1. Introduction
Natural bioactive substances, produced by various organisms, have been extensively studied for various reasons over the past decades [
1,
2,
3]. Extracellular vesicles (EVs), secreted by cells for information exchange, have received increasing attention owing to several of their properties, such as a wide range of functionalities, biocompatibility, low immunogenicity, and target specificity, for preclinical and clinical applications [
4,
5].
EVs, approximately 30–1000 nm in size, are involved in the intercellular transport of nucleic acids, proteins, lipids, and metabolites by encapsulating these within EV lipid bilayers [
5]. Importantly, the encapsulated materials and functionality of EVs can vary according to the origin, characteristics, and conditions of the cells, as well as environmental changes [
6,
7,
8]. Owing to this characteristic, EVs derived from prokaryotic and eukaryotic cells have wide applications in the areas of research, diagnostics, and therapy for various diseases. EVs isolated from living organisms can be used to control or treat various diseases. For instance, clinical studies have investigated the potential of mesenchymal stem cell-derived EVs in the treatment of immune disorders, cardiovascular diseases, neurological diseases, wounds, and COVID-19 [
4,
9]. In addition, various studies have demonstrated that EVs isolated from probiotic strains of microorganisms, such as
Lactobacillus paracasei and
Propionibacterium freudenreichii, can regulate excessive inflammatory reactions [
10,
11]. Thus, research on the biological, functional, and immunological characteristics of EVs derived from various organisms may lead to advances in the food, pharmaceutical, and medical industries.
Similar to animal-derived EVs, plant-derived EVs act as protective vessels that are involved in transporting various substances between cells and contribute to plant growth and development, protective responses, and plant–microbe coexistence [
12,
13]. Recent studies have demonstrated that EVs isolated from grapes, ginger, broccoli, and grapefruits perform antioxidant and anti-inflammatory activities inside their cells [
14,
15]. Despite the benefits of plant-derived EVs, a number of drawbacks exist because the functional characteristics of plant-derived EVs can vary depending on the growth conditions [
16]. Nevertheless, due to the advent of plant tissue culture, a valuable technique, this particular problem can be overcome. Plant tissue culture can also be used for conserving and restoring endangered plants, as well as growing a complete plant from its constituent parts (cells, tissues, organs, embryo, etc.) under sterile conditions [
17,
18]. The technique generally comprises two steps: induction of dedifferentiated calli from plant tissues and redifferentiation of a callus capable of doing so [
17]. Many bioactive substances produced by the mother plant can be produced using plant callus culture [
19]. Paclitaxel (brand name: CyviloqTM), developed by Samyang Genex, South Korea, is an example of a medication developed using callus cultures [
20,
21].
Recently, we established a callus culture system for
Aster yomena (
A. yomena) and demonstrated the various bioactivities of substances derived from
A. yomena callus, such as anti-photoaging, anti-melanogenic, and anti-inflammatory effects [
22,
23]. We found that the
A. yomena callus culture system could induce EV formation, resembling the induction of EV formation in other plant species, and that these EVs contained secondary metabolites with various immunological effects. We examined the immune functional characteristics of
A. yomena callus-derived EVs (AYC-EVs) to verify the feasibility of using them as agents for the treatment or alleviation of diseases. We also verified the therapeutic potential of AYC-EVs against allergic asthma in animal models of asthma.
2. Materials and Methods
2.1. Isolation and Characterization of Extracellular Vesicles in Aster yomena Callus Culture Supernatant
The culture methods for
A. yomena calli have been described in our previous study [
22]. The callus used in this study was established from an
A. yomena root. Briefly, surface-sterilized root segments (approximately 1 cm) from
A. yomena were incubated on agar-solidified MS1D medium (Sigma-Aldrich, St. Louis, MO, USA) in the presence of growth regulators (1 mg/L 2,4-dichlorophenoxyacetic acid (2,4-D), 3% (
w/
v) sucrose, and 0.4% (
w/
v) Gelrit) at 25 °C in the dark. At 2 weeks after incubation, calli that were formed from
A. yomena roots were collected, and a 500-mL flask containing 200 mL of liquid MS1D medium was inoculated with 20 g of callus. The inoculated calli were cultured on a rotary shaker (80 rpm) at 25 °C in the dark. At 2 weeks after incubation, AYC-EVs were isolated from culture supernatants. Non-EVs, including cells, cell debris, microparticles, and apoptotic bodies, were removed using the following process. First, callus pellets in the culture supernatant were depleted by centrifugation at 1800×
g for 20 min at 4 °C, and the supernatant fluids were centrifuged again at 3500×
g for 20 min at 4 °C. The harvested supernatants were centrifuged at 10,000×
g for 20 min at 4 °C and the pellets were depleted once again. The supernatant fluids that were depleted of non-EVs were filtered through a 0.2-µm vacuum filter (ThermoFisher Scientific, Waltham, MA, USA) to remove potential bacterial contaminants. Next, EVs in the filtered supernatants were isolated using a combination of tangential flow filtration (TFF; Pall Life Sciences, Port Washington, NY, USA) and cushioned ultracentrifugation. In the size-based particle separation method, filtered supernatants (1 L) were concentrated to 35 mL using TFF with a 500-kDa MWCO ultrafiltration membrane filter capsule (Pall Life Sciences). Next, supernatants (35 mL) mixed with soluble proteins and AYC-EVs were added to an ultracentrifuge tube (38.5 mL; Beckman Coulter, California, USA) for the SW 32 Ti Rotor (Beckman Coulter) and 3 mL Lymphoprep
TM solutions (density 1.077 g/mL; StemCell Technologies, Vancouver, BC, Canada), as cushioning buffer, were loaded using a Pasteur pipette (Sigma-Aldrich) into a tube containing 35 mL of supernatant. The tubes were centrifuged at 120,000×
g for 60 min at 4 °C using a Beckman Coulter ultracentrifuge (Opima XE-100). After ultracentrifugation, a volume of 4 mL from the bottom of the ultracentrifuge tubes consisting of 3 mL cushioning buffer and 1 mL supernatant (including AYC-EVs) was carefully collected and diluted to 100 mL with cold PBS as the dialysis buffer. A second TFF (at 500-kDa cut-off) process was then performed to concentrate the EVs and change the buffer. A total volume of 104 mL (consisting of 3 mL of cushioning buffer, 1 mL of AYC-EVs, and 100 mL of PBS) was concentrated to 5 mL to obtain AYC-EVs. Finally, 5 mL of AYC-EVs were filtered through a 0.22-μm syringe filter (Thermo Fisher Scientific) for use in in vitro and in vivo experiments and stored at −80 °C until further use.
The protein concentrations of AYC-EVs were analyzed using a BCA protein assay kit (Thermo Fisher Scientific). The size distribution of AYC-EVs was characterized using transmission electron microscopy (TEM) and dynamic light scattering (DLS). For TEM analysis, 5 μL of AYC-EVs were dropped onto a formvar-carbon-coated TEM grid (Electron Microscopy Sciences, Hatfield, PA, USA) and negatively stained using 2% uranyl acetate (Sigma-Aldrich, St. Louis, MO, USA). Images were visualized using a TEM (JEOL-2100F, Tokyo, Japan). In addition, the size of the AYC-EVs was evaluated using DLS (Malvern Instruments, Malvern, UK), according to the manufacturer’s protocol.
2.2. Isolation of Aster yomena Callus Extracts
The inoculated calli were cultured on a rotary shaker (80 rpm) at 25 °C in the dark, as described in the Materials and Methods section, for AYC-EV isolation. At 2 weeks after incubation, callus pellets were collected to isolate the pellet extracts (AYC-P-E) of A. yomena calli via centrifugation at 1800× g for 20 min at 4 °C. The supernatant fluids (AYC-S-E) were freeze-dried and resuspended in 5 mL of PBS. Then, 5 mL of AYC-S-E was filtered through a 0.22 μm syringe filter and used for in vitro experiments. The pellets were suspended in 500 mL of deionized water and incubated at 80 °C for 2 h. Next, incubated samples were centrifuged at 10,000× g for 20 min at 4 °C, and supernatants were freeze-dried and resuspended in 5 mL of PBS. Finally, 5 mL of AYC-P-E was filtered through a 0.22 μm syringe filter and used for in vitro experiments.
2.3. Characterization and Identification of the Major Metabolites in AYC-EVs
Characterization and identification of major metabolites present in AYC-EVs was carried out using ultra-performance liquid chromatography-quadrupole time-of-flight mass spectrometry (UPLC-QTOF/MS). AYC-EVs powders (1 mg) were first produced using a vacuum freeze dryer (VD-800F; Taitec, Saitama-ken, Japan) and dissolved in 80% methanol (100 μL). Subsequently, AYC-EV solutions (10 μL) dissolved in methanol were injected into an Acquity UPLC BEH C18 column (Waters, Milford, MA, USA) that was equilibrated with ultrapure water supplemented with 0.1% formic acid. Then, the column was eluted using a gradient with acetonitrile, supplemented with 0.1% formic acid, under a flow rate of 0.35 mL/min (total 9 min). The eluted metabolites were measured using QTOF/MS in positive electrospray ionization (ESI) mode as follows: capillary voltage: 3 kV, sampling cones: 30 V, source temperature: 100 °C, desolvation flow rate: 800 L/h, and sheath gas temperature: 400 °C. TOF/MS and MS/MS scan ranges were m/z 50–1500 and m/z 50–1000, respectively. Leucine-enkephalin at m/z 556.2771 for positive mode was used as the lock mass (10 s frequency). UNIFI software (Waters) was used for collection, normalization, and alignment of the MS datasets. The identification of metabolites was obtained by using web-based bioinformatics databases (Human Metabolome Database, Metabolite and Chemical Entity database, and Chemspider database, each accessed on January 3 2022), and authentic reference standards.
2.4. Differentiation of Dendritic Cells (DCs) from Mouse Bone Marrow
Bone marrow cells were isolated from 8-week-old female C57BL/6 mice (Orient Bio, Seongnam, South Korea) and cultured in an incubator at 37 °C and 5% CO2 in complete RPMI1640 medium (Gibco BRL, Grand Island, NY, USA) containing 20 ng/mL GM-CSF (JW Creagene, Gyeonggi, Korea), 2.5 ng/mL IL-4 (JW Creagene), 10% fetal bovine serum (Gibco BRL), and 1% antibiotics (penicillin/streptomycin, Gibco BRL). On the 8th day, the cells were harvested and labeled with CD11c MicroBeads (Miltenyi Biotec, San Diego, CA, USA) at 4 °C for 15 min. After incubation, CD11c-labeled cells (CD11c+ cells) were isolated using MACS LS separation columns (Miltenyi Biotec), according to the manufacturer’s protocol. The purity of the CD11c+ dendritic cells (DCs) was determined to be >90%.
2.5. Analysis of DC Viability and Cytotoxicity of AYC-EVs in DCs
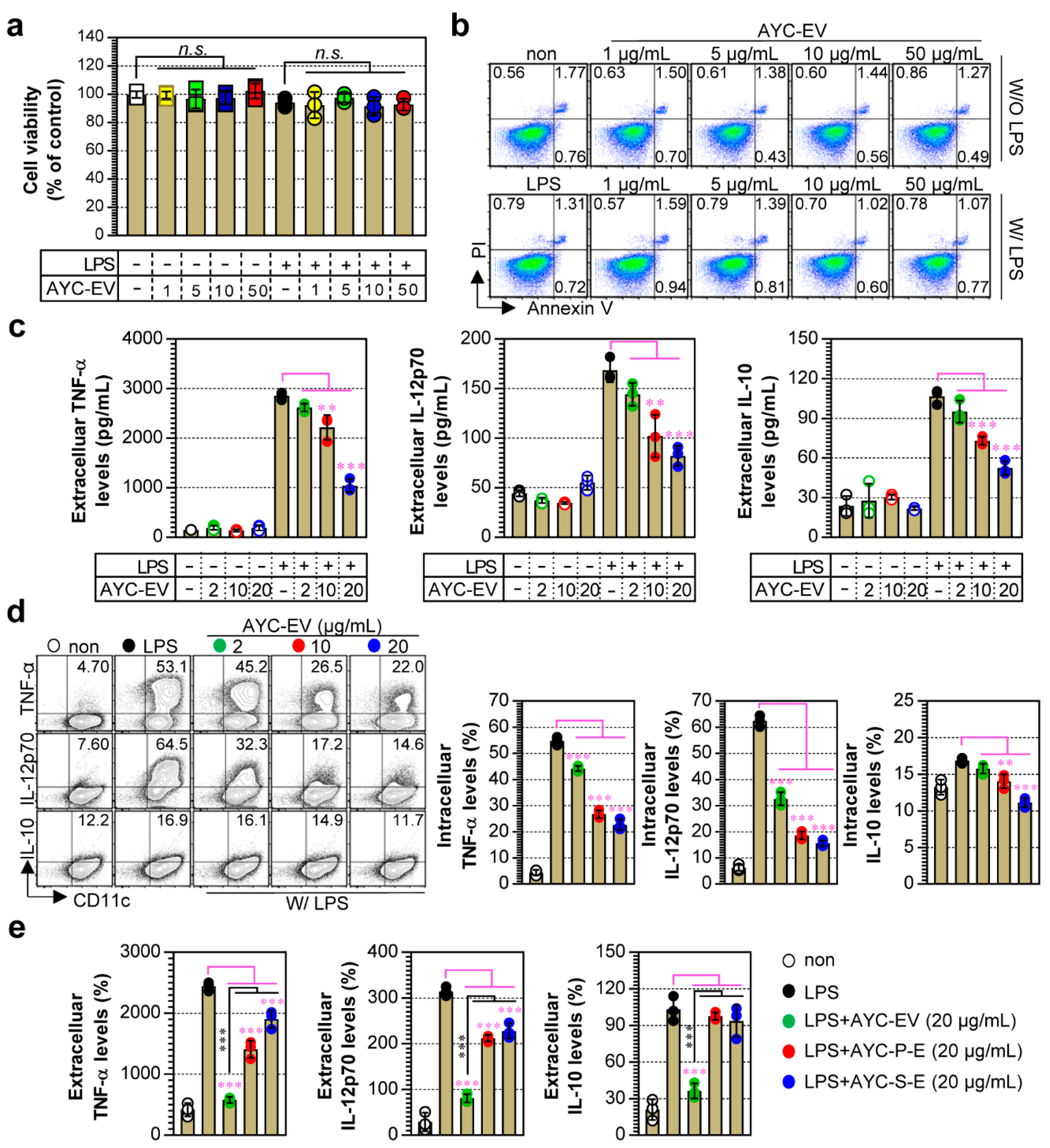
To analyze the viability of immature and mature DCs, DCs (0.5 × 106 cells per well in 500 μL complete RPMI1640 media) were incubated with or without 100 ng/mL lipopolysaccharide (LPS; Invitrogen, San Diego, CA, USA) as a positive control for DC maturation for 2 h, and then treated with various concentrations (1–50 μg/mL) of AYC-EVs for 24 h. After stimulation, cell viability was analyzed using the EZ-Cytox Cell Viability Assay Kit (DoGen, Seoul, Korea), according to the manufacturer’s protocol. Briefly, 50 μL of EZ-Cytox kit reagent was added to each well and incubated at 37 °C for 60 min. After incubation, absorbance at 450 nm was measured using a microplate reader (Molecular Devices Inc., San Jose, CA, USA). For cytotoxicity analysis, cells (non-, LPS-, AYC-EV-, AYC-EV, and LPS-treated DCs) were harvested 24 h after AYC-EV treatment and stained with Annexin V and propidium iodide (PI) using FITC Annexin V/Dead Cell Apoptosis Kit (Thermo Fisher Scientific), according to the manufacturer’s protocol. Annexin V/PI-stained cells were analyzed using a Life Launch Attune Nxt Flow Cytometer (Thermo Fisher Scientific) and FlowJo software (Version 10; Tree Star, Inc., Ashland, OR, USA).
2.6. Analysis of Extracellular and Intracellular Cytokines in DCs
To analyze extracellular cytokines, DCs were incubated with or without 100 ng/mL LPS for 2 h and then treated with AYC-EVs (2, 10, and 20 μg/mL) for 24 h. The culture supernatants were harvested 24 h after AYC-EV treatment, and extracellular cytokine levels for TNF-α, IL-12p70, and IL-10 were analyzed using a cytokine-specific sandwich ELISA kit (ThermoFisher Scientific), according to the manufacturer’s protocol. To analyze intracellular cytokines, DCs were incubated with 100 ng/mL LPS for 2 h and treated with AYC-EVs (2, 10, and 20 μg/mL) for 12 h in the presence of 1x protein transport inhibitor cocktail (Thermo Fisher Scientific). Subsequently, the cells were harvested and washed twice with cold PBS. Next, the cells were stained with anti-CD11c antibody (PE-Cy7; BD Bioscience, San Jose, CA, USA) and Live/Dead Fixable Dead Cell Stain kit (L/D; Invitrogen) reagent for 15 min at room temperature and then fixed and permeabilized using a BD Cytofix/Cytoperm kit reagent, according to the manufacturer’s protocol. Finally, the cells were washed twice with BD Perm/Wash Buffer and stained with anti-TNF-α (APC; Thermo Fisher Scientific), anti-IL-12p70 (FITC; BD Bioscience), and anti-IL-10 (PE; BD Biosciences) antibodies for 20 min at room temperature. Fluorescein-stained cells were analyzed using a Life Launch Attune Nxt flow cytometer.
2.7. Analysis of Immune-Modifying Surface Molecules on DCs
To evaluate whether the expression of surface molecules on immature and mature DCs could be regulated through AYC-EV treatment, DCs were incubated with or without 100 ng/mL LPS for 2 h, and then treated with AYC-EVs (2, 10, and 20 μg/mL). At 24 h after AYC-EVs treatment, the cells were harvested and washed twice with cold PBS. Next, cells were stained with anti-CD11c (PE-Cy7), anti-CD80 (APC; BD Biosciences), anti-CD86 (FITC; BD Biosciences), anti-MHC-I (PE; BD Biosciences), and anti-MHC-II antibodies (PerCp-Cy5.5; BD Biosciences) and L/D reagent for 15 min at room temperature. CD80-, CD86-, MHC-I-, and MHC-II-positive CD11c+ cells were analyzed using a Life Launch Attune Nxt Flow Cytometer. All isotype controls (APC-conjugated Hamster IgG2, FITC-conjugated rat IgG2a, PE-conjugated rat IgG2a, and PerCp-Cy5.5-conjugated rat IgG2b) for surface molecules were purchased from BD Biosciences.
2.8. Analysis of Antigen Uptake and Antigen Presenting Ability on DCs
To analyze their antigen uptake ability, DCs were incubated with 100 ng/mL LPS for 2 h and then treated with AYC-EVs (2, 10, and 20 μg/mL) for 24 h. After treatment, 0.5 mg/mL FITC-conjugated Dextran (40,000 Da, Sigma-Aldrich) was added to each well and then incubated at 37 °C or 4 °C for 30 min. Cell culture conditions at 4 °C were used as negative controls for those at 37 °C. Next, the cells were harvested and washed three times with cold PBS. CD11c+ dextran + cells were analyzed using a Life Launch Attune Nxt flow cytometer. For analysis of antigen presenting ability, antigen presentation induced by MHC-I was examined, based on treatment with ovalbumin (OVA) protein (Sigma-Aldrich) or OVA257–264 peptide (SIINFEKL; positive control for MHC-I-antigen presentation) in each experimental condition. In addition, antigen presentation induced by MHC-II was examined upon treatment with Eα44–76 peptide (RLEEFAKFASFEAQGALANIAVDKANLDVMKKR; underlined sequence bound to MHC-II) or Eα52–68 peptide (ASFEAQGALANIAVDKA; positive control for MHC-II-antigen presentation). Briefly, DCs were incubated with 100 ng/mL LPS for 2 h and then treated with AYC-EVs (20 μg/mL) for 24 h in the presence of OVA (500 μg/mL) or Eα44–76 peptide (25 μg/mL). The cells were harvested and washed twice with cold PBS. Next, the cells were stained with anti-CD11c (PE-Cy7) and Y-Ae (PE, Thermo Fisher Scientific) or I-Ab antibodies (FITC, Thermo Fisher Scientific). Here, the H-2Kb antibody was expected to react with the MHC-I self OVA257–264 peptide and the Y-Ae antibody was expected to react with the MHC-II self Eα52–68 peptide. CD11c+H-2Kb+ and CD11c+Y-Ae+ cells were analyzed using a Life Launch Attune Nxt flow cytometer.
2.9. Allogenic Mixed Lymphocyte Reaction Assay
An allogeneic mixed lymphocyte reaction (MLR) assay was conducted using DCs from an 8-week-old female C57BL/6 mouse and T cells from an 8-week-old female BALB/c mouse. First, DCs were incubated with 100 ng/mL LPS for 2 h and then treated with AYC-EVs (20 μg/mL) for 24 h. These cells were then used in the MLR assay. Spleen cells were isolated from the BALB/c mouse and labeled with CD4 or CD8 microbeads (Miltenyi Biotec) at 4 °C for 15 min. After incubation, CD4- or CD8-labeled cells were isolated using MACS LS separation columns, according to the manufacturer’s protocol. Isolated T cells were stained using the CellTrace CFSE Cell proliferation kit reagent (2 μM, Invitrogen) for 10 min in a 37 °C water bath. Next, 96-well plates were coated with anti-CD3e (1 μg/mL; Invitrogen) antibody for 2 h at 37 °C, and the cells (DCs; 2 × 105 cells per well and CFSE-labeled T cells; 1 × 106 cells per well) were co-cultured with anti-CD28 antibody (1 μg/mL; Invitrogen) in anti-CD3e-coated plates. After 2 days of co-culture, cell culture supernatants were harvested, and IFN-γ, TNF-α, IL-2, IL-5, and IL-17A levels were analyzed using a cytokine-specific ELISA kit. Kits were purchased from Thermo Fisher Scientific. Finally, the harvested T cells were stained with anti-CD4 or anti-CD8 antibodies, and T cell proliferation levels were analyzed using a Life Launch Attune Nxt Flow Cytometer.
2.10. Therapeutic Efficacy of AYC-EVs in a Mouse Model of Ovalbumin-Mediated Allergic Asthma
The therapeutic efficacy of AYC-EVs in a mouse model of ovalbumin (OVA)-mediated allergic asthma was evaluated in compliance with the Institutional Animal Care and Use Committee (permit number: KRIBB-AEC-21120), with approval from the Korea Research Institute of Bioscience and Biotechnology (KRIBB). Herein 10-week-old female BALB/c mice (weight:20 ± 2 g) were sourced from Orient Bio and housed at the KRIBB animal facility under specific pathogen-free conditions at constant temperature (22 ± 2 °C), light/dark cycle (12 h/12 h), and humidity (55 ± 5%). In addition, the mice were acclimated for 1 week after transport to a new facility and randomly divided as follows (n = 5 per group): control group (Group 1), OVA group (Group 2), OVA/DEX group (Group 3), and OVA/AYC-EV group (Group 4). For the control groups, mice were injected intraperitoneally with 200 μL of PBS on days 1 and 8. Next, the PBS-injected mice were orally administered 200 μL PBS once per day on days 12–17. Finally, the mice were challenged for 1 h with PBS via an ultrasonic nebulizer (NE-U12; OMRON Corp., Tokyo, Japan) once per day on days 15, 16, and 17. For the OVA groups, mice were sensitized intraperitoneally with 200 μL of PBS containing OVA (40 μg, Sigma-Aldrich) and aluminum hydroxide gel (2 mg, Sigma-Aldrich) on days 1 and 8. Next, OVA-injected mice were orally administered 200 μL of PBS once per day on days 12–17. Finally, the mice were challenged for 1 h with OVA (1%, w/v, in PBS) once per day via a nebulizer on days 15, 16, and 17. For the OVA/DEX groups, mice were sensitized intraperitoneally with 200 μL of PBS containing OVA (40 μg) and aluminum hydroxide gel (2 mg) on days 1 and 8. Next, OVA-injected mice were orally administered dexamethasone (3 mg/kg, Sigma-Aldrich) once per day on days 12–17. Finally, the mice were challenged for 1 h with OVA (1%, w/v, in PBS) once per day via a nebulizer on days 15, 16, and 17. For the OVA/AYC-EV groups, mice were sensitized intraperitoneally with 200 μL of PBS containing OVA (40 μg) and aluminum hydroxide gel (2 mg) on days 1 and 8. Next, OVA-injected mice were orally administered AYC-EVs (4 and 8 mg/kg) once per day, on days 12–17. Finally, the mice were challenged for 1 h with OVA (1%, w/v, in PBS) once per day via a nebulizer on days 15, 16, and 17. All groups were euthanized after 19 days.
2.11. Analysis of Airway Resistance in a Mouse Model of Allergic Asthma Induced by Ovalbumin Antigen
At 24 h after the last OVA challenge, airway resistance values were assessed via Flexivent (SCIREQ Scientific Respiratory Equipment Inc., Montreal, PQ, Canada) [
24]. Mice were anesthetized with an intraperitoneal injection of avertin (250 mg/kg, Sigma-Aldrich). After baseline impedance measurements, the mice were placed in a chamber and nebulized with aerosolized PBS or methacholine (10 and 40 mg/mL) via an Aeroneb nebulizer (SCIREQ) for 10 s. Airway resistance levels were measured every 30 s for 1 min and then refreshed for 2 min. The volume history of the lung was established with 6-s deep inflations to a pressure limit of 30 cm H
2O.
2.12. Analysis of ELISA and Inflammatory Cells in Bronchoalveolar Lavage Fluid (BALF)
BALF samples were obtained in accordance with a previously described method [
25]. Mice were sacrificed at 48 h after last OVA challenge under isoflurane inhalation anesthesia, and a tracheostomy was conducted. To obtain BALF, ice-cold PBS (0.7 mL) was infused into the lungs twice and withdrawn each time using a tracheal cannula (total volume of 1.4 mL). The BALF samples were centrifuged at 1000 rpm for 10 min at 4 °C, and the collected supernatants were stored at –70 °C. The collected pellets were resuspended in 250 μL PBS and centrifuged to attach the cells to a slide (1000 rpm for 5 min at 20 °C) using a Cytospin 4 centrifuge (Thermo Scientific, Waltham, MA, USA). For differential cell count, slides were dried, and cells were fixed and stained with Diff-Quik
® staining reagent (IMEB, San Marcos, CA, USA) according to the manufacturer’s instructions. Five images of each slide were captured using a Leica DM5000B microscope and Leica Application Suite acquisition software (Leica Microsystems, Wetzlar, Germany) under a 40× objective lens. Next, the total cells, eosinophils, antigen-presenting cells (APCs, including DCs and macrophages), neutrophils, and lymphocytes were enumerated. TNF-α (R&D Systems, Minneapolis, MN, USA), IL-4 (R&D Systems), IL-5 (R&D Systems), IL-13 (R&D Systems), eotaxin (R&D Systems), and mucin 5AC (MUC5AC; Cusabio Biotech Co., Wuhan, China) levels in supernatants were measured with each ELISA kit, following the manufacturer’s instructions. The level of total IgE and OVA-specific IgE in the serum were measured using ELISA kits (BioLegend, CA, USA), following the manufacturer’s instructions. Absorbance was measured at 450 nm using a microplate reader (iMarkTM; Bio-Rad Laboratories, Richmond, CA, USA).
2.13. Histopathology
After collecting BALF, the dissected lung tissues were fixed in 10% (v/v) neutral-buffered formalin for 48 h at room temperature. Fixed tissues were embedded in paraffin, and cut into 4-μm-thick sections. The slides were stained with hematoxylin and eosin (H&E) (BBC Biochemical, Mount Vernon, WA, USA) or periodic acid-Schiff (PAS), respectively, to evaluate the extent of airway inflammation or mucus production. The quantitative determination for airway inflammation or mucus production was performed and the degree of each lesion was graded in a blinded manner using a light microscope (Leica Microsystem, Wetzlar, Germany) with 10× and 20× objective lenses, on the following scale: 0, no lesions; 1, minimal; 2, mild; 3, moderate; and 4, severe.
2.14. Analysis of Splenic DC and CD4+ T Cell Responses in a Mouse Model of Ovalbumin-Mediated Allergic Asthma
After mice were euthanized, splenocytes from each group were isolated and stained with L/D reagent, lineage marker cocktails (FITC-conjugated anti-CD3, anti-CD19, and anti-NK1.1 antibodies; BD Biosciences) and anti-CD11c (PE-Cy7), anti-CD80 (APC), anti-MHC-I (PE), and MHC-II (PerCp-Cy5.5) antibodies for 15 min at room temperature. After staining, splenic DC (lineage-CD11c+MHC-II+) maturation (CD80 and MHC-I expression on lineage-CD11c+MHC-II+ cells) was analyzed using a Life Launch Attune Nxt Flow Cytometer. For the analysis of Th1, Th2, Th17, and Th2/Th17 cells, splenocytes from each group were isolated and stimulated with 1× Cell Stimulation Cocktail (Thermo Fisher Scientific) supplemented with PMA, ionomycin, and protein inhibitors for 4 h at 37 °C. For analysis of Th1, Th2, and Th17 cells, cells were harvested and stained with anti-CD3 antibody (Alexa 700; BD Biosciences), anti-CD4 antibody (PerCp-Cy5.5; BD Bioscience), and L/D reagent for 15 min at room temperature, and then fixed and permeabilized using BD Cytofix/Cytoperm kit reagents according to the manufacturer’s protocol. Cells were then washed with BD Perm/Wash Buffer and stained with anti-IFN-γ (PE; Thermo Fisher Scientific), anti-IL-5 (APC; BD Bioscience), and anit-IL-17A (PE-Cy7; Thermo Fisher Scientific) antibodies for 20 min at room temperature. For analysis of regulatory T cells, splenocytes from each group were directly stained with anti-CD3 antibody (Alexa 700), anti-CD4 antibody (PerCp-Cy5.5), anti-CD25 antibody (APC; ThermoFisher Scientific), and L/D reagent for 15 min at room temperature. Cells were washed with cold PBS, fixed, and permeabilized using Foxp3/Transcription Factor Staining Buffer Set (Thermo Fisher Scientific) reagents according to the manufacturer’s protocol. After several washes with Fixation/Per Dilunt (Thermo Fisher Scientific), cells were stained with anti-Foxp3 (PE; Thermo Fisher Scientific) antibody for 20 min at room temperature. After staining, regulatory T cells (CD3+CD4+CD25+Foxp3+) were analyzed using a Life Launch Attune Nxt Flow Cytometer. The absolute number of splenic DCs and T cell subsets was analyzed by multiplying the percentage of each cell type by the total cell number in the single-cell suspension of each group.
2.15. Statistical Analysis
Statistical differences for all data were assessed using GraphPad Prism 7.0 software (San Diego, CA, USA). Differences between groups (two or three groups) were evaluated using unpaired t-tests and one-way ANOVA, respectively. The following p-value thresholds of significance were used: * p < 0.05, ** p < 0.01, and *** p < 0.001.
4. Discussion
Although numerous studies have reported the functionalities of plant callus-derived extracts, to the best of our knowledge, no functional studies have been conducted on plant callus-derived EVs. In this study, we demonstrated that callus derived EVs can be produced using an A. yomena callus culture system, and that these EVs consist of 17 major metabolites. Importantly, we demonstrated that AYC-EVs can inhibit T-cell proliferation and activities associated with the etiology of various inflammatory diseases by converting the phenotype and functionalities of mature DCs into those of immature DCs. Based on these immunological characteristics, it may be feasible to use AYC-EVs as therapeutic agents for inflammatory diseases. To verify this hypothesis, we examined the therapeutic effects of AYC-EVs in an animal model of asthma. AYC-EVs injected into mouse models of asthma were found to reduce the levels of major factors (an increase in bronchoconstriction, Th2 cytokine levels, inflammatory cell infiltration, MUC5AC expression, eotaxin expression, IgE production, and lung inflammation), for developing asthma, and relieve pathological and immunological symptoms in animal models.
DCs are APCs that play a central role in initiating innate and acquired immune responses [
32,
33]. Pathogens or autoantigens may induce the maturation of immature DCs, thereby, directly and indirectly, regulating the activity of other immune cells [
33,
34]. Through the process of acquiring, processing, and presenting pathogen-specific antigens in times of infection by human pathogens (viruses, bacteria, etc.), DCs induce the activation and differentiation of naïve T cells and, consequently, induce pathogen-specific T cell responses to activate host defense against pathogens [
35,
36]. However, excessive induction of DC maturation leads to excessive cytokine secretion, resulting in the stimulation of normal cells, induction of cell apoptosis, and secondary stimulation of immune cells by dead cells, ultimately causing acute or chronic inflammatory diseases [
37,
38,
39]. As is evident, DCs play an important role in regulating the host immune response and are used in research aimed at understanding disease pathogenesis or therapeutic mechanisms, in addition to drug development [
35,
40,
41]. Interestingly, after inducing maturation of DCs using LPS, and treating these DCs with AYC-EVs, we observed that AYC-EVs inhibited not only LPS-induced phenotypic maturation by inhibiting the expression of surface molecules in DCs, increasing the internalization of extracellular antigens, and suppressing the antigen-presenting ability of DCs, but also the functional maturation of DCs by reducing the expression of pro- and anti-inflammatory cytokines. In addition, unlike DCs treated with LPS alone, LPS-treated DCs that were additionally treated with AYC-EVs inhibited T cell proliferation and various T cell activities (Th1, Th2, Th17, and CD8 T cells) when interacting with CD4
+ and CD8
+ T cells. These results demonstrated that treatment with AYC-EVs can inhibit pathogenic or excessive T cell responses observed during various inflammatory diseases mediated by mature DCs.
Allergic asthma, a T cell-mediated inflammatory disease, is characterized by airway hyper-responsiveness, bronchoconstriction, elevated blood IgE levels, and airway inflammation caused by genetic and environmental factors [
42,
43]. Allergic asthma caused by exposure to allergens is observed when DCs react to the recognition of allergens and induction of differentiation of naïve CD4
+ T cells into Th2 and Th17 cells, thereby promoting or inducing the secretion of factors that induce asthma [
43,
44,
45]. Th2 immunity is generally predominant in patients with mild asthma, whereas Th17, or combined Th2 and Th17, (Th2/Th17) immunity is predominant among those with severe asthma [
43]. Given the immunological characteristics of allergic asthma, regulating the maturation of DCs to control Th2 and Th17 cells may significantly relieve the symptoms of asthma [
44,
46]. Interestingly, although the AYC-EV treatment was not sufficient to decrease the infiltration of inflammatory cells (particularly eosinophils, neutrophils, and lymphocytes) in BALF of allergic asthma (unlike dexamethasone treatment), the infiltration of APCs (including DCs and macrophages) sharply decreased in the AYC-EV treatment groups, compared with the observation in the dexamethasone treatment group. In addition, we found that the oral administration of AYC-EVs in ovalbumin-sensitized asthmatic mouse models reduced OVA-induced DC maturation, Th2, and Th2/Th17-mediated immune responses, as well as the pathological symptoms (an increase in bronchoconstriction and inflammation) of allergic asthma, and the levels of the major factors (upregulation of Th2 cytokines, IgE, MUC5AC, and eotaxin expressions in serum or BALF) inducing asthma. These results indicate that the therapeutic effect of AYC-EVs against allergic asthma may be caused by down-regulation of T cell activation via the inhibition of DC maturation and infiltration.
Many studies have demonstrated that Th1 cells, which are involved in the antagonism of Th2 and IgE synthesis, are important immune cells that alleviate the symptoms of allergic asthma [
47,
48]. However, recent studies have reported that Th1-cell activation can induce lung inflammation when the symptoms of asthma worsen [
49]. The effect of Th1 cells on the onset and symptoms of asthma remains unclear. It is clear that Th1-mediated immune responses can aggravate various inflammatory diseases [
50]. Therefore, discovery and assessment of drug candidates that can not only inhibit Th2- and Th17-mediated responses, but also Th1-mediated responses, is urgently required for the development of medications for asthma, which can manifest through various symptoms and pathogenic mechanisms. In our in vivo study, Th1 immunity was reduced in ovalbumin-sensitized asthmatic mouse models in contrast to Th2 and Th2/Th17 immunity, and oral administration of AYC-EVs further inhibited Th1 immunity. In our analysis of regulatory T cells, which can inhibit various T cell activities and alleviate asthma symptoms, we observed that while the numbers of regulatory T cells were reduced in asthmatic mouse models, they were restored to a level similar to that observed in the control group upon the administration of AYC-EVs. Similar results (suppression of activated Th1, Th2, and Th17 cells) were observed after dexamethasone treatment.
Our results suggest that the secondary metabolite composition of AYC-EVs may contribute to the inhibition of excessive T cell responses by regulating DC maturation. Interestingly, AYC-EVs include various omega-3, omega-6, omega-7, and omega-fatty acids and their derivates. The functions of omega-3 and omega-6 fatty acids that can inhibit DC maturation and the omega-positive anti-inflammatory role of omega-9 fatty acids in sepsis have been reported [
51]. Furthermore, linoleoyl ethanolamide plays an important role in the anti-inflammatory action of activated macrophages, and its potential as a therapeutic agent for inflammatory skin diseases has been reported [
52]. AYC-EV treatment is expected to inhibit DC maturation because AYC-EVs contain large amounts of omega fatty acids, their derivatives, and linoleoyl ethanolamide. Despite these results, our research has several limitations. First, although 17 metabolites that were expected to have various immunological functions were identified, no studies were included to show whether any of these metabolites are involved in AYC-EV-related functions; thus, more detailed studies for metabolites involved in these functions need to be conducted. In addition, it is necessary to further identify various nucleic acids (DNA, mRNA, miRNA, etc.) and proteins contained in AYC-EVs, because EVs can induce biological activity by nucleic acids and proteins, as well as metabolites. These additional studies may yield important results for finding AYC-EV components that play a dominant role in their biological activity. Second, further studies on the molecular mechanism involved in inhibiting DC maturation are required to further elucidate the pharmacological properties of AYC-EVs.
In conclusion, in this study, we confirmed that AYC-EVs, which contain 17 metabolites, inhibited DC maturation and reduced excessive T cell responses induced by DC maturation. In addition, AYC-EVs could inhibit Th2 and mixed Th2/Th17 responses associated with the etiology of asthma in asthmatic mouse models and alleviate various asthma symptoms. These results indicate that AYC-EVs may be potential drug candidates for various inflammatory diseases. Taken together, the current results can provide various advantages for the development of plant-derived therapeutic agents for the treatment of various inflammatory diseases. Callus-derived EVs can be used as interesting materials to replace plant-derived therapeutic agents, and these systems may be an alternative technology to supplement or solve various problems (plants: change in EV characteristics according to environmental changes; mammalian cells: high cost of growth medium) that arise in the development of other sources of EVs.

 ,
, 








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}