
Regenerative and Anti-Inflammatory Potential of Regularly Fed, Starved Cells and Extracellular Vesicles In Vivo
 , , , , and
, , , , and 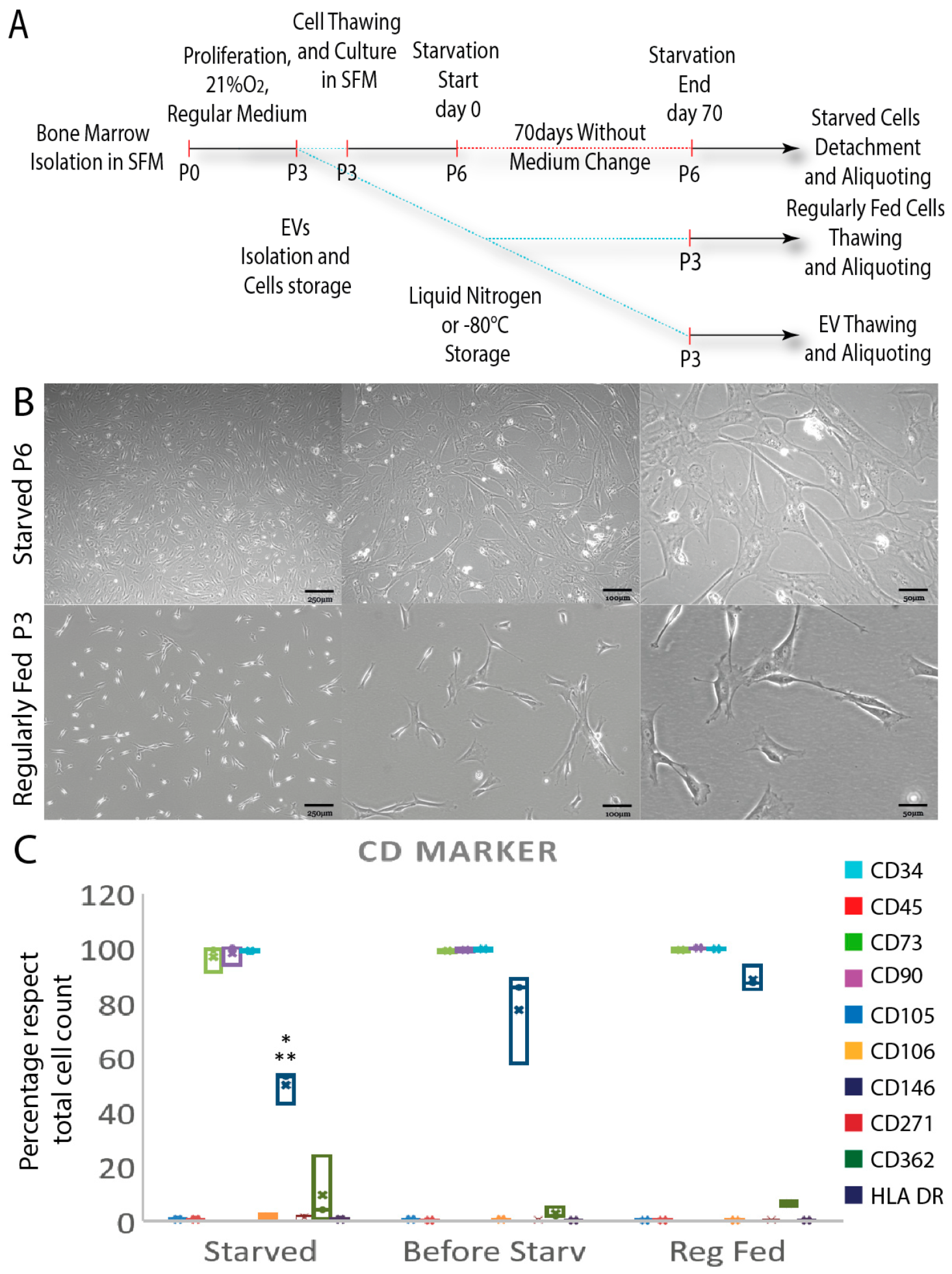{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. MSC Isolation from Human Bone Marrow
2.2. Extracellular Vesicle Isolation
2.3. Nanoparticle Tracking Analysis
2.4. Transmission Electron Microscopy
2.5. Immunoblot
2.6. Flow Cytometry
2.7. Differentiation Assays
2.8. Protein Array
2.9. Fracture Model
2.10. Cells and EVs Administration
2.11. Animal Sacrifice and Tissues Collection
2.12. Multiplex ELISA Cytokine Analysis
2.13. Micro-Computed Tomography
2.14. Statistical Analysis
3. Results
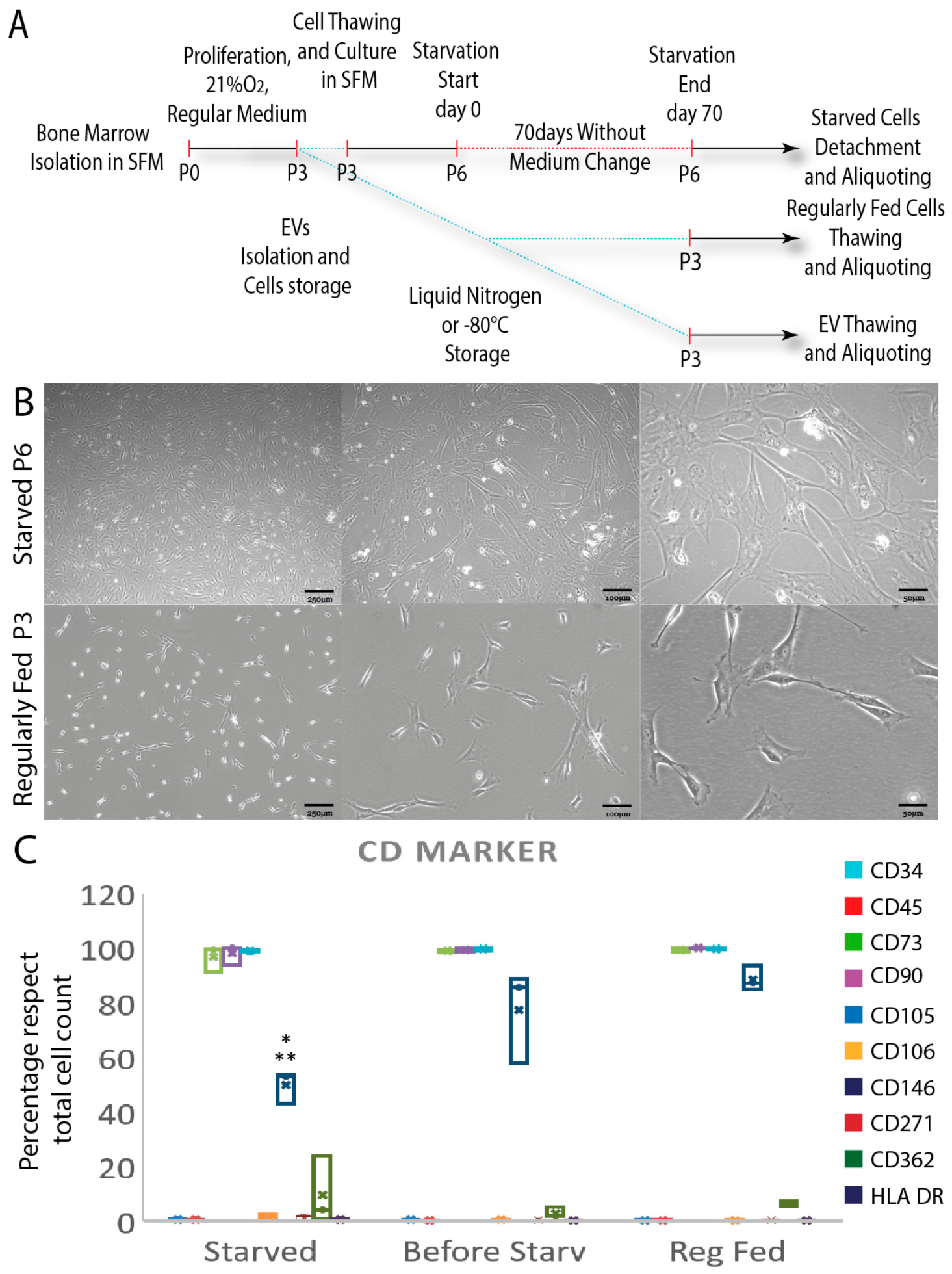
3.1. Starved and Regularly Fed Cells Characterization
3.2. EVs Characterization
3.3. Diabetic Fractured Mouse Model Shows Increased Inflammation and Reduced Mineral Matrix Content
3.4. Starved Cells Enhance Anti-Inflammatory Effects and Favor Earlier Soft Callus Formation and Mineral Matrix Deposition
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yan, W.; Li, X. Impact of Diabetes and Its Treatments on Skeletal Diseases. Front. Med. 2013, 7, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Bahney, C.S.; Zondervan, R.L.; Allison, P.; Theologis, A.; Ashley, J.W.; Ahn, J.; Miclau, T.; Marcucio, R.S.; Hankenson, K.D. Cellular Biology of Fracture Healing. J. Orthop. Res. 2019, 37, 35–50. [Google Scholar] [CrossRef] [PubMed]
- Arthur, A.; Gronthos, S. Clinical Application of Bone Marrow Mesenchymal Stem/Stromal Cells to Repair Skeletal Tissue. Int. J. Mol. Sci. 2020, 21, 9759. [Google Scholar] [CrossRef] [PubMed]
- Williams, A.R.; Hare, J.M. Mesenchymal Stem Cells: Biology, Pathophysiology, Translational Findings, and Therapeutic Implications for Cardiac Disease. Circ. Res. 2011, 109, 923–940. [Google Scholar] [CrossRef]
- Zhang, J.; Feng, Z.; Wei, J.; Yu, Y.; Luo, J.; Zhou, J.; Li, Y.; Zheng, X.; Tang, W.; Liu, L.; et al. Repair of Critical-Sized Mandible Defects in Aged Rat Using Hypoxia Preconditioned BMSC with Up-Regulation of Hif-1α. Int. J. Biol. Sci. 2018, 14, 449–460. [Google Scholar] [CrossRef]
- Lin, Y.; Anderson, J.D.; Rahnama, L.M.A.; Gu, S.V.; Knowlton, A.A. Exosomes in Disease and Regeneration: Biological Functions, Diagnostics, and Beneficial Effects. Am. J. Physiol. Heart Circ. Physiol. 2020, 319, H1162–H1180. [Google Scholar] [CrossRef]
- Galleu, A.; Riffo-Vasquez, Y.; Trento, C.; Lomas, C.; Dolcetti, L.; Cheung, T.S.; von Bonin, M.; Barbieri, L.; Halai, K.; Ward, S.; et al. Apoptosis in Mesenchymal Stromal Cells Induces in Vivo Recipient-Mediated Immunomodulation. Sci. Transl. Med. 2017, 9, eaam7828. [Google Scholar] [CrossRef]
- Toma, C.; Pittenger, M.F.; Cahill, K.S.; Byrne, B.J.; Kessler, P.D. Human Mesenchymal Stem Cells Differentiate to a Cardiomyocyte Phenotype in the Adult Murine Heart. Circulation 2002, 105, 93–98. [Google Scholar] [CrossRef]
- Wang, H.; Cao, F.; De, A.; Cao, Y.; Contag, C.; Gambhir, S.S.; Wu, J.C.; Chen, X. Trafficking Mesenchymal Stem Cell Engraftment and Differentiation in Tumor-Bearing Mice by Bioluminescence Imaging. Stem Cells 2009, 27, 1548–1558. [Google Scholar] [CrossRef]
- Moya, A.; Larochette, N.; Paquet, J.; Deschepper, M.; Bensidhoum, M.; Izzo, V.; Kroemer, G.; Petite, H.; Logeart-Avramoglou, D. Quiescence Preconditioned Human Multipotent Stromal Cells Adopt a Metabolic Profile Favorable for Enhanced Survival under Ischemia. Stem Cells 2017, 35, 181–196. [Google Scholar] [CrossRef]
- Contessotto, P.; Pandit, A. Therapies to Prevent Post-Infarction Remodelling: From Repair to Regeneration. Biomaterials 2021, 275, 120906. [Google Scholar] [CrossRef] [PubMed]
- Caplan, A.I. What’s in a Name? Tissue Eng. Part A 2010, 16, 2415–2417. [Google Scholar] [CrossRef] [PubMed]
- Amiri, F.; Jahanian-Najafabadi, A.; Roudkenar, M.H. In Vitro Augmentation of Mesenchymal Stem Cells Viability in Stressful Microenvironments: In Vitro Augmentation of Mesenchymal Stem Cells Viability. Cell Stress Chaperones 2015, 20, 237–251. [Google Scholar] [CrossRef] [PubMed]
- Crisan, M.; Yap, S.; Casteilla, L.; Chen, C.W.; Corselli, M.; Park, T.S.; Andriolo, G.; Sun, B.; Zheng, B.; Zhang, L.; et al. A Perivascular Origin for Mesenchymal Stem Cells in Multiple Human Organs. Cell Stem Cell 2008, 3, 301–313. [Google Scholar] [CrossRef]
- Nuschke, A.; Rodrigues, M.; Wells, A.W.; Sylakowski, K.; Wells, A. Mesenchymal Stem Cells/Multipotent Stromal Cells (MSCs) Are Glycolytic and Thus Glucose Is a Limiting Factor of in Vitro Models of MSC Starvation. Stem Cell Res. Ther. 2016, 7, 179. [Google Scholar] [CrossRef]
- Mylotte, L.A.; Duffy, A.M.; Murphy, M.; O’Brien, T.; Samali, A.; Barry, F.; Szegezdi, E. Metabolic Flexibility Permits Mesenchymal Stem Cell Survival in an Ischemic Environment. Stem Cells 2008, 26, 1325–1336. [Google Scholar] [CrossRef]
- Ferro, F.; Spelat, R.; Shaw, G.; Duffy, N.; Islam, M.N.; O’Shea, P.M.; O’Toole, D.; Howard, L.; Murphy, J.M. Survival/Adaptation of Bone Marrow-Derived Mesenchymal Stem Cells After Long-Term Starvation Through Selective Processes. Stem Cells 2019, 37, 813–827. [Google Scholar] [CrossRef]
- Estrada, J.C.; Albo, C.; Benguría, A.; Dopazo, A.; López-Romero, P.; Carrera-Quintanar, L.; Roche, E.; Clemente, E.P.; Enríquez, J.A.; Bernad, A.; et al. Culture of Human Mesenchymal Stem Cells at Low Oxygen Tension Improves Growth and Genetic Stability by Activating Glycolysis. Cell Death Differ. 2012, 19, 743–755. [Google Scholar] [CrossRef]
- Silva, L.H.A.; Antunes, M.A.; Dos Santos, C.C.; Weiss, D.J.; Cruz, F.F.; Rocco, P.R.M. Strategies to Improve the Therapeutic Effects of Mesenchymal Stromal Cells in Respiratory Diseases. Stem Cell Res. Ther. 2018, 9, 45. [Google Scholar] [CrossRef]
- Zhang, Q.; Yang, Y.J.; Wang, H.; Dong, Q.T.; Wang, T.J.; Qian, H.Y.; Xu, H. Autophagy Activation: A Novel Mechanism of Atorvastatin to Protect Mesenchymal Stem Cells from Hypoxia and Serum Deprivation via AMP-Activated Protein Kinase/Mammalian Target of Rapamycin Pathway. Stem Cells Dev. 2012, 21, 1321–1332. [Google Scholar] [CrossRef] [Green Version]
- Watson, L.; Chen, X.Z.; Ryan, A.E.; Fleming, Á.; Carbin, A.; O’Flynn, L.; Loftus, P.G.; Horan, E.; Connolly, D.; McDonnell, P.; et al. Administration of Human Non-Diabetic Mesenchymal Stromal Cells to a Murine Model of Diabetic Fracture Repair: A Pilot Study. Cells 2020, 9, 1394. [Google Scholar] [CrossRef] [PubMed]
- Motyl, K.; McCabe, L.R. Streptozotocin, Type I Diabetes Severity and Bone. Biol. Proced. Online 2009, 11, 296–315. [Google Scholar] [CrossRef] [PubMed]
- Davey, G.C.; Patil, S.B.; O’Loughlin, A.; O’Brien, T. Mesenchymal Stem Cell-Based Treatment for Microvascular and Secondary Complications of Diabetes Mellitus. Front. Endocrinol. 2014, 5, 86. [Google Scholar] [CrossRef]
- Maruyama, M.; Rhee, C.; Utsunomiya, T.; Zhang, N.; Ueno, M.; Yao, Z.; Goodman, S.B. Modulation of the Inflammatory Response and Bone Healing. Front. Endocrinol. 2020, 11, 386. [Google Scholar] [CrossRef] [PubMed]
- Loi, F.; Córdova, L.A.; Pajarinen, J.; Lin, T.; Yao, Z.; Goodman, S.B. Inflammation, Fracture and Bone Repair. Bone 2016, 86, 119–130. [Google Scholar] [CrossRef] [PubMed]
- Lacey, D.C.; Simmons, P.J.; Graves, S.E.; Hamilton, J.A. Proinflammatory Cytokines Inhibit Osteogenic Differentiation from Stem Cells: Implications for Bone Repair during Inflammation. Osteoarthr. Cartil. 2009, 17, 735–742. [Google Scholar] [CrossRef] [PubMed]
- Kayal, R.A.; Siqueira, M.; Alblowi, J.; McLean, J.; Krothapalli, N.; Faibish, D.; Einhorn, T.A.; Gerstenfeld, L.C.; Graves, D.T. TNF-Alpha Mediates Diabetes-Enhanced Chondrocyte Apoptosis during Fracture Healing and Stimulates Chondrocyte Apoptosis through FOXO1. J. Bone Miner. Metab. 2010, 25, 1604–1615. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wang, L.; Kikuiri, T.; Akiyama, K.; Chen, C.; Xu, X.; Yang, R.; Chen, W.; Wang, S.; Shi, S. Mesenchymal Stem Cell-Based Tissue Regeneration Is Governed by Recipient T Lymphocytes via IFN-γ and TNF-α. Nat. Med. 2011, 17, 1594–1601. [Google Scholar] [CrossRef]
- Deshpande, S.; James, A.W.; Blough, J.; Donneys, A.; Wang, S.C.; Cederna, P.S.; Buchman, S.R.; Levi, B. Reconciling the Effects of Inflammatory Cytokines on Mesenchymal Cell Osteogenic Differentiation. J. Surg. Res. 2013, 185, 278–285. [Google Scholar] [CrossRef]
- Kayal, R.A.; Tsatsas, D.; Bauer, M.A.; Allen, B.; Al-Sebaei, M.O.; Kakar, S.; Leone, C.W.; Morgan, E.F.; Gerstenfeld, L.C.; Einhorn, T.A.; et al. Diminished Bone Formation during Diabetic Fracture Healing Is Related to the Premature Resorption of Cartilage Associated with Increased Osteoclast Activity. J. Bone Miner. Res. 2007, 22, 560–568. [Google Scholar] [CrossRef] [Green Version]
- Ogasawara, A.; Nakajima, A.; Nakajima, F.; Goto, K.I.; Yamazaki, M. Molecular Basis for Affected Cartilage Formation and Bone Union in Fracture Healing of the Streptozotocin-Induced Diabetic Rat. Bone 2008, 43, 832–839. [Google Scholar] [CrossRef] [PubMed]
- McCauley, J.; Bitsaktsis, C.; Cottrell, J. Macrophage Subtype and Cytokine Expression Characterization during the Acute Inflammatory Phase of Mouse Bone Fracture Repair. J. Orthop. Res. 2020, 38, 1693–1702. [Google Scholar] [CrossRef] [PubMed]
- Gorgun, C.; Palamà, M.E.F.; Reverberi, D.; Gagliani, M.C.; Cortese, K.; Tasso, R.; Gentili, C. Role of Extracellular Vesicles from Adipose Tissue- and Bone Marrow-Mesenchymal Stromal Cells in Endothelial Proliferation and Chondrogenesis. Stem Cells Transl. Med. 2021, 10, 1680–1695. [Google Scholar] [CrossRef]
- Gorgun, C.; Reverberi, D.; Rotta, G.; Villa, F.; Quarto, R.; Tasso, R. Isolation and Flow Cytometry Characterization of Extracellular-Vesicle Subpopulations Derived from Human Mesenchymal Stromal Cells. Curr. Protoc. Stem Cell Biol. 2019, 48, e76. [Google Scholar] [CrossRef] [PubMed]
- Marturano, J.E.; Cleveland, B.C.; Byrne, M.A.; O’Connell, S.L.; Wixted, J.J.; Billiar, K.L. An Improved Murine Femur Fracture Device for Bone Healing Studies. J. Biomech. 2008, 41, 1222–1228. [Google Scholar] [CrossRef]
- Doube, M.; Kłosowski, M.M.; Arganda-Carreras, I.; Cordelières, F.P.; Dougherty, R.P.; Jackson, J.S.; Schmid, B.; Hutchinson, J.R.; Shefelbine, S.J. BoneJ: Free and Extensible Bone Image Analysis in ImageJ. Bone 2010, 47, 1076–1079. [Google Scholar] [CrossRef] [PubMed]
- Théry, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal Information for Studies of Extracellular Vesicles 2018 (MISEV2018): A Position Statement of the International Society for Extracellular Vesicles and Update of the MISEV2014 Guidelines. Null 2018, 7, 1535750. [Google Scholar] [CrossRef] [PubMed]
- Kolar, P.; Schmidt-Bleek, K.; Schell, H.; Gaber, T.; Toben, D.; Schmidmaier, G.; Perka, C.; Buttgereit, F.; Duda, G.N. The Early Fracture Hematoma and Its Potential Role in Fracture Healing. Tissue Eng. Part B Rev. 2010, 16, 427–434. [Google Scholar] [CrossRef]
- Xing, Z.; Lu, C.; Hu, D.; Yu, Y.; Wang, X.; Colnot, C.; Nakamura, M.; Wu, Y.; Miclau, T.; Marcucio, R.S. Multiple Roles for CCR2 during Fracture Healing. Dis. Models Mech. 2010, 3, 451–458. [Google Scholar] [CrossRef]
- Marsell, R.; Einhorn, T.A. The Biology of Fracture Healing. Injury 2011, 42, 551–555. [Google Scholar] [CrossRef] [Green Version]
- Nam, D.; Mau, E.; Wang, Y.; Wright, D.; Silkstone, D.; Whetstone, H.; Whyne, C.; Alman, B. T-Lymphocytes Enable Osteoblast Maturation via IL-17F during the Early Phase of Fracture Repair. PLoS ONE 2012, 7, e40044. [Google Scholar] [CrossRef]
- Gerstenfeld, L.C.; Cho, T.J.; Kon, T.; Aizawa, T.; Tsay, A.; Fitch, J.; Barnes, G.L.; Graves, D.T.; Einhorn, T.A. Impaired Fracture Healing in the Absence of TNF-Alpha Signaling: The Role of TNF-Alpha in Endochondral Cartilage Resorption. J. Bone Miner. Res. 2003, 18, 1584–1592. [Google Scholar] [CrossRef]
- Vries, M.H.M.; Wagenaar, A.; Verbruggen, S.E.L.; Molin, D.G.M.; Dijkgraaf, I.; Hackeng, T.H.; Post, M.J. CXCL1 Promotes Arteriogenesis through Enhanced Monocyte Recruitment into the Peri-Collateral Space. Angiogenesis 2015, 18, 163–171. [Google Scholar] [CrossRef] [PubMed]
- Gerstenfeld, L.C.; Cho, T.J.; Kon, T.; Aizawa, T.; Cruceta, J.; Graves, B.D.; Einhorn, T.A. Impaired Intramembranous Bone Formation during Bone Repair in the Absence of Tumor Necrosis Factor-Alpha Signaling. Cells Tissues Organs 2001, 169, 285–294. [Google Scholar] [CrossRef] [PubMed]
- Schumpp, B.; Schlaeger, E.J. Growth Study of Lactate and Ammonia Double-Resistant Clones of HL-60 Cells. Cytotechnology 1992, 8, 39–44. [Google Scholar] [CrossRef]
- Freund, N.W.; Croughan, M.S. A Simple Method to Reduce Both Lactic Acid and Ammonium Production in Industrial Animal Cell Culture. Int. J. Mol. Sci. 2018, 19, 385. [Google Scholar] [CrossRef]
- Yang, Y.-H.K.; Ogando, C.R.; Wang See, C.; Chang, T.-Y.; Barabino, G.A. Changes in Phenotype and Differentiation Potential of Human Mesenchymal Stem Cells Aging in Vitro. Stem Cell Res. Ther. 2018, 9, 131. [Google Scholar] [CrossRef]
- Gordon, S.; Martinez, F.O. Alternative Activation of Macrophages: Mechanism and Functions. Immunity 2010, 32, 593–604. [Google Scholar] [CrossRef]
- Hagiwara, H.; Mitsumata, M.; Yamane, T.; Jin, X.; Yoshida, Y. Laminar Shear Stress-Induced GRO MRNA and Protein Expression in Endothelial Cells. Circulation 1998, 98, 2584–2590. [Google Scholar] [CrossRef]
- Pulkkinen, H.H.; Kiema, M.; Lappalainen, J.P.; Toropainen, A.; Beter, M.; Tirronen, A.; Holappa, L.; Niskanen, H.; Kaikkonen, M.U.; Ylä-Herttuala, S.; et al. BMP6/TAZ-Hippo Signaling Modulates Angiogenesis and Endothelial Cell Response to VEGF. Angiogenesis 2021, 24, 129–144. [Google Scholar] [CrossRef]
- Bach, L.A. IGF-Binding Proteins. J. Mol. Endocrinol. 2018, 61, T11–T28. [Google Scholar] [CrossRef] [PubMed]
- Bach, L.A. What Happened to the IGF Binding Proteins? Endocrinology 2018, 159, 570–578. [Google Scholar] [CrossRef] [PubMed]
- Hamidouche, Z.; Fromigué, O.; Ringe, J.; Häupl, T.; Marie, P.J. Crosstalks between Integrin Alpha 5 and IGF2/IGFBP2 Signalling Trigger Human Bone Marrow-Derived Mesenchymal Stromal Osteogenic Differentiation. BMC Cell Biol. 2010, 11, 44. [Google Scholar] [CrossRef] [PubMed]
- Zhen, R.; Yang, J.; Wang, Y.; Li, Y.; Chen, B.; Song, Y.; Ma, G.; Yang, B. Hepatocyte Growth Factor Improves Bone Regeneration via the Bone Morphogenetic Protein-2-mediated NF-κB Signaling Pathway. Mol. Med. Rep. 2018, 17, 6045–6053. [Google Scholar] [CrossRef]
- Iyer, S.S.; Rojas, M. Anti-Inflammatory Effects of Mesenchymal Stem Cells: Novel Concept for Future Therapies. Expert Opin. Biol. Ther. 2008, 8, 569–581. [Google Scholar] [CrossRef]
- Ono, T.; Okamoto, K.; Nakashima, T.; Nitta, T.; Hori, S.; Iwakura, Y.; Takayanagi, H. IL-17-Producing Γδ T Cells Enhance Bone Regeneration. Nat. Commun. 2016, 7, 10928. [Google Scholar] [CrossRef]
- Wang, Y.; Kim, J.; Chan, A.; Whyne, C.; Nam, D. A Two Phase Regulation of Bone Regeneration: IL-17F Mediates Osteoblastogenesis via C/EBP-β in Vitro. Bone 2018, 116, 47–57. [Google Scholar] [CrossRef]
- Edderkaoui, B. Potential Role of Chemokines in Fracture Repair. Front. Endocrinol. 2017, 8, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ferro, F.; Spelat, R.; Shaw, G.; Coleman, C.M.; Chen, X.Z.; Connolly, D.; Palamá, E.M.F.; Gentili, C.; Contessotto, P.; Murphy, M.J. Regenerative and Anti-Inflammatory Potential of Regularly Fed, Starved Cells and Extracellular Vesicles In Vivo. Cells 2022, 11, 2696. https://doi.org/10.3390/cells11172696
Ferro F, Spelat R, Shaw G, Coleman CM, Chen XZ, Connolly D, Palamá EMF, Gentili C, Contessotto P, Murphy MJ. Regenerative and Anti-Inflammatory Potential of Regularly Fed, Starved Cells and Extracellular Vesicles In Vivo. Cells. 2022; 11(17):2696. https://doi.org/10.3390/cells11172696
Chicago/Turabian StyleFerro, Federico, Renza Spelat, Georgina Shaw, Cynthia M. Coleman, Xi Zhe Chen, David Connolly, Elisabetta M. F. Palamá, Chiara Gentili, Paolo Contessotto, and Mary J. Murphy. 2022. "Regenerative and Anti-Inflammatory Potential of Regularly Fed, Starved Cells and Extracellular Vesicles In Vivo" Cells 11, no. 17: 2696. https://doi.org/10.3390/cells11172696