
Quetiapine Ameliorates MIA-Induced Impairment of Sensorimotor Gating: Focus on Neuron-Microglia Communication and the Inflammatory Response in the Frontal Cortex of Adult Offspring of Wistar Rats

Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Prenatal Treatment with LPS
2.3. Behavioural Study—Prepulse Inhibition Test
2.4. Antipsychotic Drugs Administration
2.5. Biochemical Study
2.5.1. Tissue Collection and Preparation
2.5.2. Quantitative Real-Time Polymerase Chain Reaction
2.5.3. Enzyme-Linked Immunosorbent Assay
2.6. Statistical Data Analysis
3. Results
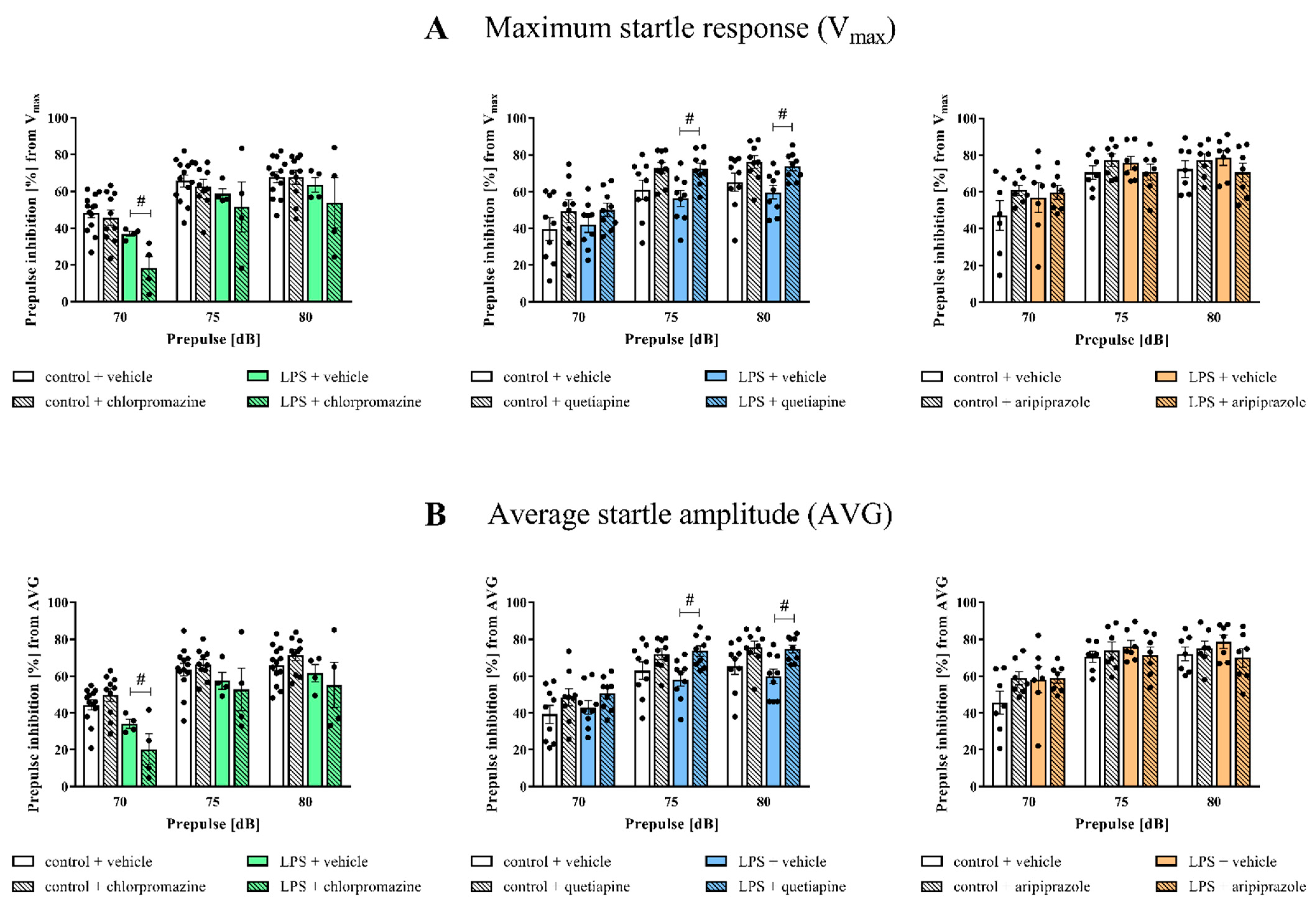
3.1. Prepulse Inhibition of the Acoustic Startle Response
3.2. Cx3cl1, Cx3cr1, Cd200 and Cd200r mRNA Expression in the Frontal Cortices of the Offspring
3.3. Cd40, Cd68, Arg1 and Igf-1 mRNA Expression in the Frontal Cortices of the Offspring
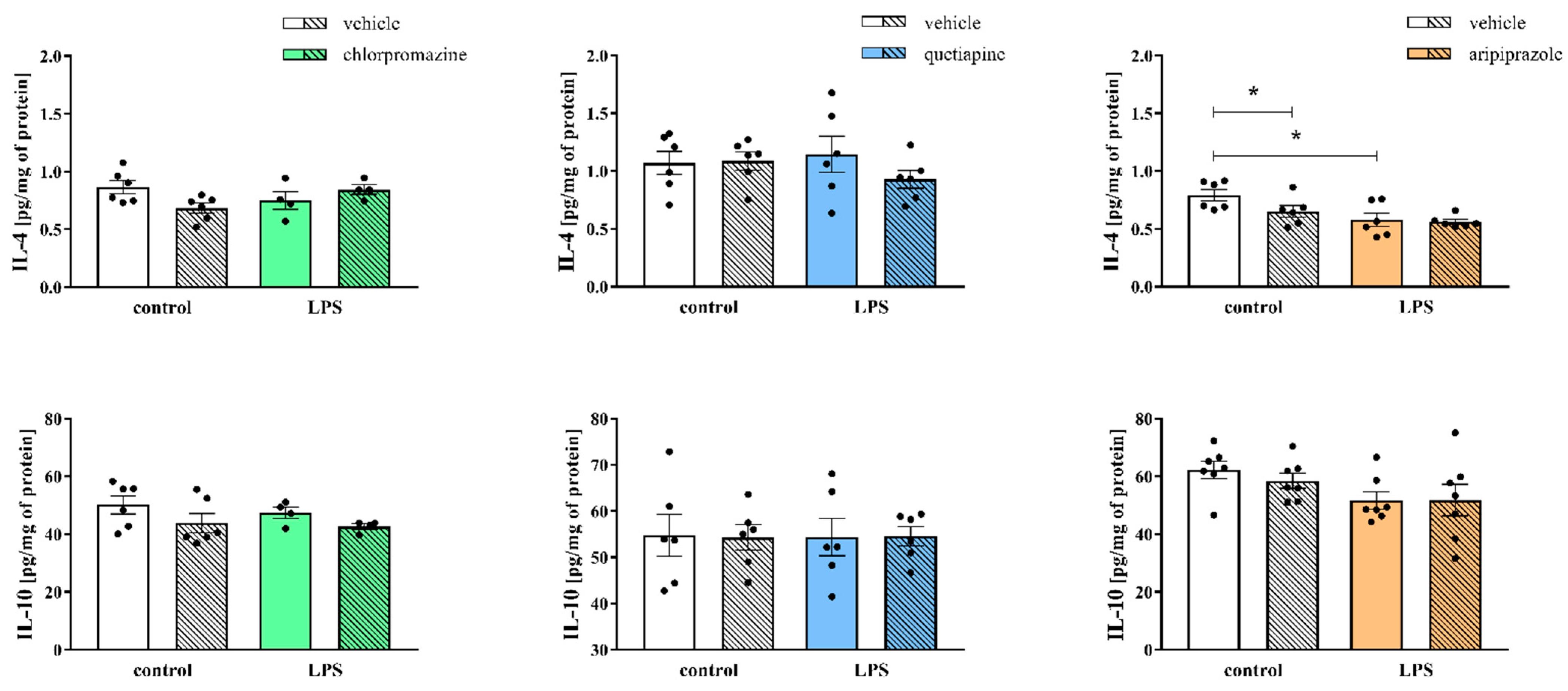
3.4. Levels of Pro- and Anti-Inflammatory Cytokines in the Frontal Cortices of the Offspring
3.5. Cx3cl1, Cx3cr1, Cd200, Cd200r, Cd40, Cd68, Arg1 and Igf-1 mRNA Expression in the Hippocampi of the Offspring
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jauhar, S.; Johnstone, M.; McKenna, P.J. Schizophrenia. Lancet 2022, 399, 473–486. [Google Scholar] [CrossRef] [PubMed]
- Mäki, P.; Veijola, J.; Jones, P.B.; Murray, G.K.; Koponen, H.; Tienari, P.; Miettunen, J.; Tanskanen, P.; Wahlberg, K.E.; Koskinen, J.; et al. Predictors of schizophrenia—A review. Br. Med. Bull. 2005, 73–74, 1–15. [Google Scholar] [CrossRef] [PubMed]
- van Os, J.; Kapur, S. Schizophrenia. Lancet 2009, 374, 635–645. [Google Scholar] [CrossRef]
- Saetre, P.; Emilsson, L.; Axelsson, E.; Kreuger, J.; Lindholm, E.; Jazin, E. Inflammation-related genes up-regulated in schizophrenia brains. BMC Psychiatry 2007, 7, 1–10. [Google Scholar] [CrossRef]
- Trépanier, M.O.; Hopperton, K.E.; Mizrahi, R.; Mechawar, N.; Bazinet, R.P. Postmortem evidence of cerebral inflammation in schizophrenia: A systematic review. Mol. Psychiatry 2016, 21, 1009–1026. [Google Scholar] [CrossRef] [PubMed]
- Barichello, T.; Simoes, L.R.; Quevedo, J.; Zhang, X.Y. Microglial Activation and Psychotic Disorders: Evidence from Pre-clinical and Clinical Studies. In Neuroinflammation and Schizophrenia; Current Topics in Behavioral Neurosciences; Springer: Cham, Switzerland, 2020; Volume 44, pp. 161–205. [Google Scholar]
- Potvin, S.; Stip, E.; Sepehry, A.A.; Gendron, A.; Bah, R.; Kouassi, E. Inflammatory Cytokine Alterations in Schizophrenia: A Systematic Quantitative Review. Biol. Psychiatry 2008, 63, 801–808. [Google Scholar] [CrossRef]
- Miller, B.J.; Bickley, P.; Seabolt, W.; Mellor, A.; Kirkpatrick, B. Meta-Analysis of Cytokine Alterations in Schizophrenia: Clinical Status and Antipsychotic Effects. Biol. Psychiatry 2011, 70, 663–671. [Google Scholar] [CrossRef] [PubMed]
- Estes, M.L.; McAllister, A.K. Maternal immune activation: Implications for neuropsychiatric disorders. Science. 2016, 353, 772–777. [Google Scholar] [CrossRef]
- Müller, N. Inflammation in schizophrenia: Pathogenetic aspects and therapeutic considerations. Schizophr. Bull. 2018, 44, 973–982. [Google Scholar] [CrossRef]
- Chamera, K.; Trojan, E.; Szuster-Głuszczak, M.; Basta-Kaim, A. The Potential Role of Dysfunctions in Neuron-Microglia Communication in the Pathogenesis of Brain Disorders. Curr. Neuropharmacol. 2020, 18, 408–430. [Google Scholar] [CrossRef]
- Romero, E.; Ali, C.; Molina-Holgado, E.; Castellano, B.; Guaza, C.; Borrell, J. Neurobehavioral and immunological consequences of prenatal immune activation in rats. Influence of antipsychotics. Neuropsychopharmacology 2007, 32, 1791–1804. [Google Scholar] [CrossRef] [PubMed]
- Chamera, K.; Kotarska, K.; Szuster-Głuszczak, M.; Trojan, E.; Skórkowska, A.; Pomierny, B.; Krzyżanowska, W.; Bryniarska, N.; Basta-Kaim, A. The prenatal challenge with lipopolysaccharide and polyinosinic:polycytidylic acid disrupts CX3CL1-CX3CR1 and CD200-CD200R signalling in the brains of male rat offspring: A link to schizophrenia-like behaviours. J. Neuroinflamm. 2020, 17, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Csatlosova, K.; Bogi, E.; Durisova, B.; Grinchii, D.; Paliokha, R.; Moravcikova, L.; Lacinova, L.; Jezova, D.; Dremencov, E. Maternal immune activation in rats attenuates the excitability of monoamine-secreting neurons in adult offspring in a sex-specific way. Eur. Neuropsychopharmacol. 2021, 43, 82–91. [Google Scholar] [CrossRef]
- Vojtechova, I.; Maleninska, K.; Kutna, V.; Klovrza, O.; Tuckova, K.; Petrasek, T.; Stuchlik, A. Behavioral Alterations and Decreased Number of Parvalbumin-Positive Interneurons in Wistar Rats after Maternal Immune Activation by Lipopolysaccharide: Sex Matters. Int. J. Mol. Sci. 2021, 22, 3274. [Google Scholar] [CrossRef]
- Basta-Kaim, A.; Szczęsny, E.; Leśkiewicz, M.; Głombik, K.; Budziszewska, B.; Regulska, M.; Kubera, M.; Nowak, W.; Wędzony, K.; Lasoń, W. Maternal immune activation leads to age-related behavioral and immunological changes in male rat offspring—The effect of antipsychotic drugs. Pharmacol. Rep. 2012, 64, 1400–1410. [Google Scholar] [CrossRef]
- Borrell, J.; Vela, J.M.; Arévalo-Martin, A.; Molina-Holgado, E.; Guaza, C. Prenatal immune challenge disrupts sensorimotor gating in adult rats: Implications for the etiopathogenesis of schizophrenia. Neuropsychopharmacology 2002, 26, 204–215. [Google Scholar] [CrossRef]
- Santos-Toscano, R.; Ucha, M.; Borcel, É.; Ambrosio, E.; Higuera-Matas, A. Maternal immune activation is associated with a lower number of dopamine receptor 3-expressing granulocytes with no alterations in cocaine reward, resistance to extinction or cue-induced reinstatement. Pharmacol. Biochem. Behav. 2020, 193, 172930. [Google Scholar] [CrossRef]
- Chamera, K.; Szuster-Głuszczak, M.; Trojan, E.; Basta-Kaim, A. Maternal Immune Activation Sensitizes Male Offspring Rats to Lipopolysaccharide-Induced Microglial Deficits Involving the Dysfunction of CD200–CD200R and CX3CL1–CX3CR1 Systems. Cells 2020, 9, 1676. [Google Scholar] [CrossRef]
- Basta-Kaim, A.; Fijał, K.; Budziszewska, B.; Regulska, M.; Leśkiewicz, M.; Kubera, M.; Gołembiowska, K.; Lasoń, W.; Wędzony, K. Prenatal lipopolysaccharide treatment enhances MK-801-induced psychotomimetic effects in rats. Pharmacol. Biochem. Behav. 2011, 98, 241–249. [Google Scholar] [CrossRef] [PubMed]
- Basta-Kaim, A.; Budziszewska, B.; Leśkiewicz, M.; Fijał, K.; Regulska, M.; Kubera, M.; Wędzony, K.; Lasoń, W. Hyperactivity of the hypothalamus–pituitary–adrenal axis in lipopolysaccharide-induced neurodevelopmental model of schizophrenia in rats: Effects of antipsychotic drugs. Eur. J. Pharmacol. 2011, 650, 586–595. [Google Scholar] [CrossRef]
- Waterhouse, U.; Roper, V.E.; Brennan, K.A.; Ellenbroek, B.A. Nicotine ameliorates cognitive deficits induced by maternal LPS exposure: A study in rats. Dis. Model. Mech. 2016, 9, 1159–1167. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, U.; Brennan, K.A.; Ellenbroek, B.A. Nicotine self-administration reverses cognitive deficits in a rat model for schizophrenia. Addict. Biol. 2018, 23, 620–630. [Google Scholar] [CrossRef]
- Chamera, K.; Szuster-Głuszczak, M.; Basta-Kaim, A. Shedding light on the role of CX3CR1 in the pathogenesis of schizophrenia. Pharmacol. Rep. 2021, 73, 1063–1078. [Google Scholar] [CrossRef] [PubMed]
- Manich, G.; Recasens, M.; Valente, T.; Almolda, B.; González, B.; Castellano, B. Role of the CD200-CD200R Axis During Homeostasis and Neuroinflammation. Neuroscience 2019, 405, 118–136. [Google Scholar] [CrossRef]
- Biber, K.; Neumann, H.; Inoue, K.; Boddeke, H.W.G.M. Neuronal ‘On’ and ‘Off’ signals control microglia. Trends Neurosci. 2007, 30, 596–602. [Google Scholar] [CrossRef] [PubMed]
- Denieffe, S.; Kelly, R.J.; McDonald, C.; Lyons, A.; Lynch, M.A. Classical activation of microglia in CD200-deficient mice is a consequence of blood brain barrier permeability and infiltration of peripheral cells. Brain. Behav. Immun. 2013, 34, 86–97. [Google Scholar] [CrossRef] [PubMed]
- Paolicelli, R.C.; Bisht, K.; Tremblay, M.-È. Fractalkine regulation of microglial physiology and consequences on the brain and behavior. Front. Cell. Neurosci. 2014, 8. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, A.; Yang, L.; Itoh, S.; Mori, K.; Tanaka, J. Microglia, a potential source of neurons, astrocytes, and oligodendrocytes. Glia 2004, 45, 96–104. [Google Scholar] [CrossRef]
- Du, W.; Bos, P.D. Tracing bone marrow-derived microglia in brain metastatic tumors. In Methods in Enzymology; Elsevier Inc.: Amsterdam, The Netherlands, 2020; Volume 635, pp. 95–110. ISBN 9780128186770. [Google Scholar]
- Gomez-Nicola, D.; Perry, V.H. Microglial Dynamics and Role in the Healthy and Diseased Brain: A Paradigm of Functional Plasticity. Neurosci. 2015, 21, 169–184. [Google Scholar] [CrossRef] [PubMed]
- Wolf, S.A.; Boddeke, H.W.G.M.; Kettenmann, H. Microglia in Physiology and Disease. Annu. Rev. Physiol. 2017, 79, 619–643. [Google Scholar] [CrossRef] [PubMed]
- Lannes, N.; Eppler, E.; Etemad, S.; Yotovski, P.; Filgueira, L. Microglia at center stage: A comprehensive review about the versatile and unique residential macrophages of the central nervous system. Oncotarget 2017, 8, 114393–114413. [Google Scholar] [CrossRef]
- Gober, R.; Ardalan, M.; Shiadeh, S.M.J.; Duque, L.; Garamszegi, S.P.; Ascona, M.; Barreda, A.; Sun, X.; Mallard, C.; Vontell, R.T. Microglia activation in postmortem brains with schizophrenia demonstrates distinct morphological changes between brain regions. Brain Pathol. 2022, 32, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Snijders, G.J.L.J.; van Zuiden, W.; Sneeboer, M.A.M.; Berdenis van Berlekom, A.; van der Geest, A.T.; Schnieder, T.; MacIntyre, D.J.; Hol, E.M.; Kahn, R.S.; de Witte, L.D. A loss of mature microglial markers without immune activation in schizophrenia. Glia 2021, 69, 1251–1267. [Google Scholar] [CrossRef]
- Uranova, N.A.; Vikhreva, O.V.; Rakhmanova, V.I. Abnormal microglial reactivity in gray matter of the prefrontal cortex in schizophrenia. Asian J. Psychiatr. 2021, 63. [Google Scholar] [CrossRef] [PubMed]
- Leucht, S.; Barnes, T.R.E.; Kissling, W.; Engel, R.R.; Correll, C.; Kane, J.M. Relapse prevention in schizophrenia with new-generation antipsychotics: A systematic review and exploratory meta-analysis of randomized, controlled trials. Am. J. Psychiatry 2003, 160, 1209–1222. [Google Scholar] [CrossRef]
- Keefe, R.S.E.; Seidman, L.J.; Christensen, B.K.; Hamer, R.M.; Sharma, T.; Sitskoorn, M.M.; Lewine, R.R.J.; Yurgelun-Todd, D.A.; Gur, R.C.; Tohen, M.; et al. Comparative effect of atypical and conventional antipsychotic drugs on neurocognition in first-episode psychosis: A randomized, double-blind trial of olanzapine versus low doses of haloperidol. Am. J. Psychiatry 2004, 161, 985–995. [Google Scholar] [CrossRef]
- Sabe, M.; Zhao, N.; Crippa, A.; Kaiser, S. Antipsychotics for negative and positive symptoms of schizophrenia: Dose-response meta-analysis of randomized controlled acute phase trials. npj Schizophr. 2021, 7, 43. [Google Scholar] [CrossRef]
- Meltzer, H.Y. Update on typical and atypical antipsychotic drugs. Annu. Rev. Med. 2013, 64, 393–406. [Google Scholar] [CrossRef] [PubMed]
- Stępnicki, P.; Kondej, M.; Kaczor, A.A. Current Concepts and Treatments of Schizophrenia. Molecules 2018, 23, 2087. [Google Scholar] [CrossRef] [PubMed]
- Remington, G.; Hahn, M.K.; Agarwal, S.M.; Chintoh, A.; Agid, O. Schizophrenia: Antipsychotics and drug development. Behav. Brain Res. 2021, 414. [Google Scholar] [CrossRef] [PubMed]
- Saha, K.B.; Bo, L.; Zhao, S.; Xia, J.; Sampson, S.; Zaman, R.U. Chlorpromazine versus atypical antipsychotic drugs for schizophrenia. Cochrane Database Syst. Rev. 2016, CD010631. [Google Scholar] [CrossRef] [PubMed]
- Aringhieri, S.; Carli, M.; Kolachalam, S.; Verdesca, V.; Cini, E.; Rossi, M.; McCormick, P.J.; Corsini, G.U.; Maggio, R.; Scarselli, M. Molecular targets of atypical antipsychotics: From mechanism of action to clinical differences. Pharmacol. Ther. 2018, 192, 20–41. [Google Scholar] [CrossRef]
- Grinchii, D.; Dremencov, E. Mechanism of action of atypical antipsychotic drugs in mood disorders. Int. J. Mol. Sci. 2020, 21, 9532. [Google Scholar] [CrossRef] [PubMed]
- Kusumi, I.; Boku, S.; Takahashi, Y. Psychopharmacology of atypical antipsychotic drugs: From the receptor binding profile to neuroprotection and neurogenesis. Psychiatry Clin. Neurosci. 2015, 69, 243–258. [Google Scholar] [CrossRef]
- Baldwin, C.M.; Scott, L.J. Quetiapine extended release: In schizophrenia. CNS Drugs 2009, 23, 261–269. [Google Scholar] [CrossRef] [PubMed]
- Mauri, M.C.; Paletta, S.; Maffini, M.; Colasanti, A.; Dragogna, F.; Di Pace, C.; Altamura, A.C. Clinical pharmacology of atypical antipsychotics: An update. EXCLI J. 2014, 13, 1163–1191. [Google Scholar] [PubMed]
- Bian, Q.; Kato, T.; Monji, A.; Hashioka, S.; Mizoguchi, Y.; Horikawa, H.; Kanba, S. The effect of atypical antipsychotics, perospirone, ziprasidone and quetiapine on microglial activation induced by interferon-γ. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2008, 32, 42–48. [Google Scholar] [CrossRef] [PubMed]
- Shao, Y.; Peng, H.; Huang, Q.; Kong, J.; Xu, H. Quetiapine mitigates the neuroinflammation and oligodendrocyte loss in the brain of C57BL/6 mouse following cuprizone exposure for one week. Eur. J. Pharmacol. 2015, 765, 249–257. [Google Scholar] [CrossRef]
- Zhu, S.; Shi, R.; Li, V.; Wang, J.; Zhang, R.; Tempier, A.; He, J.; Kong, J.; Wang, J.-F.; Li, X.-M. Quetiapine Attenuates Glial Activation and Proinflammatory Cytokines in APP/PS1 Transgenic Mice via Inhibition of Nuclear Factor- B Pathway. Int. J. Neuropsychopharmacol. 2015, 18, 1–11. [Google Scholar] [CrossRef]
- Casey, A.B.; Canal, C.E. Classics in Chemical Neuroscience: Aripiprazole. ACS Chem. Neurosci. 2017, 8, 1135–1146. [Google Scholar] [CrossRef] [Green Version]
- Kato, T.; Mizoguchi, Y.; Monji, A.; Horikawa, H.; Suzuki, S.O.; Seki, Y.; Iwaki, T.; Hashioka, S.; Kanba, S. Inhibitory effects of aripiprazole on interferon-γ-induced microglial activation via intracellular Ca2+ regulation in vitro. J. Neurochem. 2008, 106, 815–825. [Google Scholar] [CrossRef] [PubMed]
- Obuchowicz, E.; Turchan, J. Effects of acute or long-term treatment with chlorpromazine, haloperidol or sulpiride on neuropeptide Y-like immunoreactivity concentrations in the nucleus accumbens of rat. Eur. Neuropsychopharmacol. 1999, 9, 51–59. [Google Scholar] [CrossRef]
- Huang, Q.-Y.; Li, X.-F.; Liu, S.-P. E-cadherin and caveolin-1 alterations in the heart of rats having undergone chlorpromazine-induced toxicity. Mol. Med. Rep. 2012, 5, 705–709. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Liu, F.; Zu, Q.; Xu, Z.; Zheng, H.; Li, X.; Wang, W. Chronic administration of quetiapine attenuates the phencyclidine-induced recognition memory impairment and hippocampal oxidative stress in rats. Neuroreport 2018, 29, 1099–1103. [Google Scholar] [CrossRef] [PubMed]
- Murata, Y.; Matsuda, H.; Mikami, Y.; Hirose, S.; Mori, M.; Ohe, K.; Mine, K.; Enjoji, M. Chronic administration of quetiapine stimulates dorsal hippocampal proliferation and immature neurons of male rats, but does not reverse psychosocial stress-induced hyponeophagic behavior. Psychiatry Res. 2019, 272, 411–418. [Google Scholar] [CrossRef]
- Russo, E.; Citraro, R.; Davoli, A.; Gallelli, L.; Donato Di Paola, E.; De Sarro, G. Ameliorating effects of aripiprazole on cognitive functions and depressive-like behavior in a genetic rat model of absence epilepsy and mild-depression comorbidity. Neuropharmacology 2013, 64, 371–379. [Google Scholar] [CrossRef]
- Tuplin, E.W.; Stocco, M.R.; Holahan, M.R. Attenuation of MK-801-induced behavioral perseveration by typical and atypical antipsychotic pretreatment in rats. Behav. Neurosci. 2015, 129, 399–411. [Google Scholar] [CrossRef]
- Braff, D.L.; Light, G.A. The use of neurophysiological endophenotypes to understand the genetic basis of schizophrenia. Dialogues Clin. Neurosci. 2005, 7, 125–135. [Google Scholar] [CrossRef]
- Braff, D.L.; Geyer, M.A.; Light, G.A.; Sprock, J.; Perry, W.; Cadenhead, K.S.; Swerdlow, N.R. Impact of prepulse characteristics on the detection of sensorimotor gating deficits in schizophrenia. Schizophr. Res. 2001, 49, 171–178. [Google Scholar] [CrossRef]
- Moriwaki, M.; Kishi, T.; Takahashi, H.; Hashimoto, R.; Kawashima, K.; Okochi, T.; Kitajima, T.; Furukawa, O.; Fujita, K.; Takeda, M.; et al. Prepulse inhibition of the startle response with chronic schizophrenia: A replication study. Neurosci. Res. 2009, 65, 259–262. [Google Scholar] [CrossRef]
- Mena, A.; Ruiz-Salas, J.C.; Puentes, A.; Dorado, I.; Ruiz-Veguilla, M.; De la Casa, L.G. Reduced prepulse inhibition as a biomarker of schizophrenia. Front. Behav. Neurosci. 2016, 10. [Google Scholar] [CrossRef] [PubMed]
- Alam, M.; Angelov, S.; Stemmler, M.; von Wrangel, C.; Krauss, J.K.; Schwabe, K. Neuronal activity of the prefrontal cortex is reduced in rats selectively bred for deficient sensorimotor gating. Prog. Neuro-Psychopharmacology Biol. Psychiatry 2015, 56, 174–184. [Google Scholar] [CrossRef]
- Oliveras, I.; Río-Álamos, C.; Cañete, T.; Blázquez, G.; Martínez-Membrives, E.; Giorgi, O.; Corda, M.G.; Tobeña, A.; Fernández-Teruel, A. Prepulse inhibition predicts spatial working memory performance in the inbred Roman high- and low-avoidance rats and in genetically heterogeneous NIH-HS rats: Relevance for studying pre-attentive and cognitive anomalies in schizophrenia. Front. Behav. Neurosci. 2015, 9, 1–16. [Google Scholar] [CrossRef]
- Romero, E.; Guaza, C.; Castellano, B.; Borrell, J. Ontogeny of sensorimotor gating and immune impairment induced by prenatal immune challenge in rats: Implications for the etiopathology of schizophrenia. Mol. Psychiatry 2010, 15, 372–383. [Google Scholar] [CrossRef]
- Basta-Kaim, A.; Fijał, K.; Ślusarczyk, J.; Trojan, E.; Głombik, K.; Budziszewska, B.; Leśkiewicz, M.; Regulska, M.; Kubera, M.; Lasoń, W.; et al. Prenatal administration of lipopolysaccharide induces sex-dependent changes in glutamic acid decarboxylase and parvalbumin in the adult rat brain. Neuroscience 2015, 287, 78–92. [Google Scholar] [CrossRef]
- Kohl, S.; Heekeren, K.; Klosterkötter, J.; Kuhn, J. Prepulse inhibition in psychiatric disorders—Apart from schizophrenia. J. Psychiatr. Res. 2013, 47, 445–452. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.; Powell, S.B. Sensorimotor gating deficits in “two-hit” models of schizophrenia risk factors. Schizophr. Res. 2018, 198, 68–83. [Google Scholar] [CrossRef]
- Witten, L.; Bastlund, J.F.; Glenthøj, B.Y.; Bundgaard, C.; Steiniger-Brach, B.; Mørk, A.; Oranje, B. Comparing Pharmacological Modulation of Sensory Gating in Healthy Humans and Rats: The Effects of Reboxetine and Haloperidol. Neuropsychopharmacology 2016, 41, 638–645. [Google Scholar] [CrossRef] [PubMed]
- Fitch, R.H.; Threlkeld, S.W.; McClure, M.M.; Peiffer, A.M. Use of a modified prepulse inhibition paradigm to assess complex auditory discrimination in rodents. Brain Res. Bull. 2008, 76, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Stip, E.; Rizvi, T.A.; Mustafa, F.; Javaid, S.; Aburuz, S.; Ahmed, N.N.; Abdel Aziz, K.; Arnone, D.; Subbarayan, A.; Al Mugaddam, F.; et al. The Large Action of Chlorpromazine: Translational and Transdisciplinary Considerations in the Face of COVID-19. Front. Pharmacol. 2020, 11, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Basta-Kaim, A.; Budziszewska, B.; Jaworska-Feil, L.; Tetich, M.; Leśkiewicz, M.; Kubera, M.; Lasoń, W. Chlorpromazine inhibits the glucocorticoid receptor-mediated gene transcription in a calcium-dependent manner. Neuropharmacology 2002, 43, 1035–1043. [Google Scholar] [CrossRef]
- Dejanovic, B.; Stevanovic, I.; Ninkovic, M.; Stojanovic, I.; Lavrnja, I.; Radicevic, T.; Pavlovic, M. Agmatine protection against chlorpromazine-induced forebrain cortex injury in rats. J. Vet. Sci. 2016, 17, 53. [Google Scholar] [CrossRef] [PubMed]
- Le Pen, G.; Moreau, J.-L. Disruption of Prepulse Inhibition of Startle Reflex in a Neurodevelopmental Model of Schizophrenia Reversal by Clozapine, Olanzapine and Risperidone But Not by Haloperidol. Neuropsychopharmacology 2002, 27, 1–11. [Google Scholar] [CrossRef]
- Swerdlow, N.R.; Bakshi, V.; Waikar, M.; Taaid, N.; Geyer, M.A. Seroquel, clozapine and chlorpromazine restore sensorimotor gating in ketamine-treated rats. Psychopharmacology 1998, 140, 75–80. [Google Scholar] [CrossRef]
- Yamada, S.; Harano, M.; Annoh, N.; Nakamura, K.; Tanaka, M. Involvement of serotonin 2A receptors in phencyclidine-induced disruption of prepulse inhibition of the acoustic startle in rats. Biol. Psychiatry 1999, 46, 832–838. [Google Scholar] [CrossRef]
- Yamashita, N.; Takahashi, A.; Takao, K.; Yamamoto, T.; Kolattukudy, P.; Miyakawa, T.; Goshima, Y. Mice lacking collapsin response mediator protein 1 manifest hyperactivity, impaired learning and memory, and impaired prepulse inhibition. Front. Behav. Neurosci. 2013, 7, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Ratajczak, P.; Kus, K.; Skurzyńska, M.; Nowakowska, E. The influence of aripiprazole and venlafaxine on the antidepressant-like effect observed in prenatally stressed rats (animal model of depression). Hum. Exp. Toxicol. 2018, 37, 972–982. [Google Scholar] [CrossRef] [PubMed]
- Lech, M.A.; Kamińska, K.; Leśkiewicz, M.; Lorenc-Koci, E.; Rogóż, Z. Impact of repeated co-treatment with escitalopram and aripiprazole on the schizophrenia-like behaviors and BDNF mRNA expression in the adult Sprague–Dawley rats exposed to glutathione deficit during early postnatal development of the brain. Pharmacol. Rep. 2021, 73, 1712–1723. [Google Scholar] [CrossRef]
- Lech, M.A.; Kaminska, K.; Wasik, A.; Lorenc-Koci, E.; Rogoz, Z. Repeated co-treatment with mirtazapine and aripiprazole reversed the schizophrenia-like behaviors and increased the brain-derived neurotrophic factor mRNA expression in the adult Sprague-Dawley rats exposed to glutathione deficit during early postnatal brain development. J. Physiol. Pharmacol. 2021, 72, 699–709. [Google Scholar] [CrossRef]
- Richtand, N.M.; Ahlbrand, R.; Horn, P.; Tambyraja, R.; Grainger, M.; Bronson, S.L.; McNamara, R.K. Fluoxetine and aripiprazole treatment following prenatal immune activation exert longstanding effects on rat locomotor response. Physiol. Behav. 2012, 106, 171–177. [Google Scholar] [CrossRef] [Green Version]
- Hereta, M.; Kamińska, K.; Białoń, M.; Wąsik, A.; Lorenc-Koci, E.; Rogóż, Z. Effect of combined treatment with aripiprazole and antidepressants on the MK-801-induced deficits in recognition memory in novel recognition test and on the release of monoamines in the rat frontal cortex. Behav. Brain Res. 2020, 393, 112769. [Google Scholar] [CrossRef]
- Mace, S.; Taylor, D. Aripiprazole: Dose-response relationship in schizophrenia and schizoaffective disorder. CNS Drugs 2009, 23, 773–780. [Google Scholar] [CrossRef] [PubMed]
- Sajeev Kumar, P.B.; Pandey, R.S.; Thirthalli, J.; Siva Kumar, P.T.; Naveen Kumar, C. A Comparative Study of Short Term Efficacy of Aripiprazole and Risperidone in Schizophrenia. Curr. Neuropharmacol. 2017, 15, 1073–1084. [Google Scholar] [CrossRef]
- Fejgin, K.; Safonov, S.; Pålsson, E.; Wass, C.; Engel, J.A.; Svensson, L.; Klamer, D. The atypical antipsychotic, aripiprazole, blocks phencyclidine-induced disruption of prepulse inhibition in mice. Psychopharmacology 2007, 191, 377–385. [Google Scholar] [CrossRef]
- Torrisi, S.A.; Laudani, S.; Contarini, G.; De Luca, A.; Geraci, F.; Managò, F.; Papaleo, F.; Salomone, S.; Drago, F.; Leggio, G.M. Dopamine, Cognitive Impairments and Second-Generation Antipsychotics: From Mechanistic Advances to More Personalized Treatments. Pharmaceuticals 2020, 13, 365. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Zu, Q.; Wen, C.; Liu, Q.; You, P.; Li, X.; Wang, W. Quetiapine Attenuates Schizophrenia-Like Behaviors and Demyelination in a MK-801–Induced Mouse Model of Schizophrenia. Front. Psychiatry 2020, 11, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Seeman, P. Schizophrenia and dopamine receptors. Eur. Neuropsychopharmacol. 2013, 23, 999–1009. [Google Scholar] [CrossRef] [PubMed]
- Amato, D. Serotonin in antipsychotic drugs action. Behav. Brain Res. 2015, 277, 125–135. [Google Scholar] [CrossRef]
- Leza, J.C.; García-Bueno, B.; Bioque, M.; Arango, C.; Parellada, M.; Do, K.; O’Donnell, P.; Bernardo, M. Inflammation in schizophrenia: A question of balance. Neurosci. Biobehav. Rev. 2015, 55, 612–626. [Google Scholar] [CrossRef] [PubMed]
- Wohleb, E.S. Neuron–Microglia Interactions in Mental Health Disorders: “For Better, and For Worse”. Front. Immunol. 2016, 7, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szepesi, Z.; Manouchehrian, O.; Bachiller, S.; Deierborg, T. Bidirectional Microglia–Neuron Communication in Health and Disease. Front. Cell. Neurosci. 2018, 12. [Google Scholar] [CrossRef]
- Roudebush, R.E.; Berry, P.L.; Layman, N.K.; Butler, L.D.; Bryant, H.U. Dissociation of immunosuppression by chlorpromazine and trifluoperazine from pharmacologic activities as dopamine antagonists. Int. J. Immunopharmacol. 1991, 13, 961–968. [Google Scholar] [CrossRef]
- Yoo, S.; Kim, M.-Y.; Cho, J.Y. Syk and Src-targeted anti-inflammatory activity of aripiprazole, an atypical antipsychotic. Biochem. Pharmacol. 2018, 148, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Chistiakov, D.A.; Killingsworth, M.C.; Myasoedova, V.A.; Orekhov, A.N.; Bobryshev, Y.V. CD68/macrosialin: Not just a histochemical marker. Lab. Investig. 2017, 97, 4–13. [Google Scholar] [CrossRef]
- Rabinowitz, S.S.; Gordon, S. Macrosialin, a macrophage-restricted membrane sialoprotein differentially glycosylated in response to inflammatory stimuli. J. Exp. Med. 1991, 174, 827–836. [Google Scholar] [CrossRef]
- López-González, I.; Pinacho, R.; Vila, È.; Escanilla, A.; Ferrer, I.; Ramos, B. Neuroinflammation in the dorsolateral prefrontal cortex in elderly chronic schizophrenia. Eur. Neuropsychopharmacol. 2019, 29, 384–396. [Google Scholar] [CrossRef]
- De Picker, L.J.; Victoriano, G.M.; Richards, R.; Gorvett, A.J.; Lyons, S.; Buckland, G.R.; Tofani, T.; Norman, J.L.; Chatelet, D.S.; Nicoll, J.A.R.; et al. Immune environment of the brain in schizophrenia and during the psychotic episode: A human post-mortem study. Brain. Behav. Immun. 2021, 97, 319–327. [Google Scholar] [CrossRef] [PubMed]
- Monji, A.; Kato, T.; Kanba, S. Cytokines and schizophrenia: Microglia hypothesis of schizophrenia. Psychiatry Clin. Neurosci. 2009, 63, 257–265. [Google Scholar] [CrossRef]
- Müller, N.; Weidinger, E.; Leitner, B.; Schwarz, M.J. The role of inflammation in schizophrenia. Front. Neurosci. 2015, 9. [Google Scholar] [CrossRef]
- Giovanoli, S.; Weber-Stadlbauer, U.; Schedlowski, M.; Meyer, U.; Engler, H. Prenatal immune activation causes hippocampal synaptic deficits in the absence of overt microglia anomalies. Brain. Behav. Immun. 2016, 55, 25–38. [Google Scholar] [CrossRef] [Green Version]
- Shin, H.; Song, J.-H. Antipsychotics, chlorpromazine and haloperidol inhibit voltage-gated proton currents in BV2 microglial cells. Eur. J. Pharmacol. 2014, 738, 256–262. [Google Scholar] [CrossRef]
- Heikkila, R.E.; Cohen, G.; Manian, A.A. Reactivity of various phenothiazine derivatives with oxygen and oxygen radicals. Biochem. Pharmacol. 1975, 24, 363–369. [Google Scholar] [CrossRef]
- Cadet, J.L.; Perumal, A.S. Chronic treatment with prolixin causes oxidative stress in rat brain. Biol. Psychiatry 1990, 28, 738–740. [Google Scholar] [CrossRef]
- Kato, T.A.; Monji, A.; Yasukawa, K.; Mizoguchi, Y.; Horikawa, H.; Seki, Y.; Hashioka, S.; Han, Y.-H.; Kasai, M.; Sonoda, N.; et al. Aripiprazole inhibits superoxide generation from phorbol-myristate-acetate (PMA)-stimulated microglia in vitro: Implication for antioxidative psychotropic actions via microglia. Schizophr. Res. 2011, 129, 172–182. [Google Scholar] [CrossRef] [PubMed]
- Seki, Y.; Kato, T.A.; Monji, A.; Mizoguchi, Y.; Horikawa, H.; Sato-Kasai, M.; Yoshiga, D.; Kanba, S. Pretreatment of aripiprazole and minocycline, but not haloperidol, suppresses oligodendrocyte damage from interferon-γ-stimulated microglia in co-culture model. Schizophr. Res. 2013, 151, 20–28. [Google Scholar] [CrossRef]
- Sato-Kasai, M.; Kato, T.A.; Ohgidani, M.; Mizoguchi, Y.; Sagata, N.; Inamine, S.; Horikawa, H.; Hayakawa, K.; Shimokawa, N.; Kyuragi, S.; et al. Aripiprazole inhibits polyI:C-induced microglial activation possibly via TRPM7. Schizophr. Res. 2016, 178, 35–43. [Google Scholar] [CrossRef]
- Zhao, Z.; Luo, G.; Liu, M.; Guo, H.; Xue, M.; Wang, X.; Li, X.-M.; He, J. Quetiapine reduces microglial number in the hippocampus of a transgenic mouse model of Alzheimer’s disease. Neuroreport 2014, 25, 870–874. [Google Scholar] [CrossRef] [PubMed]
- Turra, B.O.; Barbisan, F.; Azzolin, V.F.; Teixeira, C.F.; Flores, T.; Braun, L.E.; de Oliveira Nerys, D.A.; Rissi, V.B.; de Oliveira Alves, A.; Assmann, C.E.; et al. Unmetabolized quetiapine exerts an in vitro effect on innate immune cells by modulating inflammatory response and neutrophil extracellular trap formation. Biomed. Pharmacother. 2020, 131, 110497. [Google Scholar] [CrossRef]
- Mantere, O.; Trontti, K.; García-González, J.; Balcells, I.; Saarnio, S.; Mäntylä, T.; Lindgren, M.; Kieseppä, T.; Raij, T.; Honkanen, J.K.; et al. Immunomodulatory effects of antipsychotic treatment on gene expression in first-episode psychosis. J. Psychiatr. Res. 2019, 109, 18–26. [Google Scholar] [CrossRef]
- Dawidowski, B.; Grelecki, G.; Biłgorajski, A.; Podwalski, P.; Misiak, B.; Samochowiec, J. Effect of Antipsychotic Treatment on Neutrophil-to-Lymphocyte Ratio during Hospitalization for Acute Psychosis in the Course of Schizophrenia—A Cross-Sectional Retrospective Study. J. Clin. Med. 2021, 11, 232. [Google Scholar] [CrossRef]
- Na, K.-S.; Jung, H.-Y.; Kim, Y.-K. The role of pro-inflammatory cytokines in the neuroinflammation and neurogenesis of schizophrenia. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2014, 48, 277–286. [Google Scholar] [CrossRef]
- Zhang, X.Y.; Zhou, D.F.; Qi, L.Y.; Chen, S.; Cao, L.Y.; Chen, D.C.; Xiu, M.H.; Wang, F.; Wu, G.Y.; Lu, L.; et al. Superoxide dismutase and cytokines in chronic patients with schizophrenia: Association with psychopathology and response to antipsychotics. Psychopharmacology 2009, 204, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Dawidowski, B.; Górniak, A.; Podwalski, P.; Lebiecka, Z.; Misiak, B.; Samochowiec, J. The Role of Cytokines in the Pathogenesis of Schizophrenia. J. Clin. Med. 2021, 10, 3849. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.A.; Burgess, S.; Suckling, J.; Lalousis, P.A.; Batool, F.; Griffiths, S.L.; Palmer, E.; Karwath, A.; Barsky, A.; Gkoutos, G.V.; et al. Inflammation and Brain Structure in Schizophrenia and Other Neuropsychiatric Disorders. JAMA Psychiatry 2022, 79, 498. [Google Scholar] [CrossRef]
- Chen, A.T.; Nasrallah, H.A. Neuroprotective effects of the second generation antipsychotics. Schizophr. Res. 2019, 208, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Gabay, C.; Lamacchia, C.; Palmer, G. IL-1 pathways in inflammation and human diseases. Nat. Rev. Rheumatol. 2010, 6, 232–241. [Google Scholar] [CrossRef]
- Coelho, F.M.; Reis, H.J.; Nicolato, R.; Romano-Silva, M.A.; Teixeira, M.M.; Bauer, M.E.; Teixeira, A.L. Increased Serum Levels of Inflammatory Markers in Chronic Institutionalized Patients with Schizophrenia. Neuroimmunomodulation 2008, 15, 140–144. [Google Scholar] [CrossRef] [PubMed]
- Bradley, J. TNF-mediated inflammatory disease. J. Pathol. 2008, 214, 149–160. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Peng, W.; Wang, J.; Zhou, W.; Zhou, Y.; Ying, B. Plasma levels of IL-1Ra are associated with schizophrenia. Psychiatry Clin. Neurosci. 2019, 73, 109–115. [Google Scholar] [CrossRef]
- Jaehne, E.J.; Corrigan, F.; Toben, C.; Jawahar, M.C.; Baune, B.T. The effect of the antipsychotic drug quetiapine and its metabolite norquetiapine on acute inflammation, memory and anhedonia. Pharmacol. Biochem. Behav. 2015, 135, 136–144. [Google Scholar] [CrossRef]
- Krause, D.; Weidinger, E.; Dippel, C.; Riedel, M.; Schwarz, M.J.; Müller, N.; Myint, A.M. Impact of different antipsychotics on cytokines and tryptophan metabolites in stimulated cultures from patients with schizophrenia. Psychiatr. Danub. 2013, 25, 389–397. [Google Scholar]
- Şimşek, Ş.; Yıldırım, V.; Çim, A.; Kaya, S. Serum IL-4 and IL-10 Levels Correlate with the Symptoms of the Drug-Naive Adolescents with First Episode, Early Onset Schizophrenia. J. Child Adolesc. Psychopharmacol. 2016, 26, 721–726. [Google Scholar] [CrossRef] [PubMed]
- Na, K.-S.; Kim, Y.-K. Monocytic, Th1 and Th2 Cytokine Alterations in the Pathophysiology of Schizophrenia. Neuropsychobiology 2007, 56, 55–63. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, S.M.; Scully, P.; Dinan, T.G. Increased tumor necrosis factor-alpha concentrations with interleukin-4 concentrations in exacerbations of schizophrenia. Psychiatry Res. 2008, 160, 256–262. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.-K.; Myint, A.-M.; Verkerk, R.; Scharpe, S.; Steinbusch, H.; Leonard, B. Cytokine Changes and Tryptophan Metabolites in Medication-Naïve and Medication-Free Schizophrenic Patients. Neuropsychobiology 2009, 59, 123–129. [Google Scholar] [CrossRef] [PubMed]
- Tourjman, V.; Kouassi, É.; Koué, M.-È.; Rocchetti, M.; Fortin-Fournier, S.; Fusar-Poli, P.; Potvin, S. Antipsychotics’ effects on blood levels of cytokines in schizophrenia: A meta-analysis. Schizophr. Res. 2013, 151, 43–47. [Google Scholar] [CrossRef] [PubMed]
- Romeo, B.; Brunet-Lecomte, M.; Martelli, C.; Benyamina, A. Kinetics of Cytokine Levels during Antipsychotic Treatment in Schizophrenia: A Meta-Analysis. Int. J. Neuropsychopharmacol. 2018, 21, 828–836. [Google Scholar] [CrossRef] [PubMed]
- Noto, C.; Ota, V.K.; Gouvea, E.S.; Rizzo, L.B.; Spindola, L.M.N.; Honda, P.H.S.; Cordeiro, Q.; Belangero, S.I.; Bressan, R.A.; Gadelha, A.; et al. Effects of Risperidone on Cytokine Profile in Drug-Naive First-Episode Psychosis. Int. J. Neuropsychopharmacol. 2015, 18, 1–8. [Google Scholar] [CrossRef]
- Szymona, K.; Zdzisińska, B.; Karakuła-Juchnowicz, H.; Kocki, T.; Kandefer-Szerszeń, M.; Flis, M.; Rosa, W.; Urbańska, E.M. Correlations of Kynurenic Acid, 3-Hydroxykynurenine, sIL-2R, IFN-α, and IL-4 with Clinical Symptoms During Acute Relapse of Schizophrenia. Neurotox. Res. 2017, 32, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Marcinowicz, P.; Więdłocha, M.; Zborowska, N.; Dębowska, W.; Podwalski, P.; Misiak, B.; Tyburski, E.; Szulc, A. A Meta-Analysis of the Influence of Antipsychotics on Cytokines Levels in First Episode Psychosis. J. Clin. Med. 2021, 10, 2488. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cohort 1 | Cohort 2 | Cohort 3 |
|---|---|---|
| chlorpromazine | quetiapine | aripiprazole |
| control + vehicle to chlorpromazine (n = 13) | control + vehicle to quetiapine (n = 9) | control + vehicle to aripiprazole (n = 7) |
| control + chlorpromazine (n = 10) | control + quetiapine (n = 9) | control + aripiprazole (n = 7) |
| LPS + vehicle to chlorpromazine (n = 4) | LPS + vehicle to quetiapine (n = 9) | LPS + vehicle to aripiprazole (n = 7) |
| LPS + chlorpromazine (n = 4) | LPS + quetiapine (n = 9) | LPS + aripiprazole (n = 7) |
| (A) Cohort 1—chlorpromazine | ||||
|---|---|---|---|---|
| Factor | Gene Expression in the Frontal Cortices | |||
| Control | LPS | |||
| vehicle | chlorpromazine | vehicle | chlorpromazine | |
| Cx3cl1 | 1.38 ± 0.57 | 1.75 ± 0.71 | 5.27 ± 1.20 * | 7.16 ± 1.16 |
| Cx3cr1 | 1.28 ± 0.46 | 1.46 ± 0.60 | 5.39 ± 1.83 * | 8.29 ± 1.82 |
| Cd200 | 1.28 ± 0.43 | 1.67 ± 0.71 | 4.34 ± 1.23 * | 4.75 ± 1.11 |
| Cd200r | 1.33 ± 0.46 | 1.74 ± 0.50 | 13.24 ± 4.58 * | 11.45 ± 3.03 |
| (B) Cohort 2—quetiapine | ||||
| Factor | Gene Expression in the Frontal Cortices | |||
| Control | LPS | |||
| vehicle | quetiapine | vehicle | quetiapine | |
| Cx3cl1 | 1.01 ± 0.06 | 0.94 ± 0.12 | 1.09 ± 0.10 | 1.12 ± 0.10 |
| Cx3cr1 | 1.01 ± 0.04 | 1.27 ± 0.12 | 1.51 ± 0.23 | 1.48 ± 0.25 |
| Cd200 | 1.05 ± 0.16 | 0.42 ± 0.12 | 1.33 ± 0.41 | 1.50 ± 0.32 |
| Cd200r | 1.12 ± 0.22 | 0.60 ± 0.24 | 2.95 ± 0.65 * | 2.28 ± 0.71 |
| (C) Cohort 3—aripiprazole | ||||
| Factor | Gene Expression in the Frontal Cortices | |||
| Control | LPS | |||
| vehicle | aripiprazole | vehicle | aripiprazole | |
| Cx3cl1 | 1.05 ± 0.15 | 1.20 ± 0.21 | 1.58 ± 0.28 | 1.87 ± 0.26 |
| Cx3cr1 | 1.04 ± 0.13 | 0.92 ± 0.12 | 1.00 ± 0.20 | 1.19 ± 0.16 |
| Cd200 | 1.04 ± 0.12 | 1.16 ± 0.15 | 1.20 ± 0.17 | 1.29 ± 0.13 |
| Cd200r | 1.10 ± 0.22 | 1.79 ± 0.43 | 2.61 ± 0.53 * | 2.14 ± 0.34 |
| (A) Cohort 1—chlorpromazine | ||||
|---|---|---|---|---|
| Factor | Gene Expression in the Frontal Cortices | |||
| control | LPS | |||
| vehicle | chlorpromazine | vehicle | chlorpromazine | |
| Cd40 | 1.41 ± 0.53 | 1.14 ± 0.45 | 1.41 ± 0.53 | 1.14 ± 0.45 |
| Cd68 | 1.33 ± 0.37 | 1.19 ± 0.40 | 1.33 ± 0.37 | 1.19 ± 0.40 |
| Arg1 | 1.77 ± 0.71 | 1.96 ± 0.76 | 1.77 ± 0.71 | 1.96 ± 0.76 |
| Igf-1 | 1.12 ± 0.19 | 1.30 ± 0.25 | 1.12 ± 0.19 | 1.30 ± 0.25 |
| (B) Cohort 2—quetiapine | ||||
| Factor | Gene Expression in the Frontal Cortices | |||
| control | LPS | |||
| vehicle | quetiapine | vehicle | quetiapine | |
| Cd40 | 1.02 ± 0.06 | 0.86 ± 0.11 | 1.27 ± 0.14 | 1.02 ± 0.16 |
| Cd68 | 0.87 ± 0.05 | 0.73 ± 0.09 | 1.18 ± 0.11 * | 0.86 ± 0.09 # |
| Arg1 | 1.12 ± 0.23 | 0.36 ± 0.11 | 2.04 ± 0.52 | 2.29 ± 0.47 |
| Igf-1 | 1.00 ± 0.03 | 0.75 ± 0.09 | 1.15 ± 0.19 | 1.27 ± 0.22 |
| (C) Cohort 3—aripiprazole | ||||
| Factor | Gene Expression in the Frontal Cortices | |||
| control | LPS | |||
| vehicle | aripiprazole | vehicle | aripiprazole | |
| Cd40 | 1.01 ± 0.06 | 0.88 ± 0.11 | 1.30 ± 0.17 | 1.35 ± 0.24 |
| Cd68 | 1.01 ± 0.07 | 0.96 ± 0.09 | 0.96 ± 0.16 | 1.10 ± 0.22 |
| Arg1 | 1.06 ± 0.15 | 1.48 ± 0.22 | 1.39 ± 0.16 | 1.56 ± 0.17 |
| Igf-1 | 1.01 ± 0.05 | 1.06 ± 0.02 | 1.27 ± 0.08 * | 1.26 ± 0.07 |
| (A) | ||||
|---|---|---|---|---|
| Factor | Gene Expression in the Hippocampi | |||
| control | LPS | |||
| vehicle | quetiapine | vehicle | quetiapine | |
| Cx3cl1 | 1.02 ± 0.08 | 1.56 ± 0.27 | 0.97 ± 0.16 | 1.31 ± 0.25 |
| Cx3cr1 | 1.10 ± 0.18 | 1.91 ± 0.37 | 1.17 ± 0.26 | 1.47 ± 0.28 |
| Cd200 | 1.02 ± 0.07 | 1.12 ± 0.09 | 1.03 ± 0.11 | 1.13 ± 0.14 |
| Cd200r | 1.20 ± 0.29 | 1.90 ± 0.43 | 1.28 ± 0.34 | 1.64 ± 0.25 |
| (B) | ||||
| Factor | Gene Expression in the Hippocampi | |||
| control | LPS | |||
| vehicle | quetiapine | vehicle | quetiapine | |
| Cd40 | 1.05 ± 0.13 | 1.42 ± 0.23 | 1.07 ± 0.20 | 1.34 ± 0.24 |
| Cd68 | 1.02 ± 0.07 | 1.26 ± 0.06 | 1.02 ± 0.14 | 1.20 ± 0.17 |
| Arg1 | 1.04 ± 0.11 | 1.27 ± 0.14 | 1.11 ± 0.09 | 1.32 ± 0.11 |
| Igf-1 | 1.02 ± 0.08 | 1.27 ± 0.12 | 1.04 ± 0.10 | 1.35 ± 0.19 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chamera, K.; Curzytek, K.; Kamińska, K.; Trojan, E.; Basta-Kaim, A. Quetiapine Ameliorates MIA-Induced Impairment of Sensorimotor Gating: Focus on Neuron-Microglia Communication and the Inflammatory Response in the Frontal Cortex of Adult Offspring of Wistar Rats. Cells 2022, 11, 2788. https://doi.org/10.3390/cells11182788
Chamera K, Curzytek K, Kamińska K, Trojan E, Basta-Kaim A. Quetiapine Ameliorates MIA-Induced Impairment of Sensorimotor Gating: Focus on Neuron-Microglia Communication and the Inflammatory Response in the Frontal Cortex of Adult Offspring of Wistar Rats. Cells. 2022; 11(18):2788. https://doi.org/10.3390/cells11182788
Chicago/Turabian StyleChamera, Katarzyna, Katarzyna Curzytek, Kinga Kamińska, Ewa Trojan, and Agnieszka Basta-Kaim. 2022. "Quetiapine Ameliorates MIA-Induced Impairment of Sensorimotor Gating: Focus on Neuron-Microglia Communication and the Inflammatory Response in the Frontal Cortex of Adult Offspring of Wistar Rats" Cells 11, no. 18: 2788. https://doi.org/10.3390/cells11182788