
Integrated Analysis of Transcriptome, microRNAs, and Chromatin Accessibility Revealed Potential Early B-Cell Factor1-Regulated Transcriptional Networks during the Early Development of Fetal Brown Adipose Tissues in Rabbits

 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics Approval
2.2. Experimental Animals and Histological Observation
2.3. Library Preparation, Sequencing, and Analysis of RNA-Seq
2.4. Library Preparation, Sequencing, and Analysis of miRNA-Seq
2.5. Library Preparation, Sequencing, and Analysis of ATAC-Seq
2.6. Functional Annotation and Pathway Enrichment
2.7. Quantitative Real-Time PCR (RT-qPCR)
2.8. Statistical Analysis
3. Results
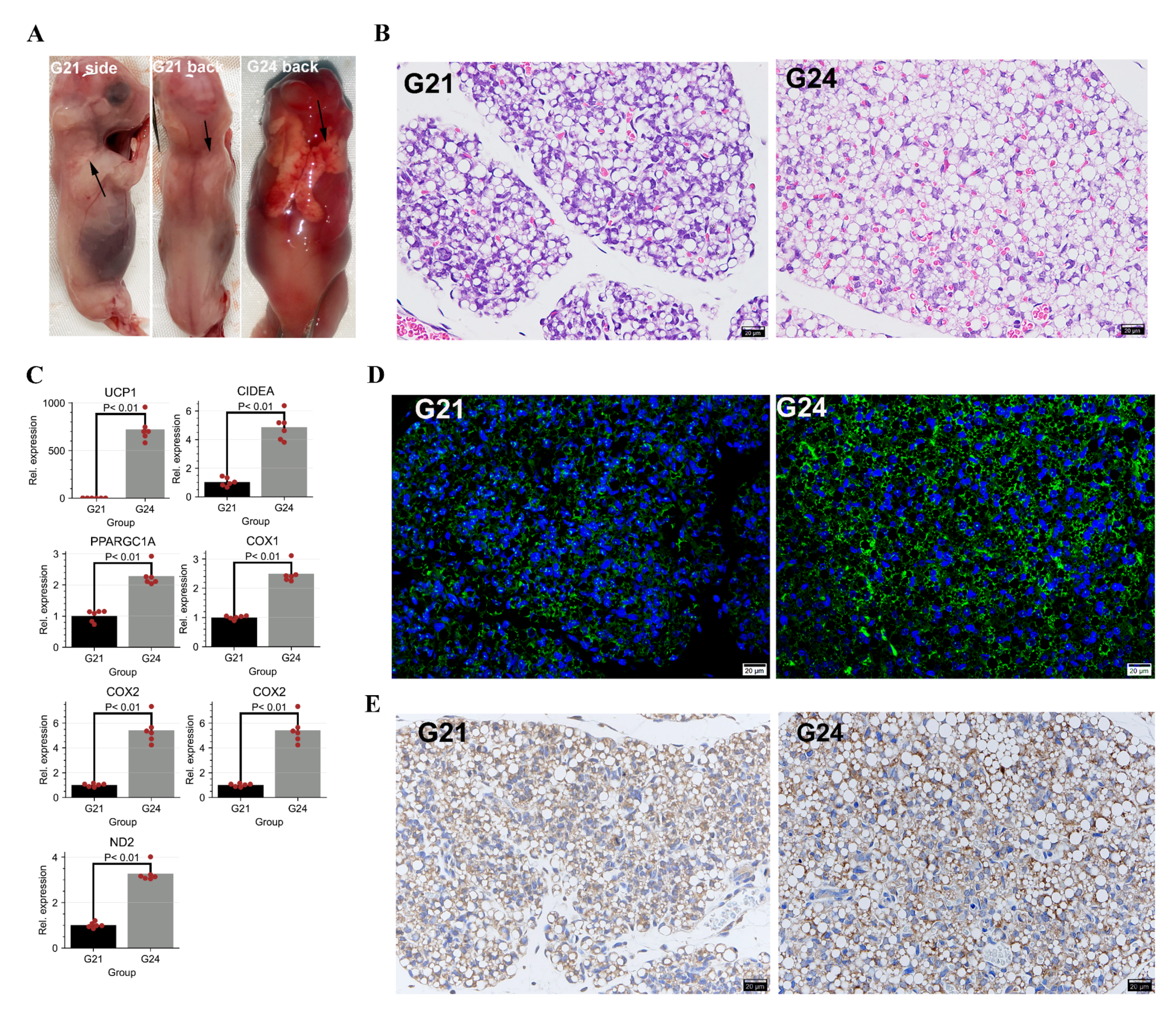
3.1. Changes of Histological Characterization and Genome-Wide Gene Expression during the Early FBATs Development in Rabbits
3.2. Identification of miRNAs during Early FBAT Development
3.3. Chromatin Accessibility Changes during the Early FBAT Development in Rabbits
3.4. Proximal Regulation of Chromatin Accessibility and the miRNA Regulation during the Early Development of FBAT
3.5. Genome-Wide Footprinting Analysis Revealed EBF1 Is Important for the Early Development of FBAT
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kasari, T.R. Weakness in the newborn calf. Vet. Clin. N. Am. Food Anim. Pract. 1994, 10, 167–180. [Google Scholar] [CrossRef]
- Carstens, G.E. Cold thermoregulation in the newborn calf. Vet. Clin. N. Am. Food Anim. Pract. 1994, 10, 69–106. [Google Scholar] [CrossRef]
- Trevisanuto, D.; Testoni, D.; de Almeida, M.F.B. Maintaining normothermia: Why and how? Semin. Fetal Neonatal Med. 2018, 23, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Q.; Lin, J.; Huang, J.; Zhang, H.; Zhang, R.; Zhang, X.; Cao, C.; Hambly, C.; Qin, G.; Yao, J.; et al. Reconstitution of UCP1 using CRISPR/Cas9 in the white adipose tissue of pigs decreases fat deposition and improves thermogenic capacity. Proc. Natl. Acad. Sci. USA 2017, 114, E9474–E9482. [Google Scholar] [CrossRef]
- Young, B.A. Cold stress as it affects animal production. J. Anim. Sci. 1981, 52, 154–163. [Google Scholar] [CrossRef]
- Luo, N.; Wang, J.; Hu, Y.; Zhao, Z.; Zhao, Y.; Chen, X. Cold and heat climatic variations reduce indigenous goat birth weight and enhance pre-weaning mortality in subtropical monsoon region of China. Trop. Anim. Health Prod. 2020, 52, 1385–1394. [Google Scholar] [CrossRef]
- Yang, Y.; Chen, N.; Sun, L.; Zhang, Y.; Wu, Y.; Wang, Y.; Liao, X.; Mi, J. Short-term cold stress can reduce the abundance of antibiotic resistance genes in the cecum and feces in a pig model. J. Hazard. Mater. 2021, 416, 125868. [Google Scholar] [CrossRef]
- Ogunbameru, B.O.; Kornegay, E.T.; Wood, C.M. Evaluation of methods of providing supplemental heat to newborn pigs during and after farrowing. J. Anim. Sci. 1991, 69, 3939–3944. [Google Scholar] [CrossRef]
- Zhang, Y.; Sun, L.; Zhu, R.; Zhang, S.; Liu, S.; Wang, Y.; Wu, Y.; Xing, S.; Liao, X.; Mi, J. Porcine gut microbiota in mediating host metabolic adaptation to cold stress. NPJ Biofilms Microbiomes 2022, 8, 18. [Google Scholar] [CrossRef]
- Djimènou, D.; Adoukonou-Sagbadja, H.; Dayo, G.K.; Chrysostome, C.; Koudande, D.O. Genetic diversity and phylogenetic relationships within local pigs in southern Benin. Trop. Anim. Health Prod. 2021, 53, 434. [Google Scholar] [CrossRef]
- Symonds, M.E.; Pope, M.; Sharkey, D.; Budge, H. Adipose tissue and fetal programming. Diabetologia 2012, 55, 1597–1606. [Google Scholar] [CrossRef] [PubMed]
- Cypess, A.M.; Lehman, S.; Williams, G.; Tal, I.; Rodman, D.; Goldfine, A.B.; Kuo, F.C.; Palmer, E.L.; Tseng, Y.H.; Doria, A.; et al. Identification and importance of brown adipose tissue in adult humans. N. Engl. J. Med. 2009, 360, 1509–1517. [Google Scholar] [CrossRef] [PubMed]
- van Marken Lichtenbelt, W.D.; Vanhommerig, J.W.; Smulders, N.M.; Drossaerts, J.M.; Kemerink, G.J.; Bouvy, N.D.; Schrauwen, P.; Teule, G.J. Cold-activated brown adipose tissue in healthy men. N. Engl. J. Med. 2009, 360, 1500–1508. [Google Scholar] [CrossRef] [PubMed]
- Saito, M.; Okamatsu-Ogura, Y.; Matsushita, M.; Watanabe, K.; Yoneshiro, T.; Nio-Kobayashi, J.; Iwanaga, T.; Miyagawa, M.; Kameya, T.; Nakada, K.; et al. High incidence of metabolically active brown adipose tissue in healthy adult humans: Effects of cold exposure and adiposity. Diabetes 2009, 58, 1526–1531. [Google Scholar] [CrossRef] [PubMed]
- Virtanen, K.A.; Lidell, M.E.; Orava, J.; Heglind, M.; Westergren, R.; Niemi, T.; Taittonen, M.; Laine, J.; Savisto, N.J.; Enerbäck, S. Functional brown adipose tissue in healthy adults. N. Engl. J. Med. 2009, 360, 1518–1525. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.J.; Ihara, T.; Yoshioka, H.; Itoyama, E.; Kitamura, S.; Nagase, H.; Murakami, H.; Hoshino, Y.; Murakami, M.; Tomonaga, S.; et al. Expression levels of brown/beige adipocyte-related genes in fat depots of vitamin A-restricted fattening cattle1. J. Anim. Sci. 2018, 96, 3884–3896. [Google Scholar] [CrossRef] [PubMed]
- Dawkins, M.J.; Hull, D. Brown Adipose Tissue and the response of New-Born Rabbits to Cold. J. Physiol. 1964, 172, 216–238. [Google Scholar] [CrossRef]
- Symonds, M.E.; Pope, M.; Budge, H. The Ontogeny of Brown Adipose Tissue. Annu. Rev. Nutr. 2015, 35, 295–320. [Google Scholar] [CrossRef]
- Inagaki, T.; Sakai, J.; Kajimura, S. Transcriptional and epigenetic control of brown and beige adipose cell fate and function. Nat. Rev. Mol. Cell Biol. 2016, 17, 480–495. [Google Scholar] [CrossRef]
- Alvarez-Dominguez, J.R.; Bai, Z.; Xu, D.; Yuan, B.; Lo, K.A.; Yoon, M.J.; Lim, Y.C.; Knoll, M.; Slavov, N.; Chen, S.; et al. De Novo Reconstruction of Adipose Tissue Transcriptomes Reveals Long Non-coding RNA Regulators of Brown Adipocyte Development. Cell Metab. 2015, 21, 764–776. [Google Scholar] [CrossRef] [Green Version]
- Lai, B.; Lee, J.E.; Jang, Y.; Wang, L.; Peng, W.; Ge, K. MLL3/MLL4 are required for CBP/p300 binding on enhancers and super-enhancer formation in brown adipogenesis. Nucleic Acids Res. 2017, 45, 6388–6403. [Google Scholar] [CrossRef]
- Sambeat, A.; Gulyaeva, O.; Dempersmier, J.; Tharp, K.M.; Stahl, A.; Paul, S.M.; Sul, H.S. LSD1 Interacts with Zfp516 to Promote UCP1 Transcription and Brown Fat Program. Cell Rep. 2016, 15, 2536–2549. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Gao, M.; Zhu, F.; Li, X.; Yang, Y.; Yan, Q.; Jia, L.; Xie, L.; Chen, Z. METTL3 is essential for postnatal development of brown adipose tissue and energy expenditure in mice. Nat. Commun. 2020, 11, 1648. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Mi, L.; Xiong, J.; Orchard, P.; Yu, Q.; Yu, L.; Zhao, X.Y.; Meng, Z.X.; Parker, S.C.J.; Lin, J.D.; et al. BAF60a deficiency uncouples chromatin accessibility and cold sensitivity from white fat browning. Nat. Commun. 2020, 11, 2379. [Google Scholar] [CrossRef] [PubMed]
- Hiraike, Y.; Waki, H.; Yu, J.; Nakamura, M.; Miyake, K.; Nagano, G.; Nakaki, R.; Suzuki, K.; Kobayashi, H.; Yamamoto, S.; et al. NFIA co-localizes with PPARγ and transcriptionally controls the brown fat gene program. Nat. Cell Biol. 2017, 19, 1081–1092. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Zhang, J.; Zou, Y.; Wang, F.; Li, J.; Sun, F.; Luo, X.; Zhang, M.; Guo, Y.; Yu, Q.; et al. Kdm2a deficiency in macrophages enhances thermogenesis to protect mice against HFD-induced obesity by enhancing H3K36me2 at the Pparg locus. Cell Death Differ. 2021, 28, 1880–1899. [Google Scholar] [CrossRef]
- Klemm, S.L.; Shipony, Z.; Greenleaf, W.J. Chromatin accessibility and the regulatory epigenome. Nat. Rev. Genet. 2019, 20, 207–220. [Google Scholar] [CrossRef]
- Wang, L.; Yang, X.; Zhu, Y.; Zhan, S.; Chao, Z.; Zhong, T.; Guo, J.; Wang, Y.; Li, L.; Zhang, H. Genome-Wide Identification and Characterization of Long Noncoding RNAs of Brown to White Adipose Tissue Transformation in Goats. Cells 2019, 8, 904. [Google Scholar] [CrossRef]
- Du, K.; Bai, X.; Yang, L.; Shi, Y.; Chen, L.; Wang, H.; Cai, M.; Wang, J.; Chen, S.; Jia, X.; et al. De Novo Reconstruction of Transcriptome Identified Long Non-Coding RNA Regulator of Aging-Related Brown Adipose Tissue Whitening in Rabbits. Biology 2021, 10, 1176. [Google Scholar] [CrossRef]
- Heim, T. Thermogenesis in the newborn infant. Clin. Obstet. Gynecol. 1971, 14, 790–820. [Google Scholar] [CrossRef]
- Wang, G.Z.; Du, K.; Hu, S.Q.; Chen, S.Y.; Jia, X.B.; Cai, M.C.; Shi, Y.; Wang, J.; Lai, S.J. Genome-wide identification and characterization of long non-coding RNAs during postnatal development of rabbit adipose tissue. Lipids Health Dis. 2018, 17, 271. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Kozomara, A.; Birgaoanu, M.; Griffiths-Jones, S. miRBase: From microRNA sequences to function. Nucleic Acids Res. 2019, 47, D155–D162. [Google Scholar] [CrossRef]
- Friedländer, M.R.; Mackowiak, S.D.; Li, N.; Chen, W.; Rajewsky, N. miRDeep2 accurately identifies known and hundreds of novel microRNA genes in seven animal clades. Nucleic Acids Res. 2012, 40, 37–52. [Google Scholar] [CrossRef]
- Pu, M.; Chen, J.; Tao, Z.; Miao, L.; Qi, X.; Wang, Y.; Ren, J. Regulatory network of miRNA on its target: Coordination between transcriptional and post-transcriptional regulation of gene expression. Cell Mol. Life Sci. 2019, 76, 441–451. [Google Scholar] [CrossRef]
- Enright, A.J.; John, B.; Gaul, U.; Tuschl, T.; Sander, C.; Marks, D.S. MicroRNA targets in Drosophila. Genome Biol. 2003, 5, R1. [Google Scholar] [CrossRef] [Green Version]
- Krüger, J.; Rehmsmeier, M. RNAhybrid: microRNA target prediction easy, fast and flexible. Nucleic Acids Res. 2006, 34, W451–W454. [Google Scholar] [CrossRef] [PubMed]
- Buenrostro, J.D.; Giresi, P.G.; Zaba, L.C.; Chang, H.Y.; Greenleaf, W.J. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods 2013, 10, 1213–1218. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Pireddu, L.; Leo, S.; Zanetti, G. SEAL: A distributed short read mapping and duplicate removal tool. Bioinformatics 2011, 27, 2159–2160. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, T.; Meyer, C.A.; Eeckhoute, J.; Johnson, D.S.; Bernstein, B.E.; Nusbaum, C.; Myers, R.M.; Brown, M.; Li, W.; et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 2008, 9, R137. [Google Scholar] [CrossRef] [PubMed]
- Ross-Innes, C.S.; Stark, R.; Teschendorff, A.E.; Holmes, K.A.; Ali, H.R.; Dunning, M.J.; Brown, G.D.; Gojis, O.; Ellis, I.O.; Green, A.R.; et al. Differential oestrogen receptor binding is associated with clinical outcome in breast cancer. Nature 2012, 481, 389–393. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.G.; He, Q.Y. ChIPseeker: An R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics 2015, 31, 2382–2383. [Google Scholar] [CrossRef] [PubMed]
- Xie, H.; Zhang, W.; Zhang, M.; Akhtar, T.; Li, Y.; Yi, W.; Sun, X.; Zuo, Z.; Wei, M.; Fang, X.; et al. Chromatin accessibility analysis reveals regulatory dynamics of developing human retina and hiPSC-derived retinal organoids. Sci. Adv. 2020, 6, eaay5247. [Google Scholar] [CrossRef]
- Fornes, O.; Castro-Mondragon, J.A.; Khan, A.; van der Lee, R.; Zhang, X.; Richmond, P.A.; Modi, B.P.; Correard, S.; Gheorghe, M.; Baranašić, D.; et al. JASPAR 2020: Update of the open-access database of transcription factor binding profiles. Nucleic Acids Res. 2020, 48, D87–D92. [Google Scholar] [CrossRef]
- Bentsen, M.; Goymann, P.; Schultheis, H.; Klee, K.; Petrova, A.; Wiegandt, R.; Fust, A.; Preussner, J.; Kuenne, C.; Braun, T.; et al. ATAC-seq footprinting unravels kinetics of transcription factor binding during zygotic genome activation. Nat. Commun. 2020, 11, 4267. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. Omics 2012, 16, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Delgado, G.; Martinez-Tellez, B.; Acosta, F.M.; Virtue, S.; Vidal-Puig, A.; Gil, A.; Llamas-Elvira, J.M.; Ruiz, J.R. Brown Adipose Tissue Volume and Fat Content Are Positively Associated with Whole-Body Adiposity in Young Men-Not in Women. Diabetes 2021, 70, 1473–1485. [Google Scholar] [CrossRef] [PubMed]
- Cristancho, A.G.; Lazar, M.A. Forming functional fat: A growing understanding of adipocyte differentiation. Nat. Rev. Mol. Cell Biol. 2011, 12, 722–734. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Pan, R.; Pfeifer, A. Regulation of brown and beige fat by microRNAs. Pharmacol. Ther. 2017, 170, 1–7. [Google Scholar] [CrossRef]
- Pan, D.; Mao, C.; Quattrochi, B.; Friedline, R.H.; Zhu, L.J.; Jung, D.Y.; Kim, J.K.; Lewis, B.; Wang, Y.X. MicroRNA-378 controls classical brown fat expansion to counteract obesity. Nat. Commun. 2014, 5, 4725. [Google Scholar] [CrossRef]
- Sun, L.; Xie, H.; Mori, M.A.; Alexander, R.; Yuan, B.; Hattangadi, S.M.; Liu, Q.; Kahn, C.R.; Lodish, H.F. Mir193b-365 is essential for brown fat differentiation. Nat. Cell Biol. 2011, 13, 958–965. [Google Scholar] [CrossRef]
- Hu, F.; Wang, M.; Xiao, T.; Yin, B.; He, L.; Meng, W.; Dong, M.; Liu, F. miR-30 promotes thermogenesis and the development of beige fat by targeting RIP140. Diabetes 2015, 64, 2056–2068. [Google Scholar] [CrossRef]
- Yue, J.; Hou, X.; Liu, X.; Wang, L.; Gao, H.; Zhao, F.; Shi, L.; Shi, L.; Yan, H.; Deng, T.; et al. The landscape of chromatin accessibility in skeletal muscle during embryonic development in pigs. J. Anim. Sci. Biotechnol. 2021, 12, 56. [Google Scholar] [CrossRef]
- Alexandre, P.A.; Naval-Sánchez, M.; Menzies, M.; Nguyen, L.T.; Porto-Neto, L.R.; Fortes, M.R.S.; Reverter, A. Chromatin accessibility and regulatory vocabulary across indicine cattle tissues. Genome Biol. 2021, 22, 273. [Google Scholar] [CrossRef]
- Gross, D.S.; Garrard, W.T. Nuclease hypersensitive sites in chromatin. Annu. Rev. Biochem. 1988, 57, 159–197. [Google Scholar] [CrossRef]
- Hu, S.; Yang, S.; Lu, Y.; Deng, Y.; Li, L.; Zhu, J.; Zhang, Y.; Hu, B.; Hu, J.; Xia, L.; et al. Dynamics of the Transcriptome and Accessible Chromatin Landscapes During Early Goose Ovarian Development. Front. Cell Dev. Biol. 2020, 8, 196. [Google Scholar] [CrossRef]
- Yan, F.; Powell, D.R.; Curtis, D.J.; Wong, N.C. From reads to insight: A hitchhiker’s guide to ATAC-seq data analysis. Genome Biol. 2020, 21, 22. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, A.M.; Wang, Z.; Schug, J.; Naji, A.; Kaestner, K.H. Integration of ATAC-seq and RNA-seq identifies human alpha cell and beta cell signature genes. Mol. Metab. 2016, 5, 233–244. [Google Scholar] [CrossRef]
- Gao, H.; Mejhert, N.; Fretz, J.A.; Arner, E.; Lorente-Cebrián, S.; Ehrlund, A.; Dahlman-Wright, K.; Gong, X.; Strömblad, S.; Douagi, I.; et al. Early B cell factor 1 regulates adipocyte morphology and lipolysis in white adipose tissue. Cell Metab. 2014, 19, 981–992. [Google Scholar] [CrossRef] [PubMed]
- Angueira, A.R.; Shapira, S.N.; Ishibashi, J.; Sampat, S.; Sostre-Colón, J.; Emmett, M.J.; Titchenell, P.M.; Lazar, M.A.; Lim, H.W.; Seale, P. Early B Cell Factor Activity Controls Developmental and Adaptive Thermogenic Gene Programming in Adipocytes. Cell Rep. 2020, 30, 2869–2878.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Du, K.; Shi, Y.; Bai, X.; Chen, L.; Sun, W.; Chen, S.; Wang, J.; Jia, X.; Lai, S. Integrated Analysis of Transcriptome, microRNAs, and Chromatin Accessibility Revealed Potential Early B-Cell Factor1-Regulated Transcriptional Networks during the Early Development of Fetal Brown Adipose Tissues in Rabbits. Cells 2022, 11, 2675. https://doi.org/10.3390/cells11172675
Du K, Shi Y, Bai X, Chen L, Sun W, Chen S, Wang J, Jia X, Lai S. Integrated Analysis of Transcriptome, microRNAs, and Chromatin Accessibility Revealed Potential Early B-Cell Factor1-Regulated Transcriptional Networks during the Early Development of Fetal Brown Adipose Tissues in Rabbits. Cells. 2022; 11(17):2675. https://doi.org/10.3390/cells11172675
Chicago/Turabian StyleDu, Kun, Yu Shi, Xue Bai, Li Chen, Wenqiang Sun, Shiyi Chen, Jie Wang, Xianbo Jia, and Songjia Lai. 2022. "Integrated Analysis of Transcriptome, microRNAs, and Chromatin Accessibility Revealed Potential Early B-Cell Factor1-Regulated Transcriptional Networks during the Early Development of Fetal Brown Adipose Tissues in Rabbits" Cells 11, no. 17: 2675. https://doi.org/10.3390/cells11172675