

Tetraspanin 8 Subfamily Members Regulate Substrate-Specificity of a Disintegrin and Metalloprotease 17

 , and
, and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and Transfection
2.2. Generation of Stable Inducible Tetraspanin-Overexpressing HEK293T Cell Lines
2.3. Generation of Tspan8-Deficient Cells
2.4. Flow Cytometry
2.5. Exosome Isolation
2.6. Gaussia Princeps Luciferase-Based Protein Complementation Assay
2.7. AP-Shedding Assay
2.8. ADAM17 Cell Surface Activity Assay
2.9. Enzyme-Linked Immunosorbent Assay (ELISA)
2.10. Immunofluorescence
2.11. Red Fluorescent Protein Dimerization Assay
2.12. Fluorescence Energy-Transfer (FRET) Microscopy
2.13. Stimulated Emission Depletion (STED) Super-Resolution Microscopy
2.14. Sucrose Gradient Centrifugation
2.15. Co-Immunoprecipitation Experiments
2.16. SDS-PAGE and Immunoblotting
2.17. Molecular Modeling
2.18. Gene Expression Meta-Analysis
2.19. Statistical Analysis
3. Results
3.1. Tetraspanins CD9, CD81 and Tspan8 Are Co-Expressed with ADAM17 in Tumor Cells
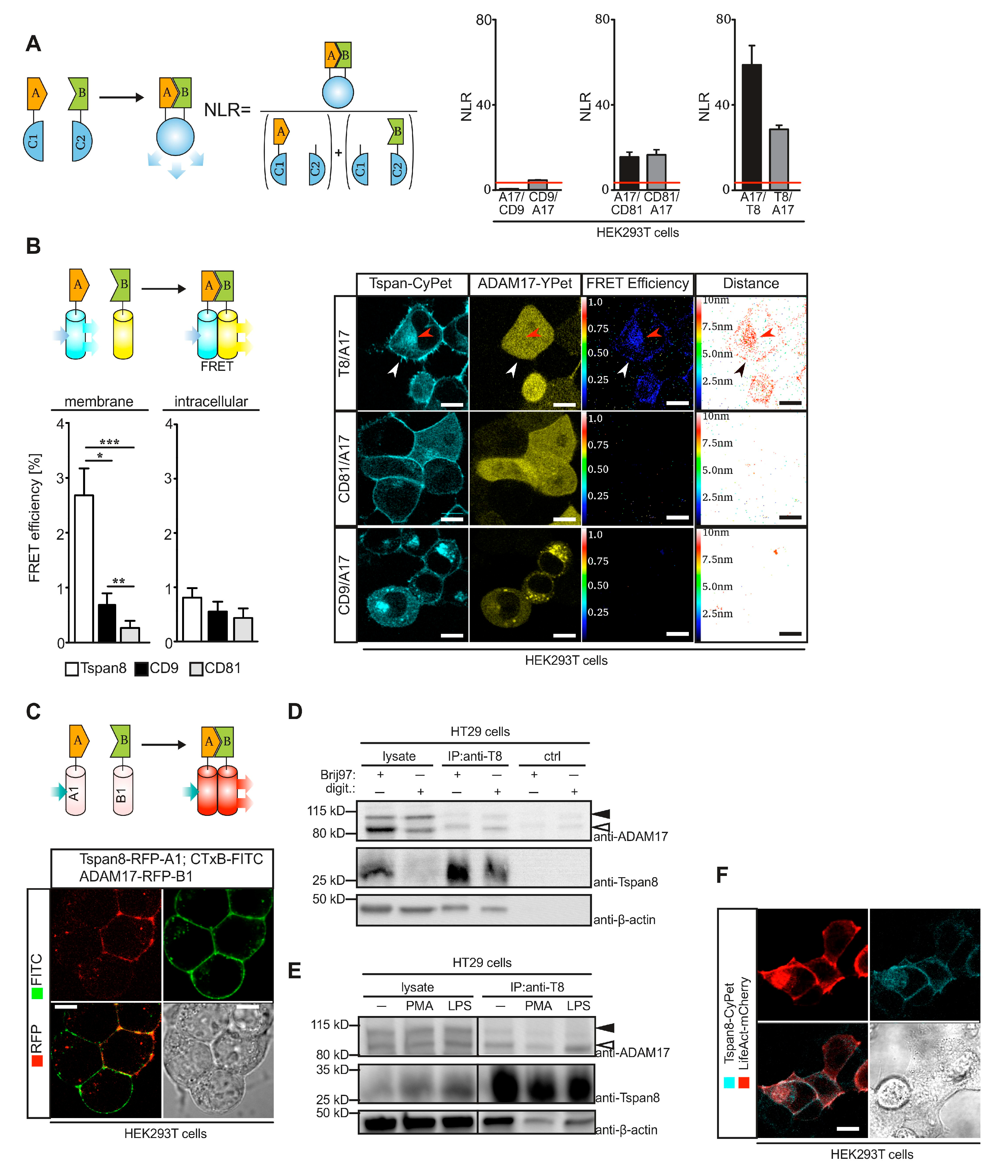
3.2. Tspan8 Physically Interacts with ADAM17 at the Plasma Membrane
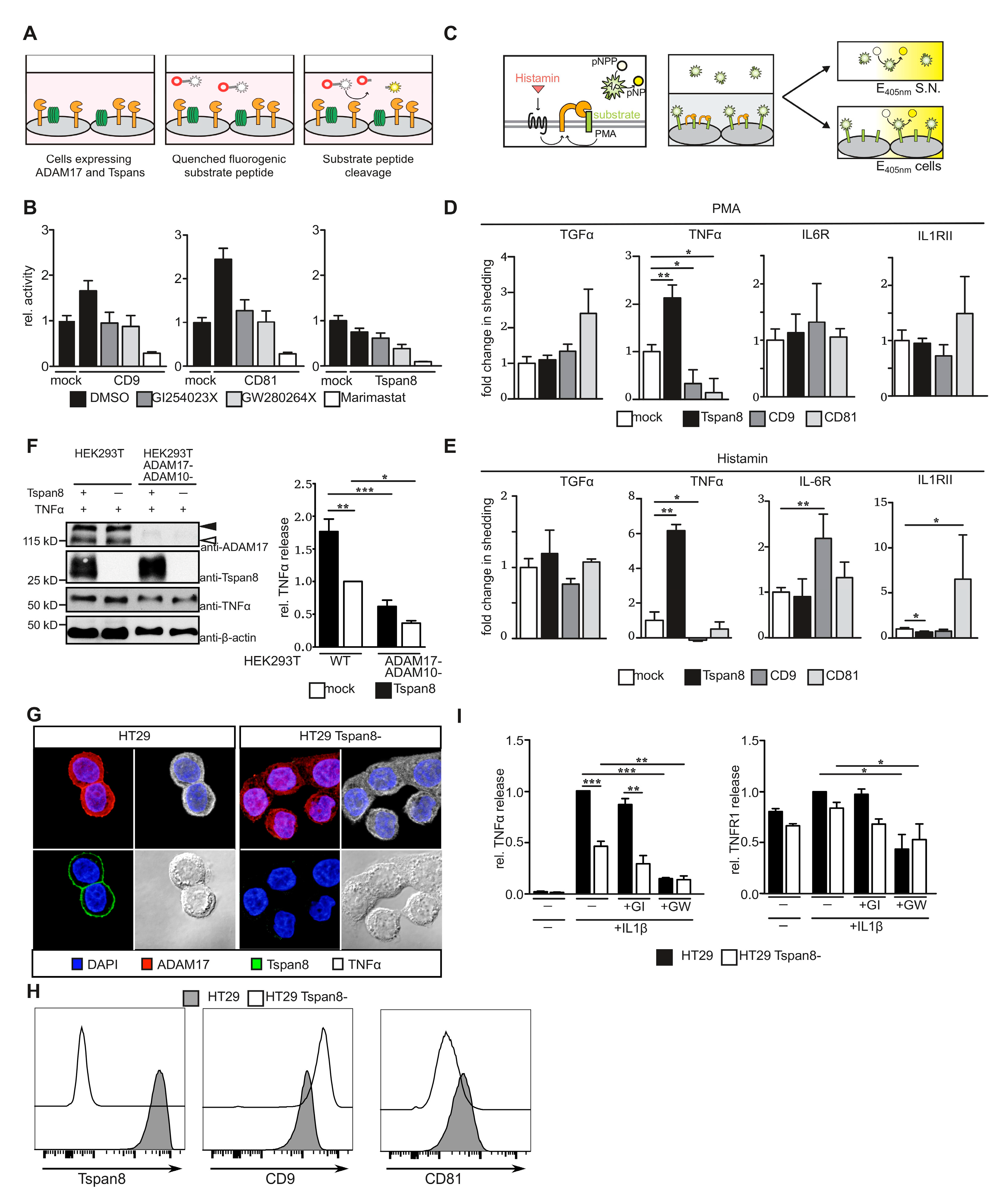
3.3. Tetraspanins CD9, CD81 and Tspan8 Alter Substrate Specificity of ADAM17
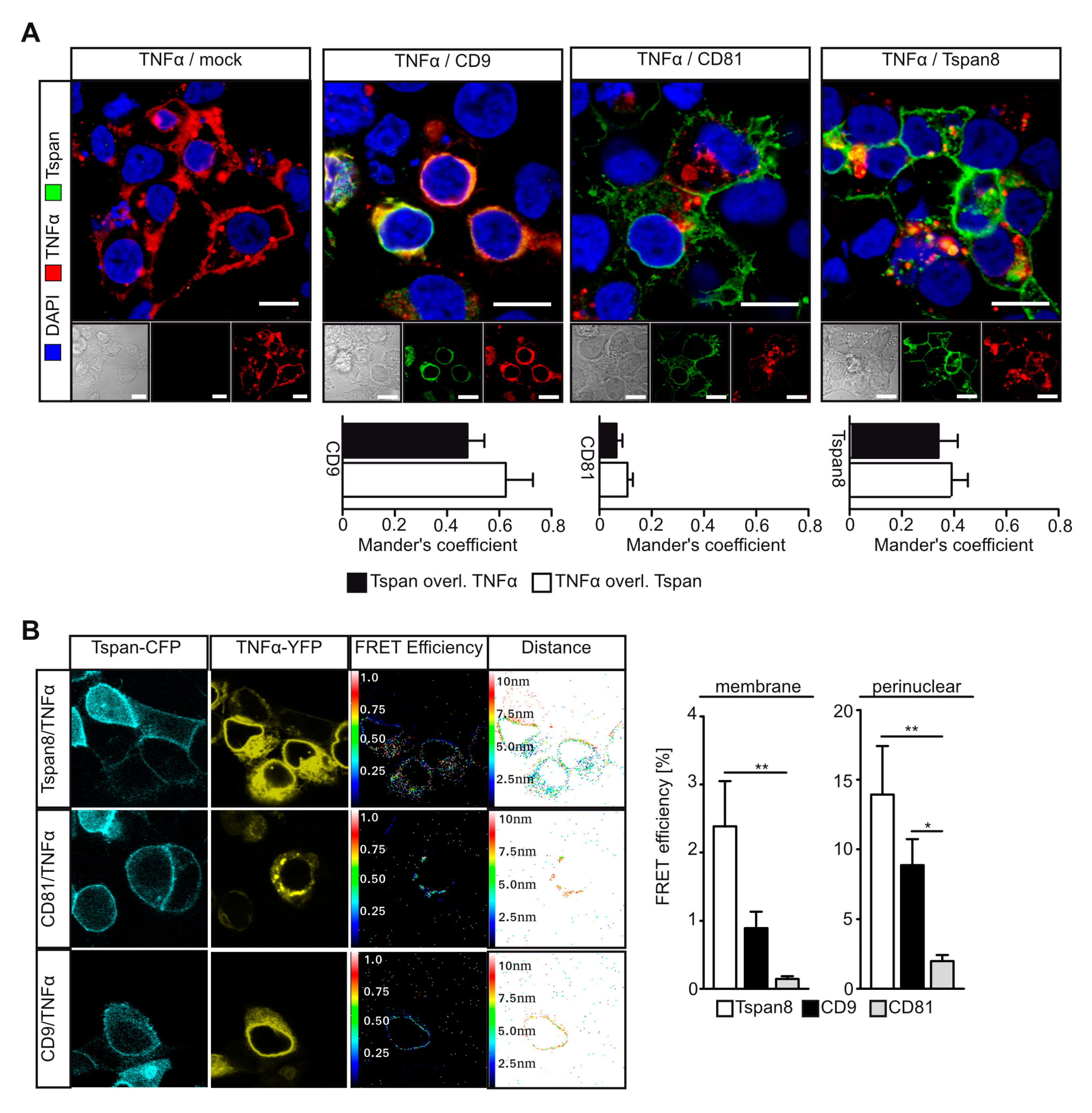
3.4. Tetraspanins CD9 and Tspan8 Associate with TNFα at Different Subcellular Compartments
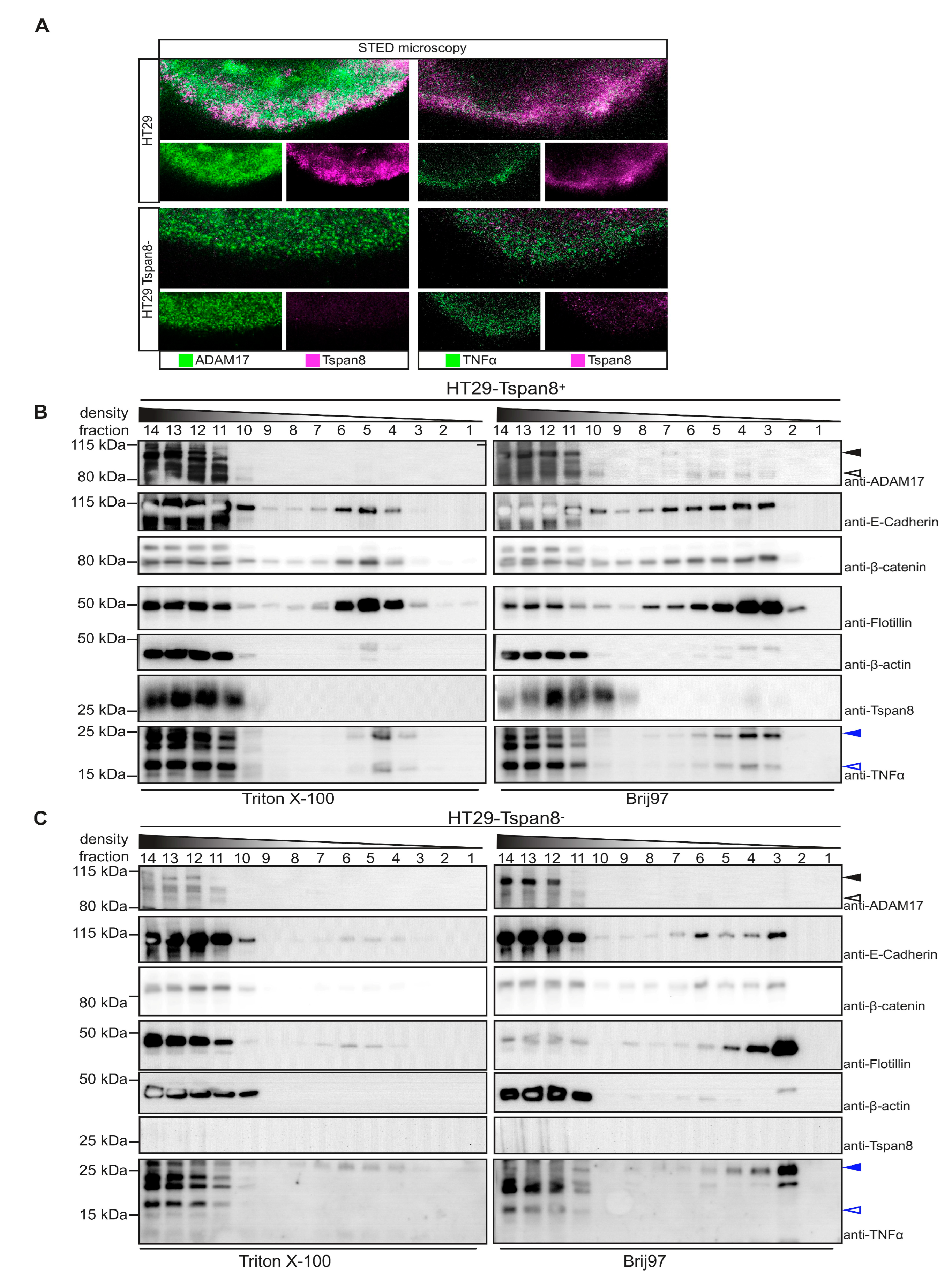
3.5. Tspan8 Promotes ADAM17-Mediated TNF α Processing through Recruitment of ADAM17 to Tetraspanin-Enriched Micro-Domains (TEMs)
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Edwards, D.R.; Handsley, M.M.; Pennington, C.J. The ADAM metalloproteinases. Mol. Asp. Med. 2008, 29, 258–289. [Google Scholar] [CrossRef]
- Reiss, K.; Saftig, P. The “A Disintegrin And Metalloprotease” (ADAM) family of sheddases: Physiological and cellular functions. Semin. Cell Dev. Biol. 2009, 20, 126–137. [Google Scholar] [CrossRef] [PubMed]
- Black, R.A.; Rauch, C.T.; Kozlosky, C.J.; Peschon, J.J.; Slack, J.L.; Wolfson, M.F.; Castner, B.J.; Stocking, K.L.; Reddy, P.; Srinivasan, S.; et al. A metalloproteinase disintegrin that releases tumour-necrosis factor-α from cells. Nature 1997, 385, 729–733. [Google Scholar] [CrossRef] [PubMed]
- Peschon, J.J.; Slack, J.L.; Reddy, P.; Stocking, K.L.; Sunnarborg, S.W.; Lee, D.C.; Russell, W.E.; Castner, B.J.; Johnson, R.S.; Fitzner, J.N.; et al. An Essential Role for Ectodomain Shedding in Mammalian Development. Science 1998, 282, 1281–1284. [Google Scholar] [CrossRef] [PubMed]
- Bolik, J.; Tirnitz-Parker, J.E.E.; Schmidt-Arras, D. ADAM and ADAMTS Proteases in Hepatic Disorders. J. Ren. Hepatic Disord. 2019, 3, 23–32. [Google Scholar] [CrossRef]
- Sommer, A.; Kordowski, F.; Büch, J.; Maretzky, T.; Evers, A.; Andrä, J.; Düsterhöft, S.; Michalek, M.; Lorenzen, I.; Somasundaram, P.; et al. Phosphatidylserine exposure is required for ADAM17 sheddase function. Nat. Commun. 2016, 7, 11523. [Google Scholar] [CrossRef]
- Yáñez-Mó, M.; Gutiérrez-López, M.D.; Cabañas, C. Functional interplay between tetraspanins and proteases. Cell. Mol. Life Sci. CMLS 2011, 68, 3323–3335. [Google Scholar] [CrossRef]
- Matthews, A.L.; Noy, P.J.; Reyat, J.S.; Tomlinson, M.G. Regulation of A disintegrin and metalloproteinase (ADAM) family sheddases ADAM10 and ADAM17: The emerging role of tetraspanins and rhomboids. Platelets 2017, 28, 333–341. [Google Scholar] [CrossRef]
- Charrin, S.; Jouannet, S.; Boucheix, C.; Rubinstein, E. Tetraspanins at a glance. J. Cell Sci. 2014, 127, 3641–3648. [Google Scholar] [CrossRef]
- Charrin, S.; Manié, S.; Thiele, C.; Billard, M.; Gerlier, D.; Boucheix, C.; Rubinstein, E. A physical and functional link between cholesterol and tetraspanins. Eur. J. Immunol. 2003, 33, 2479–2489. [Google Scholar] [CrossRef]
- Odintsova, E.; Butters, T.D.; Monti, E.; Sprong, H.; van Meer, G.; Berditchevski, F. Gangliosides play an important role in the organization of CD82-enriched microdomains. Biochem. J. 2006, 400, 315–325. [Google Scholar] [CrossRef] [PubMed]
- Espenel, C.; Margeat, E.; Dosset, P.; Arduise, C.; Le Grimellec, C.; Royer, C.A.; Boucheix, C.; Rubinstein, E.; Milhiet, P.-E. Single-molecule analysis of CD9 dynamics and partitioning reveals multiple modes of interaction in the tetraspanin web. J. Cell Biol. 2008, 182, 765–776. [Google Scholar] [CrossRef] [PubMed]
- Hemler, M.E. Specific tetraspanin functions. J. Cell Biol. 2001, 155, 1103–1107. [Google Scholar] [CrossRef]
- Kuhn, S.; Koch, M.; Nuübel, T.; Ladwein, M.; Antolovic, D.; Klingbeil, P.; Hildebrand, D.; Moldenhauer, G.; Langbein, L.; Franke, W.W.; et al. A Complex of EpCAM, Claudin-7, CD44 Variant Isoforms, and Tetraspanins Promotes Colorectal Cancer Progression. Mol. Cancer Res. 2007, 5, 553–567. [Google Scholar] [CrossRef]
- He, G.; Dhar, D.; Nakagawa, H.; Font-Burgada, J.; Ogata, H.; Jiang, Y.; Shalapour, S.; Seki, E.; Yost, S.E.; Jepsen, K.; et al. Identification of Liver Cancer Progenitors Whose Malignant Progression Depends on Autocrine IL-6 Signaling. Cell 2013, 155, 384–396. [Google Scholar] [CrossRef] [PubMed]
- Hemler, M.E. Tetraspanin proteins promote multiple cancer stages. Nat. Cancer 2014, 14, 49–60. [Google Scholar] [CrossRef]
- Park, C.S.; Kim, T.-K.; Kim, H.G.; Kim, Y.-J.; Jeoung, M.H.; Lee, W.R.; Go, N.K.; Heo, K.; Lee, S. Therapeutic targeting of tetraspanin8 in epithelial ovarian cancer invasion and metastasis. Oncogene 2016, 35, 4540–4548. [Google Scholar] [CrossRef]
- Berthier-Vergnes, O.; El Kharbili, M.; De La Fouchardière, A.; Pointecouteau, T.; Verrando, P.; Wierinckx, A.; Lachuer, J.; Le Naour, F.; Lamartine, J. Gene expression profiles of human melanoma cells with different invasive potential reveal TSPAN8 as a novel mediator of invasion. Br. J. Cancer 2011, 104, 155–165. [Google Scholar] [CrossRef]
- El Kharbili, M.; Agaësse, G.; Barbollat-Boutrand, L.; Pommier, R.; De La Fouchardière, A.; LaRue, L.; Caramel, J.; Puisieux, A.; Berthier-Vergnes, O.; Masse, I. Tspan8-β-catenin positive feedback loop promotes melanoma invasion. Oncogene 2019, 38, 3781–3793. [Google Scholar] [CrossRef]
- Fujiwara, K.; Ohuchida, K.; Sada, M.; Horioka, K.; Ulrich, C.D., III; Shindo, K.; Ohtsuka, T.; Takahata, S.; Mizumoto, K.; Oda, Y.; et al. CD166/ALCAM Expression Is Characteristic of Tumorigenicity and Invasive and Migratory Activities of Pancreatic Cancer Cells. PLoS ONE 2014, 9, e107247. [Google Scholar] [CrossRef] [Green Version]
- Yue, S.; Mu, W.; Erb, U.; Zöller, M. The tetraspanins CD151 and Tspan8 are essential exosome components for the crosstalk between cancer initiating cells and their surrounding. Oncotarget 2015, 6, 2366–2384. [Google Scholar] [CrossRef] [PubMed]
- Dornier, E.; Coumailleau, F.; Ottavi, J.-F.; Moretti, J.; Boucheix, C.; Mauduit, P.; Schweisguth, F.; Rubinstein, E. TspanC8 tetraspanins regulate ADAM10/Kuzbanian trafficking and promote Notch activation in flies and mammals. J. Cell Biol. 2012, 199, 481–496. [Google Scholar] [CrossRef] [PubMed]
- Haining, E.J.; Yang, J.; Bailey, R.L.; Khan, K.; Collier, R.; Tsai, S.; Watson, S.P.; Frampton, J.; Garcia, P.; Tomlinson, M.G. The TspanC8 Subgroup of Tetraspanins Interacts with A Disintegrin and Metalloprotease 10 (ADAM10) and Regulates Its Maturation and Cell Surface Expression. J. Biol. Chem. 2012, 287, 39753–39765. [Google Scholar] [CrossRef]
- Prox, J.; Willenbrock, M.; Weber, S.; Lehmann, T.; Schmidt-Arras, D.; Schwanbeck, R.; Saftig, P.; Schwake, M. Tetraspanin15 regulates cellular trafficking and activity of the ectodomain sheddase ADAM10. Cell. Mol. Life Sci. CMLS 2012, 69, 2919–2932. [Google Scholar] [CrossRef] [PubMed]
- Seipold, L.; Altmeppen, H.; Koudelka, T.; Tholey, A.; Kasparek, P.; Sedlacek, R.; Schweizer, M.; Bär, J.; Mikhaylova, M.; Glatzel, M.; et al. In vivo regulation of the A disintegrin and metalloproteinase 10 (ADAM10) by the tetraspanin 15. Cell. Mol. Life Sci. CMLS 2018, 75, 3251–3267. [Google Scholar] [CrossRef]
- Xu, D.; Sharma, C.; Hemler, M.E. Tetraspanin12 regulates ADAM10-dependent cleavage of amyloid precursor protein. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2009, 23, 3674–3681. [Google Scholar] [CrossRef]
- Seipold, L.; Damme, M.; Prox, J.; Rabe, B.; Kasparek, P.; Sedlacek, R.; Altmeppen, H.; Willem, M.; Boland, B.; Glatzel, M.; et al. Tetraspanin 3: A central endocytic membrane component regulating the expression of ADAM10, presenilin and the amyloid precursor protein. BBA Mol. Cell. Res. 2017, 1864, 217–230. [Google Scholar] [CrossRef]
- Arduise, C.; Abache, T.; Li, L.; Billard, M.; Chabanon, A.; Ludwig, A.; Mauduit, P.; Boucheix, C.; Rubinstein, E.; Le Naour, F. Tetraspanins Regulate ADAM10-Mediated Cleavage of TNF-α and Epidermal Growth Factor. J. Immunol. 2008, 181, 7002–7013. [Google Scholar] [CrossRef]
- Gutiérrez-López, M.D.; Gilsanz, A.; Yáñez-Mó, M.; Ovalle, S.; Lafuente, E.M.; Domínguez, C.; Monk, P.N.; González-Alvaro, I.; Sánchez-Madrid, F.; Cabañas, C. The sheddase activity of ADAM17/TACE is regulated by the tetraspanin CD9. Cell. Mol. Life Sci. CMLS 2011, 68, 3275–3292. [Google Scholar] [CrossRef]
- Tsukamoto, S.; Takeuchi, M.; Kawaguchi, T.; Togasaki, E.; Yamazaki, A.; Sugita, Y.; Muto, T.; Sakai, S.; Takeda, Y.; Ohwada, C.; et al. Tetraspanin CD9 modulates ADAM17-mediated shedding of LR11 in leukocytes. Exp. Mol. Med. 2014, 46, e89. [Google Scholar] [CrossRef] [Green Version]
- Machado-Pineda, Y.; Cardeñes, B.; Reyes, R.; López-Martín, S.; Toribio, V.; Sánchez-Organero, P.; Suarez, H.; Grötzinger, J.; Lorenzen, I.; Yáñez-Mó, M.; et al. CD9 Controls Integrin α5β1-Mediated Cell Adhesion by Modulating Its Association With the Metalloproteinase ADAM17. Front. Immunol. 2018, 9, 2474. [Google Scholar] [CrossRef]
- Liu, J.; Zhu, G.; Jia, N.; Wang, W.; Wang, Y.; Yin, M.; Jiang, X.; Huang, Y.; Zhang, J. CD9 regulates keratinocyte migration by negatively modulating the sheddase activity of ADAM17. Int. J. Biol. Sci. 2019, 15, 493–506. [Google Scholar] [CrossRef] [PubMed]
- Murphy, G. The ADAMs: Signalling scissors in the tumour microenvironment. Nat. Cancer 2008, 8, 929–941. [Google Scholar] [CrossRef] [PubMed]
- Balkwill, F. Tumour necrosis factor and cancer. Nat. Cancer 2009, 9, 361–371. [Google Scholar] [CrossRef]
- Schmidt-Arras, D.; Rose-John, S. IL-6 pathway in the liver: From physiopathology to therapy. J. Hepatol. 2016, 64, 1403–1415. [Google Scholar] [CrossRef] [PubMed]
- Bergmann, J.; Müller, M.; Baumann, N.; Reichert, M.; Heneweer, C.; Bolik, J.; Lücke, K.; Gruber, S.; Carambia, A.; Boretius, S.; et al. IL-6 trans-signaling is essential for the development of hepatocellular carcinoma in mice. Hepatology 2017, 65, 89–103. [Google Scholar] [CrossRef]
- Schmidt, S.; Schumacher, N.; Schwarz, J.; Tangermann, S.; Kenner, L.; Schlederer, M.; Sibilia, M.; Linder, M.; Altendorf-Hofmann, A.; Knösel, T.; et al. ADAM17 is required for EGF-R–induced intestinal tumors via IL-6 trans-signaling. J. Exp. Med. 2018, 215, 1205–1225. [Google Scholar] [CrossRef]
- Sanjana, N.E.; Shalem, O.; Zhang, F. Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 2014, 11, 783–784. [Google Scholar] [CrossRef]
- Cassonnet, P.; Rolloy, C.; Neveu, G.; Vidalain, P.-O.; Chantier, T.; Pellet, J.; Jones, L.; Muller, M.; Demeret, C.; Gaud, G.; et al. Benchmarking a luciferase complementation assay for detecting protein complexes. Nat. Chem. Biol. 2011, 8, 990–992. [Google Scholar] [CrossRef]
- Inoue, A.; Ishiguro, J.; Kitamura, H.; Arima, N.; Okutani, M.; Shuto, A.; Higashiyama, S.; Ohwada, T.; Arai, H.; Makide, K.; et al. TGFα shedding assay: An accurate and versatile method for detecting GPCR activation. Nat. Methods 2012, 9, 1021–1029. [Google Scholar] [CrossRef]
- Bolte, S.; Cordelières, F.P. A guided tour into subcellular colocalization analysis in light microscopy. J. Microsc. 2006, 224, 213–232. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, F.; Müller, M.; Prox, J.; Arnold, P.; Schönherr, C.; Tredup, C.; Minder, P.; Ebsen, H.; Janssen, O.; Annaert, W.; et al. Tetraspanin 8 is an interactor of the metalloprotease meprin β within tetraspanin-enriched microdomains. Biol. Chem. 2016, 397, 857–869. [Google Scholar] [CrossRef] [PubMed]
- Ebsen, H.; Lettau, M.; Kabelitz, D.; Janssen, O. Subcellular localization and activation of ADAM proteases in the context of FasL shedding in T lymphocytes. Mol. Immunol. 2015, 65, 416–428. [Google Scholar] [CrossRef] [PubMed]
- Zimmerman, B.; Kelly, B.; McMillan, B.J.; Seegar, T.C.; Dror, R.O.; Kruse, A.C.; Blacklow, S.C. Crystal Structure of a Full-Length Human Tetraspanin Reveals a Cholesterol-Binding Pocket. Cell 2016, 167, 1041–1051.e11. [Google Scholar] [CrossRef]
- Webb, B.; Sali, A. Protein Structure Modeling with MODELLER. Methods Mol. Biol. 2014, 1137, 145–159. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas Research Network; Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.R.M.; Ozenberger, B.A.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J.M. The Cancer Genome Atlas Pan-Cancer analysis project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar] [CrossRef]
- Kosinski, M.; Biecek, P. RTCGA: The Cancer Genome Atlas Data Integration. 2019. R package version 1.26.0. Available online: https:/rctga.github.io/RCTGA (accessed on 14 November 2019).
- Kassambara, A. ggpubr: ‘ggplot2’ Based Publication Ready Plots. 2019. R package version 0.4.0. Available online: https://rpkgs.datanovia.com/ggpubr (accessed on 14 November 2019).
- Nguyen, A.; Daugherty, P.S. Evolutionary optimization of fluorescent proteins for intracellular FRET. Nat. Biotechnol. 2005, 23, 355–360. [Google Scholar] [CrossRef]
- Alford, S.C.; Abdelfattah, A.S.; Ding, Y.; Campbell, R.E. A Fluorogenic Red Fluorescent Protein Heterodimer. Chem. Biol. 2012, 19, 353–360. [Google Scholar] [CrossRef]
- Kenworthy, A.K.; Petranova, N.; Edidin, M. High-Resolution FRET Microscopy of Cholera Toxin B-Subunit and GPI-anchored Proteins in Cell Plasma Membranes. Mol. Biol. Cell 2000, 11, 1645–1655. [Google Scholar] [CrossRef]
- Yáñez-Mó, M.; Barreiro, O.; Gordon-Alonso, M.; Sala-Valdés, M.; Sánchez-Madrid, F. Tetraspanin-enriched microdomains: A functional unit in cell plasma membranes. Trends Cell Biol. 2009, 19, 434–446. [Google Scholar] [CrossRef]
- Tsukamoto, H.; Tanida, S.; Ozeki, K.; Ebi, M.; Mizoshita, T.; Shimura, T.; Mori, Y.; Kataoka, H.; Kamiya, T.; Fukuda, S.; et al. Annexin A2 Regulates A Disintegrin and Metalloproteinase 17–mediated Ectodomain Shedding of Pro–Tumor Necrosis Factor-α in Monocytes and Colon Epithelial Cells. Inflamm. Bowel Dis. 2013, 19, 1365–1373. [Google Scholar] [CrossRef] [PubMed]
- Moss, M.L.; Sklair-Tavron, L.; Nudelman, R. Drug Insight: Tumor necrosis factor-converting enzyme as a pharmaceutical target for rheumatoid arthritis. Nat. Clin. Pract. Rheumatol. 2008, 4, 300–309. [Google Scholar] [CrossRef]
- Moss, M.L.; Minond, D. Recent Advances in ADAM17 Research: A Promising Target for Cancer and Inflammation. Mediat. Inflamm. 2017, 2017, 9673537. [Google Scholar] [CrossRef] [PubMed]
- Noack, M.; Miossec, P. Selected cytokine pathways in rheumatoid arthritis. Semin. Immunopathol. 2017, 39, 365–383. [Google Scholar] [CrossRef] [PubMed]
- Moss, M.L.; Jin, S.-L.C.; Milla, M.E.; Burkhart, W.; Carter, H.L.; Chen, W.-J.; Clay, W.C.; Didsbury, J.R.; Hassler, D.; Hoffman, C.R.; et al. Cloning of a disintegrin metalloproteinase that processes precursor tumour-necrosis factor-α. Nature 1997, 385, 733–736. [Google Scholar] [CrossRef]
- Zunke, F.; Rose-John, S. The shedding protease ADAM17: Physiology and pathophysiology. BBA Mol. Cell. Res. 2017, 1864, 2059–2070. [Google Scholar] [CrossRef]
- Adrain, C.; Zettl, M.; Christova, Y.; Taylor, N.; Freeman, M. Tumor Necrosis Factor Signaling Requires iRhom2 to Promote Trafficking and Activation of TACE. Science 2012, 335, 225–228. [Google Scholar] [CrossRef]
- McIlwain, D.R.; Lang, P.A.; Maretzky, T.; Hamada, K.; Ohishi, K.; Maney, S.K.; Berger, T.; Murthy, A.; Duncan, G.; Xu, H.C.; et al. iRhom2 Regulation of TACE Controls TNF-Mediated Protection Against Listeria and Responses to LPS. Science 2012, 335, 229–232. [Google Scholar] [CrossRef]
- Weskamp, G.; Tüshaus, J.; Li, D.; Feederle, R.; Maretzky, T.; Swendemann, S.; Falck-Pedersen, E.; McIlwain, D.R.; Mak, T.W.; Salmon, J.E.; et al. ADAM17 stabilizes its interacting partner inactive Rhomboid 2 (iRhom2) but not inactive Rhomboid 1 (iRhom1). J. Biol. Chem. 2020, 295, 4350–4358. [Google Scholar] [CrossRef]
- Maretzky, T.; McIlwain, D.R.; Issuree, P.D.A.; Li, X.; Malapeira, J.; Amin, S.; Lang, P.A.; Mak, T.W.; Blobel, C.P. iRhom2 controls the substrate selectivity of stimulated ADAM17-dependent ectodomain shedding. Proc. Natl. Acad. Sci. USA 2013, 110, 11433–11438. [Google Scholar] [CrossRef] [Green Version]
- Tang, B.; Li, X.; Maretzky, T.; Perez-Aguilar, J.M.; McIlwain, D.; Xie, Y.; Zheng, Y.; Mak, T.W.; Weinstein, H.; Blobel, C.P. Substrate-selective protein ectodomain shedding by ADAM17 and iRhom2 depends on their juxtamembrane and transmembrane domains. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2020, 34, 4956–4969. [Google Scholar] [CrossRef] [PubMed]
- Matthews, V.; Schuster, B.; Schütze, S.; Bussmeyer, I.; Ludwig, A.; Hundhausen, C.; Sadowski, T.; Saftig, P.; Hartmann, D.; Kallen, K.-J.; et al. Cellular Cholesterol Depletion Triggers Shedding of the Human Interleukin-6 Receptor by ADAM10 and ADAM17 (TACE). J. Biol. Chem. 2003, 278, 38829–38839. [Google Scholar] [CrossRef] [PubMed]
- D’Alessio, A.; Esposito, B.; Giampietri, C.; Ziparo, E.; Pober, J.S.; Filippini, A. Plasma membrane microdomains regulate TACE-dependent TNFR1 shedding in human endothelial cells. J. Cell. Mol. Med. 2012, 16, 627–636. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Càceres, J.; Caja, L.; Mainez, J.; Mayoral, R.; Martín-Sanz, P.; Moreno-Vicente, R.; del Pozo, M..; Dooley, S.; Egea, G.; Fabregat, I. Caveolin-1 is required for TGF-β-induced transactivation of the EGF receptor pathway in hepatocytes through the activation of the metalloprotease TACE/ADAM17. Cell Death Dis. 2014, 5, e1326. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Müller, M.; Saunders, C.; Senftleben, A.; Heidbuechel, J.P.W.; Halwachs, B.; Bolik, J.; Hedemann, N.; Röder, C.; Bauerschlag, D.; Rose-John, S.; et al. Tetraspanin 8 Subfamily Members Regulate Substrate-Specificity of a Disintegrin and Metalloprotease 17. Cells 2022, 11, 2683. https://doi.org/10.3390/cells11172683
Müller M, Saunders C, Senftleben A, Heidbuechel JPW, Halwachs B, Bolik J, Hedemann N, Röder C, Bauerschlag D, Rose-John S, et al. Tetraspanin 8 Subfamily Members Regulate Substrate-Specificity of a Disintegrin and Metalloprotease 17. Cells. 2022; 11(17):2683. https://doi.org/10.3390/cells11172683
Chicago/Turabian StyleMüller, Miryam, Claire Saunders, Anke Senftleben, Johannes P. W. Heidbuechel, Birgit Halwachs, Julia Bolik, Nina Hedemann, Christian Röder, Dirk Bauerschlag, Stefan Rose-John, and et al. 2022. "Tetraspanin 8 Subfamily Members Regulate Substrate-Specificity of a Disintegrin and Metalloprotease 17" Cells 11, no. 17: 2683. https://doi.org/10.3390/cells11172683
APA StyleMüller, M., Saunders, C., Senftleben, A., Heidbuechel, J. P. W., Halwachs, B., Bolik, J., Hedemann, N., Röder, C., Bauerschlag, D., Rose-John, S., & Schmidt-Arras, D. (2022). Tetraspanin 8 Subfamily Members Regulate Substrate-Specificity of a Disintegrin and Metalloprotease 17. Cells, 11(17), 2683. https://doi.org/10.3390/cells11172683