
Angiotensin II-Induced Signal Transduction Mechanisms for Cardiac Hypertrophy

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
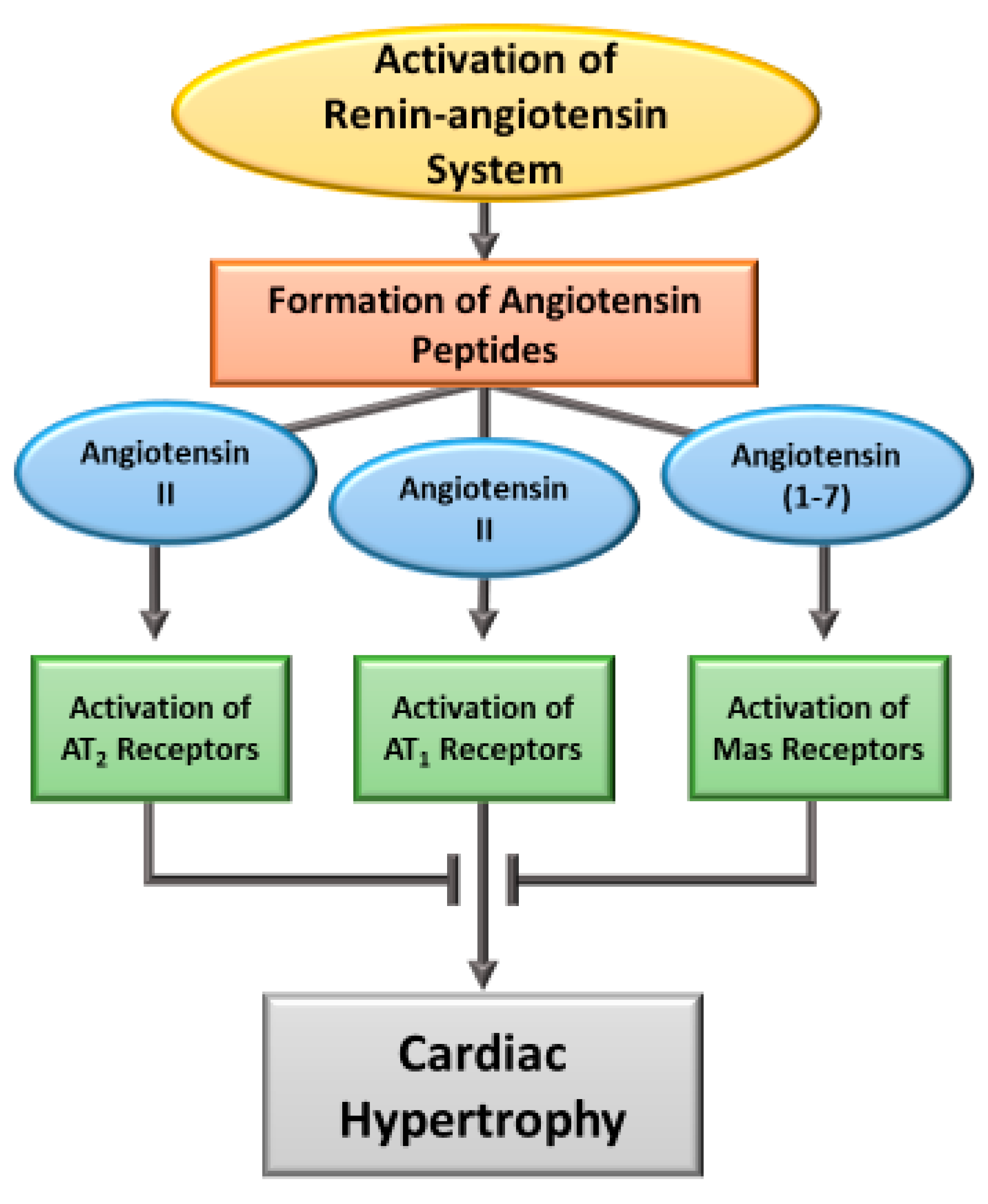
2. Induction of Cardiac Hypertrophy by Angiotensin
3. Angiotensin-Induced Signal Transduction Pathways for Hypertrophic Responses
3.1. Ang II-AT1R Activated Signaling and Cardiac Hypertrophy
3.2. Ang II-ATIR/ROS/Redox Signaling and Cardiac Hypertrophy
3.3. Ang II-AT1R Induced ROS—Mitochondrial Dysfunction
3.4. Ang II-AT1R Induced ROS and Nuclear Factor Erythroid-2 Elated Factor 2 (Nrf2)
3.5. Ang II-AT2R and Ang (1-7)-Mas Receptor Activated Signaling Mechanisms in Cardiac Hypertrophy
4. Therapeutic Strategies for Preventing or Regression of Ang II-Induced Cardiac Hypertrophy
5. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dostal, D.E.; Baker, K.M. The cardiac renin-angiotensin system: Conceptual, or a regulator of cardiac function? Circ. Res. 1999, 85, 643–650. [Google Scholar] [CrossRef] [PubMed]
- Jin, M.; Wilhelm, M.J.; Lang, R.E.; Unger, T.; Lindpaintner, K.; Ganten, D. Endogenous tissue renin-angiotensin systems: From molecular biology to therapy. Am. J. Med. 1988, 84, 28–36. [Google Scholar] [CrossRef]
- Matsuda, S.; Umemoto, S.; Yoshimura, K.; Itoh, S.; Murata, T.; Fukai, T.; Matsuzaki, M. Angiotensin II activates MCP-1 and induces cardiac hypertrophy and dysfunction via Toll-like receptor 4. J. Atheroscler. Thromb. 2015, 22, 833–844. [Google Scholar] [CrossRef] [Green Version]
- Singh, M.V.; Cicha, M.Z.; Nunez, S.; Meyerholz, D.K.; Chapleau, M.W.; Abboud, F.M. Angiotensin II-induced hypertension and cardiac hypertrophy are differentially mediated by TLR3-and TLR4-dependent pathways. Am. J. Physiol. Circ. Physiol. 2019, 316, H1027–H1038. [Google Scholar] [CrossRef] [PubMed]
- Touyz, R.M.; Berry, C. Recent advances in angiotensin II signaling. Braz. J. Med. Biol. Res. 2002, 35, 1001–1015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindpaintner, K.; Ganten, D. The cardiac renin-angiotensin system. An appraisal of present experimental and clinical evidence. Circ. Res. 1991, 68, 905–921. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Iwao, H. Molecular and cellular mechanisms of angiotensin II-mediated cardiovascular and renal diseases. Pharmacol. Rev. 2000, 52, 11–34. [Google Scholar]
- Dzau, V.J. Circulating versus local renin-angiotensin system in cardiovascular homeostasis. Circulation 1988, 77, I4-13. [Google Scholar]
- Mehta, P.K.; Griendling, K.K. Angiotensin II cell signaling: Physiological and pathological effects in the cardiovascular system. Am. J. Physiol. Physiol. 2007, 292, C82–C97. [Google Scholar] [CrossRef]
- Dzau, V. The cardiovascular continuum and renin–angiotensin–aldosterone system blockade. J. Hypertens. 2005, 23, S9–S17. [Google Scholar] [CrossRef]
- Petroff, M.G.V.; Aiello, E.A.; Palomeque, J.; Salas, M.A.; Mattiazzi, A. Subcellular mechanisms of the positive inotropic effect of angiotensin II in cat myocardium. J. Physiol. 2000, 529, 189–203. [Google Scholar] [CrossRef] [PubMed]
- Shao, Q.; Saward, L.; Zahradka, P.; Dhalla, N.S. Ca2+ mobilization in adult rat cardiomyocytes by angiotensin type 1 and 2 receptors. Biochem. Pharmacol. 1998, 55, 1413–1418. [Google Scholar] [CrossRef]
- Guan, X.; Hong, X.; Zhao, N.; Liu, X.; Chen, Y.; Deng, T.; Wang, X.; Wang, J.; Ji, G. CD38 promotes angiotensin II-induced cardiac hypertrophy. J. Cell. Mol. Med. 2017, 21, 1492–1502. [Google Scholar] [CrossRef] [PubMed]
- Wenzel, S.; Taimor, G.; Piper, H.M.; Schlüeter, K. Redox-sensitve intermediates mediate angiotensin II-induced p38 MAP kinase activation, AP-1 binding activity, and TGF-β expression in adult ventricular cardiomyocytes. FASEB J. 2001, 15, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Zhou, N.; Li, L.; Wu, J.; Gong, H.; Niu, Y.; Sun, A.; Ge, J.; Zou, Y. Mechanical stress-evoked but angiotensin II-independent activation of angiotensin II type 1 receptor induces cardiac hypertrophy through calcineurin pathway. Biochem. Biophys. Res. Commun. 2010, 397, 263–269. [Google Scholar] [CrossRef] [PubMed]
- Miyata, S.; Haneda, T. Hypertrophic growth of cultured neonatal rat heart cells mediated by type 1 angiotensin II receptor. Am. J. Physiol. Circ. Physiol. 1994, 266, H2443–H2451. [Google Scholar] [CrossRef]
- De Mello, W.C.; Danser, A.H.J. Angiotensin II and the heart: On the intracrine renin-angiotensin system. Hypertension 2000, 35, 1183–1188. [Google Scholar] [CrossRef] [Green Version]
- Schlüter, K.-D.; Wenzel, S. Angiotensin II: A hormone involved in and contributing to pro-hypertrophic cardiac networks and target of anti-hypertrophic cross-talks. Pharmacol. Ther. 2008, 119, 311–325. [Google Scholar] [CrossRef]
- Baker, K.M.; Dostal, D.E. Angiotensin II stimulation of left ventricular hypertrophy in adult rat heart: Mediation by the AT1 receptor. Am. J. Hypertens. 1992, 5, 276–280. [Google Scholar] [CrossRef]
- Sadoshima, J.; Izumo, S. Molecular Characterization of Angiotensin II- Induced Hypertrophy of Cardiac Myocytes and Hyperplasia of Cardiac Fibroblasts. Circ. Res. 1993, 73, 413–423. [Google Scholar] [CrossRef] [Green Version]
- Diaz, L.S.P.; Schuman, M.L.; Aisicovich, M.; Toblli, J.E.; Pirola, C.J.; Landa, M.S.; García, S.I. Angiotensin II requires an intact cardiac thyrotropin-releasing hormone (TRH) system to induce cardiac hypertrophy in mouse. J. Mol. Cell. Cardiol. 2018, 124, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sadoshima, J.; Izumo, S. Signal transduction pathways of angiotensin II--induced c-fos gene expression in cardiac myocytes in vitro. Roles of phospholipid-derived second messengers. Circ. Res. 1993, 73, 424–438. [Google Scholar] [CrossRef] [PubMed]
- Das, D.K.; Maulik, N.; Engelam, R.M. Redox regulation of angiotensin II siganling in the heart. J. Cell. Mol. Med. 2004, 8, 144–152. [Google Scholar] [CrossRef] [PubMed]
- Baker, K.M.; Campanile, C.P.; Trachte, G.J.; Peach, M.J. Identification and characterization of the rabbit angiotensin II myocardial receptor. Circ. Res. 1984, 54, 286–293. [Google Scholar] [CrossRef] [Green Version]
- Hunyady, L.; Catt, K.J. Pleiotropic AT1 receptor signaling pathways mediating physiological and pathogenic actions of angiotensin II. Mol. Endocrinol. 2006, 20, 953–970. [Google Scholar] [CrossRef]
- Gavras, I.; Gavras, H. Angiotensin II as a cardiovascular risk factor. J. Hum. Hypertens. 2002, 16, S2–S6. [Google Scholar] [CrossRef]
- Bhullar, S.K.; Shah, A.K.; Dhalla, N.S. Mechanisms for the development of heart failure and improvement of cardiac function by angiotensin-converting enzyme inhibitors. Scr. Med. 2022, 53, 51–76. [Google Scholar] [CrossRef]
- Sriramula, S.; Haque, M.; Majid, D.S.A.; Francis, J. Involvement of tumor necrosis factor-α in angiotensin II–mediated effects on salt appetite, hypertension, and cardiac hypertrophy. Hypertension 2008, 51, 1345–1351. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.-W.; Zheng, R.-H.; Bai, F.; Sturdivant, K.; Wang, N.-P.; James, E.A.; Bose, H.S.; Zhao, Z.-Q. Steroidogenic acute regulatory protein/aldosterone synthase mediates angiotensin II-induced cardiac fibrosis and hypertrophy. Mol. Biol. Rep. 2020, 47, 1207–1222. [Google Scholar] [CrossRef]
- Matsubara, H. Pathophysiological role of angiotensin II type 2 receptor in cardiovascular and renal diseases. Circ. Res. 1998, 83, 1182–1191. [Google Scholar] [CrossRef]
- Dhalla, N.S.; Heyliger, C.E.; Beamish, R.E.; Innes, I.R. Pathophysiological aspects of myocardial hypertrophy. Can. J. Cardiol. 1987, 3, 183. [Google Scholar] [PubMed]
- Frey, N.; Olson, E.N. Cardiac hypertrophy: The good, the bad, and the ugly. Annu. Rev. Physiol. 2003, 65, 45–79. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, I.; Minamino, T. Physiological and pathological cardiac hypertrophy. J. Mol. Cell. Cardiol. 2016, 97, 245–262. [Google Scholar] [CrossRef]
- Esposito, G.; Rapacciuolo, A.; Naga Prasad, S.V.; Takaoka, H.; Thomas, S.A.; Koch, W.J.; Rockman, H.A. Genetic alterations that inhibit in vivo pressure-overload hypertrophy prevent cardiac dysfunction despite increased wall stress. Circulation 2002, 105, 85–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maillet, M.; Van Berlo, J.H.; Molkentin, J.D. Molecular basis of physiological heart growth: Fundamental concepts and new players. Nat. Rev. Mol. Cell. Biol. 2013, 14, 38–48. [Google Scholar] [CrossRef] [Green Version]
- Bernardo, B.C.; Weeks, K.L.; Pretorius, L.; McMullen, J.R. Molecular distinction between physiological and pathological cardiac hypertrophy: Experimental findings and therapeutic strategies. Pharmacol. Ther. 2010, 128, 191–227. [Google Scholar] [CrossRef]
- Balakumar, P.; Singh, M. Possible role of poly (ADP-ribose) polymerase in pathological and physiological cardiac hypertrophy. Methods Find. Exp. Clin. Pharmacol. 2006, 28, 683–690. [Google Scholar] [CrossRef]
- Oldfield, C.J.; Duhamel, T.A.; Dhalla, N.S. Mechanisms for the transition from physiological to pathological cardiac hypertrophy. Can. J. Physiol. Pharmacol. 2020, 98, 74–84. [Google Scholar] [CrossRef] [Green Version]
- Sugden, P.H.; Clerk, A. Cellular mechanisms of cardiac hypertrophy. J. Mol. Med. 1998, 76, 725–746. [Google Scholar] [CrossRef] [Green Version]
- Tham, Y.K.; Bernardo, B.C.; Ooi, J.Y.Y.; Weeks, K.L.; McMullen, J.R. Pathophysiology of cardiac hypertrophy and heart failure: Signaling pathways and novel therapeutic targets. Arch. Toxicol. 2015, 89, 1401–1438. [Google Scholar] [CrossRef]
- Simões e Silva, A.C.; Silveira, K.D.; Ferreira, A.J.; Teixeira, M.M. ACE2, angiotensin-(1-7) and Mas receptor axis in inflammation and fibrosis. Br. J. Pharmacol. 2013, 169, 477–492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santos, R.A.S.; Sampaio, W.O.; Alzamora, A.C.; Motta-Santos, D.; Alenina, N.; Bader, M.; Campagnole-Santos, M.J. The ACE2/angiotensin-(1–7)/MAS axis of the renin-angiotensin system: Focus on angiotensin-(1–7). Physiol. Rev. 2018, 98, 505–553. [Google Scholar] [CrossRef] [PubMed]
- Mercure, C.; Yogi, A.; Callera, G.E.; Aranha, A.B.; Bader, M.; Ferreira, A.J.; Santos, R.A.S.; Walther, T.; Touyz, R.M.; Reudelhuber, T.L. Angiotensin (1-7) blunts hypertensive cardiac remodeling by a direct effect on the heart. Circ. Res. 2008, 103, 1319–1326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steckelings, U.M.; de Kloet, A.; Sumners, C. Centrally mediated cardiovascular actions of the angiotensin II type 2 receptor. Trends Endocrinol. Metab. 2017, 28, 684–693. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Li, X.; Yuan, H. Angiotensin II type-2 receptor-specific effects on the cardiovascular system. Cardiovasc. Diagn. Ther. 2012, 2, 56. [Google Scholar] [PubMed]
- Sandberg, K. Structural analysis and regulation of angiotensin II receptors. Trends Endocrinol. Metab. 1994, 5, 28–35. [Google Scholar] [CrossRef]
- De Gasparo, M.; Catt, K.J.; Inagami, T.; Wright, J.W.; Unger, T.H. International union of pharmacology. XXIII. The angiotensin II receptors. Pharmacol. Rev. 2000, 52, 415–472. [Google Scholar]
- Hao, J.; Wang, B.; Jones, S.C.; Jassal, D.S.; Dixon, I.M.C. Interaction between angiotensin II and Smad proteins in fibroblasts in failing heart and in vitro. Am. J. Physiol. Circ. Physiol. 2000, 279, H3020–H3030. [Google Scholar] [CrossRef] [Green Version]
- Wollert, K.C.; Drexler, H. The renin–angiotensin system and experimental heart failure. Cardiovasc. Res. 1999, 43, 838–849. [Google Scholar] [CrossRef] [Green Version]
- Verma, K.; Pant, M.; Paliwal, S.; Dwivedi, J.; Sharma, S. An insight on multicentric signaling of angiotensin II in cardiovascular system: A recent update. Front. Pharmacol. 2021, 12, 734917. [Google Scholar] [CrossRef]
- Prisant, L.M. Management of hypertension in patients with cardiac disease: Use of renin-angiotensin blocking agents. Am. J. Med. 2008, 121, S8–S15. [Google Scholar] [CrossRef] [PubMed]
- Escobar, C.; Barrios, V.; Calderón, A.; Barrios, S.; Echarri, R.; Navarro-Cid, J.; Ferrer, E.; Fernandez, R. Electrocardiographic left ventricular hypertrophy regression induced by an angiotensin receptor blocker-based regimen in hypertensive patients with the metabolic syndrome: Data from the SARA study. J. Clin. Hypertens. 2008, 10, 208–214. [Google Scholar] [CrossRef] [PubMed]
- Okin, P.M.; Devereux, R.B.; Jern, S.; Kjeldsen, S.E.; Julius, S.; Nieminen, M.S.; Snapinn, S.; Harris, K.E.; Aurup, P.; Edelman, J.M. Regression of electrocardiographic left ventricular hypertrophy during antihypertensive treatment and the prediction of major cardiovascular events. JAMA 2004, 292, 2343–2349. [Google Scholar] [CrossRef] [PubMed]
- Dasgupta, C.; Zhang, L. Angiotensin II receptors and drug discovery in cardiovascular disease. Drug Discov. Today 2011, 16, 22–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luedde, M.; Katus, H.A.; Frey, N. Novel molecular targets in the treatment of cardiac hypertrophy. Recent Pat. Cardiovasc. Drug Discov. 2006, 1, 1–20. [Google Scholar] [CrossRef]
- Gasc, J.-M.; Shanmugam, S.; Sibony, M.; Corvol, P. Tissue-specific expression of type 1 angiotensin II receptor subtypes. An insitu hybridization study. Hypertension 1994, 24, 531–537. [Google Scholar] [CrossRef] [Green Version]
- Bhullar, S.K.; Shah, A.K.; Dhalla, N.S. Role of angiotensin II in the development of subcellular remodeling in heart failure. Explor. Med. 2021, 2, 352–371. [Google Scholar] [CrossRef]
- Santos, R.A.S.; Simoes e Silva, A.C.; Maric, C.; Silva, D.M.R.; Machado, R.P.; De Buhr, I.; Heringer-Walther, S.; Pinheiro, S.V.B.; Lopes, M.T.; Bader, M.; et al. Angiotensin-(1-7) is an endogenous ligand for the G protein-coupled receptor Mas. Proc. Natl. Acad. Sci. USA 2003, 100, 8258–8263. [Google Scholar] [CrossRef] [Green Version]
- Sadoshima, J. Versatility of the angiotensin II type 1 receptor. Circ. Res. 1998, 82, 1352–1355. [Google Scholar] [CrossRef]
- Van Kesteren, C.A.M.; Van Heugten, H.A.A.; Lamers, J.M.J.; Saxena, P.R.; Schalekamp, M.; Danser, A.H.J. Angiotensin II-mediated growth and antigrowth effects in cultured neonatal rat cardiac myocytes and fibroblasts. J. Mol. Cell. Cardiol. 1997, 29, 2147–2157. [Google Scholar] [CrossRef]
- Shah, A.K.; Bhullar, S.K.; Elimban, V.; Dhalla, N.S. Oxidative stress as a mechanism for functional alterations in cardiac hypertrophy and heart failure. Antioxidants 2021, 10, 931. [Google Scholar] [CrossRef]
- Zhai, P.; Galeotti, J.; Liu, J.; Holle, E.; Yu, X.; Wagner, T.; Sadoshima, J. An angiotensin II type 1 receptor mutant lacking epidermal growth factor receptor transactivation does not induce angiotensin II–mediated cardiac hypertrophy. Circ. Res. 2006, 99, 528–536. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wang, S.; Wang, C.; Song, H.; Han, H.; Hang, P.; Jiang, Y.; Wei, L.; Huo, R.; Sun, L. Upregulation of M3 muscarinic receptor inhibits cardiac hypertrophy induced by angiotensin II. J. Transl. Med. 2013, 11, 209. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.Y.; Kuo, W.W.; Yeh, Y.L.; Ho, T.J.; Lin, J.Y.; Lin, D.Y.; Chu, C.H.; Tsai, F.J.; Tsai, C.H. ANG II promotes IGF-IIR expression and cardiomyocyte apoptosis by inhibiting HSF1 via JNK activation and SIRT1 degradation. Cell Death Differ. 2014, 21, 1262–1274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, L.; Ma, B.; Han, X. The role of autophagy in angiotensin II-induced pathological cardiac hypertrophy. J. Mol. Endocrinol. 2016, 57, R143–R152. [Google Scholar] [CrossRef] [Green Version]
- Wenzel, S.; Abdallah, Y.; Helmig, S.; Schäfer, C.; Piper, H.M.; Schlüter, K.-D. Contribution of PI 3-kinase isoforms to angiotensin II-and α-adrenoceptor-mediated signalling pathways in cardiomyocytes. Cardiovasc. Res. 2006, 71, 352–362. [Google Scholar] [CrossRef]
- Zhong, T.; Wang, Z.; Niloy, S.I.; Shen, Y.; O’Rourke, S.T.; Sun, C. Role of PI3-kinase in angiotensin induced cardiac hypertrophy: Class I Versus Class III. Front. Pharmacol. 2021, 12, 103. [Google Scholar] [CrossRef]
- Blankesteijn, W.M.; van de Schans, V.A.M.; ter Horst, P.; Smits, J.F.M. The Wnt/frizzled/GSK-3β pathway: A novel therapeutic target for cardiac hypertrophy. Trends Pharmacol. Sci. 2008, 29, 175–180. [Google Scholar] [CrossRef]
- Griendling, K.K.; Ushio-Fukai, M.; Lassègue, B.; Alexander, R.W. Angiotensin II signaling in vascular smooth muscle: New concepts. Hypertension 1997, 29, 366–370. [Google Scholar] [CrossRef]
- Miura, S.-I.; Saku, K.; Karnik, S.S. Molecular analysis of the structure and function of the angiotensin II type 1 receptor. Hypertens. Res. 2003, 26, 937–943. [Google Scholar] [CrossRef] [Green Version]
- Aikawa, R.; Komuro, I.; Yamazaki, T.; Zou, Y.; Kudoh, S.; Zhu, W.; Kadowaki, T.; Yazaki, Y. Rho family small G proteins play critical roles in mechanical stress–induced hypertrophic responses in cardiac myocytes. Circ. Res. 1999, 84, 458–466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forrester, S.J.; Booz, G.W.; Sigmund, C.D.; Coffman, T.M. Angiotensin II signal transduction: An update on mechanisms of physiology and pathophysiology. Physiol. Rev. 2018, 98, 1627–1738. [Google Scholar] [CrossRef] [PubMed]
- Touyz, R.M.; Schiffrin, E.L. Signal transduction mechanisms mediating the physiological and pathophysiological actions of angiotensin II in vascular smooth muscle cells. Pharmacol. Rev. 2000, 52, 639–672. [Google Scholar] [PubMed]
- Zhai, P.; Yamamoto, M.; Galeotti, J.; Liu, J.; Masurekar, M.; Thaisz, J.; Irie, K.; Holle, E.; Yu, X.; Kupershmidt, S. Cardiac-specific overexpression of AT1 receptor mutant lacking Gαq/Gαi coupling causes hypertrophy and bradycardia in transgenic mice. J. Clin. Investig. 2005, 115, 3045–3056. [Google Scholar] [CrossRef] [Green Version]
- Higuchi, S.; Ohtsu, H.; Suzuki, H.; Shirai, H.; Frank, G.D.; Eguchi, S. Angiotensin II signal transduction through the AT1 receptor: Novel insights into mechanisms and pathophysiology. Clin. Sci. 2007, 112, 417–428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, H.; Motley, E.D.; Frank, G.D.; Utsunomiya, H.; Eguchi, S. Recent progress in signal transduction research of the angiotensin II type-1 receptor: Protein kinases, vascular dysfunction and structural requirement. Curr. Med. Chem. Hematol. Agents 2005, 3, 305–322. [Google Scholar] [CrossRef]
- Rohini, A.; Agrawal, N.; Koyani, C.N.; Singh, R. Molecular targets and regulators of cardiac hypertrophy. Pharmacol. Res. 2010, 61, 269–280. [Google Scholar] [CrossRef] [PubMed]
- Sopontammarak, S.; Aliharoob, A.; Ocampo, C.; Arcilla, R.A.; Gupta, M.P.; Gupta, M. Mitogen-activated protein kinases (p38 and c-Jun NH2-terminal kinase) are differentially regulated during cardiac volume and pressure overload hypertrophy. Cell Biochem. Biophys. 2005, 43, 61–76. [Google Scholar] [CrossRef]
- Miura, S.; Zhang, J.; Matsuo, Y.; Saku, K.; Karnik, S.S. Activation of extracellular signal-activated kinase by angiotensin II-induced Gq-independent epidermal growth factor receptor transactivation. Hypertens. Res. 2004, 27, 765–770. [Google Scholar] [CrossRef] [Green Version]
- Sadoshima, J.; Qiu, Z.; Morgan, J.P.; Izumo, S. Angiotensin II and other hypertrophic stimuli mediated by G-protein–coupled receptors activate tyrosine kinase, mitogen-activated protein kinase, and 90-kd s6 kinase in cardiac myocytes: The critical role of Ca2+-dependent signaling. Circ. Res. 1995, 76, 1–15. [Google Scholar] [CrossRef]
- Takeishi, Y.; Huang, Q.; Abe, J.; Glassman, M.; Che, W.; Lee, J.-D.; Kawakatsu, H.; Lawrence, E.G.; Hoit, B.D.; Berk, B.C. Src and multiple MAP kinase activation in cardiac hypertrophy and congestive heart failure under chronic pressure-overload: Comparison with acute mechanical stretch. J. Mol. Cell. Cardiol. 2001, 33, 1637–1648. [Google Scholar] [CrossRef] [PubMed]
- Purcell, N.H.; Wilkins, B.J.; York, A.; Saba-El-Leil, M.K.; Meloche, S.; Robbins, J.; Molkentin, J.D. Genetic inhibition of cardiac ERK1/2 promotes stress-induced apoptosis and heart failure but has no effect on hypertrophy in vivo. Proc. Natl. Acad. Sci. USA 2007, 104, 14074–14079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, D.; Li, Z.; Bao, P.; Chen, M.; Zhang, M.; Yan, F.; Xu, Y.; Ji, C.; Hu, X.; Sanchis, D. Nrf2 deficiency aggravates Angiotensin II-induced cardiac injury by increasing hypertrophy and enhancing IL-6/STAT3-dependent inflammation. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2019, 1865, 1253–1264. [Google Scholar] [CrossRef]
- Dorn, G.W.; Force, T. Protein kinase cascades in the regulation of cardiac hypertrophy. J. Clin. Investig. 2005, 115, 527–537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molkentin, J.D.; Dorn II, G.W. Cytoplasmic signaling pathways that regulate cardiac hypertrophy. Annu. Rev. Physiol. 2001, 63, 391–426. [Google Scholar] [CrossRef]
- Li, N.; Wang, H.-X.; Han, Q.-Y.; Li, W.-J.; Zhang, Y.-L.; Du, J.; Xia, Y.-L.; Li, H.-H. Activation of the cardiac proteasome promotes angiotension II-induced hypertrophy by down-regulation of ATRAP. J. Mol. Cell. Cardiol. 2015, 79, 303–314. [Google Scholar] [CrossRef]
- Mori, J.; Basu, R.; McLean, B.A.; Das, S.K.; Zhang, L.; Patel, V.B.; Wagg, C.S.; Kassiri, Z.; Lopaschuk, G.D.; Oudit, G.Y. Agonist-induced hypertrophy and diastolic dysfunction are associated with selective reduction in glucose oxidation: A metabolic contribution to heart failure with normal ejection fraction. Circ. Heart. Fail. 2012, 5, 493–503. [Google Scholar] [CrossRef] [Green Version]
- Foäldes, G.; Vajda, S.; Lakoé-Futoé, Z.; Saérmaén, B.; Skoumal, R.; Ilves, M.; Dechâtel, R.; Karaédi, I.; Toéth, M.; Ruskoaho, H. Distinct modulation of angiotensin II-induced early left ventricular hypertrophic gene programming by dietary fat type. J. Lipid Res. 2006, 47, 1219–1226. [Google Scholar] [CrossRef] [Green Version]
- Lin, L.; Wang, H.-X.; Han, Q.-Y.; Li, W.-J.; Zhang, Y.-L.; Du, J.; Xia, Y.-L.; Li, H.-H. High-density lipoprotein inhibits mechanical stress-induced cardiomyocyte autophagy and cardiac hypertrophy through angiotensin II type 1 receptor-mediated PI 3K/Akt pathway. J. Cell. Mol. Med. 2015, 19, 1929–1938. [Google Scholar] [CrossRef]
- Thomas, W.G.; Brandenburger, Y.; Autelitano, D.J.; Pham, T.; Qian, H.; Hannan, R.D. Adenoviral-directed expression of the type 1A angiotensin receptor promotes cardiomyocyte hypertrophy via transactivation of the epidermal growth factor receptor. Circ. Res. 2002, 90, 135–142. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.Y.; Lee, F.; Peng, S.; Lin, K.; Chen, R.; Ho, T.; Tsai, F.; Padma, V.V.; Kuo, W.; Huang, C. HSF1 phosphorylation by ERK/GSK3 suppresses RNF126 to sustain IGF-IIR expression for hypertension-induced cardiomyocyte hypertrophy. J. Cell. Physiol. 2018, 233, 979–989. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.; Yang, R.; Wang, M.; Liu, J.; Wang, Y.; Zhang, H.; Li, Y. Angiotensin II type-1 receptor-JAK/STAT pathway mediates the induction of visfatin in angiotensin II-induced cardiomyocyte hypertrophy. Am. J. Med. Sci. 2012, 343, 220–226. [Google Scholar] [CrossRef] [PubMed]
- Shanmugam, P.; Valente, A.J.; Prabhu, S.D.; Venkatesan, B.; Yoshida, T.; Delafontaine, P.; Chandrasekar, B. Angiotensin-II type 1 receptor and NOX2 mediate TCF/LEF and CREB dependent WISP1 induction and cardiomyocyte hypertrophy. J. Mol. Cell. Cardiol. 2011, 50, 928–938. [Google Scholar] [CrossRef]
- Bendall, J.K.; Cave, A.C.; Heymes, C.; Gall, N.; Shah, A.M. Pivotal role of a gp91phox-containing NADPH oxidase in angiotensin II-induced cardiac hypertrophy in mice. Circulation 2002, 105, 293–296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, M.Y.; Griendling, K.K. Redox signaling, vascular function, and hypertension. Antioxid. Redox Signal. 2008, 10, 1045–1059. [Google Scholar] [CrossRef] [PubMed]
- Gupta, M.; Singal, P.K. Higher antioxidative capacity during a chronic stable heart hypertrophy. Circ. Res. 1989, 64, 398–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sag, C.M.; Santos, C.X.C.; Shah, A.M. Redox regulation of cardiac hypertrophy. J. Mol. Cell. Cardiol. 2014, 73, 103–111. [Google Scholar] [CrossRef] [Green Version]
- Burgoyne, J.R.; Mongue-Din, H.; Eaton, P.; Shah, A.M. Redox signaling in cardiac physiology and pathology. Circ. Res. 2012, 111, 1091–1106. [Google Scholar] [CrossRef]
- Madamanchi, N.R.; Runge, M.S. Redox signaling in cardiovascular health and disease. Free Radic. Biol. Med. 2013, 61, 3–501. [Google Scholar] [CrossRef] [Green Version]
- Maejima, Y.; Kuroda, J.; Matsushima, S.; Ago, T.; Sadoshima, J. Regulation of myocardial growth and death by NADPH oxidase. J. Mol. Cell. Cardiol. 2011, 50, 408–416. [Google Scholar] [CrossRef] [Green Version]
- Murdoch, C.E.; Zhang, M.; Cave, A.C.; Shah, A.M. NADPH oxidase-dependent redox signalling in cardiac hypertrophy, remodelling and failure. Cardiovasc. Res. 2006, 71, 208–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.-M.; Gall, N.P.; Grieve, D.J.; Chen, M.; Shah, A.M. Activation of NADPH oxidase during progression of cardiac hypertrophy to failure. Hypertension 2002, 40, 477–484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sirker, A.; Zhang, M.; Shah, A.M. NADPH oxidases in cardiovascular disease: Insights from in vivo models and clinical studies. Basic Res. Cardiol. 2011, 106, 735–747. [Google Scholar] [CrossRef] [PubMed]
- Griendling, K.K.; Sorescu, D.; Ushio-Fukai, M. NAD(P)H oxidase: Role in cardiovascular biology and disease. Circ. Res. 2000, 86, 494–501. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Murugesan, P.; Huang, K.; Cai, H. NADPH oxidases and oxidase crosstalk in cardiovascular diseases: Novel therapeutic targets. Nat. Rev. Cardiol. 2020, 17, 170–194. [Google Scholar] [CrossRef]
- Sadoshima Lassègue, B.; San Martín, A.; Griendling, K.K. Biochemistry, physiology, and pathophysiology of NADPH oxidases in the cardiovascular system. Circ. Res. 2012, 110, 1364–1390. [Google Scholar] [CrossRef]
- Brown, D.I.; Griendling, K.K. Regulation of signal transduction by reactive oxygen species in the cardiovascular system. Circ. Res. 2015, 116, 531–549. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Q.D.; Viswanadhapalli, S.; Williams, P.; Shi, Q.; Tan, C.; Yi, X.; Bhandari, B.; Abboud, H.E. NADPH oxidase 4 induces cardiac fibrosis and hypertrophy through activating Akt/mTOR and NFκB signaling pathways. Circulation 2015, 131, 643–655. [Google Scholar] [CrossRef] [Green Version]
- Ago, T.; Sadoshima, J. From contractile enhancement to pathological hypertrophy: Angiotensin II–induced Nox2-mediated reactive oxygen species. J. Am. Coll. Cardiol. 2015, 66, 273–277. [Google Scholar] [CrossRef] [Green Version]
- Hingtgen, S.D.; Tian, X.; Yang, J.; Dunlay, S.M.; Peek, A.S.; Wu, Y.; Sharma, R.V.; Engelhardt, J.F.; Davisson, R.L. Nox2-containing NADPH oxidase and Akt activation play a key role in angiotensin II-induced cardiomyocyte hypertrophy. Physiol. Genom. 2006, 26, 180–191. [Google Scholar] [CrossRef] [Green Version]
- Looi, Y.H.; Grieve, D.J.; Siva, A.; Walker, S.J.; Anilkumar, N.; Cave, A.C.; Marber, M.; Monaghan, M.J.; Shah, A.M. Involvement of NOX2 NADPH oxidase in adverse cardiac remodeling after myocardial infarction. Hypertension 2008, 51, 319–325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bánfi, B.; Tirone, F.; Durussel, I.; Knisz, J.; Moskwa, P.; Molnár, G.Z.; Krause, K.-H.; Cox, J.A. Mechanism of Ca2+ activation of the NADPH oxidase 5 (NOX5). J. Biol. Chem. 2004, 279, 18583–18591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Touyz, R.M.; Anagnostopoulou, A.; Rios, F.; Montezano, A.C.; Camargo, L.L. NOX5: Molecular biology and pathophysiology. Exp. Physiol. 2019, 104, 605–616. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.-J.; Zhao, C.-L.; Ouyang, S.; Deng, K.-Q.; Zhu, L.; Montezano, A.C.; Zhang, C.; Hu, F.; Zhu, X.-Y.; Tian, S. Ca2+-dependent NOX5 (NADPH Oxidase 5) exaggerates cardiac hypertrophy through reactive oxygen species production. Hypertension 2020, 76, 827–838. [Google Scholar] [CrossRef] [PubMed]
- Doughan, A.K.; Harrison, D.G.; Dikalov, S.I. Molecular mechanisms of angiotensin II–mediated mitochondrial dysfunction: Linking mitochondrial oxidative damage and vascular endothelial dysfunction. Circ. Res. 2008, 102, 488–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, J.-X.; Yin, J.-X.; Yang, R.-F.; Li, S.; Renshaw, A.O.; Li, Y.-L.; Schultz, H.D.; Zimmerman, M.C. Mitochondria-produced superoxide mediates angiotensin II-induced inhibition of neuronal potassium current. Am. J. Physiol. Physiol. 2010, 298, C857–C865. [Google Scholar] [CrossRef] [Green Version]
- Ide, T.; Tsutsui, H.; Hayashidani, S.; Kang, D.; Suematsu, N.; Nakamura, K.; Utsumi, H.; Hamasaki, N.; Takeshita, A. Mitochondrial DNA damage and dysfunction associated with oxidative stress in failing hearts after myocardial infarction. Circ. Res. 2001, 88, 529–535. [Google Scholar] [CrossRef] [Green Version]
- Zinkevich, N.S.; Gutterman, D.D. ROS-induced ROS release in vascular biology: Redox-redox signaling. Am. J. Physiol. Circ. Physiol. 2011, 301, H647–H653. [Google Scholar] [CrossRef] [Green Version]
- Dai, D.-F.; Johnson, S.C.; Villarin, J.J.; Chin, M.T.; Nieves-Cintrón, M.; Chen, T.; Marcinek, D.J.; Dorn, G.W.; Kang, Y.J.; Prolla, T.A. Mitochondrial oxidative stress mediates angiotensin II–induced cardiac hypertrophy and Gαq overexpression–induced heart failure. Circ. Res. 2011, 108, 837–846. [Google Scholar] [CrossRef] [Green Version]
- Maack, C.; Böhm, M. Targeting mitochondrial oxidative stress in heart failure: Throttling the afterburner. J. Am. Coll. Cardiol. 2011, 58, 83–86. [Google Scholar] [CrossRef] [Green Version]
- Dikalov, S.I.; Nazarewicz, R.R.; Bikineyeva, A.; Hilenski, L.; Lassegue, B.; Griendling, K.K.; Harrison, D.G.; Dikalova, A.E. Nox2-induced production of mitochondrial superoxide in angiotensin II-mediated endothelial oxidative stress and hypertension. Antioxid. Redox Signal. 2014, 20, 281–294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zimmerman, M.C.; Sharma, R.V.; Davisson, R.L. Superoxide mediates angiotensin II–induced influx of extracellular calcium in neural cells. Hypertension 2005, 45, 717–723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, Y.S.; de Mattos, A.B.M.; Shao, D.; Li, T.; Nabben, M.; Kim, M.; Wang, W.; Tian, R.; Kolwicz Jr, S.C. Preservation of myocardial fatty acid oxidation prevents diastolic dysfunction in mice subjected to angiotensin II infusion. J. Mol. Cell. Cardiol. 2016, 100, 64–71. [Google Scholar] [CrossRef] [PubMed]
- Shiomi, T.; Tsutsui, H.; Matsusaka, H.; Murakami, K.; Hayashidani, S.; Ikeuchi, M.; Wen, J.; Kubota, T.; Utsumi, H.; Takeshita, A. Overexpression of glutathione peroxidase prevents left ventricular remodeling and failure after myocardial infarction in mice. Circulation 2004, 109, 544–549. [Google Scholar] [CrossRef] [Green Version]
- Schreckenberg, R.; Rebelo, M.; Deten, A.; Weber, M.; Rohrbach, S.; Pipicz, M.; Csonka, C.; Ferdinandy, P.; Schulz, R.; Schlüter, K.-D. Specific mechanisms underlying right heart failure: The missing upregulation of superoxide dismutase-2 and its decisive role in antioxidative defense. Antioxid. Redox Signal. 2015, 23, 1220–1232. [Google Scholar] [CrossRef]
- Seddon, M.; Looi, Y.H.; Shah, A.M. Oxidative stress and redox signalling in cardiac hypertrophy and heart failure. Heart 2007, 93, 903–907. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Ichikawa, T.; Villacorta, L.; Janicki, J.S.; Brower, G.L.; Yamamoto, M.; Cui, T. Nrf2 protects against maladaptive cardiac responses to hemodynamic stress. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 1843–1850. [Google Scholar] [CrossRef]
- Li, J.; Zhang, C.; Xing, Y.; Janicki, J.S.; Yamamoto, M.; Wang, X.L.; Tang, D.-Q.; Cui, T. Up-regulation of p27kip1 contributes to Nrf2-mediated protection against angiotensin II-induced cardiac hypertrophy. Cardiovasc. Res. 2011, 90, 315–324. [Google Scholar] [CrossRef]
- Vashi, R.; Patel, B.M. NRF2 in cardiovascular diseases: A ray of hope! J. Cardiovasc. Transl. Res. 2021, 14, 573–586. [Google Scholar] [CrossRef]
- Li, J.; Ichikawa, T.; Janicki, J.S.; Cui, T. Targeting the Nrf2 pathway against cardiovascular disease. Expert Opin. Ther. Targets 2009, 13, 785–794. [Google Scholar] [CrossRef]
- Ma, Q. Role of Nrf2 in oxidative stress and toxicity. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 401–426. [Google Scholar] [CrossRef] [Green Version]
- Nie, P.; Meng, F.; Zhang, J.; Wei, X.; Shen, C. Astragaloside IV exerts a myocardial protective effect against cardiac hypertrophy in rats, partially via activating the Nrf2/HO-1 signaling pathway. Oxid. Med. Cell. Longev. 2019, 2019, 4625912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castoldi, G.; di Gioia, C.R.T.; Roma, F.; Carletti, R.; Manzoni, G.; Stella, A.; Zerbini, G.; Perseghin, G. Activation of angiotensin type 2 (AT2) receptors prevents myocardial hypertrophy in Zucker diabetic fatty rats. Acta Diabetol. 2019, 56, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Matavelli, L.C.; Siragy, H.M. AT2 receptor activities and pathophysiological implications. J. Cardiovasc. Pharmacol. 2015, 65, 226–232. [Google Scholar] [CrossRef] [Green Version]
- Bartunek, J.; Weinberg, E.O.; Tajima, M.; Rohrbach, S.; Lorell, B.H. Angiotensin II type 2 receptor blockade amplifies the early signals of cardiac growth response to angiotensin II in hypertrophied hearts. Circulation 1999, 99, 22–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chow, B.S.M.; Allen, T.J. Angiotensin II type 2 receptor (AT2R) in renal and cardiovascular disease. Clin. Sci. 2016, 130, 1307–1326. [Google Scholar] [CrossRef]
- Padia, S.H.; Carey, R.M. AT2 receptors: Beneficial counter-regulatory role in cardiovascular and renal function. Pflugers Arch. Eur. J. Physiol. 2013, 465, 99–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sumners, C.; de Kloet, A.D.; Krause, E.G.; Unger, T.; Steckelings, U.M. Angiotensin type 2 receptors: Blood pressure regulation and end organ damage. Curr. Opin. Pharmacol. 2015, 21, 115–121. [Google Scholar] [CrossRef] [Green Version]
- Carneiro-Ramos, M.S.; Diniz, G.P.; Nadu, A.P.; Almeida, J.; Vieira, R.L.P.; Santos, R.A.S.; Barreto-Chaves, M.L.M. Blockade of angiotensin II type 2 receptor prevents thyroxine-mediated cardiac hypertrophy by blocking Akt activation. Basic Res. Cardiol. 2010, 105, 325–335. [Google Scholar] [CrossRef]
- Tsutsumi, Y.; Matsubara, H.; Masaki, H.; Kurihara, H.; Murasawa, S.; Takai, S.; Miyazaki, M.; Nozawa, Y.; Ozono, R.; Nakagawa, K. Angiotensin II type 2 receptor overexpression activates the vascular kinin system and causes vasodilation. J. Clin. Investig. 1999, 104, 925–935. [Google Scholar] [CrossRef] [Green Version]
- Nakajima, M.; Hutchinson, H.G.; Fujinaga, M.; Hayashida, W.; Morishita, R.; Zhang, L.; Horiuchi, M.; Pratt, R.E.; Dzau, V.J. The angiotensin II type 2 (AT2) receptor antagonizes the growth effects of the AT1 receptor: Gain-of-function study using gene transfer. Proc. Natl. Acad. Sci. USA. 1995, 92, 10663–10667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, H.; Yu, D.; Hu, Y.; Zhang, P.; Yang, Y.; Hu, Q.; Li, M. Angiotensin II upregulates cyclophilin A by enhancing ROS production in rat cardiomyocytes. Mol. Med. Rep. 2018, 18, 4349–4355. [Google Scholar] [CrossRef] [Green Version]
- Santos, R.A.S.; Ferreira, A.J.; Verano-Braga, T.; Bader, M. Angiotensin-converting enzyme 2, angiotensin-(1-7) and Mas: New players of the renin-angiotensin system. J. Endocrinol. 2013, 216, R1–R17. [Google Scholar] [CrossRef] [PubMed]
- McKinney, C.A.; Fattah, C.; Loughrey, C.M.; Milligan, G.; Nicklin, S.A. Angiotensin-(1–7) and angiotensin-(1–9): Function in cardiac and vascular remodelling. Clin. Sci. 2014, 126, 815–827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chappell, M.C. The non-classical renin-angiotensin system and renal function. Compr. Physiol. 2012, 2, 2733. [Google Scholar] [PubMed] [Green Version]
- Liang, B.; Li, Y.; Han, Z.; Xue, J.; Zhang, Y.; Jia, S.; Wang, C. ACE2-Ang (1-7) axis is induced in pressure overloaded rat model. Int. J. Clin. Exp. Pathol. 2015, 8, 1443. [Google Scholar]
- Gomes, E.R.M.; Santos, R.A.S.; Guatimosim, S. Angiotensin-(1-7)-mediated signaling in cardiomyocytes. Int. J. Hypertens. 2012, 2012, 493129. [Google Scholar] [CrossRef] [Green Version]
- Jeuthe, S.; Dietrich, T.; Berger, F.; Kuehne, T.; Kozerke, S.; Messroghli, D.R. Closed-chest small animal model to study myocardial infarction in an MRI environment in real time. Int. J. Cardiovasc. Imaging 2015, 31, 115–121. [Google Scholar]
- Dias-Peixoto, M.F.; Santos, R.A.S.; Gomes, E.R.M.; Alves, M.N.M.; Almeida, P.W.M.; Greco, L.; Rosa, M.; Fauler, B.; Bader, M.; Alenina, N. Molecular mechanisms involved in the angiotensin-(1-7)/Mas signaling pathway in cardiomyocytes. Hypertension 2008, 52, 542–548. [Google Scholar] [CrossRef] [Green Version]
- Walther, T.; Falk, V.; Langebartels, G.; Krüger, M.; Bernhardt, U.; Diegeler, A.; Gummert, J.; Autschbach, R.; Mohr, F.W. Prospectively randomized evaluation of stentless versus conventional biological aortic valves: Impact on early regression of left ventricular hypertrophy. Circulation 1999, 100, 6–10. [Google Scholar] [CrossRef]
- Karnik, S.S.; Singh, K.D.; Tirupula, K.; Unal, H. Significance of angiotensin 1–7 coupling with MAS1 receptor and other GPCRs to the renin-angiotensin system: IUPHAR Review 22. Br. J. Pharmacol. 2017, 174, 737–753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, V.B.; Zhong, J.-C.; Grant, M.B.; Oudit, G.Y. Role of the ACE2/angiotensin 1–7 axis of the renin–angiotensin system in heart failure. Circ. Res. 2016, 118, 1313–1326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dahlöf, B.; Devereux, R.B.; Kjeldsen, S.E.; Julius, S.; Beevers, G.; de Faire, U.; Fyhrquist, F.; Ibsen, H.; Kristiansson, K.; Lederballe-Pedersen, O. Cardiovascular morbidity and mortality in the losartan intervention for endpoint reduction in hypertension study (LIFE): A randomised trial against atenolol. Lancet 2002, 359, 995–1003. [Google Scholar] [CrossRef]
- Tashiro, K.; Kuwano, T.; Ideishi, A.; Morita, H.; Idemoto, Y.; Goto, M.; Suematsu, Y.; Miura, S. sacubitril/valsartan inhibits cardiomyocyte hypertrophy in angiotensin II-anduced aypertensive mice independent of a blood pressure-lowering effect. Cardiol. Res. 2020, 11, 376. [Google Scholar] [CrossRef]
- Wang, X.; Ye, Y.; Gong, H.; Wu, J.; Yuan, J.; Wang, S.; Yin, P.; Ding, Z.; Kang, L.; Jiang, Q. The effects of different angiotensin II type 1 receptor blockers on the regulation of the ACE-AngII-AT1 and ACE2-Ang (1–7)-Mas axes in pressure overload-induced cardiac remodeling in male mice. J. Mol. Cell. Cardiol. 2016, 97, 180–190. [Google Scholar] [CrossRef]
- Diniz, G.P.; Carneiro-Ramos, M.S.; Barreto-Chaves, M.L.M. Angiotensin type 1 receptor mediates thyroid hormone-induced cardiomyocyte hypertrophy through the Akt/GSK-3β/mTOR signaling pathway. Basic Res. Cardiol. 2009, 104, 653–667. [Google Scholar] [CrossRef]
- Kang, B.-Y.; Khan, J.A.; Ryu, S.; Shekhar, R.; Seung, K.-B.; Mehta, J.L. Curcumin reduces angiotensin II-mediated cardiomyocyte growth via LOX-1 inhibition. J. Cardiovasc. Pharmacol. 2010, 55, 176–183. [Google Scholar] [CrossRef]
- Takano, A.P.C.; Senger, N.; Munhoz, C.D.; Barreto-Chaves, M.L.M. AT1 receptor blockage impairs NF-κB activation mediated by thyroid hormone in cardiomyocytes. Pflügers Arch. J. Physiol. 2018, 470, 549–558. [Google Scholar] [CrossRef]
- Lee, C.Y.; Park, H.K.; Lee, B.-S.; Jeong, S.; Hyun, S.; Choi, J.-W.; Kim, S.W.; Lee, S.; Lim, S.; Hwang, K.-C. Novel therapeutic effects of pterosin B on Ang II-induced cardiomyocyte hypertrophy. Molecules 2020, 25, 5279. [Google Scholar] [CrossRef]
- Zheng, R.-H.; Bai, X.-J.; Zhang, W.-W.; Wang, J.; Bai, F.; Yan, C.-P.; James, E.A.; Bose, H.S.; Wang, N.-P.; Zhao, Z.-Q. Liraglutide attenuates cardiac remodeling and improves heart function after abdominal aortic constriction through blocking angiotensin II type 1 receptor in rats. Drug Des. Devel. Ther. 2019, 13, 2745. [Google Scholar] [CrossRef] [Green Version]
- Sabharwal, N.K.; Swinburn, J.; Lahiri, A.; Senior, R. Effect of imidapril and nifedipine on left ventricular hypertrophy in untreated hypertension. Clin. Drug Investig. 2005, 25, 367–375. [Google Scholar] [CrossRef]
- Ferrari, R.; Pasanisi, G.; Notarstefano, P.; Campo, G.; Gardini, E.; Ceconi, C. Specific properties and effect of perindopril in controlling the renin–angiotensin system. Am. J. Hypertens. 2005, 18, 142S–154S. [Google Scholar] [CrossRef] [Green Version]
- Kongstad-Rasmussen, O.; Blomstrand, P.; Broqvist, M.; Dahlström, U.; Wranne, B. Treatment with ramipril improves systolic function even in patients with mild systolic dysfunction and symptoms of heart failure after acute myocardial infarction. Clin. Cardiol. 1998, 21, 807–811. [Google Scholar] [CrossRef]
- Anderson, V.R.; Perry, C.M.; Robinson, D.M. Ramipril. Am. J. Cardiovasc. Drugs 2006, 6, 417–432. [Google Scholar] [CrossRef] [PubMed]
- Pfeffer, J.M.; Pfeffer, M.A.; Braunwald, E. Hemodynamic benefits and prolonged survival with long-term captopril therapy in rats with myocardial infarction and heart failure. Circulation 1987, 75, I149-55. [Google Scholar]
- Cleary, J.D.; Taylor, J.W. Enalapril: A new angiotensin converting enzyme inhibitor. Drug Intell. Clin. Pharm. 1986, 20, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Gremmler, B.; Kunert, M.; Schleiting, H.; Ulbricht, L.J. Improvement of cardiac output in patients with severe heart failure by use of ACE-inhibitors combined with the AT1-antagonist eprosartan. Eur. J. Heart Fail 2000, 2, 183–187. [Google Scholar] [CrossRef] [Green Version]
- Touyz, R.M.; Briones, A.M. Reactive oxygen species and vascular biology: Implications in human hypertension. Hypertens. Res. 2011, 34, 5–14. [Google Scholar] [CrossRef] [Green Version]
- Takimoto, E.; Kass, D.A. Role of oxidative stress in cardiac hypertrophy and remodeling. Hypertension 2007, 49, 241–248. [Google Scholar] [CrossRef] [PubMed]
- Custodis, F.; Eberl, M.; Kilter, H.; Böhm, M.; Laufs, U. Association of RhoGDIα with Rac1 GTPase mediates free radical production during myocardial hypertrophy. Cardiovasc. Res. 2006, 71, 342–351. [Google Scholar] [CrossRef]
- Lyu, L.; Chen, J.; Wang, W.; Yan, T.; Lin, J.; Gao, H.; Li, H.; Lv, R.; Xu, F.; Fang, L. Scoparone alleviates Ang II-induced pathological myocardial hypertrophy in mice by inhibiting oxidative stress. J. Cell. Mol. Med. 2021, 25, 3136–3148. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.-M.; Chen, Y.-H.; Chiang, M.-T.; Chau, L.-Y. Heme oxygenase-1 inhibits angiotensin II-induced cardiac hypertrophy in vitro and in vivo. Circulation 2004, 110, 309–316. [Google Scholar] [CrossRef] [Green Version]
- Widder, J.D.; Fraccarollo, D.; Galuppo, P.; Hansen, J.M.; Jones, D.P.; Ertl, G.; Bauersachs, J. Attenuation of angiotensin II–induced vascular dysfunction and hypertension by overexpression of thioredoxin 2. Hypertension 2009, 54, 338–344. [Google Scholar] [CrossRef] [PubMed]
- Rosenbaugh, E.G.; Savalia, K.K.; Manickam, D.S.; Zimmerman, M.C. Antioxidant-based therapies for angiotensin II-associated cardiovascular diseases. Am. J. Physiol. Integr. Comp. Physiol. 2013, 304, R917–R928. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Puyol, M.; Griera-Merino, M.; Perez-Rivero, G.; Diez-Marques, M.L.; Ruiz-Torres, M.P.; Rodriguez-Puyol, D. Angiotensin II induces a rapid and transient increase of reactive oxygen species. Antioxid. Redox Signal. 2002, 4, 869–875. [Google Scholar] [CrossRef] [PubMed]
- Hanna, I.R.; Taniyama, Y.; Szöcs, K.; Rocic, P.; Griendling, K.K. NAD (P) H oxidase-derived reactive oxygen species as mediators of angiotensin II signaling. Antioxid. Redox Signal. 2002, 4, 899–914. [Google Scholar] [CrossRef]
- Higuchi, Y.; Otsu, K.; Nishida, K.; Hirotani, S.; Nakayama, H.; Yamaguchi, O.; Hikoso, S.; Kashiwase, K.; Takeda, T.; Watanabe, T. The small GTP-binding protein Rac1 induces cardiac myocyte hypertrophy through the activation of apoptosis signal-regulating kinase 1 and nuclear factor-κB. J. Biol. Chem. 2003, 278, 20770–20777. [Google Scholar] [CrossRef] [Green Version]
- Jobs, A.; Abdin, A.; de Waha-Thiele, S.; Eitel, I.; Thiele, H.; de Wit, C.; Vonthein, R. Angiotensin-converting-enzyme inhibitors in hemodynamic congestion: A meta-analysis of early studies. Clin. Res. Cardiol. 2019, 108, 1240–1248. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bhullar, S.K.; Dhalla, N.S. Angiotensin II-Induced Signal Transduction Mechanisms for Cardiac Hypertrophy. Cells 2022, 11, 3336. https://doi.org/10.3390/cells11213336
Bhullar SK, Dhalla NS. Angiotensin II-Induced Signal Transduction Mechanisms for Cardiac Hypertrophy. Cells. 2022; 11(21):3336. https://doi.org/10.3390/cells11213336
Chicago/Turabian StyleBhullar, Sukhwinder K., and Naranjan S. Dhalla. 2022. "Angiotensin II-Induced Signal Transduction Mechanisms for Cardiac Hypertrophy" Cells 11, no. 21: 3336. https://doi.org/10.3390/cells11213336
APA StyleBhullar, S. K., & Dhalla, N. S. (2022). Angiotensin II-Induced Signal Transduction Mechanisms for Cardiac Hypertrophy. Cells, 11(21), 3336. https://doi.org/10.3390/cells11213336