L.A.; Kurakata, S.; Fujiwara, K.; Wong, A.K.; Kreinest, P.A.; Williams, S.F.; Haugen, B.R.; Klopper, J.P.; Smallridge, R.C. Novel high-affinity PPARγ agonist alone and in combination with paclitaxel inhibits human anaplastic thyroid carcinoma tumor growth via p21WAF1/CIP1. Oncogene 2006, 25, 2304–2317. [Google Scholar] [CrossRef] [Green Version]
- Xin, B.; Yokoyama, Y.; Shigeto, T.; Futagami, M.; Mizunuma, H. Inhibitory effect of meloxicam, a selective cyclooxygenase-2 inhibitor, and ciglitazone, a peroxisome proliferator-activated receptor gamma ligand, on the growth of human ovarian cancers. Cancer 2007, 110, 791–800. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, Y.; Xin, B.; Shigeto, T.; Mizunuma, H. Combination of ciglitazone, a peroxisome proliferator-activated receptor γ ligand, and cisplatin enhances the inhibition of growth of human ovarian cancers. J. Cancer Res. Clin. Oncol. 2011, 137, 1219–1228. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.W.; Wang, X.P.; Wu, K. Suppression of pancreatic carcinoma growth by activating peroxisome proliferator-activated receptor γ involves angiogenesis inhibition. World J. Gastroenterol. 2009, 15, 441–448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, G.; Yin, L.; Lan, J.; Tong, R.; Li, M.; Na, F.; Mo, X.; Chen, C.; Xue, J.; Lu, Y. Synergy between peroxisome proliferator-activated receptor γ agonist and radiotherapy in cancer. Cancer Sci. 2018, 109, 2243–2255. [Google Scholar] [CrossRef]
- Kramer, K.; Wu, J.; Crowe, D.L. Tumor suppressor control of the cancer stem cell niche. Oncogene 2016, 35, 4165–4178. [Google Scholar] [CrossRef] [Green Version]
- Tian, L.; Zhou, J.; Casimiro, M.C.; Liang, B.; Ojeifo, J.O.; Wang, M.; Hyslop, T.; Wang, C.; Pestell, R.G. Activating peroxisome proliferator-activated receptor γ mutant promotes tumor growth in vivo by enhancing angiogenesis. Cancer Res. 2009, 69, 9236–9244. [Google Scholar] [CrossRef] [Green Version]
- Pich, C.; Meylan, P.; Mastelic-Gavillet, B.; Nguyen, T.N.; Loyon, R.; Trang, B.K.; Moser, H.; Moret, C.; Goepfert, C.; Hafner, J.; et al. Induction of Paracrine Signaling in Metastatic Melanoma Cells by PPARγ Agonist Rosiglitazone Activates Stromal Cells and Enhances Tumor Growth. Cancer Res. 2018, 78, 6447–6461. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, J.A.; Bishop-Bailey, D. PPARβ/δ a potential target in pulmonary hypertension blighted by cancer risk. Pulm. Circ. 2019, 9, 1–2. [Google Scholar] [CrossRef] [Green Version]
- Cai, Y.; Liu, H.; Song, E.; Wang, L.; Xu, J.; He, Y.; Zhang, D.; Zhang, L.; Cheng, K.K.; Jin, L.; et al. Deficiency of telomere-associated repressor activator protein 1 precipitates cardiac aging in mice. Theranostics 2021, 11, 4710–4727. [Google Scholar] [CrossRef]
- Di Leo, L.; Vegliante, R.; Ciccarone, F.; Salvatori, I.; Scimeca, M.; Bonanno, E.; Sagnotta, A.; Grazi, G.L.; Aquilano, K.; Ciriolo, M.R. Forcing ATGL expression in hepatocarcinoma cells imposes glycolytic rewiring through PPAR-α/p300-mediated acetylation of p53. Oncogene 2019, 38, 1860–1875. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Zhang, Y.; Sun, S.; Wang, K.; Qian, J.; Cui, Z.; Tao, T.; Zhou, J. ACOX2 is a prognostic marker and impedes the progression of hepatocellular carcinoma via PPARα pathway. Cell Death Dis. 2021, 12, 15. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Zhong, L.; Yu, L.; Xiong, L.; Dan, W.; Li, J.; Ye, J.; Chu, X.; Liu, C.; Liu, B. TRIB3 destabilizes tumor suppressor PPARα expression through ubiquitin-mediated proteasome degradation in acute myeloid leukemia. Life Sci. 2020, 257, 118021. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Xie, C.; Brocker, C.N.; Fan, J.; Wu, X.; Feng, L.; Wang, Q.; Zhao, J.; Lu, D.; Tandon, M.; et al. Intestinal PPARα Protects Against Colon Carcinogenesis via Regulation of Methyltransferases DNMT1 and PRMT6. Gastroenterology 2019, 157, 744–759.e744. [Google Scholar] [CrossRef]
- Lopez-Guadamillas, E.; Fernandez-Marcos, P.J.; Pantoja, C.; Muñoz-Martin, M.; Martínez, D.; Gómez-López, G.; Campos-Olivas, R.; Valverde, A.M.; Serrano, M. p21Cip1 plays a critical role in the physiological adaptation to fasting through activation of PPARα. Sci. Rep. 2016, 6, 34542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pozzi, S.; Boergesen, M.; Sinha, S.; Mandrup, S.; Mantovani, R. Peroxisome proliferator-activated receptor-α is a functional target of p63 in adult human keratinocytes. J. Investig. Dermatol. 2009, 129, 2376–2385. [Google Scholar] [CrossRef] [Green Version]
- Gizard, F.; Amant, C.; Barbier, O.; Bellosta, S.; Robillard, R.; Percevault, F.; Sevestre, H.; Krimpenfort, P.; Corsini, A.; Rochette, J.; et al. PPAR α inhibits vascular smooth muscle cell proliferation underlying intimal hyperplasia by inducing the tumor suppressor p16INK4a. J. Clin. Investig. 2005, 115, 3228–3238. [Google Scholar] [CrossRef] [PubMed]
- Sherr, C.J. Cancer cell cycles. Science 1996, 274, 1672–1677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamasaki, D.; Kawabe, N.; Nakamura, H.; Tachibana, K.; Ishimoto, K.; Tanaka, T.; Aburatani, H.; Sakai, J.; Hamakubo, T.; Kodama, T.; et al. Fenofibrate suppresses growth of the human hepatocellular carcinoma cell via PPARα-independent mechanisms. Eur. J. Cell Biol. 2011, 90, 657–664. [Google Scholar] [CrossRef]
- Hann, S.S.; Zheng, F.; Zhao, S. Targeting 3-phosphoinositide-dependent protein kinase 1 by N-acetyl-cysteine through activation of peroxisome proliferators activated receptor alpha in human lung cancer cells, the role of p53 and p65. J. Exp. Clin. Cancer Res. 2013, 32, 43. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Zhang, S.; Xue, J.; Avery, J.; Wu, J.; Lind, S.E.; Ding, W.Q. Activation of peroxisome proliferator-activated receptor α (PPARα) suppresses hypoxia-inducible factor-1α (HIF-1α) signaling in cancer cells. J. Biol. Chem. 2012, 287, 35161–35169. [Google Scholar] [CrossRef] [Green Version]
- Cheng, P.; Yang, S.S.; Hu, X.G.; Zhou, X.Y.; Zhang, Y.J.; Jin, G.; Zhou, Y.Q. Menin prevents liver steatosis through co-activation of peroxisome proliferator-activated receptor alpha. FEBS Lett. 2011, 585, 3403–3408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Deguchi, Y.; Wei, D.; Liu, F.; Moussalli, M.J.; Deguchi, E.; Li, D.; Wang, H.; Valentin, L.A.; Colby, J.K.; et al. Rapid acceleration of KRAS-mutant pancreatic carcinogenesis via remodeling of tumor immune microenvironment by PPARδ. Nat. Commun. 2022, 13, 2665. [Google Scholar] [CrossRef] [PubMed]
- Bapat, S.P.; Whitty, C.; Mowery, C.T.; Liang, Y.; Yoo, A.; Jiang, Z.; Peters, M.C.; Zhang, L.J.; Vogel, I.; Zhou, C.; et al. Obesity alters pathology and treatment response in inflammatory disease. Nature 2022, 604, 337–342. [Google Scholar] [CrossRef] [PubMed]
- Shao, J.; Sheng, H.; DuBois, R.N. Peroxisome proliferator-activated receptors modulate K-Ras-mediated transformation of intestinal epithelial cells. Cancer Res. 2002, 62, 3282–3288. [Google Scholar]
- Li, Z.; Li, H.; Zhao, Z.B.; Zhu, W.; Feng, P.P.; Zhu, X.W.; Gong, J.P. SIRT4 silencing in tumor-associated macrophages promotes HCC development via PPARδ signalling-mediated alternative activation of macrophages. J. Exp. Clin. Cancer Res. 2019, 38, 469. [Google Scholar] [CrossRef]
- Rangel-Sánchez, I.Y.; Salas-Treviño, D.; Soto-Domínguez, A.; Garza-Rodríguez, O.I.; Saucedo-Cárdenas, O.; Zapata-Benavides, P.; Zarate-Ramos, J.J.; Cedillo-Rosales, S.; Zamora-Ávila, D.E. Expression of the Wilms’ tumour gene and its association with PPARβ/δ in healthy skin and melanoma of horses. Acta Vet. Hung. 2021, 68, 374–379. [Google Scholar] [CrossRef]
- Wagner, K.D.; El Maï, M.; Ladomery, M.; Belali, T.; Leccia, N.; Michiels, J.F.; Wagner, N. Altered VEGF Splicing Isoform Balance in Tumor Endothelium Involves Activation of Splicing Factors Srpk1 and Srsf1 by the Wilms’ Tumor Suppressor Wt1. Cells 2019, 8, 41. [Google Scholar] [CrossRef] [Green Version]
- El Maï, M.; Wagner, K.D.; Michiels, J.F.; Ambrosetti, D.; Borderie, A.; Destree, S.; Renault, V.; Djerbi, N.; Giraud-Panis, M.J.; Gilson, E.; et al. The Telomeric Protein TRF2 Regulates Angiogenesis by Binding and Activating the PDGFRβ Promoter. Cell Rep. 2014, 9, 1047–1060. [Google Scholar] [CrossRef] [Green Version]
- Wagner, N.; Michiels, J.F.; Schedl, A.; Wagner, K.D. The Wilms’ tumour suppressor WT1 is involved in endothelial cell proliferation and migration: Expression in tumour vessels in vivo. Oncogene 2008, 27, 3662–3672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanada, S.; Tsuruta, T.; Haraguchi, K.; Okamoto, M.; Sugiyama, H.; Koido, S. Long-term survival of pancreatic cancer patients treated with multimodal therapy combined with WT1-targeted dendritic cell vaccines. Hum. Vaccin. Immunother. 2019, 15, 397–406. [Google Scholar] [CrossRef] [Green Version]
- Sugiyama, H. WT1 (Wilms’ tumor gene 1): Biology and cancer immunotherapy. Jpn. J. Clin. Oncol. 2010, 40, 377–387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oka, Y.; Tsuboi, A.; Oji, Y.; Kawase, I.; Sugiyama, H. WT1 peptide vaccine for the treatment of cancer. Curr. Opin. Immunol. 2008, 20, 211–220. [Google Scholar] [CrossRef] [PubMed]
- Oji, Y.; Miyoshi, S.; Maeda, H.; Hayashi, S.; Tamaki, H.; Nakatsuka, S.; Yao, M.; Takahashi, E.; Nakano, Y.; Hirabayashi, H.; et al. Overexpression of the Wilms’ tumor gene WT1 in de novo lung cancers. Int. J. Cancer 2002, 100, 297–303. [Google Scholar] [CrossRef]
- Lim, H.J.; Lee, S.; Park, J.H.; Lee, K.S.; Choi, H.E.; Chung, K.S.; Lee, H.H.; Park, H.Y. PPAR δ agonist L-165041 inhibits rat vascular smooth muscle cell proliferation and migration via inhibition of cell cycle. Atherosclerosis 2009, 202, 446–454. [Google Scholar] [CrossRef]
- Zhai, Y.; Wu, R.; Schwartz, D.R.; Darrah, D.; Reed, H.; Kolligs, F.T.; Nieman, M.T.; Fearon, E.R.; Cho, K.R. Role of β-catenin/T-cell factor-regulated genes in ovarian endometrioid adenocarcinomas. Am. J. Pathol. 2002, 160, 1229–1238. [Google Scholar] [CrossRef]
- Kundu, J.; Wahab, S.M.; Kundu, J.K.; Choi, Y.L.; Erkin, O.C.; Lee, H.S.; Park, S.G.; Shin, Y.K. Tob1 induces apoptosis and inhibits proliferation, migration and invasion of gastric cancer cells by activating Smad4 and inhibiting β-catenin signaling. Int. J. Oncol. 2012, 41, 839–848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kannan-Thulasiraman, P.; Seachrist, D.D.; Mahabeleshwar, G.H.; Jain, M.K.; Noy, N. Fatty acid-binding protein 5 and PPARβ/δ are critical mediators of epidermal growth factor receptor-induced carcinoma cell growth. J. Biol. Chem. 2010, 285, 19106–19115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pseftogas, A.; Gonidas, C.; Mosialos, G. Activation of peroxisome proliferator-activated receptor gamma in mammary epithelial cells upregulates the expression of tumor suppressor Cyld to mediate growth inhibition and anti-inflammatory effects. Int. J. Biochem. Cell Biol. 2017, 82, 49–56. [Google Scholar] [CrossRef]
- Avasarala, S.; Bikkavilli, R.K.; Van Scoyk, M.; Zhang, W.; Lapite, A.; Hostetter, L.; Byers, J.T.; Heasley, L.E.; Sohn, J.W.; Winn, R.A. Heterotrimeric G-protein, Gα16, is a critical downstream effector of non-canonical Wnt signaling and a potent inhibitor of transformed cell growth in non small cell lung cancer. PLoS ONE 2013, 8, e76895. [Google Scholar] [CrossRef]
- Calo, E.; Quintero-Estades, J.A.; Danielian, P.S.; Nedelcu, S.; Berman, S.D.; Lees, J.A. Rb regulates fate choice and lineage commitment in vivo. Nature 2010, 466, 1110–1114. [Google Scholar] [CrossRef] [Green Version]
- Yasmeen, R.; Meyers, J.M.; Alvarez, C.E.; Thomas, J.L.; Bonnegarde-Bernard, A.; Alder, H.; Papenfuss, T.L.; Benson, D.M.; Boyaka, P.N.; Ziouzenkova, O. Aldehyde dehydrogenase-1a1 induces oncogene suppressor genes in B cell populations. Biochim. Biophys. Acta 2013, 1833, 3218–3227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheung, K.F.; Zhao, J.; Hao, Y.; Li, X.; Lowe, A.W.; Cheng, A.S.; Sung, J.J.; Yu, J. CITED2 is a novel direct effector of peroxisome proliferator-activated receptor γ in suppressing hepatocellular carcinoma cell growth. Cancer 2013, 119, 1217–1226. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Huang, G.; Mei, S.; Qian, J.; Ji, J.; Zhang, J. Over-expression of C/EBP-α induces apoptosis in cultured rat hepatic stellate cells depending on p53 and peroxisome proliferator-activated receptor-γ. Biochem. Biophys. Res. Commun. 2009, 380, 286–291. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, L.; Heyes, E.; Grebien, F. Gain-of-Function Effects of N-Terminal CEBPA Mutations in Acute Myeloid Leukemia. Bioessays 2020, 42, e1900178. [Google Scholar] [CrossRef] [Green Version]
- Gery, S.; Tanosaki, S.; Bose, S.; Bose, N.; Vadgama, J.; Koeffler, H.P. Down-regulation and growth inhibitory role of C/EBPα in breast cancer. Clin. Cancer Res. 2005, 11, 3184–3190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonofiglio, D.; Gabriele, S.; Aquila, S.; Catalano, S.; Gentile, M.; Middea, E.; Giordano, F.; Andò, S. Estrogen receptor α binds to peroxisome proliferator-activated receptor response element and negatively interferes with peroxisome proliferator-activated receptor γ signaling in breast cancer cells. Clin. Cancer Res. 2005, 11, 6139–6147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grau, R.; Punzón, C.; Fresno, M.; Iñiguez, M.A. Peroxisome-proliferator-activated receptor α agonists inhibit cyclo-oxygenase 2 and vascular endothelial growth factor transcriptional activation in human colorectal carcinoma cells via inhibition of activator protein-1. Biochem. J. 2006, 395, 81–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meissner, M.; Berlinski, B.; Gille, J.; Doll, M.; Kaufmann, R. Peroxisome proliferator activated receptor-α agonists suppress transforming growth factor-α-induced matrix metalloproteinase-9 expression in human keratinocytes. Clin. Exp. Dermatol. 2011, 36, 911–914. [Google Scholar] [CrossRef] [PubMed]
- Grabacka, M.; Placha, W.; Plonka, P.M.; Pajak, S.; Urbanska, K.; Laidler, P.; Slominski, A. Inhibition of melanoma metastases by fenofibrate. Arch. Dermatol. Res. 2004, 296, 54–58. [Google Scholar] [CrossRef]
- Grabacka, M.; Plonka, P.M.; Urbanska, K.; Reiss, K. Peroxisome proliferator-activated receptor α activation decreases metastatic potential of melanoma cells in vitro via down-regulation of Akt. Clin. Cancer Res. 2006, 12, 3028–3036. [Google Scholar] [CrossRef] [Green Version]
- Wejksza, K.; Lee-Chang, C.; Bodogai, M.; Bonzo, J.; Gonzalez, F.J.; Lehrmann, E.; Becker, K.; Biragyn, A. Cancer-produced metabolites of 5-lipoxygenase induce tumor-evoked regulatory B cells via peroxisome proliferator-activated receptor α. J. Immunol. 2013, 190, 2575–2584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.K.; Jeong, S.H.; Jang, C.; Bae, H.; Kim, Y.H.; Park, I.; Kim, S.K.; Koh, G.Y. Tumor metastasis to lymph nodes requires YAP-dependent metabolic adaptation. Science 2019, 363, 644–649. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.W.; Chou, C.T.; Chang, C.C.; Li, Y.J.; Chen, S.T.; Lin, I.C.; Kok, S.H.; Cheng, S.J.; Lee, J.J.; Wu, T.S.; et al. HMGCS2 enhances invasion and metastasis via direct interaction with PPARα to activate Src signaling in colorectal cancer and oral cancer. Oncotarget 2017, 8, 22460–22476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leng, J.; Li, H.; Niu, Y.; Chen, K.; Yuan, X.; Chen, H.; Fu, Z.; Zhang, L.; Wang, F.; Chen, C.; et al. Low-dose mono(2-ethylhexyl) phthalate promotes ovarian cancer development through PPARα-dependent PI3K/Akt/NF-κB pathway. Sci. Total Environ. 2021, 790, 147990. [Google Scholar] [CrossRef] [PubMed]
- Coleman, J.D.; Thompson, J.T.; Smith, R.W.; Prokopczyk, B.; Vanden Heuvel, J.P. Role of Peroxisome Proliferator-Activated Receptor β/δ and B-Cell Lymphoma-6 in Regulation of Genes Involved in Metastasis and Migration in Pancreatic Cancer Cells. PPAR Res. 2013, 2013, 121956. [Google Scholar] [CrossRef] [Green Version]
- Ham, S.A.; Yoo, T.; Lee, W.J.; Hwang, J.S.; Hur, J.; Paek, K.S.; Lim, D.S.; Han, S.G.; Lee, C.H.; Seo, H.G. ADAMTS1-mediated targeting of TSP-1 by PPARδ suppresses migration and invasion of breast cancer cells. Oncotarget 2017, 8, 94091–94103. [Google Scholar] [CrossRef] [Green Version]
- Lim, J.C.W.; Kwan, Y.P.; Tan, M.S.; Teo, M.H.Y.; Chiba, S.; Wahli, W.; Wang, X. The Role of PPARβ/δ in Melanoma Metastasis. Int. J. Mol. Sci. 2018, 19, 2860. [Google Scholar] [CrossRef] [Green Version]
- Elie-Caille, C.; Lascombe, I.; Péchery, A.; Bittard, H.; Fauconnet, S. Molecular and nanoscale evaluation of N-cadherin expression in invasive bladder cancer cells under control conditions or GW501516 exposure. Mol. Cell. Biochem. 2020, 471, 113–127. [Google Scholar] [CrossRef]
- Woutersen, R.A.; Appel, M.J.; van Garderen-Hoetmer, A.; Wijnands, M.V. Dietary fat and carcinogenesis. Mutat. Res. 1999, 443, 111–127. [Google Scholar] [CrossRef]
- Wang, D.; Fu, L.; Wei, J.; Xiong, Y.; DuBois, R.N. PPARδ Mediates the Effect of Dietary Fat in Promoting Colorectal Cancer Metastasis. Cancer Res. 2019, 79, 4480–4490. [Google Scholar] [CrossRef] [Green Version]
- Sunami, E.; Tsuno, N.H.; Kitayama, J.; Saito, S.; Osada, T.; Yamaguchi, H.; Tomozawa, S.; Tsuruo, T.; Shibata, Y.; Nagawa, H. Decreased synthesis of matrix metalloproteinase-7 and adhesion to the extracellular matrix proteins of human colon cancer cells treated with troglitazone. Surg. Today 2002, 32, 343–350. [Google Scholar] [CrossRef] [PubMed]
- Yoshizumi, T.; Ohta, T.; Ninomiya, I.; Terada, I.; Fushida, S.; Fujimura, T.; Nishimura, G.; Shimizu, K.; Yi, S.; Miwa, K. Thiazolidinedione, a peroxisome proliferator-activated receptor-γ ligand, inhibits growth and metastasis of HT-29 human colon cancer cells through differentiation-promoting effects. Int. J. Oncol. 2004, 25, 631–639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sasaki, T.; Fujii, K.; Yoshida, K.; Shimura, H.; Sasahira, T.; Ohmori, H.; Kuniyasu, H. Peritoneal metastasis inhibition by linoleic acid with activation of PPARγ in human gastrointestinal cancer cells. Virchows Arch. 2006, 448, 422–427. [Google Scholar] [CrossRef] [PubMed]
- Richard, C.L.; Blay, J. Thiazolidinedione drugs down-regulate CXCR4 expression on human colorectal cancer cells in a peroxisome proliferator activated receptor γ-dependent manner. Int. J. Oncol. 2007, 30, 1215–1222. [Google Scholar] [CrossRef] [Green Version]
- Richard, C.L.; Lowthers, E.L.; Blay, J. 15-Deoxy-Δ12,14-prostaglandin J2 down-regulates CXCR4 on carcinoma cells through PPARγ- and NFkappaB-mediated pathways. Exp. Cell Res. 2007, 313, 3446–3458. [Google Scholar] [CrossRef]
- Rovito, D.; Gionfriddo, G.; Barone, I.; Giordano, C.; Grande, F.; De Amicis, F.; Lanzino, M.; Catalano, S.; Andò, S.; Bonofiglio, D. Ligand-activated PPARγ downregulates CXCR4 gene expression through a novel identified PPAR response element and inhibits breast cancer progression. Oncotarget 2016, 7, 65109–65124. [Google Scholar] [CrossRef] [Green Version]
- Pancione, M.; Forte, N.; Sabatino, L.; Tomaselli, E.; Parente, D.; Febbraro, A.; Colantuoni, V. Reduced β-catenin and peroxisome proliferator-activated receptor-γ expression levels are associated with colorectal cancer metastatic progression: Correlation with tumor-associated macrophages, cyclooxygenase 2, and patient outcome. Hum. Pathol. 2009, 40, 714–725. [Google Scholar] [CrossRef]
- Moon, C.M.; Kwon, J.H.; Kim, J.S.; Oh, S.H.; Jin Lee, K.; Park, J.J.; Pil Hong, S.; Cheon, J.H.; Kim, T.I.; Kim, W.H. Nonsteroidal anti-inflammatory drugs suppress cancer stem cells via inhibiting PTGS2 (cyclooxygenase 2) and NOTCH/HES1 and activating PPARG in colorectal cancer. Int. J. Cancer 2014, 134, 519–529. [Google Scholar] [CrossRef]
- Magenta, G.; Borenstein, X.; Rolando, R.; Jasnis, M.A. Rosiglitazone inhibits metastasis development of a murine mammary tumor cell line LMM3. BMC Cancer 2008, 8, 47. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.R.; Kim, H.J.; Lee, S.K.; Ma, G.T.; Park, K.K.; Chung, W.Y. 15-deoxy-δ12,14-prostaglandin J2 inhibits osteolytic breast cancer bone metastasis and estrogen deficiency-induced bone loss. PLoS ONE 2015, 10, e0122764. [Google Scholar] [CrossRef]
- Bren-Mattison, Y.; Van Putten, V.; Chan, D.; Winn, R.; Geraci, M.W.; Nemenoff, R.A. Peroxisome proliferator-activated receptor-γ (PPAR(γ)) inhibits tumorigenesis by reversing the undifferentiated phenotype of metastatic non-small-cell lung cancer cells (NSCLC). Oncogene 2005, 24, 1412–1422. [Google Scholar] [CrossRef] [Green Version]
- Reka, A.K.; Kurapati, H.; Narala, V.R.; Bommer, G.; Chen, J.; Standiford, T.J.; Keshamouni, V.G. Peroxisome proliferator-activated receptor-γ activation inhibits tumor metastasis by antagonizing Smad3-mediated epithelial-mesenchymal transition. Mol. Cancer Ther. 2010, 9, 3221–3232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Q.Y.; Huang, X.B.; Zhao, Y.J.; Wang, H.G.; Wang, J.B.; Liu, L.C.; Wang, L.Q.; Zhong, Q.; Xie, J.W.; Lin, J.X.; et al. The peroxisome proliferator-activated receptor agonist rosiglitazone specifically represses tumour metastatic potential in chromatin inaccessibility-mediated FABP4-deficient gastric cancer. Theranostics 2022, 12, 1904–1920. [Google Scholar] [CrossRef] [PubMed]
- Sawai, H.; Liu, J.; Reber, H.A.; Hines, O.J.; Eibl, G. Activation of peroxisome proliferator-activated receptor-γ decreases pancreatic cancer cell invasion through modulation of the plasminogen activator system. Mol. Cancer Res. 2006, 4, 159–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, H.T.; Sung, M.T.; Lee, C.C.; Kuo, Y.J.; Chi, C.W.; Lee, H.C.; Hsia, C.Y. Peroxisome Proliferator-Activated Receptor γ Expression Is Inversely Associated with Macroscopic Vascular Invasion in Human Hepatocellular Carcinoma. Int. J. Mol. Sci. 2016, 17, 1226. [Google Scholar] [CrossRef] [Green Version]
- Shen, B.; Chu, E.S.; Zhao, G.; Man, K.; Wu, C.W.; Cheng, J.T.; Li, G.; Nie, Y.; Lo, C.M.; Teoh, N.; et al. PPARγ inhibits hepatocellular carcinoma metastases in vitro and in mice. Br. J. Cancer 2012, 106, 1486–1494. [Google Scholar] [CrossRef] [Green Version]
- Tu, K.; Zheng, X.; Dou, C.; Li, C.; Yang, W.; Yao, Y.; Liu, Q. MicroRNA-130b promotes cell aggressiveness by inhibiting peroxisome proliferator-activated receptor γ in human hepatocellular carcinoma. Int. J. Mol. Sci. 2014, 15, 20486–20499. [Google Scholar] [CrossRef] [Green Version]
- Zuo, Q.; He, J.; Zhang, S.; Wang, H.; Jin, G.; Jin, H.; Cheng, Z.; Tao, X.; Yu, C.; Li, B.; et al. PPARγ Coactivator-1α Suppresses Metastasis of Hepatocellular Carcinoma by Inhibiting Warburg Effect by PPARγ-Dependent WNT/β-Catenin/Pyruvate Dehydrogenase Kinase Isozyme 1 Axis. Hepatology 2021, 73, 644–660. [Google Scholar] [CrossRef]
- Kim, K.R.; Choi, H.N.; Lee, H.J.; Baek, H.A.; Park, H.S.; Jang, K.Y.; Chung, M.J.; Moon, W.S. A peroxisome proliferator-activated receptor γ antagonist induces vimentin cleavage and inhibits invasion in high-grade hepatocellular carcinoma. Oncol. Rep. 2007, 18, 825–832. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Liu, F.; Mao, X.; Huang, J.; Yang, J.; Yin, X.; Wu, L.; Zheng, L.; Wang, Q. Elevation of miR-27b by HPV16 E7 inhibits PPARγ expression and promotes proliferation and invasion in cervical carcinoma cells. Int. J. Oncol. 2015, 47, 1759–1766. [Google Scholar] [CrossRef] [Green Version]
- Ahn, Y.H.; Yang, Y.; Gibbons, D.L.; Creighton, C.J.; Yang, F.; Wistuba, I.I.; Lin, W.; Thilaganathan, N.; Alvarez, C.A.; Roybal, J.; et al. Map2k4 functions as a tumor suppressor in lung adenocarcinoma and inhibits tumor cell invasion by decreasing peroxisome proliferator-activated receptor γ2 expression. Mol. Cell. Biol. 2011, 31, 4270–4285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Sorenson, A.L.; Poczobutt, J.; Amin, J.; Joyal, T.; Sullivan, T.; Crossno, J.T.; Weiser-Evans, M.C.; Nemenoff, R.A. Activation of PPARγ in myeloid cells promotes lung cancer progression and metastasis. PLoS ONE 2011, 6, e28133. [Google Scholar] [CrossRef] [PubMed]
- Sippel, T.R.; Johnson, A.M.; Li, H.Y.; Hanson, D.; Nguyen, T.T.; Bullock, B.L.; Poczobutt, J.M.; Kwak, J.W.; Kleczko, E.K.; Weiser-Evans, M.C.; et al. Activation of PPARγ in Myeloid Cells Promotes Progression of Epithelial Lung Tumors through TGFβ1. Mol. Cancer Res. 2019, 17, 1748–1758. [Google Scholar] [CrossRef] [PubMed]
- Herroon, M.K.; Rajagurubandara, E.; Hardaway, A.L.; Powell, K.; Turchick, A.; Feldmann, D.; Podgorski, I. Bone marrow adipocytes promote tumor growth in bone via FABP4-dependent mechanisms. Oncotarget 2013, 4, 2108–2123. [Google Scholar] [CrossRef] [Green Version]
- Niu, Z.; Shi, Q.; Zhang, W.; Shu, Y.; Yang, N.; Chen, B.; Wang, Q.; Zhao, X.; Chen, J.; Cheng, N.; et al. Caspase-1 cleaves PPARγ for potentiating the pro-tumor action of TAMs. Nat. Commun. 2017, 8, 766. [Google Scholar] [CrossRef] [Green Version]
- Shu, Y.; Qin, M.; Song, Y.; Tang, Q.; Huang, Y.; Shen, P.; Lu, Y. M2 polarization of tumor-associated macrophages is dependent on integrin β3 via peroxisome proliferator-activated receptor-γ up-regulation in breast cancer. Immunology 2020, 160, 345–356. [Google Scholar] [CrossRef]
- Zou, Y.; Watters, A.; Cheng, N.; Perry, C.E.; Xu, K.; Alicea, G.M.; Parris, J.L.D.; Baraban, E.; Ray, P.; Nayak, A.; et al. Polyunsaturated Fatty Acids from Astrocytes Activate PPARγ Signaling in Cancer Cells to Promote Brain Metastasis. Cancer Discov. 2019, 9, 1720–1735. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, I.; Mui, E.; Galbraith, L.; Patel, R.; Tan, E.H.; Salji, M.; Rust, A.G.; Repiscak, P.; Hedley, A.; Markert, E.; et al. Sleeping Beauty screen reveals Pparg activation in metastatic prostate cancer. Proc. Natl. Acad. Sci. USA 2016, 113, 8290–8295. [Google Scholar] [CrossRef] [Green Version]
- Liu, R.Z.; Choi, W.S.; Jain, S.; Dinakaran, D.; Xu, X.; Han, W.H.; Yang, X.H.; Glubrecht, D.D.; Moore, R.B.; Lemieux, H.; et al. The FABP12/PPARγ pathway promotes metastatic transformation by inducing epithelial-to-mesenchymal transition and lipid-derived energy production in prostate cancer cells. Mol. Oncol. 2020, 14, 3100–3120. [Google Scholar] [CrossRef]
- Galbraith, L.C.A.; Mui, E.; Nixon, C.; Hedley, A.; Strachan, D.; MacKay, G.; Sumpton, D.; Sansom, O.J.; Leung, H.Y.; Ahmad, I. PPAR-gamma induced AKT3 expression increases levels of mitochondrial biogenesis driving prostate cancer. Oncogene 2021, 40, 2355–2366. [Google Scholar] [CrossRef]
- Yang, D.R.; Lin, S.J.; Ding, X.F.; Miyamoto, H.; Messing, E.; Li, L.Q.; Wang, N.; Chang, C. Higher expression of peroxisome proliferator-activated receptor γ or its activation by agonist thiazolidinedione-rosiglitazone promotes bladder cancer cell migration and invasion. Urology 2013, 81, e1101–e1106. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Luo, H.Y.; Liu, G.L.; Wang, D.S.; Wang, Z.Q.; Zeng, Z.L.; Xu, R.H. Prognostic significance and therapeutic implications of peroxisome proliferator-activated receptor γ overexpression in human pancreatic carcinoma. Int. J. Oncol. 2015, 46, 175–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeuchi, A.; Yamamoto, N.; Shirai, T.; Hayashi, K.; Miwa, S.; Munesue, S.; Yamamoto, Y.; Tsuchiya, H. Clinical relevance of peroxisome proliferator-activated receptor-gamma expression in myxoid liposarcoma. BMC Cancer 2016, 16, 442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pentinmikko, N.; Iqbal, S.; Mana, M.; Andersson, S.; Cognetta, A.B.; Suciu, R.M.; Roper, J.; Luopajärvi, K.; Markelin, E.; Gopalakrishnan, S.; et al. Notum produced by Paneth cells attenuates regeneration of aged intestinal epithelium. Nature 2019, 571, 398–402. [Google Scholar] [CrossRef]
- Howroyd, P.; Swanson, C.; Dunn, C.; Cattley, R.C.; Corton, J.C. Decreased longevity and enhancement of age-dependent lesions in mice lacking the nuclear receptor peroxisome proliferator-activated receptor α (PPARα). Toxicol. Pathol. 2004, 32, 591–599. [Google Scholar] [CrossRef] [Green Version]
- Youssef, J.; Badr, M. Enhanced hepatocarcinogenicity due to agonists of peroxisome proliferator-activated receptors in senescent rats: Role of peroxisome proliferation, cell proliferation, and apoptosis. Sci. World J. 2002, 2, 1491–1500. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.J.; Ham, S.A.; Kim, M.Y.; Hwang, J.S.; Lee, H.; Kang, E.S.; Yoo, T.; Woo, I.S.; Yabe-Nishimura, C.; Paek, K.S.; et al. PPARδ coordinates angiotensin II-induced senescence in vascular smooth muscle cells through PTEN-mediated inhibition of superoxide generation. J. Biol. Chem. 2011, 286, 44585–44593. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.Y.; Kang, E.S.; Ham, S.A.; Hwang, J.S.; Yoo, T.S.; Lee, H.; Paek, K.S.; Park, C.; Lee, H.T.; Kim, J.H.; et al. The PPARδ-mediated inhibition of angiotensin II-induced premature senescence in human endothelial cells is SIRT1-dependent. Biochem. Pharmacol. 2012, 84, 1627–1634. [Google Scholar] [CrossRef]
- Ham, S.A.; Hwang, J.S.; Yoo, T.; Lee, H.; Kang, E.S.; Park, C.; Oh, J.W.; Lee, H.T.; Min, G.; Kim, J.H.; et al. Ligand-activated PPARδ inhibits UVB-induced senescence of human keratinocytes via PTEN-mediated inhibition of superoxide production. Biochem. J. 2012, 444, 27–38. [Google Scholar] [CrossRef] [Green Version]
- Altieri, P.; Spallarossa, P.; Barisione, C.; Garibaldi, S.; Garuti, A.; Fabbi, P.; Ghigliotti, G.; Brunelli, C. Inhibition of doxorubicin-induced senescence by PPARδ activation agonists in cardiac muscle cells: Cooperation between PPARδ and Bcl6. PLoS ONE 2012, 7, e46126. [Google Scholar] [CrossRef] [Green Version]
- Zhu, B.; Ferry, C.H.; Blazanin, N.; Bility, M.T.; Khozoie, C.; Kang, B.H.; Glick, A.B.; Gonzalez, F.J.; Peters, J.M. PPARβ/δ promotes HRAS-induced senescence and tumor suppression by potentiating p-ERK and repressing p-AKT signaling. Oncogene 2014, 33, 5348–5359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, B.; Ferry, C.H.; Markell, L.K.; Blazanin, N.; Glick, A.B.; Gonzalez, F.J.; Peters, J.M. The nuclear receptor peroxisome proliferator-activated receptor-β/δ (PPARβ/δ) promotes oncogene-induced cellular senescence through repression of endoplasmic reticulum stress. J. Biol. Chem. 2014, 289, 20102–20119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riahi, Y.; Kaiser, N.; Cohen, G.; Abd-Elrahman, I.; Blum, G.; Shapira, O.M.; Koler, T.; Simionescu, M.; Sima, A.V.; Zarkovic, N.; et al. Foam cell-derived 4-hydroxynonenal induces endothelial cell senescence in a TXNIP-dependent manner. J. Cell Mol. Med. 2015, 19, 1887–1899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernal, C.; Araya, C.; Palma, V.; Bronfman, M. PPARβ/δ and PPARγ maintain undifferentiated phenotypes of mouse adult neural precursor cells from the subventricular zone. Front. Cell. Neurosci. 2015, 9, 78. [Google Scholar] [CrossRef] [Green Version]
- Scholtysek, C.; Katzenbeisser, J.; Fu, H.; Uderhardt, S.; Ipseiz, N.; Stoll, C.; Zaiss, M.M.; Stock, M.; Donhauser, L.; Böhm, C.; et al. PPARβ/δ governs Wnt signaling and bone turnover. Nat. Med. 2013, 19, 608–613. [Google Scholar] [CrossRef]
- Lecot, P.; Alimirah, F.; Desprez, P.Y.; Campisi, J.; Wiley, C. Context-dependent effects of cellular senescence in cancer development. Br. J. Cancer 2016, 114, 1180–1184. [Google Scholar] [CrossRef] [Green Version]
- Milanovic, M.; Fan, D.N.Y.; Belenki, D.; Däbritz, J.H.M.; Zhao, Z.; Yu, Y.; Dörr, J.R.; Dimitrova, L.; Lenze, D.; Monteiro Barbosa, I.A.; et al. Senescence-associated reprogramming promotes cancer stemness. Nature 2018, 553, 96–100. [Google Scholar] [CrossRef] [Green Version]
- Alimirah, F.; Pulido, T.; Valdovinos, A.; Alptekin, S.; Chang, E.; Jones, E.; Diaz, D.A.; Flores, J.; Velarde, M.C.; Demaria, M.; et al. Cellular Senescence Promotes Skin Carcinogenesis through p38MAPK and p44/42MAPK Signaling. Cancer Res. 2020, 80, 3606–3619. [Google Scholar] [CrossRef]
- Abdul-Aziz, A.M.; Sun, Y.; Hellmich, C.; Marlein, C.R.; Mistry, J.; Forde, E.; Piddock, R.E.; Shafat, M.S.; Morfakis, A.; Mehta, T.; et al. Acute myeloid leukemia induces protumoral p16INK4a-driven senescence in the bone marrow microenvironment. Blood 2019, 133, 446–456. [Google Scholar] [CrossRef] [Green Version]
- Wu, L.; Li, X.; Lin, Q.; Chowdhury, F.; Mazumder, M.H.; Du, W. FANCD2 and HES1 suppress inflammation-induced PPARɣ to prevent haematopoietic stem cell exhaustion. Br. J. Haematol. 2021, 192, 652–663. [Google Scholar] [CrossRef]
- Helman, A.; Klochendler, A.; Azazmeh, N.; Gabai, Y.; Horwitz, E.; Anzi, S.; Swisa, A.; Condiotti, R.; Granit, R.Z.; Nevo, Y.; et al. p16(Ink4a)-induced senescence of pancreatic beta cells enhances insulin secretion. Nat. Med. 2016, 22, 412–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, L.; Zhou, R.; Niu, J.; McNutt, M.A.; Wang, P.; Tong, T. SIRT1 is regulated by a PPAR{γ}-SIRT1 negative feedback loop associated with senescence. Nucleic Acids Res. 2010, 38, 7458–7471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gan, Q.; Huang, J.; Zhou, R.; Niu, J.; Zhu, X.; Wang, J.; Zhang, Z.; Tong, T. PPAR{γ} accelerates cellular senescence by inducing p16INK4{α} expression in human diploid fibroblasts. J. Cell Sci. 2008, 121, 2235–2245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Theocharis, S.; Giaginis, C.; Parasi, A.; Margeli, A.; Kakisis, J.; Agapitos, E.; Kouraklis, G. Expression of peroxisome proliferator-activated receptor-γ in colon cancer: Correlation with histopathological parameters, cell cycle-related molecules, and patients’ survival. Dig. Dis. Sci. 2007, 52, 2305–2311. [Google Scholar] [CrossRef] [PubMed]
- Hasan, A.U.; Ohmori, K.; Hashimoto, T.; Kamitori, K.; Hirata, Y.; Ishihara, Y.; Okamoto, N.; Noma, T.; Kosaka, H.; Tokuda, M.; et al. Pioglitazone promotes preadipocyte proliferation by downregulating p16(Ink4a). Biochem. Biophys. Res. Commun. 2011, 411, 375–380. [Google Scholar] [CrossRef]
- Werner, C.; Gensch, C.; Pöss, J.; Haendeler, J.; Böhm, M.; Laufs, U. Pioglitazone activates aortic telomerase and prevents stress-induced endothelial apoptosis. Atherosclerosis 2011, 216, 23–34. [Google Scholar] [CrossRef]
- Mello, T.; Materozzi, M.; Galli, A. PPARs and Mitochondrial Metabolism: From NAFLD to HCC. PPAR Res. 2016, 2016, 7403230. [Google Scholar] [CrossRef]
- Vegliante, R.; Di Leo, L.; Ciccarone, F.; Ciriolo, M.R. Hints on ATGL implications in cancer: Beyond bioenergetic clues. Cell Death Dis. 2018, 9, 316. [Google Scholar] [CrossRef]
- Warburg, O.; Wind, F.; Negelein, E. The metabolism of tumors in the body. J. Gen. Physiol. 1927, 8, 519–530. [Google Scholar] [CrossRef] [Green Version]
- Maxwell, P.H.; Dachs, G.U.; Gleadle, J.M.; Nicholls, L.G.; Harris, A.L.; Stratford, I.J.; Hankinson, O.; Pugh, C.W.; Ratcliffe, P.J. Hypoxia-inducible factor-1 modulates gene expression in solid tumors and influences both angiogenesis and tumor growth. Proc. Natl. Acad. Sci. USA 1997, 94, 8104–8109. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.L.; Semenza, G.L. General involvement of hypoxia-inducible factor 1 in transcriptional response to hypoxia. Proc. Natl. Acad. Sci. USA 1993, 90, 4304–4308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forsythe, J.A.; Jiang, B.H.; Iyer, N.V.; Agani, F.; Leung, S.W.; Koos, R.D.; Semenza, G.L. Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol. Cell. Biol. 1996, 16, 4604–4613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagner, K.D.; Wagner, N.; Wellmann, S.; Schley, G.; Bondke, A.; Theres, H.; Scholz, H. Oxygen-regulated expression of the Wilms’ tumor suppressor Wt1 involves hypoxia-inducible factor-1 (HIF-1). FASEB J. 2003, 17, 1364–1366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narravula, S.; Colgan, S.P. Hypoxia-inducible factor 1-mediated inhibition of peroxisome proliferator-activated receptor α expression during hypoxia. J. Immunol. 2001, 166, 7543–7548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balamurugan, K. HIF-1 at the crossroads of hypoxia, inflammation, and cancer. Int. J. Cancer 2016, 138, 1058–1066. [Google Scholar] [CrossRef]
- Reddy, J.K.; Rao, S.; Moody, D.E. Hepatocellular carcinomas in acatalasemic mice treated with nafenopin, a hypolipidemic peroxisome proliferator. Cancer Res. 1976, 36, 1211–1217. [Google Scholar]
- Drukala, J.; Urbanska, K.; Wilk, A.; Grabacka, M.; Wybieralska, E.; Del Valle, L.; Madeja, Z.; Reiss, K. ROS accumulation and IGF-IR inhibition contribute to fenofibrate/PPARα -mediated inhibition of glioma cell motility in vitro. Mol. Cancer 2010, 9, 159. [Google Scholar] [CrossRef] [Green Version]
- Misra, P.; Reddy, J.K. Peroxisome proliferator-activated receptor-α activation and excess energy burning in hepatocarcinogenesis. Biochimie 2014, 98, 63–74. [Google Scholar] [CrossRef]
- Wilk, A.; Wyczechowska, D.; Zapata, A.; Dean, M.; Mullinax, J.; Marrero, L.; Parsons, C.; Peruzzi, F.; Culicchia, F.; Ochoa, A.; et al. Molecular mechanisms of fenofibrate-induced metabolic catastrophe and glioblastoma cell death. Mol. Cell. Biol. 2015, 35, 182–198. [Google Scholar] [CrossRef] [Green Version]
- Chekaoui, A.; Ertl, H.C.J. PPARα Agonist Fenofibrate Enhances Cancer Vaccine Efficacy. Cancer Res. 2021, 81, 4431–4440. [Google Scholar] [CrossRef]
- Bahrambeigi, S.; Molaparast, M.; Sohrabi, F.; Seifi, L.; Faraji, A.; Fani, S.; Shafiei-Irannejad, V. Targeting PPAR ligands as possible approaches for metabolic reprogramming of T cells in cancer immunotherapy. Immunol. Lett. 2020, 220, 32–37. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, P.S.; Chamoto, K.; Kumar, A.; Honjo, T. PPAR-Induced Fatty Acid Oxidation in T Cells Increases the Number of Tumor-Reactive CD8. Cancer Immunol. Res. 2018, 6, 1375–1387. [Google Scholar] [CrossRef] [PubMed]
- Stokes, W.A.; Behera, M.; Jiang, R.; Gutman, D.A.; Huang, Z.; Burns, A.; Sebastian, N.T.; Sukhatme, V.; Lowe, M.C.; Ramalingam, S.S.; et al. Impact of concomitant fibrates on immunotherapy outcomes for advanced non-small cell lung cancer. Cancer Med. 2022, 00, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Haakinson, D.J.; Leeds, S.G.; Dueck, A.C.; Gray, R.J.; Wasif, N.; Stucky, C.C.; Northfelt, D.W.; Apsey, H.A.; Pockaj, B. The impact of obesity on breast cancer: A retrospective review. Ann. Surg. Oncol. 2012, 19, 3012–3018. [Google Scholar] [CrossRef]
- La Vecchia, C.; Negri, E.; Franceschi, S.; D’Avanzo, B.; Boyle, P. A case-control study of diabetes mellitus and cancer risk. Br. J. Cancer 1994, 70, 950–953. [Google Scholar] [CrossRef]
- Blücher, C.; Iberl, S.; Schwagarus, N.; Müller, S.; Liebisch, G.; Höring, M.; Hidrobo, M.S.; Ecker, J.; Spindler, N.; Dietrich, A.; et al. Secreted Factors from Adipose Tissue Reprogram Tumor Lipid Metabolism and Induce Motility by Modulating PPARα/ANGPTL4 and FAK. Mol. Cancer Res. 2020, 18, 1849–1862. [Google Scholar] [CrossRef]
- Naiman, S.; Huynh, F.K.; Gil, R.; Glick, Y.; Shahar, Y.; Touitou, N.; Nahum, L.; Avivi, M.Y.; Roichman, A.; Kanfi, Y.; et al. SIRT6 Promotes Hepatic Beta-Oxidation via Activation of PPARα. Cell Rep. 2019, 29, 4127–4143.e4128. [Google Scholar] [CrossRef] [Green Version]
- Senni, N.; Savall, M.; Cabrerizo Granados, D.; Alves-Guerra, M.C.; Sartor, C.; Lagoutte, I.; Gougelet, A.; Terris, B.; Gilgenkrantz, H.; Perret, C.; et al. β-catenin-activated hepatocellular carcinomas are addicted to fatty acids. Gut 2019, 68, 322–334. [Google Scholar] [CrossRef]
- Andrejeva, D.; Kugler, J.M.; Nguyen, H.T.; Malmendal, A.; Holm, M.L.; Toft, B.G.; Loya, A.C.; Cohen, S.M. Metabolic control of PPAR activity by aldehyde dehydrogenase regulates invasive cell behavior and predicts survival in hepatocellular and renal clear cell carcinoma. BMC Cancer 2018, 18, 1180. [Google Scholar] [CrossRef]
- Gou, Q.; Dong, C.; Jin, J.; Liu, Q.; Lu, W.; Shi, J.; Hou, Y. PPARα agonist alleviates tumor growth and chemo-resistance associated with the inhibition of glucose metabolic pathway. Eur. J. Pharmacol. 2019, 863, 172664. [Google Scholar] [CrossRef]
- You, M.; Jin, J.; Liu, Q.; Xu, Q.; Shi, J.; Hou, Y. PPARα Promotes Cancer Cell Glut1 Transcription Repression. J. Cell. Biochem. 2017, 118, 1556–1562. [Google Scholar] [CrossRef] [PubMed]
- Han, D.; Wei, W.; Chen, X.; Zhang, Y.; Wang, Y.; Zhang, J.; Wang, X.; Yu, T.; Hu, Q.; Liu, N.; et al. NF-κB/RelA-PKM2 mediates inhibition of glycolysis by fenofibrate in glioblastoma cells. Oncotarget 2015, 6, 26119–26128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, M.; Zuo, X.; Shureiqi, I. Targeting peroxisome proliferator-activated receptor-β/δ in colon cancer: How to aim? Biochem. Pharmacol. 2013, 85, 607–611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagner, N.; Jehl-Piétri, C.; Lopez, P.; Murdaca, J.; Giordano, C.; Schwartz, C.; Gounon, P.; Hatem, S.N.; Grimaldi, P.; Wagner, K.D. Peroxisome proliferator-activated receptor β stimulation induces rapid cardiac growth and angiogenesis via direct activation of calcineurin. Cardiovasc. Res. 2009, 83, 61–71. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.V.; Hubbi, M.E.; Pan, F.; McDonald, K.R.; Mansharamani, M.; Cole, R.N.; Liu, J.O.; Semenza, G.L. Calcineurin promotes hypoxia-inducible factor 1α expression by dephosphorylating RACK1 and blocking RACK1 dimerization. J. Biol. Chem. 2007, 282, 37064–37073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeong, E.; Koo, J.E.; Yeon, S.H.; Kwak, M.K.; Hwang, D.H.; Lee, J.Y. PPARδ deficiency disrupts hypoxia-mediated tumorigenic potential of colon cancer cells. Mol. Carcinog. 2014, 53, 926–937. [Google Scholar] [CrossRef]
- Pudakalakatti, S.; Titus, M.; Enriquez, J.S.; Ramachandran, S.; Zacharias, N.M.; Shureiqi, I.; Liu, Y.; Yao, J.C.; Zuo, X.; Bhattacharya, P.K. Identifying the Metabolic Signatures of PPARD-Overexpressing Gastric Tumors. Int. J. Mol. Sci. 2022, 23, 1645. [Google Scholar] [CrossRef]
- Mana, M.D.; Hussey, A.M.; Tzouanas, C.N.; Imada, S.; Barrera Millan, Y.; Bahceci, D.; Saiz, D.R.; Webb, A.T.; Lewis, C.A.; Carmeliet, P.; et al. High-fat diet-activated fatty acid oxidation mediates intestinal stemness and tumorigenicity. Cell Rep. 2021, 35, 109212. [Google Scholar] [CrossRef]
- Hodge, A.M.; Williamson, E.J.; Bassett, J.K.; MacInnis, R.J.; Giles, G.G.; English, D.R. Dietary and biomarker estimates of fatty acids and risk of colorectal cancer. Int. J. Cancer 2015, 137, 1224–1234. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Zhao, X.; Toline, E.C.; Siegal, G.P.; Evans, L.M.; Ibrahim-Hashim, A.; Desmond, R.A.; Hardy, R.W. Prevention of carcinogenesis and inhibition of breast cancer tumor burden by dietary stearate. Carcinogenesis 2011, 32, 1251–1258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardy, S.; Langelier, Y.; Prentki, M. Oleate activates phosphatidylinositol 3-kinase and promotes proliferation and reduces apoptosis of MDA-MB-231 breast cancer cells, whereas palmitate has opposite effects. Cancer Res. 2000, 60, 6353–6358. [Google Scholar] [PubMed]
- Levi, L.; Wang, Z.; Doud, M.K.; Hazen, S.L.; Noy, N. Saturated fatty acids regulate retinoic acid signalling and suppress tumorigenesis by targeting fatty acid-binding protein 5. Nat. Commun. 2015, 6, 8794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Wang, J.; Li, X.; Xiong, X.; Zhou, Z.; Zhu, X.; Gu, Y.; Dominissini, D.; He, L.; Tian, Y.; et al. N1-methyladenosine methylation in tRNA drives liver tumourigenesis by regulating cholesterol metabolism. Nat. Commun. 2021, 12, 6314. [Google Scholar] [CrossRef]
- Zhang, W.; Xu, Y.; Xu, Q.; Shi, H.; Shi, J.; Hou, Y. PPARδ promotes tumor progression via activation of Glut1 and SLC1-A5 transcription. Carcinogenesis 2017, 38, 748–755. [Google Scholar] [CrossRef]
- Kim, M.J.; Choi, Y.K.; Park, S.Y.; Jang, S.Y.; Lee, J.Y.; Ham, H.J.; Kim, B.G.; Jeon, H.J.; Kim, J.H.; Kim, J.G.; et al. PPARδ Reprograms Glutamine Metabolism in Sorafenib-Resistant HCC. Mol. Cancer Res. 2017, 15, 1230–1242. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.H.; Song, J.; Park, K.W. The multifaceted factor peroxisome proliferator-activated receptor γ (PPARγ) in metabolism, immunity, and cancer. Arch. Pharm. Res. 2015, 38, 302–312. [Google Scholar] [CrossRef]
- Sakharkar, M.K.; Shashni, B.; Sharma, K.; Dhillon, S.K.; Ranjekar, P.R.; Sakharkar, K.R. Therapeutic implications of targeting energy metabolism in breast cancer. PPAR Res. 2013, 2013, 109285. [Google Scholar] [CrossRef] [Green Version]
- Asp, M.L.; Tian, M.; Kliewer, K.L.; Belury, M.A. Rosiglitazone delayed weight loss and anorexia while attenuating adipose depletion in mice with cancer cachexia. Cancer Biol. Ther. 2011, 12, 957–965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beluzi, M.; Peres, S.B.; Henriques, F.S.; Sertié, R.A.; Franco, F.O.; Santos, K.B.; Knobl, P.; Andreotti, S.; Shida, C.S.; Neves, R.X.; et al. Pioglitazone treatment increases survival and prevents body weight loss in tumor-bearing animals: Possible anti-cachectic effect. PLoS ONE 2015, 10, e0122660. [Google Scholar] [CrossRef] [Green Version]
- Qian, F.; Arner, B.E.; Kelly, K.M.; Annageldiyev, C.; Sharma, A.; Claxton, D.F.; Paulson, R.F.; Prabhu, K.S. Interleukin-4 treatment reduces leukemia burden in acute myeloid leukemia. FASEB J. 2022, 36, e22328. [Google Scholar] [CrossRef]
- Ning, Z.; Guo, X.; Liu, X.; Lu, C.; Wang, A.; Wang, X.; Wang, W.; Chen, H.; Qin, W.; Zhou, L.; et al. USP22 regulates lipidome accumulation by stabilizing PPARγ in hepatocellular carcinoma. Nat. Commun. 2022, 13, 2187. [Google Scholar] [CrossRef] [PubMed]
- Edwards, I.J.; Sun, H.; Hu, Y.; Berquin, I.M.; O’Flaherty, J.T.; Cline, J.M.; Rudel, L.L.; Chen, Y.Q. In vivo and in vitro regulation of syndecan 1 in prostate cells by n-3 polyunsaturated fatty acids. J. Biol. Chem. 2008, 283, 18441–18449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anagnostopoulos, G.; Motiño, O.; Li, S.; Carbonnier, V.; Chen, H.; Sica, V.; Durand, S.; Bourgin, M.; Aprahamian, F.; Nirmalathasan, N.; et al. An obesogenic feedforward loop involving PPARγ, acyl-CoA binding protein and GABA. Cell Death Dis. 2022, 13, 356. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Shen, L.; Zhan, Z.; Liu, Y.; Zhang, C.; Guo, R.; Luo, Y.; Xie, Z.; Feng, Y.; Wu, G. The long noncoding RNA MALAT1 modulates adipose loss in cancer-associated cachexia by suppressing adipogenesis through PPAR-γ. Nutr. Metab. 2021, 18, 27. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Zhou, B.; Yang, Q.; Pan, Y.; Yang, W.; Freedland, S.J.; Ding, L.W.; Freeman, M.R.; Breunig, J.J.; Bhowmick, N.A.; et al. A Transcriptional Regulatory Loop of Master Regulator Transcription Factors, PPARG, and Fatty Acid Synthesis Promotes Esophageal Adenocarcinoma. Cancer Res. 2021, 81, 1216–1229. [Google Scholar] [CrossRef] [PubMed]
- Forootan, F.S.; Forootan, S.S.; Gou, X.; Yang, J.; Liu, B.; Chen, D.; Al Fayi, M.S.; Al-Jameel, W.; Rudland, P.S.; Hussain, S.A.; et al. Fatty acid activated PPARγ promotes tumorigenicity of prostate cancer cells by up regulating VEGF via PPAR responsive elements of the promoter. Oncotarget 2016, 7, 9322–9339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.; Li, G.; Yang, Z.; Wang, L.; Zhang, L.; Wang, T.; Zhang, Y.; Zhang, S.; Han, Y.; Jia, L. Uncoupling protein 2 downregulation by hypoxia through repression of peroxisome proliferator-activated receptor γ promotes chemoresistance of non-small cell lung cancer. Oncotarget 2017, 8, 8083–8094. [Google Scholar] [CrossRef] [Green Version]
- Han, S.; Inoue, H.; Flowers, L.C.; Sidell, N. Control of COX-2 gene expression through peroxisome proliferator-activated receptor γ in human cervical cancer cells. Clin. Cancer Res. 2003, 9, 4627–4635. [Google Scholar]
- Bren-Mattison, Y.; Meyer, A.M.; Van Putten, V.; Li, H.; Kuhn, K.; Stearman, R.; Weiser-Evans, M.; Winn, R.A.; Heasley, L.E.; Nemenoff, R.A. Antitumorigenic effects of peroxisome proliferator-activated receptor-γ in non-small-cell lung cancer cells are mediated by suppression of cyclooxygenase-2 via inhibition of nuclear factor-κB. Mol. Pharmacol. 2008, 73, 709–717. [Google Scholar] [CrossRef] [Green Version]
- Hazra, S.; Dubinett, S.M. Ciglitazone mediates COX-2 dependent suppression of PGE2 in human non-small cell lung cancer cells. Prostaglandins Leukot. Essent. Fat. Acids 2007, 77, 51–58. [Google Scholar] [CrossRef] [Green Version]
- Gottfried, E.; Rogenhofer, S.; Waibel, H.; Kunz-Schughart, L.A.; Reichle, A.; Wehrstein, M.; Peuker, A.; Peter, K.; Hartmannsgruber, G.; Andreesen, R.; et al. Pioglitazone modulates tumor cell metabolism and proliferation in multicellular tumor spheroids. Cancer Chemother. Pharmacol. 2011, 67, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.; Kulp, S.K.; Chen, C.S. Energy restriction as an antitumor target of thiazolidinediones. J. Biol. Chem. 2010, 285, 9780–9791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Devchand, P.R.; Keller, H.; Peters, J.M.; Vazquez, M.; Gonzalez, F.J.; Wahli, W. The PPARα-leukotriene B4 pathway to inflammation control. Nature 1996, 384, 39–43. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.C.; Ding, X.; Daynes, R.A. Nuclear receptor peroxisome proliferator-activated receptor α (PPARα) is expressed in resting murine lymphocytes. The PPARα in T and B lymphocytes is both transactivation and transrepression competent. J. Biol. Chem. 2002, 277, 6838–6845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chinetti, G.; Griglio, S.; Antonucci, M.; Torra, I.P.; Delerive, P.; Majd, Z.; Fruchart, J.C.; Chapman, J.; Najib, J.; Staels, B. Activation of proliferator-activated receptors α and γ induces apoptosis of human monocyte-derived macrophages. J. Biol. Chem. 1998, 273, 25573–25580. [Google Scholar] [CrossRef] [Green Version]
- Yang, Q.; Xie, Y.; Depierre, J.W. Effects of peroxisome proliferators on the thymus and spleen of mice. Clin. Exp. Immunol. 2000, 122, 219–226. [Google Scholar] [CrossRef]
- Yang, Q.; Xie, Y.; Alexson, S.E.; Nelson, B.D.; DePierre, J.W. Involvement of the peroxisome proliferator-activated receptor α in the immunomodulation caused by peroxisome proliferators in mice. Biochem. Pharmacol. 2002, 63, 1893–1900. [Google Scholar] [CrossRef]
- Yang, Q.; Gonzalez, F.J. Peroxisome proliferator-activated receptor α regulates B lymphocyte development via an indirect pathway in mice. Biochem. Pharmacol. 2004, 68, 2143–2150. [Google Scholar] [CrossRef]
- Roberts, R.A. Peroxisome proliferators: Mechanisms of adverse effects in rodents and molecular basis for species differences. Arch. Toxicol. 1999, 73, 413–418. [Google Scholar] [CrossRef]
- Stienstra, R.; Mandard, S.; Patsouris, D.; Maass, C.; Kersten, S.; Müller, M. Peroxisome proliferator-activated receptor α protects against obesity-induced hepatic inflammation. Endocrinology 2007, 148, 2753–2763. [Google Scholar] [CrossRef] [Green Version]
- Shiri-Sverdlov, R.; Wouters, K.; van Gorp, P.J.; Gijbels, M.J.; Noel, B.; Buffat, L.; Staels, B.; Maeda, N.; van Bilsen, M.; Hofker, M.H. Early diet-induced non-alcoholic steatohepatitis in APOE2 knock-in mice and its prevention by fibrates. J. Hepatol. 2006, 44, 732–741. [Google Scholar] [CrossRef] [PubMed]
- Riccardi, L.; Mazzon, E.; Bruscoli, S.; Esposito, E.; Crisafulli, C.; Di Paola, R.; Caminiti, R.; Riccardi, C.; Cuzzocrea, S. Peroxisome proliferator-activated receptor-α modulates the anti-inflammatory effect of glucocorticoids in a model of inflammatory bowel disease in mice. Shock 2009, 31, 308–316. [Google Scholar] [CrossRef] [PubMed]
- Azuma, Y.T.; Nishiyama, K.; Matsuo, Y.; Kuwamura, M.; Morioka, A.; Nakajima, H.; Takeuchi, T. PPARα contributes to colonic protection in mice with DSS-induced colitis. Int. Immunopharmacol. 2010, 10, 1261–1267. [Google Scholar] [CrossRef] [PubMed]
- Manoharan, I.; Suryawanshi, A.; Hong, Y.; Ranganathan, P.; Shanmugam, A.; Ahmad, S.; Swafford, D.; Manicassamy, B.; Ramesh, G.; Koni, P.A.; et al. Homeostatic PPARα Signaling Limits Inflammatory Responses to Commensal Microbiota in the Intestine. J. Immunol. 2016, 196, 4739–4749. [Google Scholar] [CrossRef] [PubMed]
- Michalik, L.; Desvergne, B.; Tan, N.S.; Basu-Modak, S.; Escher, P.; Rieusset, J.; Peters, J.M.; Kaya, G.; Gonzalez, F.J.; Zakany, J.; et al. Impaired skin wound healing in peroxisome proliferator-activated receptor (PPAR)α and PPARβ mutant mice. J. Cell. Biol. 2001, 154, 799–814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poynter, M.E.; Daynes, R.A. Peroxisome proliferator-activated receptor α activation modulates cellular redox status, represses nuclear factor-kappaB signaling, and reduces inflammatory cytokine production in aging. J. Biol. Chem. 1998, 273, 32833–32841. [Google Scholar] [CrossRef] [Green Version]
- Mandard, S.; Patsouris, D. Nuclear control of the inflammatory response in mammals by peroxisome proliferator-activated receptors. PPAR Res. 2013, 2013, 613864. [Google Scholar] [CrossRef] [Green Version]
- Daynes, R.A.; Jones, D.C. Emerging roles of PPARs in inflammation and immunity. Nat. Rev. Immunol. 2002, 2, 748–759. [Google Scholar] [CrossRef]
- Hermanowski-Vosatka, A.; Gerhold, D.; Mundt, S.S.; Loving, V.A.; Lu, M.; Chen, Y.; Elbrecht, A.; Wu, M.; Doebber, T.; Kelly, L.; et al. PPARα agonists reduce 11β-hydroxysteroid dehydrogenase type 1 in the liver. Biochem. Biophys. Res. Commun. 2000, 279, 330–336. [Google Scholar] [CrossRef]
- Hill, M.R.; Clarke, S.; Rodgers, K.; Thornhill, B.; Peters, J.M.; Gonzalez, F.J.; Gimble, J.M. Effect of peroxisome proliferator-activated receptor alpha activators on tumor necrosis factor expression in mice during endotoxemia. Infect. Immun. 1999, 67, 3488–3493. [Google Scholar] [CrossRef] [Green Version]
- Jackson, S.M.; Parhami, F.; Xi, X.P.; Berliner, J.A.; Hsueh, W.A.; Law, R.E.; Demer, L.L. Peroxisome proliferator-activated receptor activators target human endothelial cells to inhibit leukocyte-endothelial cell interaction. Arterioscler. Thromb. Vasc. Biol. 1999, 19, 2094–2104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marx, N.; Sukhova, G.K.; Collins, T.; Libby, P.; Plutzky, J. PPARα activators inhibit cytokine-induced vascular cell adhesion molecule-1 expression in human endothelial cells. Circulation 1999, 99, 3125–3131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Staels, B.; Koenig, W.; Habib, A.; Merval, R.; Lebret, M.; Torra, I.P.; Delerive, P.; Fadel, A.; Chinetti, G.; Fruchart, J.C.; et al. Activation of human aortic smooth-muscle cells is inhibited by PPARα but not by PPARγ activators. Nature 1998, 393, 790–793. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Shi, W.; Tontonoz, P.; Wang, S.; Subbanagounder, G.; Hedrick, C.C.; Hama, S.; Borromeo, C.; Evans, R.M.; Berliner, J.A.; et al. Role for peroxisome proliferator-activated receptor α in oxidized phospholipid-induced synthesis of monocyte chemotactic protein-1 and interleukin-8 by endothelial cells. Circ. Res. 2000, 87, 516–521. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Kurupati, R.; Liu, L.; Zhou, X.Y.; Zhang, G.; Hudaihed, A.; Filisio, F.; Giles-Davis, W.; Xu, X.; Karakousis, G.C.; et al. Enhancing CD8. Cancer Cell 2017, 32, 377–391.e379. [Google Scholar] [CrossRef] [Green Version]
- Yin, X.; Zeng, W.; Wu, B.; Wang, L.; Wang, Z.; Tian, H.; Jiang, Y.; Clay, R.; Wei, X.; Qin, Y.; et al. PPARα Inhibition Overcomes Tumor-Derived Exosomal Lipid-Induced Dendritic Cell Dysfunction. Cell Rep. 2020, 33, 108278. [Google Scholar] [CrossRef]
- Bishop-Bailey, D.; Bystrom, J. Emerging roles of peroxisome proliferator-activated receptor-β/δ in inflammation. Pharmacol. Ther. 2009, 124, 141–150. [Google Scholar] [CrossRef]
- Peters, J.M.; Lee, S.S.; Li, W.; Ward, J.M.; Gavrilova, O.; Everett, C.; Reitman, M.L.; Hudson, L.D.; Gonzalez, F.J. Growth, adipose, brain, and skin alterations resulting from targeted disruption of the mouse peroxisome proliferator-activated receptor β(δ). Mol. Cell. Biol. 2000, 20, 5119–5128. [Google Scholar] [CrossRef] [Green Version]
- Tan, N.S.; Michalik, L.; Noy, N.; Yasmin, R.; Pacot, C.; Heim, M.; Flühmann, B.; Desvergne, B.; Wahli, W. Critical roles of PPAR β/δ in keratinocyte response to inflammation. Genes Dev. 2001, 15, 3263–3277. [Google Scholar] [CrossRef] [Green Version]
- Bishop-Bailey, D.; Hla, T. Endothelial cell apoptosis induced by the peroxisome proliferator-activated receptor (PPAR) ligand 15-deoxy-Δ12,14-prostaglandin J2. J. Biol. Chem. 1999, 274, 17042–17048. [Google Scholar] [CrossRef] [Green Version]
- Rival, Y.; Benéteau, N.; Taillandier, T.; Pezet, M.; Dupont-Passelaigue, E.; Patoiseau, J.F.; Junquéro, D.; Colpaert, F.C.; Delhon, A. PPARα and PPARδ activators inhibit cytokine-induced nuclear translocation of NF-κB and expression of VCAM-1 in EAhy926 endothelial cells. Eur. J. Pharmacol. 2002, 435, 143–151. [Google Scholar] [CrossRef]
- Fan, Y.; Wang, Y.; Tang, Z.; Zhang, H.; Qin, X.; Zhu, Y.; Guan, Y.; Wang, X.; Staels, B.; Chien, S.; et al. Suppression of pro-inflammatory adhesion molecules by PPAR-δ in human vascular endothelial cells. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 315–321. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.H.; Chawla, A.; Urbiztondo, N.; Liao, D.; Boisvert, W.A.; Evans, R.M.; Curtiss, L.K. Transcriptional repression of atherogenic inflammation: Modulation by PPARδ. Science 2003, 302, 453–457. [Google Scholar] [CrossRef] [PubMed]
- Piqueras, L.; Sanz, M.J.; Perretti, M.; Morcillo, E.; Norling, L.; Mitchell, J.A.; Li, Y.; Bishop-Bailey, D. Activation of PPARβ/δ inhibits leukocyte recruitment, cell adhesion molecule expression, and chemokine release. J. Leukoc. Biol. 2009, 86, 115–122. [Google Scholar] [CrossRef]
- Welch, J.S.; Ricote, M.; Akiyama, T.E.; Gonzalez, F.J.; Glass, C.K. PPARγ and PPARδ negatively regulate specific subsets of lipopolysaccharide and IFN-γ target genes in macrophages. Proc. Natl. Acad. Sci. USA 2003, 100, 6712–6717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, K.; Reilly, S.M.; Karabacak, V.; Gangl, M.R.; Fitzgerald, K.; Hatano, B.; Lee, C.H. Adipocyte-derived Th2 cytokines and myeloid PPARδ regulate macrophage polarization and insulin sensitivity. Cell Metab. 2008, 7, 485–495. [Google Scholar] [CrossRef] [Green Version]
- Odegaard, J.I.; Ricardo-Gonzalez, R.R.; Red Eagle, A.; Vats, D.; Morel, C.R.; Goforth, M.H.; Subramanian, V.; Mukundan, L.; Ferrante, A.W.; Chawla, A. Alternative M2 activation of Kupffer cells by PPARδ ameliorates obesity-induced insulin resistance. Cell Metab. 2008, 7, 496–507. [Google Scholar] [CrossRef] [Green Version]
- Gallardo-Soler, A.; Gómez-Nieto, C.; Campo, M.L.; Marathe, C.; Tontonoz, P.; Castrillo, A.; Corraliza, I. Arginase I induction by modified lipoproteins in macrophages: A peroxisome proliferator-activated receptor-γ/δ-mediated effect that links lipid metabolism and immunity. Mol. Endocrinol. 2008, 22, 1394–1402. [Google Scholar] [CrossRef]
- Mukundan, L.; Odegaard, J.I.; Morel, C.R.; Heredia, J.E.; Mwangi, J.W.; Ricardo-Gonzalez, R.R.; Goh, Y.P.; Eagle, A.R.; Dunn, S.E.; Awakuni, J.U.; et al. PPAR-δ senses and orchestrates clearance of apoptotic cells to promote tolerance. Nat. Med. 2009, 15, 1266–1272. [Google Scholar] [CrossRef]
- Adhikary, T.; Wortmann, A.; Schumann, T.; Finkernagel, F.; Lieber, S.; Roth, K.; Toth, P.M.; Diederich, W.E.; Nist, A.; Stiewe, T.; et al. The transcriptional PPARβ/δ network in human macrophages defines a unique agonist-induced activation state. Nucleic Acids Res. 2015, 43, 5033–5051. [Google Scholar] [CrossRef] [Green Version]
- Schumann, T.; Adhikary, T.; Wortmann, A.; Finkernagel, F.; Lieber, S.; Schnitzer, E.; Legrand, N.; Schober, Y.; Nockher, W.A.; Toth, P.M.; et al. Deregulation of PPARβ/δ target genes in tumor-associated macrophages by fatty acid ligands in the ovarian cancer microenvironment. Oncotarget 2015, 6, 13416–13433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schote, A.B.; Turner, J.D.; Schiltz, J.; Muller, C.P. Nuclear receptors in human immune cells: Expression and correlations. Mol. Immunol. 2007, 44, 1436–1445. [Google Scholar] [CrossRef] [PubMed]
- al Yacoub, N.; Romanowska, M.; Krauss, S.; Schweiger, S.; Foerster, J. PPARδ is a type 1 IFN target gene and inhibits apoptosis in T cells. J. Investig. Dermatol. 2008, 128, 1940–1949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saibil, S.D.; St Paul, M.; Laister, R.C.; Garcia-Batres, C.R.; Israni-Winger, K.; Elford, A.R.; Grimshaw, N.; Robert-Tissot, C.; Roy, D.G.; Jones, R.G.; et al. Activation of Peroxisome Proliferator-Activated Receptors α and δ Synergizes with Inflammatory Signals to Enhance Adoptive Cell Therapy. Cancer Res. 2019, 79, 445–451. [Google Scholar] [CrossRef] [Green Version]
- Jakobsen, M.A.; Petersen, R.K.; Kristiansen, K.; Lange, M.; Lillevang, S.T. Peroxisome proliferator-activated receptor α, δ, γ1 and γ2 expressions are present in human monocyte-derived dendritic cells and modulate dendritic cell maturation by addition of subtype-specific ligands. Scand. J. Immunol. 2006, 63, 330–337. [Google Scholar] [CrossRef] [PubMed]
- Maciel, T.T.; Moura, I.C.; Hermine, O. The role of mast cells in cancers. F1000Prime Rep. 2015, 7, 09. [Google Scholar] [CrossRef]
- Yao, P.L.; Morales, J.L.; Gonzalez, F.J.; Peters, J.M. Peroxisome proliferator-activated receptor-β/δ modulates mast cell phenotype. Immunology 2017, 150, 456–467. [Google Scholar] [CrossRef] [Green Version]
- Michelet, X.; Dyck, L.; Hogan, A.; Loftus, R.M.; Duquette, D.; Wei, K.; Beyaz, S.; Tavakkoli, A.; Foley, C.; Donnelly, R.; et al. Metabolic reprogramming of natural killer cells in obesity limits antitumor responses. Nat. Immunol. 2018, 19, 1330–1340. [Google Scholar] [CrossRef]
- Yang, X.Y.; Wang, L.H.; Chen, T.; Hodge, D.R.; Resau, J.H.; DaSilva, L.; Farrar, W.L. Activation of human T lymphocytes is inhibited by peroxisome proliferator-activated receptor γ (PPARγ) agonists. PPARγ co-association with transcription factor NFAT. J. Biol. Chem. 2000, 275, 4541–4544. [Google Scholar] [CrossRef] [Green Version]
- Clark, R.B.; Bishop-Bailey, D.; Estrada-Hernandez, T.; Hla, T.; Puddington, L.; Padula, S.J. The nuclear receptor PPAR γ and immunoregulation: PPAR γ mediates inhibition of helper T cell responses. J. Immunol. 2000, 164, 1364–1371. [Google Scholar] [CrossRef] [Green Version]
- Desreumaux, P.; Dubuquoy, L.; Nutten, S.; Peuchmaur, M.; Englaro, W.; Schoonjans, K.; Derijard, B.; Desvergne, B.; Wahli, W.; Chambon, P.; et al. Attenuation of colon inflammation through activators of the retinoid X receptor (RXR)/peroxisome proliferator-activated receptor γ (PPARγ) heterodimer. A basis for new therapeutic strategies. J. Exp. Med. 2001, 193, 827–838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, C.G.; Wen, X.; Bailey, S.T.; Jiang, W.; Rangwala, S.M.; Keilbaugh, S.A.; Flanigan, A.; Murthy, S.; Lazar, M.A.; Wu, G.D. A novel therapy for colitis utilizing PPAR-γ ligands to inhibit the epithelial inflammatory response. J. Clin. Investig. 1999, 104, 383–389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hontecillas, R.; Bassaganya-Riera, J. Peroxisome proliferator-activated receptor γ is required for regulatory CD4+ T cell-mediated protection against colitis. J. Immunol. 2007, 178, 2940–2949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harris, S.G.; Phipps, R.P. The nuclear receptor PPAR γ is expressed by mouse T lymphocytes and PPAR γ agonists induce apoptosis. Eur. J. Immunol. 2001, 31, 1098–1105. [Google Scholar] [CrossRef]
- Wang, Y.L.; Frauwirth, K.A.; Rangwala, S.M.; Lazar, M.A.; Thompson, C.B. Thiazolidinedione activation of peroxisome proliferator-activated receptor γ can enhance mitochondrial potential and promote cell survival. J. Biol. Chem. 2002, 277, 31781–31788. [Google Scholar] [CrossRef] [Green Version]
- Jo, S.H.; Yang, C.; Miao, Q.; Marzec, M.; Wasik, M.A.; Lu, P.; Wang, Y.L. Peroxisome proliferator-activated receptor γ promotes lymphocyte survival through its actions on cellular metabolic activities. J. Immunol. 2006, 177, 3737–3745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klotz, L.; Burgdorf, S.; Dani, I.; Saijo, K.; Flossdorf, J.; Hucke, S.; Alferink, J.; Nowak, N.; Novak, N.; Beyer, M.; et al. The nuclear receptor PPAR γ selectively inhibits Th17 differentiation in a T cell-intrinsic fashion and suppresses CNS autoimmunity. J. Exp. Med. 2009, 206, 2079–2089. [Google Scholar] [CrossRef]
- Tosolini, M.; Kirilovsky, A.; Mlecnik, B.; Fredriksen, T.; Mauger, S.; Bindea, G.; Berger, A.; Bruneval, P.; Fridman, W.H.; Pagès, F.; et al. Clinical impact of different classes of infiltrating T cytotoxic and helper cells (Th1, th2, treg, th17) in patients with colorectal cancer. Cancer Res. 2011, 71, 1263–1271. [Google Scholar] [CrossRef] [Green Version]
- Tobiasova, Z.; Zhang, L.; Yi, T.; Qin, L.; Manes, T.D.; Kulkarni, S.; Lorber, M.I.; Rodriguez, F.C.; Choi, J.M.; Tellides, G.; et al. Peroxisome proliferator-activated receptor-γ agonists prevent in vivo remodeling of human artery induced by alloreactive T cells. Circulation 2011, 124, 196–205. [Google Scholar] [CrossRef] [Green Version]
- Guri, A.J.; Mohapatra, S.K.; Horne, W.T.; Hontecillas, R.; Bassaganya-Riera, J. The role of T cell PPAR γ in mice with experimental inflammatory bowel disease. BMC Gastroenterol. 2010, 10, 60. [Google Scholar] [CrossRef] [Green Version]
- Wohlfert, E.A.; Nichols, F.C.; Nevius, E.; Clark, R.B. Peroxisome proliferator-activated receptor γ (PPARγ) and immunoregulation: Enhancement of regulatory T cells through PPARγ-dependent and -independent mechanisms. J. Immunol. 2007, 178, 4129–4135. [Google Scholar] [CrossRef] [PubMed]
- Feuerer, M.; Hill, J.A.; Mathis, D.; Benoist, C. Foxp3+ regulatory T cells: Differentiation, specification, subphenotypes. Nat. Immunol. 2009, 10, 689–695. [Google Scholar] [CrossRef] [PubMed]
- Cipolletta, D.; Feuerer, M.; Li, A.; Kamei, N.; Lee, J.; Shoelson, S.E.; Benoist, C.; Mathis, D. PPAR-γ is a major driver of the accumulation and phenotype of adipose tissue Treg cells. Nature 2012, 486, 549–553. [Google Scholar] [CrossRef] [PubMed]
- Ercolano, G.; Gomez-Cadena, A.; Dumauthioz, N.; Vanoni, G.; Kreutzfeldt, M.; Wyss, T.; Michalik, L.; Loyon, R.; Ianaro, A.; Ho, P.C.; et al. PPARɣ drives IL-33-dependent ILC2 pro-tumoral functions. Nat. Commun. 2021, 12, 2538. [Google Scholar] [CrossRef] [PubMed]
- Faveeuw, C.; Fougeray, S.; Angeli, V.; Fontaine, J.; Chinetti, G.; Gosset, P.; Delerive, P.; Maliszewski, C.; Capron, M.; Staels, B.; et al. Peroxisome proliferator-activated receptor γ activators inhibit interleukin-12 production in murine dendritic cells. FEBS Lett. 2000, 486, 261–266. [Google Scholar] [CrossRef]
- Szatmari, I.; Gogolak, P.; Im, J.S.; Dezso, B.; Rajnavolgyi, E.; Nagy, L. Activation of PPARγ specifies a dendritic cell subtype capable of enhanced induction of iNKT cell expansion. Immunity 2004, 21, 95–106. [Google Scholar] [CrossRef] [Green Version]
- Fu, S.; He, K.; Tian, C.; Sun, H.; Zhu, C.; Bai, S.; Liu, J.; Wu, Q.; Xie, D.; Yue, T.; et al. Impaired lipid biosynthesis hinders anti-tumor efficacy of intratumoral iNKT cells. Nat. Commun. 2020, 11, 438. [Google Scholar] [CrossRef] [Green Version]
- Kardos, J.; Chai, S.; Mose, L.E.; Selitsky, S.R.; Krishnan, B.; Saito, R.; Iglesia, M.D.; Milowsky, M.I.; Parker, J.S.; Kim, W.Y.; et al. Claudin-low bladder tumors are immune infiltrated and actively immune suppressed. JCI Insight 2016, 1, e85902. [Google Scholar] [CrossRef] [Green Version]
- Tate, T.; Xiang, T.; Wobker, S.E.; Zhou, M.; Chen, X.; Kim, H.; Batourina, E.; Lin, C.S.; Kim, W.Y.; Lu, C.; et al. Pparg signaling controls bladder cancer subtype and immune exclusion. Nat. Commun. 2021, 12, 6160. [Google Scholar] [CrossRef]
- Korpal, M.; Puyang, X.; Jeremy Wu, Z.; Seiler, R.; Furman, C.; Oo, H.Z.; Seiler, M.; Irwin, S.; Subramanian, V.; Julie Joshi, J.; et al. Evasion of immunosurveillance by genomic alterations of PPARγ/RXRα in bladder cancer. Nat. Commun. 2017, 8, 103. [Google Scholar] [CrossRef] [Green Version]
- Gyöngyösi, A.; Nagy, L. Potential Therapeutic Use of PPARγ-Programed Human Monocyte-Derived Dendritic Cells in Cancer Vaccination Therapy. PPAR Res. 2008, 2008, 473804. [Google Scholar] [CrossRef] [PubMed]
- Zhao, F.; Xiao, C.; Evans, K.S.; Theivanthiran, T.; DeVito, N.; Holtzhausen, A.; Liu, J.; Liu, X.; Boczkowski, D.; Nair, S.; et al. Paracrine Wnt5a-β-Catenin Signaling Triggers a Metabolic Program that Drives Dendritic Cell Tolerization. Immunity 2018, 48, 147–160.e147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, P.H.; Tyrrell, H.E.; Gao, L.; Xu, D.; Quan, J.; Gill, D.; Rai, L.; Ding, Y.; Plant, G.; Chen, Y.; et al. Adiponectin receptor signaling on dendritic cells blunts antitumor immunity. Cancer Res. 2014, 74, 5711–5722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Sui, S.; Wang, L.; Li, H.; Zhang, L.; Xu, S.; Zheng, X. Inhibition of tumor propellant glutathione peroxidase 4 induces ferroptosis in cancer cells and enhances anticancer effect of cisplatin. J. Cell. Physiol. 2020, 235, 3425–3437. [Google Scholar] [CrossRef] [PubMed]
- Han, L.; Bai, L.; Qu, C.; Dai, E.; Liu, J.; Kang, R.; Zhou, D.; Tang, D.; Zhao, Y. PPARG-mediated ferroptosis in dendritic cells limits antitumor immunity. Biochem. Biophys. Res. Commun. 2021, 576, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Rodriguez-Galán, M.C.; Subleski, J.J.; Ortaldo, J.R.; Hodge, D.L.; Wang, J.M.; Shimozato, O.; Reynolds, D.A.; Young, H.A. Peroxisome proliferator-activated receptor-γ and its ligands attenuate biologic functions of human natural killer cells. Blood 2004, 104, 3276–3284. [Google Scholar] [CrossRef] [Green Version]
- Kliewer, S.A.; Forman, B.M.; Blumberg, B.; Ong, E.S.; Borgmeyer, U.; Mangelsdorf, D.J.; Umesono, K.; Evans, R.M. Differential expression and activation of a family of murine peroxisome proliferator-activated receptors. Proc. Natl. Acad. Sci. USA 1994, 91, 7355–7359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tontonoz, P.; Nagy, L.; Alvarez, J.G.; Thomazy, V.A.; Evans, R.M. PPARγ promotes monocyte/macrophage differentiation and uptake of oxidized LDL. Cell 1998, 93, 241–252. [Google Scholar] [CrossRef] [Green Version]
- Ricote, M.; Huang, J.; Fajas, L.; Li, A.; Welch, J.; Najib, J.; Witztum, J.L.; Auwerx, J.; Palinski, W.; Glass, C.K. Expression of the peroxisome proliferator-activated receptor γ (PPARγ) in human atherosclerosis and regulation in macrophages by colony stimulating factors and oxidized low density lipoprotein. Proc. Natl. Acad. Sci. USA 1998, 95, 7614–7619. [Google Scholar] [CrossRef] [Green Version]
- Jiang, C.; Ting, A.T.; Seed, B. PPAR-γ agonists inhibit production of monocyte inflammatory cytokines. Nature 1998, 391, 82–86. [Google Scholar] [CrossRef]
- Ricote, M.; Li, A.C.; Willson, T.M.; Kelly, C.J.; Glass, C.K. The peroxisome proliferator-activated receptor-γ is a negative regulator of macrophage activation. Nature 1998, 391, 79–82. [Google Scholar] [CrossRef] [PubMed]
- Thieringer, R.; Fenyk-Melody, J.E.; Le Grand, C.B.; Shelton, B.A.; Detmers, P.A.; Somers, E.P.; Carbin, L.; Moller, D.E.; Wright, S.D.; Berger, J. Activation of peroxisome proliferator-activated receptor γ does not inhibit IL-6 or TNF-α responses of macrophages to lipopolysaccharide in vitro or in vivo. J. Immunol. 2000, 164, 1046–1054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chawla, A.; Barak, Y.; Nagy, L.; Liao, D.; Tontonoz, P.; Evans, R.M. PPAR-γ dependent and independent effects on macrophage-gene expression in lipid metabolism and inflammation. Nat. Med. 2001, 7, 48–52. [Google Scholar] [CrossRef] [PubMed]
- Moore, K.J.; Rosen, E.D.; Fitzgerald, M.L.; Randow, F.; Andersson, L.P.; Altshuler, D.; Milstone, D.S.; Mortensen, R.M.; Spiegelman, B.M.; Freeman, M.W. The role of PPAR-γ in macrophage differentiation and cholesterol uptake. Nat. Med. 2001, 7, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Bouhlel, M.A.; Derudas, B.; Rigamonti, E.; Dièvart, R.; Brozek, J.; Haulon, S.; Zawadzki, C.; Jude, B.; Torpier, G.; Marx, N.; et al. PPARγ activation primes human monocytes into alternative M2 macrophages with anti-inflammatory properties. Cell Metab. 2007, 6, 137–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Ginderachter, J.A.; Meerschaut, S.; Liu, Y.; Brys, L.; De Groeve, K.; Hassanzadeh Ghassabeh, G.; Raes, G.; De Baetselier, P. Peroxisome proliferator-activated receptor γ (PPARγ) ligands reverse CTL suppression by alternatively activated (M2) macrophages in cancer. Blood 2006, 108, 525–535. [Google Scholar] [CrossRef] [PubMed]
- Cheng, W.Y.; Huynh, H.; Chen, P.; Peña-Llopis, S.; Wan, Y. Macrophage PPARγ inhibits Gpr132 to mediate the anti-tumor effects of rosiglitazone. Elife 2016, 5, e18501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gionfriddo, G.; Plastina, P.; Augimeri, G.; Catalano, S.; Giordano, C.; Barone, I.; Morelli, C.; Giordano, F.; Gelsomino, L.; Sisci, D.; et al. Modulating Tumor-Associated Macrophage Polarization by Synthetic and Natural PPARγ Ligands as a Potential Target in Breast Cancer. Cells 2020, 9, 174. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.B.; Ahn, Y.H.; Jung, J.H.; Lee, Y.J.; Lee, J.H.; Kang, J.L. Programming of macrophages by UV-irradiated apoptotic cancer cells inhibits cancer progression and lung metastasis. Cell Mol. Immunol. 2019, 16, 851–867. [Google Scholar] [CrossRef] [Green Version]
- Penas, F.; Mirkin, G.A.; Vera, M.; Cevey, Á.; González, C.D.; Gómez, M.I.; Sales, M.E.; Goren, N.B. Treatment in vitro with PPARα and PPARγ ligands drives M1-to-M2 polarization of macrophages from T. cruzi-infected mice. Biochim. Biophys. Acta 2015, 1852, 893–904. [Google Scholar] [CrossRef] [Green Version]
- Moreira, T.G.; Horta, L.S.; Gomes-Santos, A.C.; Oliveira, R.P.; Queiroz, N.M.G.P.; Mangani, D.; Daniel, B.; Vieira, A.T.; Liu, S.; Rodrigues, A.M.; et al. CLA-supplemented diet accelerates experimental colorectal cancer by inducing TGF-β-producing macrophages and T cells. Mucosal Immunol. 2019, 12, 188–199. [Google Scholar] [CrossRef] [PubMed] [Green Version]

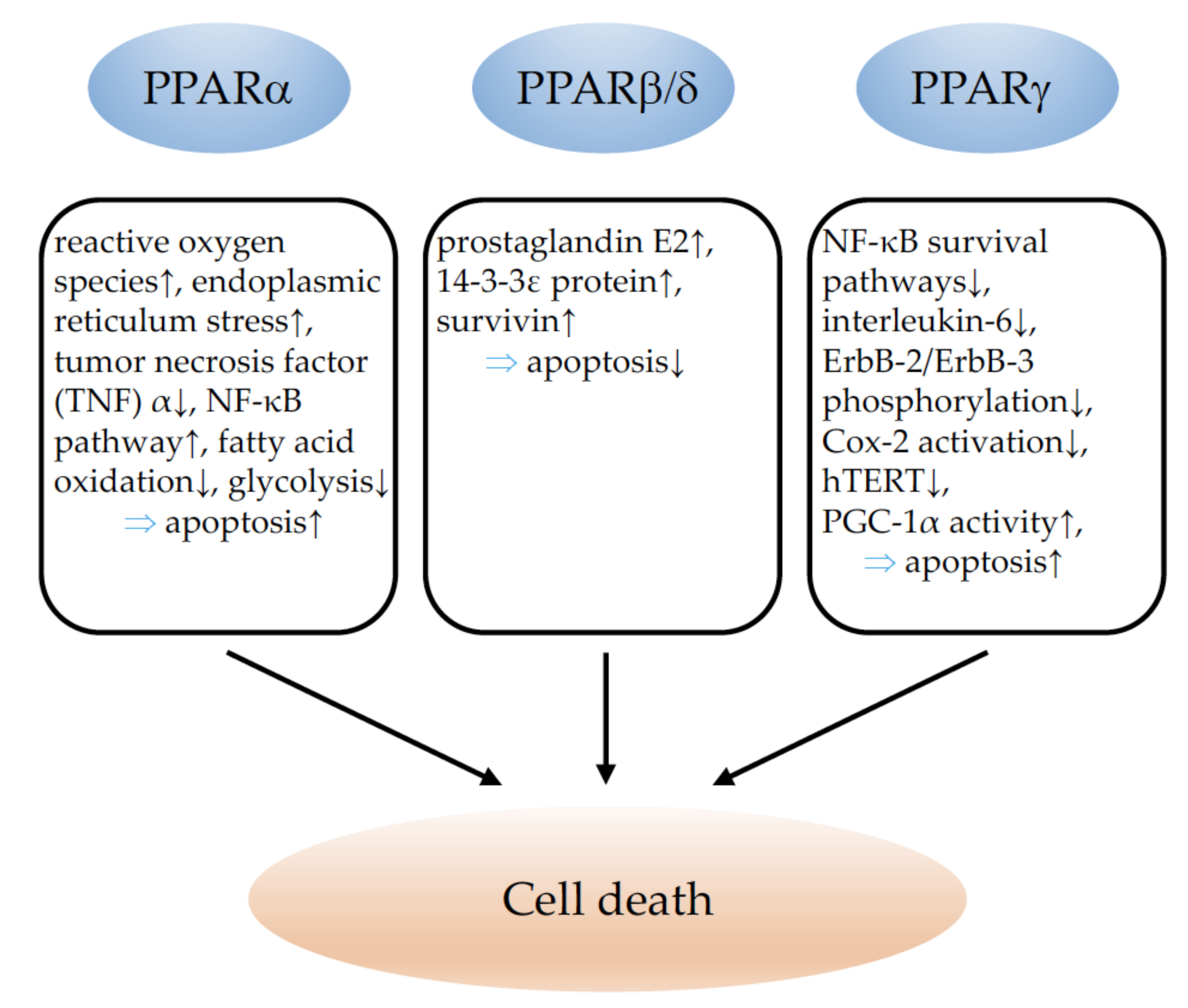

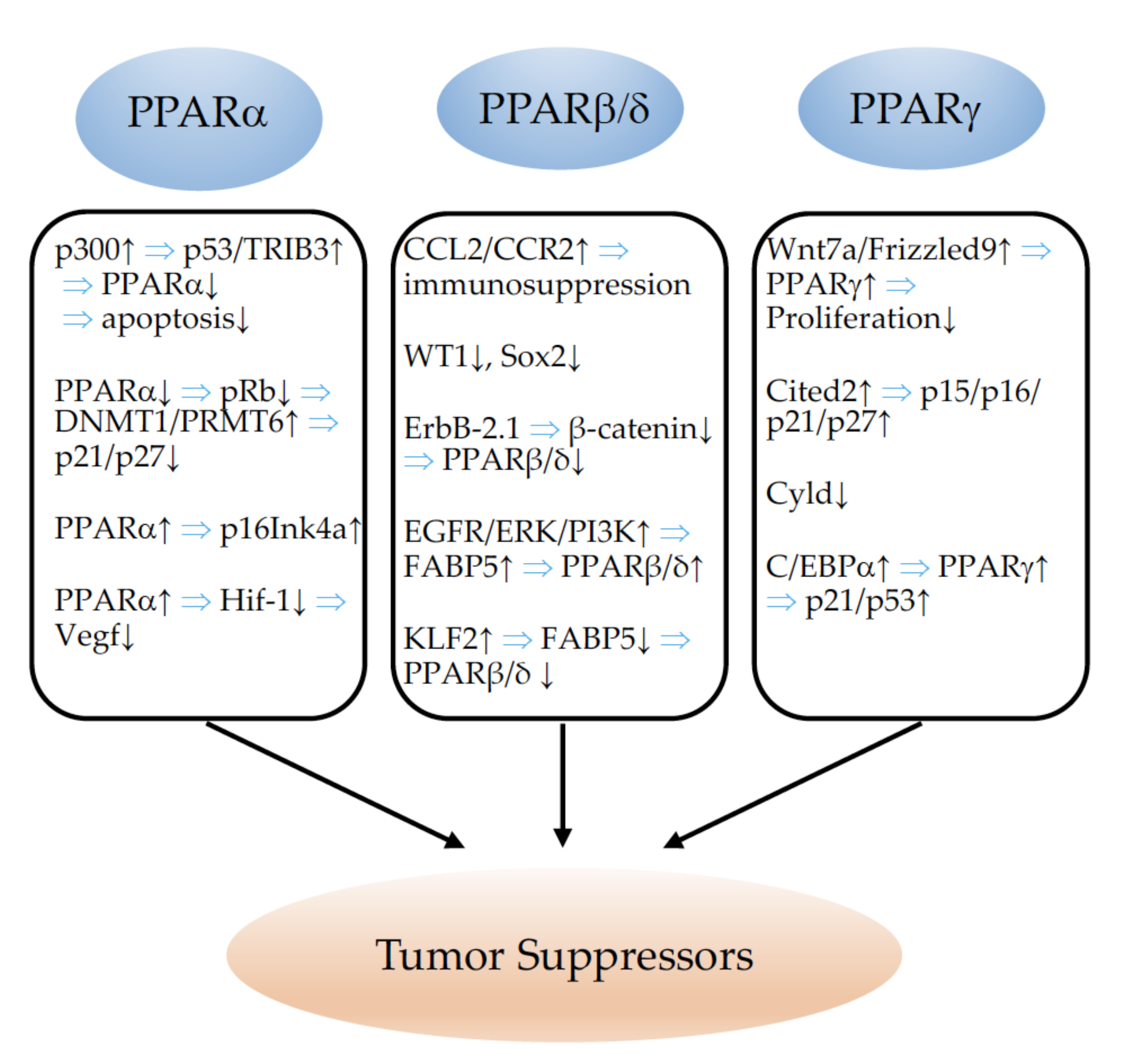





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model | Intervention | Outcome | References |
|---|---|---|---|
| In vitro | |||
| MCF-7, MDA-MB-231 breast cancer cell lines | Clofibrate, Wy-14,643 | Proliferation⇧ | [39] |
| MCF-7 breast cancer cell line | Leptin, glucose | Proliferation⇧ | [40] |
| MDA-MB-231, MCF-7, BT-474 breast cancer cell lines | AA | Proliferation⇧ | [41] |
| MDA-MB-231, MCF-7 breast cancer cell line | AA | Proliferation⇩ | [42] |
| Triple-negative breast cancer cell lines | Fenofibrate | Proliferation⇩ | [43] |
| SUM149PT and SUM1315MO2 inflammatory breast cancer cell lines | Clofibrate | Proliferation⇩ | [44] |
| Ishikawa endometrial cancer cells | Fenofibrate | Proliferation⇩, tumor growth≈ | [55] |
| BsB8 mouse medulloblastoma cells, human D384, and Daoy medulloblastoma cells | Fenofibrate | Proliferation⇩ | [57] |
| U87 glioblastoma cell line | Fenofibrate | Proliferation⇩ | [58] |
| Neuroblastoma cell line | Fenofibrate | Proliferation⇩ | [59] |
| MDA-MB-231 breast, Panc-1 pancreatic cancer cell line | GW6471 (antagonist), Wy-14,643 | Proliferation⇩ upon antagonist, proliferation⇧ upon agonist | [61] |
| A549 and SK-MES-1 lung cancer cell lines | Fenofibrate | Proliferation⇩ | [64] |
| In vivo | |||
| Mouse xenograft models | Fenofibrate | Tumor growth⇩ | [43] |
| Wildtype mice | Wy-14,643 | Liver tumorigenesis⇧ | [47] |
| Hepatitis C virus transgenic mice with activated PPARα | Liver tumorigenesis⇧ | [48] | |
| Transgenic mice with PPARα activation in hepatocytes | Hepatocytic overexpression | Proliferation⇧ | [49] |
| PPARα-knockout mice | Diethylnitrosamine-induced hepatocarcinoma | Liver tumorigenesis⇧ | [52] |
| PPARα-knockout mice | Syngenic MEF/RS tumors, LLC1 lung cancer, B16 melanoma | Tumor growth⇩ | [53] |
| PPARα-knockout mice | B16 melanoma | Tumor growth⇩ | [69] |
| Ovcar-3 and Diss ovarian cancer cell lines, implanted tumors in nude mice | Clofibrate | Proliferation⇩, tumor growth⇩ | [56] |
| PPARα knockdown in glioma stem cells, xenograft models | PPARα knockdown | Proliferation⇩, tumor growth⇩ | [60] |
| Wildtype mice with LLC1 lung, B16 melanoma, or SKOV-3 ovarian cancer | NXT969 antagonist | Tumor growth⇩ | [62] |
| KRasLA2 mouse model of spontaneous primary NSCLC, orthotopic lung cancer cell injection | Wy-14,643, bezafibrate | Tumor growth⇩ | [65] |
| Wildtype and PPARα-knockout mice injected with Bcr/Abl-transformed B cells | Fenofibrate | Tumor growth⇩ | [67] |
| HCT-116 colon cancer cell line, Xenograft model | Fenofibrate | Proliferation⇩, tumor growth⇩ | [68] |
| Model | Intervention | Outcome | References |
|---|---|---|---|
| In vitro | |||
| Colon cancer cell lines | BRL 49653 activator | Proliferation⇩ | [124] |
| Colon cancer cell lines | Troglitazone | Proliferation⇩ | [126] |
| Liposarcoma cell lines | Pioglitazone | Proliferation⇩ | [131] |
| Gastric cancer cell lines | Troglitazone, pioglitazone | Proliferation⇩ | [132] |
| Gastric cancer cell lines | Troglitazone, 15d-PGJ2 | Proliferation⇩ | [133] |
| LA-N-5 nb neuroblastoma cell line | 15d-PGJ2, GW1929 | Proliferation⇩ | [135] |
| SK-N-AS, SH-SY5Y neuroblastoma cell lines | Rosiglitazone | Proliferation⇩ | [136] |
| U87MG, T98G glioblastoma cell lines | 15d-PGJ2 | Proliferation⇩ | [137] |
| U87, U251 glioblastoma cell lines | Rosiglitazone | Proliferation⇩ | [138] |
| A375 melanoma cell line | 15d-PGJ2, ciglitazone | Proliferation⇩ | [142] |
| Different melanoma cell lines | Multiple thiazolidinediones | Proliferation⇩ | [140] |
| A375 melanoma cell line, xenograft model | Ciglitazone | Proliferation⇩ | [141] |
| H1792 and H1838 NSCLC lines | Rosiglitazone | Proliferation⇩ | [144] |
| H295R adrenocortical cancer cell line | Rosiglitazone, pioglitazone | Proliferation⇩ | [145,146] |
| MCF-7 breast cancer cell line | 15d-PGJ2 | Proliferation⇩ | [159] |
| MCF-7 breast cancer cell line | 15d-PGJ2, ciglitazone | Proliferation⇩ | [160] |
| MDA-MB-231, MDA-MB-453 breast cancer cell lines | C-DIM | Proliferation⇩ | [164] |
| MDA-MB-231 breast cancer cells | Overexpression of PPARγ | Tumor growth⇩ | [166] |
| MDA-MB-231 breast cancer cells | + PPARγ-overexpressing fibroblasts | Tumor growth⇧ | [166] |
| Leiomyoma cell line | Ciglitazone, troglitazone | Proliferation⇩ | [161] |
| Ishikawa, Sawano, RL95-2 endometrial carcinoma cell lines | 15d-PGJ2 | Proliferation⇩ | [148] |
| SKOV3 ovarian cancer cell line | C-DIM | Proliferation⇩ | [149] |
| A2780, OVCAR3, OVCAR5, OVCAR8, OVCAR432, SKOV3, IGROV1 ovarian cancer cell lines | Ciglitazone, PPAR-γ antagonist GW9662 | Proliferation⇩(agonist), proliferation⇧ (antagonist) | [150] |
| RPMI 8226 multiple-myeloma cell line | Overexpression of PPAR-γ | Proliferation⇩ | [151] |
| B-cell lymphoma cell line | Silencing, overexpression of PPAR-γ | Proliferation⇧ (silencing), proliferation⇩ (overexpression) | [152] |
| G292, MG63, SAOS and U2OS osteosarcoma cell lines | Troglitazone | Proliferation⇧ | [171] |
| 143B, MNNG/HOS, MG-63, and TE-85 osteosarcoma cell lines | Troglitazone, ciglitazone | Proliferation⇩ | [172] |
| H292, H3118, HMC1, HMC3A, HMC3B mucoepidermoid carcinoma cell lines | SR10221, SR2595, T0070907 inverse agonists | Proliferation⇩, tumor growth⇩ | [174] |
| In vivo | |||
| C57BL/6J-APCMin/+ mice | BRL-49,653, troglitazone | Tumor growth⇧ | [122] |
| C57BL/6J-APCMin/+ mice | Troglitazone | Tumor growth⇧ | [123] |
| Colon cancer cell lines, xenograft mouse model | Troglitazone | Proliferation⇩, tumor growth⇩ | [125] |
| SW480 colon cancer cell line, xenograft model | C-DIM | Proliferation⇩, tumor growth⇩ | [127] |
| A549 NSCLC line, xenograft models | Troglitazone, pioglitazone | Proliferation⇩, tumor growth⇩ | [143] |
| NCI-H2347, NCI-H1993 lung adenocarcinoma cell lines, xenograft models | Pioglitazone | Proliferation⇩, tumor growth⇩ | [173] |
| Huh7 and Hep3B hepatocellular cancer cell lines, xenograft models | Troglitazone | Proliferation⇩, tumor growth⇩ | [147] |
| Dominant-negative mutant thyroid hormone receptor beta (TRbetaPV/PV mice) | Rosiglitazone | Tumor growth⇩ | [157] |
| MMTV-VpPPARγ animals | Breeding with MMTV-PyV strain | Tumor growth⇧ | [165] |
| MSE cell-specific PPARγ knockout (PPARγ-MSE KO) | 7,12-dimethylbenz[a]anthracene (DMBA)-induced breast tumorigenesis | Tumor growth⇧ | [167] |
| Thirty-eight patients with early-stage breast cancer | Rosiglitazone | Proliferation≈, tumor growth≈ | [168] |
| UV and chemically induced skin carcinogenesis | Rosiglitazone, troglitazone | Tumor growth≈ | [169] |
| CML LSCs Three patients with CML | Pioglitazone in combination with imatinib | Proliferation⇩, CMR≈5 years | [155] |
| EHMES-10, MSTO-211H mesothelioma cell lines, xenograft models | Troglitazone | Proliferation⇩, tumor growth⇩ | [153] |
| Esophageal squamous-cell carcinoma line, xenograft model | Efatutazone; troglitazone | Proliferation⇩, tumor growth⇩; proliferation≈, tumor growth≈ | [154] |
| Overexpression of dn PPAR-γ in myeloid lineage cells | Tumor growth⇧ | [158] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wagner, N.; Wagner, K.-D. Peroxisome Proliferator-Activated Receptors and the Hallmarks of Cancer. Cells 2022, 11, 2432. https://doi.org/10.3390/cells11152432
Wagner N, Wagner K-D. Peroxisome Proliferator-Activated Receptors and the Hallmarks of Cancer. Cells. 2022; 11(15):2432. https://doi.org/10.3390/cells11152432
Chicago/Turabian StyleWagner, Nicole, and Kay-Dietrich Wagner. 2022. "Peroxisome Proliferator-Activated Receptors and the Hallmarks of Cancer" Cells 11, no. 15: 2432. https://doi.org/10.3390/cells11152432