
Upregulation of Phospholipase C Gene Expression Due to Norepinephrine-Induced Hypertrophic Response


Abstract
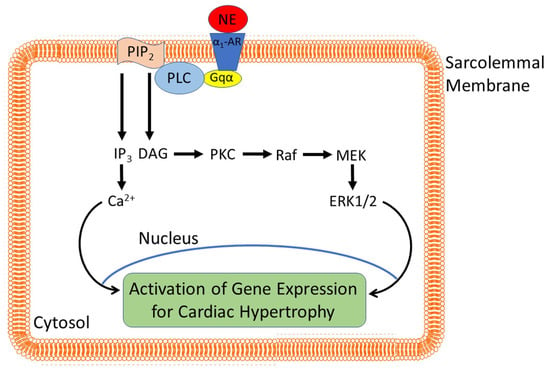
:
1. Introduction
2. Regulation of Cardiac PLC Isozymes
3. Upregulation of PLC Isozymes in Pathological Cardiomyocyte Growth
Transgenic and In Vivo Models of Cardiac Hypertrophy
4. Isolated Cardiomyocytes for Assessing the Hypertrophic Response
5. Evidence for Regression of Abnormal Cardiomyocyte Growth by α1-AR Blockade
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fiedler, B.; Wollert, K.C. Targeting calcineurin and associated pathways in cardiac hypertrophy and failure. Expert Opin. Ther. Targets 2005, 9, 963–973. [Google Scholar] [CrossRef] [PubMed]
- Balakumar, P.; Jagadeesh, G. Multifarious molecular signaling cascades of cardiac hypertrophy: Can the muddy waters be cleared? Pharmacol. Res. 2010, 62, 365–383. [Google Scholar] [CrossRef] [PubMed]
- Clerk, A.; Cullingford, T.E.; Fuller, S.J.; Giraldo, A.; Markou, T.; Pikkarainen, S.; Sugden, P.H. Signaling pathways mediating cardiac myocyte gene expression in physiological and stress responses. J. Cell Physiol. 2007, 212, 311–322. [Google Scholar] [CrossRef] [PubMed]
- De Jonge, H.W.; Dekkers, D.H.; Houtsmuller, A.B.; Sharma, H.S.; Lamers, J.M. Differential signaling and hypertrophic responses in cyclically stretched vs endothelin-1 stimulated neonatal rat cardiomyocytes. Cell Biochem. Biophys. 2007, 47, 21–32. [Google Scholar] [CrossRef] [Green Version]
- Haque, Z.K.; Wang, D.Z. How cardiomyocytes sense pathophysiological stresses for cardiac remodeling. Cell. Mol. Life Sci. 2017, 74, 983–1000. [Google Scholar] [CrossRef]
- Hefti, M.A.; Harder, B.A.; Eppenberger, H.M.; Schaub, M.C. Signaling pathways in cardiac myocyte hypertrophy. J. Mol. Cell. Cardiol. 1997, 29, 2873–2892. [Google Scholar] [CrossRef]
- Heineke, J.; Molkentin, J.D. Regulation of cardiac hypertrophy by intracellular signalling pathways. Nat. Rev. Mol. Cell Biol. 2006, 7, 589–600. [Google Scholar] [CrossRef]
- Peter, A.K.; Bjerke, M.A.; Leinwand, L.A. Biology of the cardiac myocyte in heart disease. Mol. Biol. Cell 2016, 27, 2149–2160. [Google Scholar] [CrossRef]
- Takano, A.P.C.; Senger, N.; Barreto-Chaves, M.L.M. The endocrinological component and signaling pathways associated to cardiac hypertrophy. Mol. Cell. Endocrinol. 2020, 518, 110972. [Google Scholar] [CrossRef]
- Winkle, A.J.; Nassal, D.M.; Shaheen, R.; Thomas, E.; Mohta, S.; Gratz, D.; Weinberg, S.H.; Hund, T.J. Emerging therapeutic targets for cardiac hypertrophy. Expert Opin. Ther. Targets 2022, 26, 29–40. [Google Scholar] [CrossRef]
- Freire, G.; Ocampo, C.; Ilbawi, N.; Griffin, A.J.; Gupta, M. Overt expression of AP-1 reduces alpha myosin heavy chain expression and contributes to heart failure from chronic volume overload. J. Mol. Cell. Cardiol. 2007, 43, 465–478. [Google Scholar] [CrossRef] [PubMed]
- Jeong, M.Y.; Kinugawa, K.; Vinson, C.; Long, C.S. AFos dissociates cardiac myocyte hypertrophy and expression of the pathological gene program. Circulation 2005, 111, 1645–1651. [Google Scholar] [CrossRef] [PubMed]
- Montessuit, C.; Thorburn, A. Transcriptional activation of the glucose transporter GLUT1 in ventricular cardiac myocytes by hypertrophic agonists. J. Biol. Chem. 1999, 274, 9006–9012. [Google Scholar] [CrossRef] [Green Version]
- Colella, M.; Grisan, F.; Robert, V.; Turner, J.D.; Thomas, A.P.; Pozzan, T. Ca2+ oscillation frequency decoding in cardiac cell hypertrophy: Role of calcineurin/NFAT as Ca2+ signal integrators. Proc. Natl. Acad. Sci. USA 2008, 105, 2859–2864. [Google Scholar] [CrossRef] [Green Version]
- Gupta, S.; Young, D.; Maitra, R.K.; Gupta, A.; Popovic, Z.B.; Yong, S.L.; Mahajan, A.; Wang, Q.; Sen, S. Prevention of cardiac hypertrophy and heart failure by silencing of NF-kappaB. J. Mol. Biol. 2008, 375, 637–649. [Google Scholar] [CrossRef] [Green Version]
- Robinson, E.L.; Drawnel, F.M.; Mehdi, S.; Archer, C.R.; Liu, W.; Okkenhaug, H.; Alkass, K.; Aronsen, J.M.; Nagaraju, C.K.; Sjaastad, I.; et al. MSK-mediated phosphorylation of histone H3 Ser28 couples MAPK signalling with early gene induction and cardiac hypertrophy. Cells 2022, 11, 604. [Google Scholar] [CrossRef]
- Rosenzweig, A.; Halazonetis, T.D.; Seidman, J.G.; Seidman, C.E. Proximal regulatory domains of rat atrial natriuretic factor gene. Circulation 1991, 84, 1256–1265. [Google Scholar] [CrossRef] [Green Version]
- Saadane, N.; Alpert, L.; Chalifour, L.E. Altered molecular response to adrenoreceptor-induced cardiac hypertrophy in Egr-1-deficient mice. Am. J. Physiol. Heart Circ. Physiol. 2000, 278, H796–H805. [Google Scholar] [CrossRef] [Green Version]
- Aoki, H.; Richmond, M.; Izumo, S.; Sadoshima, J. Specific role of the extracellular signal-regulated kinase pathway in angiotensin II-induced cardiac hypertrophy in vitro. Biochem. J. 2000, 347, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Hannan, R.D.; West, A.K. Adrenergic agents, but not triiodo-L-thyronine induce c-fos and c-myc expression in the rat heart. Basic Res. Cardiol. 1991, 86, 154–164. [Google Scholar] [CrossRef]
- Hannan, R.D.; Stennard, F.A.; West, A.K. Expression of c-fos and related genes in the rat heart in response to norepinephrine. J. Mol. Cell. Cardiol. 1993, 25, 1137–1148. [Google Scholar] [CrossRef] [PubMed]
- Schunkert, H.; Jahn, L.; Izumo, S.; Apstein, C.S.; Lorell, B.H. Localization and regulation of c-fos and c-jun protooncogene induction by systolic wall stress in normal and hypertrophied rat hearts. Proc. Natl. Acad. Sci. USA 1991, 88, 11480–11484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, D.J.; Rho, H.W.; Rhee, S.G. CD3 stimulation causes phosphorylation of phospholipase C-γ 1 on serine and tyrosine residues in a human T-cell line. Proc. Natl. Acad. Sci. USA 1991, 88, 5453–5456. [Google Scholar] [CrossRef] [Green Version]
- Schnabel, P.; Mies, F.; Nohr, T.; Geisler, M.; Böhm, M. Differential regulation of phospholipase C-β isozymes in cardiomyocyte hypertrophy. Biochem. Biophys. Res. Commun. 2000, 275, 1–6. [Google Scholar] [CrossRef]
- Katan, M.; Cockcroft, S. Phospholipase C families: Common themes and versatility in physiology and pathology. Prog. Lipid. Res. 2020, 80, 101065. [Google Scholar] [CrossRef]
- Vines, C.M. Phospholipase C. Adv. Exp. Med. Biol. 2012, 740, 235–254. [Google Scholar]
- Bai, H.; Wu, L.L.; Xing, D.Q.; Liu, J.; Zhao, Y.L. Angiotensin II induced upregulation of G αq/11, phospholipase C β3 and extracellular signal-regulated kinase 1/2 via angiotensin II type 1 receptor. Chin. Med. J. 2004, 117, 88–93. [Google Scholar]
- Lamers, J.M.; Eskildsen-Helmond, Y.E.; Resink, A.M.; de Jonge, H.W.; Bezstarosti, K.; Sharma, H.S.; van Heugten, H.A. Endothelin-1-induced phospholipase C-beta and D and protein kinase C isoenzyme signaling leading to hypertrophy in rat cardiomyocytes. J. Cardiovasc. Pharmacol. 1995, 26, S100–S103. [Google Scholar] [CrossRef]
- Ruwhof, C.; Egas, J.M.; van Wamel, A.E.; van der Laarse, A. Signal transduction of mechanical stress in myocytes and fibroblasts derived from neonatal rat ventricles. Neth. Heart J. 2001, 9, 372–378. [Google Scholar]
- Ruwhof, C.; van Wamel, J.T.; Noordzij, L.A.; Aydin, S.; Harper, J.C.; van der Laarse, A. Mechanical stress stimulates phospholipase C activity and intracellular calcium ion levels in neonatal rat cardiomyocytes. Cell Calcium 2001, 29, 73–83. [Google Scholar] [CrossRef]
- Woodcock, E.A.; Grubb, D.R.; Filtz, T.M.; Marasco, S.; Luo, J.; McLeod-Dryden, T.J.; Kaye, D.M.; Sadoshima, J.; Du, X.J.; Wong, C.; et al. Selective activation of the “b” splice variant of phospholipase Cβ1 in chronically dilated human and mouse atria. J. Mol. Cell. Cardiol. 2009, 47, 676–683. [Google Scholar] [CrossRef] [PubMed]
- Yan, K.; Wang, K.; Li, P. The role of post-translational modifications in cardiac hypertrophy. J. Cell. Mol. Med. 2019, 23, 3795–3807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, J.-H.; Zhang, H.-G. Transcription factors involved in the development and prognosis of cardiac remodeling. Front. Pharmacol. 2022, 13, 828549. [Google Scholar] [CrossRef] [PubMed]
- Harden, T.K.; Sondek, J. Regulation of phospholipase C isozymes by ras superfamily GTPases. Annu. Rev. Pharmacol. Toxicol. 2006, 46, 355–379. [Google Scholar] [CrossRef] [Green Version]
- Suh, P.-G.; Ryu, S.O.; Choi, W.C.; Lee, K.Y.; Rhee, S.G. Monoclonal antibodies to three phospholipase C isozymes from bovine brain. J. Biol. Chem. 1988, 263, 14497–14504. [Google Scholar] [CrossRef]
- Gresset, A.; Sondek, J.; Harden, T.K. The phospholipase C isozymes and their regulation. Subcell. Biochem. 2012, 58, 61–94. [Google Scholar]
- Fukami, K.; Inanobe, S.; Kanemaru, K.; Nakamura, Y. Phospholipase C is a key enzyme regulating intracellular calcium and modulating the phosphoinositide balance. Prog. Lipid Res. 2010, 49, 429–437. [Google Scholar] [CrossRef]
- Miao, L.N.; Pan, D.; Shi, J.; Du, J.P.; Chen, P.F.; Gao, J.; Yu, Y.; Shi, D.Z.; Guo, M. Role and mechanism of PKC-δ for cardiovascular disease: Current status and perspective. Front. Cardiovasc. Med. 2022, 9, 816369. [Google Scholar] [CrossRef]
- Singh, R.M.; Cummings, E.; Pantos, C.; Singh, J. Protein kinase C and cardiac dysfunction: A review. Heart Fail. Rev. 2017, 22, 843–859. [Google Scholar] [CrossRef] [Green Version]
- Barac, Y.D.; Zeevi-Levin, N.; Yaniv, G.; Reiter, I.; Milman, F.; Shilkrut, M.; Coleman, R.; Abassi, Z.; Binah, O. The 1,4,5-inositol trisphosphate pathway is a key component in Fas-mediated hypertrophy in neonatal rat ventricular myocytes. Cardiovasc. Res. 2005, 68, 75–86. [Google Scholar] [CrossRef] [Green Version]
- Kockskämper, J.; Zima, A.V.; Roderick, H.L.; Pieske, B.; Blatter, L.A.; Bootman, M.D. Emerging roles of inositol 1,4,5-trisphosphate signaling in cardiac myocytes. J. Mol. Cell. Cardiol. 2008, 45, 128–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansen, C.A.; Schroering, A.G.; Robishaw, J.D. Subunit expression of signal transducing G proteins in cardiac tissue: Implications for phospholipase C-β regulation. J. Mol. Cell. Cardiol. 1995, 27, 471–484. [Google Scholar] [CrossRef]
- Schnabel, P.; Gäs, H.; Nohr, T.; Camps, M.; Böhm, M. Identification and characterization of G protein-regulated phospholipase C in human myocardium. J. Mol. Cell. Cardiol. 1996, 28, 2419–2427. [Google Scholar] [CrossRef]
- Smrcka, A.V.; Brown, J.H.; Holz, G.G. Role of phospholipase Cε in physiological phosphoinositide signaling networks. Cell Signal. 2012, 24, 1333–1343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suh, P.G.; Ryu, S.H.; Moon, K.H.; Suh, H.W.; Rhee, S.G. Cloning and sequence of multiple forms of phospholipase C. Cell. 1988, 54, 161–169. [Google Scholar] [CrossRef]
- Tappia, P.S.; Liu, S.Y.; Shatadal, S.; Takeda, N.; Dhalla, N.S.; Panagia, V. Changes in sarcolemmal PLC isoenzymes in postinfarct congestive heart failure: Partial correction by imidapril. Am. J. Physiol. 1999, 277, H40–H49. [Google Scholar] [CrossRef]
- Wolf, R.A. Specific expression of phospholipase C-δ1 and γ1 by adult cardiac ventricular Myocytes. Circulation 1993, 88 (Suppl. S1), I-241. [Google Scholar]
- Nakamura, Y.; Fukami, K. Regulation and physiological functions of mammalian phospholipase C. J. Biochem. 2017, 161, 315–321. [Google Scholar] [CrossRef] [Green Version]
- Otaegui, D.; Querejeta, R.; Arrieta, A.; Lazkano, A.; Bidaurrazaga, A.; Arriandiaga, J.R.; Aldazabal, P.; Garro, M.A. Phospholipase Cβ4 isozyme is expressed in human, rat, and murine heart left ventricles and in HL-1 cardiomyocytes. Mol. Cell. Biochem. 2010, 337, 167–173. [Google Scholar] [CrossRef]
- Rhee, S.G. Regulation of phosphoinositide-specific phospholipase C. Annu. Rev. Biochem. 2001, 70, 281–312. [Google Scholar] [CrossRef]
- Lee, C.W.; Lee, K.H.; Lee, S.B.; Park, D.; Rhee, S.G. Regulation of phospholipase C-β4 by ribonucleotides and the alpha subunit of Gq. J. Biol. Chem. 1994, 269, 25335–25338. [Google Scholar] [CrossRef]
- Madukwe, J.C.; Garland-Kuntz, E.E.; Lyon, A.M.; Smrcka, A.V. G protein βγ subunits directly interact with and activate phospholipase Cϵ. J. Biol. Chem. 2018, 293, 6387–6397. [Google Scholar] [CrossRef] [Green Version]
- Cockcroft, S.; Thomas, G.M. Inositol-lipid-specific phospholipase C isoenzymes and their differential regulation by receptors. Biochem. J. 1992, 288, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Sekiya, F.; Bae, Y.S.; Rhee, S.G. Regulation of phospholipase C isozymes: Activation of phospholipase C-γ in the absence of tyrosine-phosphorylation. Chem. Phys. Lipids. 1999, 98, 3–11. [Google Scholar] [CrossRef]
- Tappia, P.S.; Padua, R.R.; Panagia, V.; Kardami, E. Fibroblast growth factor-2 stimulates phospholipase Cβ in adult cardiomyocytes. Biochem. Cell. Biol. 1999, 77, 569–575. [Google Scholar] [CrossRef]
- Tall, E.; Dormán, G.; Garcia, P.; Runnels, L.; Shah, S.; Chen, J.; Profit, A.; Gu, Q.M.; Chaudhary, A.; Prestwich, G.D.; et al. Phosphoinositide binding specificity among phospholipase C isozymes as determined by photo-cross-linking to novel substrate and product analogs. Biochemistry 1997, 36, 7239–7248. [Google Scholar] [CrossRef]
- Yagisawa, H.; Sakuma, K.; Paterson, H.F.; Cheung, R.; Allen, V.; Hirata, H.; Watanabe, Y.; Hirata, M.; Williams, R.L.; Katan, M. Replacements of single basic amino acids in the pleckstrin homology domain of phospholipase C-δ1 alter the ligand binding, phospholipase activity, and interaction with the plasma membrane. J. Biol. Chem. 1998, 273, 417–424. [Google Scholar] [CrossRef] [Green Version]
- Dupuis, M.; Houdeau, E.; Mhaouty-Kodja, S. Increased potency of α1-adrenergic receptors to induce inositol phosphates production correlates with the up-regulation of α1d/Gh α/phospholipase C δ1 signaling pathway in term rat myometrium. Reproduction 2008, 135, 55–62. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.F.; Yeh, T.S.; Chen, S.F.; Tsai, Y.H.; Chou, C.M.; Yang, Y.Y.; Huang, H.M. Nonmuscle myosin IIA (myosin heavy polypeptide 9): A novel class of signal transducer mediating the activation of Gαh/phospholipase C-δ1 pathway. Endocrinology 2010, 151, 876–885. [Google Scholar] [CrossRef] [Green Version]
- Kawaguchi, H.; Sano, H.; Iizuka, K.; Okada, H.; Kudo, T.; Kageyama, K.; Muramoto, S.; Murakami, T.; Okamoto, H.; Mochizuki, N. Phosphatidylinositol metabolism in hypertrophic rat heart. Circ. Res. 1993, 72, 966–972. [Google Scholar] [CrossRef] [Green Version]
- Shoki, M.; Kawaguchi, H.; Okamoto, H.; Sano, H.; Sawa, H.; Kudo, T.; Hirao, N.; Sakata, Y.; Yasuda, H. Phosphatidylinositol and inositolphosphatide metabolism in hypertrophied rat heart. Jpn. Circ. J. 1992, 56, 142–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dent, M.R.; Aroutiounova, N.; Dhalla, N.S.; Tappia, P.S. Losartan attenuates phospholipase C isozyme gene expression in hypertrophied hearts due to volume overload. J. Cell. Mol. Med. 2006, 10, 470–479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dent, M.R.; Dhalla, N.S.; Tappia, P.S. Phospholipase C gene expression, protein content and activities in cardiac hypertrophy and heart failure due to volume overload. Am. J. Physiol. 2004, 282, H719–H727. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Zhang, C.; Liu, C.; Zhang, A.; Li, A.; Zhang, J.; Zhang, H. Aortic constriction induces hypertension and cardiac hypertrophy via (pro)renin receptor activation and the PLC-β3 signaling pathway. Mol. Med. Rep. 2019, 19, 573–580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jalili, T.; Takeishi, Y.; Song, G.; Ball, N.A.; Howles, G.; Walsh, R.A. PKC translocation without changes in Gαq and PLC-β protein abundance in cardiac hypertrophy and failure. Am. J. Physiol. 1999, 277, H2298–H2304. [Google Scholar] [PubMed]
- Wang, H.; Oestreich, E.A.; Maekawa, N.; Bullard, T.A.; Vikstrom, K.L.; Dirksen, R.T.; Kelley, G.G.; Blaxall, B.C.; Smrcka, A.V. Phospholipase C ε modulates β-adrenergic receptor-dependent cardiac contraction and inhibits cardiac hypertrophy. Circ. Res. 2005, 97, 1305–1313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ganguly, P.K.; Lee, S.L.; Beamish, R.E.; Dhalla, N.S. Altered sympathetic system and adrenoceptors during the development of cardiac hypertrophy. Am. Heart J. 1989, 118, 520–525. [Google Scholar] [CrossRef]
- Adams, J.W.; Sakata, Y.; Davis, M.G.; Sah, V.P.; Wang, Y.; Liggett, S.B.; Chien, K.R.; Brown, J.H.; Dorn, G.W., 2nd. Enhanced Gαq signaling: A common pathway mediates cardiac hypertrophy and apoptotic heart failure. Proc. Natl. Acad. Sci. USA 1998, 95, 10140–10145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Angelo, D.D.; Sakata, Y.; Lorenz, J.N.; Boivin, G.P.; Walsh, R.A.; Liggett, S.B.; Dorn, G.W., 2nd. Transgenic Gαq overexpression induces cardiac contractile failure in mice. Proc. Natl. Acad. Sci. USA 1997, 94, 8121–8126. [Google Scholar] [CrossRef] [Green Version]
- Sakata, Y.; Hoit, B.D.; Liggett, S.B.; Walsh, R.A.; Dorn, G.W., 2nd. Decompensation of pressure-overload hypertrophy in Gαq-overexpressing mice. Circulation 1998, 97, 1488–1495. [Google Scholar] [CrossRef] [Green Version]
- Sussman, M.A.; Welch, S.; Walker, A.; Klevitsky, R.; Hewett, T.E.; Price, R.L.; Schaefer, E.; Yager, K. Altered focal adhesion regulation correlates with cardiomyopathy in mice expressing constitutively active rac1. J. Clin. Investig. 2000, 105, 875–886. [Google Scholar] [CrossRef] [Green Version]
- Mende, U.; Kagen, A.; Cohen, A.; Aramburu, J.; Schoen, F.J.; Neer, E.J. Transient cardiac expression of constitutively active Gαq leads to hypertrophy and dilated cardiomyopathy by calcineurin-dependent and independent pathways. Proc. Natl. Acad. Sci. USA 1998, 95, 13893–13898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mende, U.; Semsarian, C.; Martins, D.C.; Kagen, A.; Duffy, C.; Schoen, F.J.; Neer, E.J. Dilated cardiomyopathy in two transgenic mouse lines expressing activated G protein αq: Lack of correlation between phospholipase C activation and the phenotype. J. Mol. Cell. Cardiol. 2001, 33, 1477–14791. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Malik, S.; Kelley, G.G.; Kapiloff, M.S.; Smrcka, A.V. Phospholipase C ε scaffolds to muscle-specific A kinase anchoring protein (mAKAPβ) and integrates multiple hypertrophic stimuli in cardiac myocytes. J. Biol. Chem. 2011, 286, 23012–23021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arthur, J.F.; Matkovich, S.J.; Mitchell, C.J.; Biden, T.J.; Woodcock, E.A. Evidence for selective coupling of α1-adrenergic receptors to phospholipase C-β1 in rat neonatal cardiomyocytes. J. Biol. Chem. 2001, 276, 37341–37346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grubb, D.R.; Vasilevski, O.; Huynh, H.; Woodcock, E.A. The extreme C-terminal region of phospholipase Cβ1 determines subcellular localization and function; the “b” splice variant mediates alpha1-adrenergic receptor responses in cardiomyocytes. FASEB J. 2008, 22, 2768–2774. [Google Scholar] [CrossRef]
- Filtz, T.M.; Grubb, D.R.; McLeod-Dryden, T.J.; Luo, J.; Woodcock, E.A. Gq-initiated cardiomyocyte hypertrophy is mediated by phospholipase Cβ1b. FASEB J. 2009, 23, 3564–3570. [Google Scholar] [CrossRef]
- Singal, T.; Dhalla, N.S.; Tappia, P.S. Regulation of c-Fos and c-Jun gene expression by phospholipase C activity in adult cardiomyocytes. Mol. Cell. Biochem. 2009, 327, 229–239. [Google Scholar] [CrossRef]
- Singal, T.; Dhalla, N.S.; Tappia, P.S. Reciprocal regulation of transcription factors and PLC isozyme gene expression in adult cardiomyocytes. J. Cell. Mol. Med. 2010, 14, 1824–1835. [Google Scholar] [CrossRef] [Green Version]
- Singal, T.; Dhalla, N.S.; Tappia, P.S. Norepinephrine-induced changes in gene expression of phospholipase C in cardiomyocytes. J. Mol. Cell. Cardiol. 2006, 41, 126–137. [Google Scholar] [CrossRef]
- Giles, T.D.; Sander, G.E.; Thomas, M.G.; Quiroz, A.C. α-adrenergic mechanisms in the pathophysiology of left ventricular heart failure-An analysis of their role in systolic and diastolic dysfunction. J. Mol. Cell. Cardiol. 1996, 18, 33–43. [Google Scholar] [CrossRef]
- Motz, W.; Klepzig, M.; Strauer, B.E. Regression of cardiac hypertrophy: Experimental and clinical results. J. Cardiovasc. Pharmacol. 1987, 10, S148–S152. [Google Scholar] [CrossRef] [PubMed]
- Prasad, K.; O’Neil, C.L.; Bharadwaj, B. Effect of prolonged prazosin treatment on hemodynamic and biochemical changes in the dog heart due to chronic pressure overload. Jpn. Heart J. 1984, 25, 461–476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strauer, B.E.; Bayer, F.; Brecht, H.M.; Motz, W. The influence of sympathetic nervous activity on regression of cardiac hypertrophy. J. Hypertens. Suppl. 1985, 3, S39–S44. [Google Scholar]
- Strauer, B.E. Progression and regression of heart hypertrophy in arterial hypertension: Pathophysiology and clinical aspects. Z. Kardiol. 1995, 74, 171–178. [Google Scholar]
- Babick, A.; Elimban, V.; Dhalla, N.S. Reversal of cardiac remodeling and subcellular defects by prazosin in heart failure due to myocardial infarction. J. Clin. Exp. Cardiol. 2012, 5, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Babick, A.; Elimban, V.; Zieroth, S.; Dhalla, N.S. Reversal of cardiac dysfunction and subcellular alterations by metoprolol in heart failure due to myocardial infarction. J. Cell Physiol. 2013, 228, 2063–2070. [Google Scholar] [CrossRef]
- Massart, P.E.; Donckier, J.; Kyselovic, J.; Godfraind, T.; Heyndrickx, G.R.; Wibo, M. Carvedilol and lacidipine prevent cardiac hypertrophy and endothelin-1 gene expression after aortic banding. Hypertension 1999, 34, 1197–1201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosendorff, C. Beta-blocking agents with vasodilator activity. J. Hypertens. Suppl. 1993, 11, S37–S40. [Google Scholar] [CrossRef] [PubMed]
- Antani, J.A.; Antani, N.J.; Nanivadekar, A.S. Prazosin in chronic congestive heart failure due to ischemic heart disease. Clin. Cardiol. 1991, 14, 495–500. [Google Scholar] [CrossRef]
- Colucci, W.S.; Wynne, J.; Holman, B.L.; Braunwald, E. Long-term therapy of heart failure with prazosin: A randomized double blind trial. Am. J. Cardiol. 1980, 45, 337–344. [Google Scholar] [CrossRef]
- Goldman, S.A.; Johnson, L.L.; Escala, E.; Cannon, P.J.; Weiss, M.B. Improved exercise ejection fraction with long-term prazosin therapy in patients with heart failure. Am. J. Med. 1980, 68, 36–42. [Google Scholar] [CrossRef]
- Gomes, M.E.; Mulder, A.; Bellersen, L.; Verheugt, F.W.; Smits, P.; Tack, C.J. α-receptor blockade improves muscle perfusion and glucose uptake in heart failure. Eur. J. Heart Fail. 2010, 12, 1061–1066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kukin, M.L.; Kalman, J.; Mannino, M.; Freudenberger, R.; Buchholz, C.; Ocampo, O. Combined α-β blockade (doxazosin plus metoprolol) compared with beta blockade alone in chronic congestive heart failure. Am. J. Cardiol. 1996, 77, 486–491. [Google Scholar] [CrossRef]
- Poole-Wilson, P.A.; Swedberg, K.; Cleland, J.G.; Di Lenarda, A.; Hanrath, P.; Komajda, M.; Lubsen, J.; Lutiger, B.; Metra, M.; Remme, W.J.; et al. Comparison of carvedilol and metoprolol on clinical outcomes in patients with chronic heart failure in the Carvedilol or Metoprolol European Trial (COMET): Randomised controlled trial. Lancet 2003, 362, 7–13. [Google Scholar] [CrossRef] [Green Version]
- ALLHAT Collaborative Research Group. Major cardiovascular events in hypertensive patients randomized to doxazosin vs. chlorthalidone: The antihypertensive and lipid-lowering treatment to prevent heart attack trial (ALLHAT). JAMA 2000, 283, 1967–1975. [Google Scholar] [CrossRef] [Green Version]
- Dhaliwal, A.S.; Habib, G.; Deswal, A.; Verduzco, M.; Souchek, J.; Ramasubbu, K.; Aguilar, D.; Ma, T.S.; Jneid, H.M.; Bolos, M.; et al. Impact of α1-adrenergic antagonist use for benign prostatic hypertrophy on outcomes in patients with heart failure. Am. J. Cardiol. 2009, 104, 270–275. [Google Scholar] [CrossRef]
- Kokoz, Y.M.; Evdokimovskii, E.V.; Maltsev, A.V.; Nenov, M.N.; Nakipova, O.V.; Averin, A.S.; Pimenov, O.Y.; Teplov, I.Y.; Berezhnov, A.V.; Reyes, S.; et al. Sarcolemmal α2-adrenoceptors control protective cardiomyocyte-delimited sympathoadrenal response. J. Mol. Cell. Cardiol. 2016, 100, 9–20. [Google Scholar] [CrossRef]
- Alekseev, A.E.; Park, S.; Pimenov, O.Y.; Reyes, S.; Terzic, A. Sarcolemmal α2-adrenoceptors in feedback control of myocardial response to sympathetic challenge. Pharmacol. Ther. 2019, 197, 179–190. [Google Scholar] [CrossRef]
- Maltsev, A.V.; Kokoz, Y.M.; Evdokimovskii, E.V.; Pimenov, O.Y.; Reyes, S.; Alekseev, A.E. Alpha-2 adrenoceptors and imidazoline receptors in cardiomyocytes mediate counterbalancing effect of agmatine on NO synthesis and intracellular calcium handling. J. Mol. Cell. Cardiol. 2014, 68, 66–74. [Google Scholar] [CrossRef] [PubMed]
- Dhalla, N.S.; Xu, Y.J.; Sheu, S.S.; Tappia, P.S.; Panagia, V. Phosphatidic acid: A potential signal transducer for cardiac hypertrophy. J. Mol. Cell. Cardiol. 1997, 29, 2865–2871. [Google Scholar] [CrossRef] [PubMed]
- Tappia, P.S. Phospholipid-mediated signaling systems as novel targets for treatment of heart disease. Can. J. Physiol. Pharmacol. 2007, 85, 25–41. [Google Scholar] [CrossRef] [PubMed]
- Tappia, P.S.; Asemu, G.; Rodriguez-Leyva, D. Phospholipase C as a potential target for cardioprotection during oxidative stress. Can. J. Physiol. Pharmacol. 2010, 88, 249–263. [Google Scholar] [CrossRef]
- Tappia, P.S.; Dent, M.R.; Dhalla, N.S. Oxidative stress and redox regulation of phospholipase D in myocardial disease. Free Radic. Biol. Med. 2006, 41, 349–361. [Google Scholar] [CrossRef] [PubMed]
- Tappia, P.S.; Dhalla, N.S. Mechanisms for the defects in phospholipid signal transduction in diabetic cardiomyopathy. Indian J. Biochem. Biophys. 2014, 51, 431–440. [Google Scholar] [PubMed]
- Tappia, P.S.; Singal, T. Phospholipid-mediated signaling and heart disease. Subcell. Biochem. 2008, 49, 299–324. [Google Scholar] [PubMed]
- Tappia, P.S.; Elimban, V.; Dhalla, N.S. Involvement of phospholipase C in the norepinephrine—Induced hypertrophic response in cardiomyocytes. Scr. Med. 2022, 53, 149–157. [Google Scholar] [CrossRef]
- Tappia, P.S.; Ramjiawan, B.; Dhalla, N.S. Role of phospholipase C in catecholamine-induced increase in myocardial protein synthesis. Can. J. Physiol. Pharmacol. 2022; in press. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Transcription Factor mRNA Level (% of Control) | ||||||
|---|---|---|---|---|---|---|
| NFAT3 (99 bp) | NFκB (124 bp) | MEF2C (92 bp) | MEF2D (105 bp) | c-Fos (74 bp) | c-Jun (163 bp) | |
| Agonist | ||||||
| NE | 110 ± 8 | 112 ± 9 | 121 ± 11 | 113 ± 6 | 268 ± 12 * | 217 ± 6 * |
| PhE | 125 ± 11 | 109 ± 8 | 127 ± 8 | 130 ± 13 | 261 ± 7 * | 225 ± 8 * |
| A: | PLC mRNA Levels (% of Control) | |||
| β1 (114 bp) | β3 (230 bp) | γ1 (123 bp) | δ1 (190 bp) | |
| NE | 219 ± 11 * | 182 ± 18 * | 168 ± 18 * | 221 ± 18 * |
| NE + cFos siRNA | 80 ± 21 # | 89 ± 11 # | 160 ± 15 # | 218 ± 17 # |
| NE + cJun siRNA | 75 ± 19 # | 91 ± 9 # | 170 ± 14 # | 74 ± 8 # |
| B: | Inositol Phosphates (pmol/min/mg Protein) | |||
| PLC β1 | PLC β3 | PLC δ1 | ||
| Control | 2.8 ± 0.6 | 4.1 ± 0.8 | 10.0 ± 1.4 | |
| NE | 6.8 ± 1.0 * | 7.0 ± 1.3 * | 17.1 ± 3.0 * | |
| NE + cFos siRNA | 3.5 ± 0.7 # | 4.5 ± 0.8 # | 16.6 ± 2.4 | |
| NE + cJun siRNA | 3.0 ± 0.8 # | 3.9 ± 0.9 # | 11.8 ± 2.6 # | |
| Condition | c-Fos mRNA Expression Levels | c-Jun mRNA Expression Levels |
|---|---|---|
| NE | 214 ± 23 * | 198 ± 20 * |
| +Prazosin | 114 ± 11# | 100 ± 8 # |
| +U73122 | 83 ± 8 # | 95 ± 9 # |
| +PLC β1 siRNA | 110 ± 8 # | 85 ± 8 # |
| +PLC β3 siRNA | 105 ± 7 # | 95 ± 11 # |
| +PLC γ1 siRNA | 175 ± 25 * | 180 ± 13 * |
| +PLC δ1 siRNA | 108 ± 7 # | 80 ± 7 # |
| PLC Isozyme mRNA Levels (% of Control) | ||||
|---|---|---|---|---|
| Condition | β1 | β3 | γ1 | δ1 |
| NE | 201 ± 9 * | 188 ± 8 * | 181 ± 9 * | 159 ± 8 * |
| +Prazosin | 99 ± 11 # | 102 ± 4 # | 120 ± 5 # | 90 ± 4 # |
| +U73122 | 68 ± 5 # | 80 ± 4 # | 103 ± 11 # | 67 ± 12 # |
| +PLC β1 siRNA | 90 ± 8 # | - | - | - |
| +PLC β3 siRNA | - | 80 ± 9 # | - | - |
| +PLC γ1 siRNA | - | - | 61 ± 7 * | - |
| +PLC δ1 siRNA | - | - | - | 60 ± 8 # |
| PLC mRNA Levels (% of Control) | ||||
|---|---|---|---|---|
| Treatment | β1 | β3 | γ1 | δ1 |
| PMA (µM) | ||||
| 10.1 | 118 ± 11 | 131 ± 8 | 110 ± 10 | 108 ± 10 |
| 1.0 | 190 ± 12 * | 183 ± 9 * | 176 ± 13 * | 161 ± 15 * |
| 10.0 | 175 ± 11 * | 171 ± 10 * | 130 ± 12 * | 123 ± 12 * |
| NE (µM) | ||||
| 5.0 | 223 ± 11 * | 192 ± 19 * | 186 ± 15 * | 193 ± 15 * |
| +Bis-1 (nM) | ||||
| 50 | 95 ± 10 # | 96 ± 9 # | 111 ± 14 # | 100 ± 8 # |
| 100 | 83 ± 8 # | 80 ± 12 # | 100 ± 10 # | 82 ± 9 # |
| 200 | 82 ± 9 # | 75 ± 6 # | 82 ± 9 # | 77 ± 9 # |
| +PD98059 (nM) | ||||
| 2 | 107 ± 11 # | 90 ± 9 # | 117 ± 14 # | 120 ± 11 # |
| 10 | 84 ± 9 # | 87 ± 11 # | 103 ± 9 # | 94 ± 8 # |
| 25 | 77 ± 8 # | 82 ± 7 # | 82 ± 11 # | 90 ± 12 # |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tappia, P.S.; Dhalla, N.S. Upregulation of Phospholipase C Gene Expression Due to Norepinephrine-Induced Hypertrophic Response. Cells 2022, 11, 2488. https://doi.org/10.3390/cells11162488
Tappia PS, Dhalla NS. Upregulation of Phospholipase C Gene Expression Due to Norepinephrine-Induced Hypertrophic Response. Cells. 2022; 11(16):2488. https://doi.org/10.3390/cells11162488
Chicago/Turabian StyleTappia, Paramjit S., and Naranjan S. Dhalla. 2022. "Upregulation of Phospholipase C Gene Expression Due to Norepinephrine-Induced Hypertrophic Response" Cells 11, no. 16: 2488. https://doi.org/10.3390/cells11162488
APA StyleTappia, P. S., & Dhalla, N. S. (2022). Upregulation of Phospholipase C Gene Expression Due to Norepinephrine-Induced Hypertrophic Response. Cells, 11(16), 2488. https://doi.org/10.3390/cells11162488