
Regulation of cGAS Activity and Downstream Signaling


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. cGAS
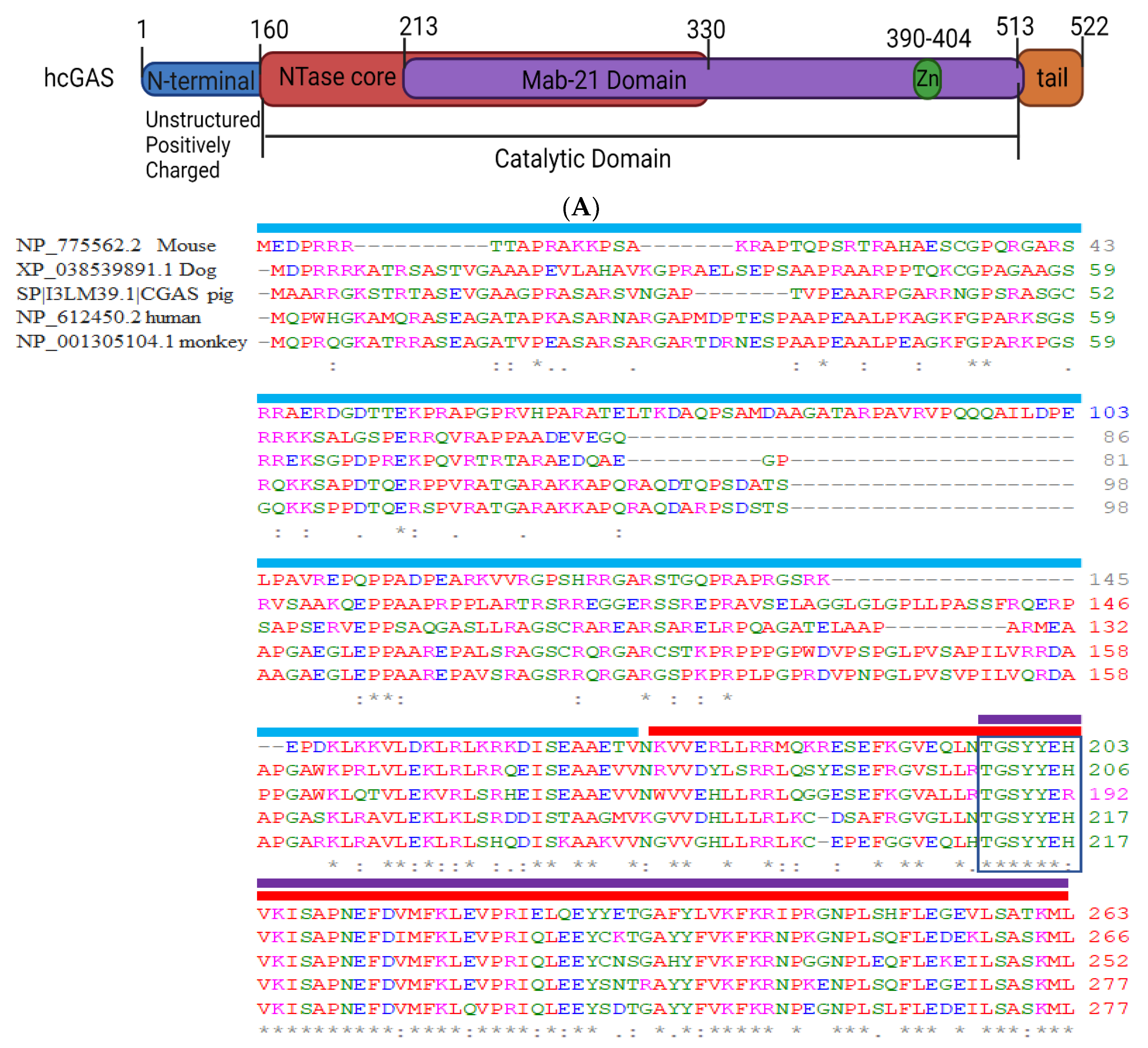
2.1. Structural Domain of cGAS
2.2. cGAS Localization
2.3. cGAS Droplet Formation
3. Regulation of cGAS Activity
3.1. Transcriptional Regulation of cGAS
3.2. Translational Regulations of cGAS Activation
3.2.1. Phosphorylation
3.2.2. Ubiquitination
3.2.3. SUMOylation
3.2.4. Acetylation
4. cGAS Activation of STING
4.1. Role of Gasdermin D
4.2. Role of Ca2+ Signaling
5. cGAS-STING Pathway and Pathologies
5.1. Acute Lung Injury (ALI) and COVID-19
5.2. Adaptive Immunity
5.3. Cancer
5.4. Role of cGAS in Other Diseases
6. Clinical Aspect of cGAS
7. Conclusions
8. Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ahn, J.; Barber, G.N. Self-DNA, STING-dependent signaling and the origins of autoinflammatory disease. Curr. Opin. Immunol. 2014, 31, 121–126. [Google Scholar] [CrossRef] [PubMed]
- Janeway, C.A., Jr.; Medzhitov, R. Innate immune recognition. Annu. Rev. Immunol. 2002, 20, 197–216. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Akira, S. The role of pattern-recognition receptors in innate immunity: Update on Toll-like receptors. Nat. Immunol. 2010, 11, 373–384. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Wu, J.; Du, F.; Chen, X.; Chen, Z.J. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 2013, 339, 786–791. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Tian, S.; Liang, J.; Fan, J.; Lai, J.; Chen, Q. Therapeutic Development by Targeting the cGAS-STING Pathway in Autoimmune Disease and Cancer. Front. Pharmacol. 2021, 12, 779425. [Google Scholar] [CrossRef]
- Hopfner, K.P.; Hornung, V. Molecular mechanisms and cellular functions of cGAS-STING signalling. Nat. Rev. Mol. Cell Biol. 2020, 21, 501–521. [Google Scholar] [CrossRef]
- George, R.D.; McVicker, G.; Diederich, R.; Ng, S.B.; MacKenzie, A.P.; Swanson, W.J.; Shendure, J.; Thomas, J.H. Trans genomic capture and sequencing of primate exomes Rev..eals new targets of positive selection. Genome. Res. 2011, 21, 1686–1694. [Google Scholar] [CrossRef]
- Hancks, D.C.; Hartley, M.K.; Hagan, C.; Clark, N.L.; Elde, N.C. Overlapping Patterns of Rapid Evolution in the Nucleic Acid Sensors cGAS and OAS1 Suggest a Common Mechanism of Pathogen Antagonism and Escape. PLoS Genet. 2015, 11, e1005203. [Google Scholar] [CrossRef]
- Kranzusch, P.J.; Lee, A.S.; Berger, J.M.; Doudna, J.A. Structure of human cGAS reveals a conserved family of second-messenger enzymes in innate immunity. Cell Rep. 2013, 3, 1362–1368. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wu, J.; Du, F.; Xu, H.; Sun, L.; Chen, Z.; Brautigam, C.A.; Zhang, X.; Chen, Z.J. The cytosolic DNA sensor cGAS forms an oligomeric complex with DNA and undergoes switch-like conformational changes in the activation loop. Cell Rep. 2014, 6, 421–430. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Shu, C.; Yi, G.; Chaton, C.T.; Shelton, C.L.; Diao, J.; Zuo, X.; Kao, C.C.; Herr, A.B.; Li, P. Cyclic GMP-AMP synthase is activated by double-stranded DNA-induced oligomerization. Immunity 2013, 39, 1019–1031. [Google Scholar] [CrossRef] [PubMed]
- Andreeva, L.; Hiller, B.; Kostrewa, D.; Lassig, C.; de Oliveira Mann, C.C.; Drexler, D.J.; Maiser, A.; Gaidt, M.; Leonhardt, H.; Hornung, V.; et al. cGAS senses long and HMGB/TFAM-bound U-turn DNA by forming protein-DNA ladders. Nature 2017, 549, 394–398. [Google Scholar] [CrossRef] [PubMed]
- Civril, F.; Deimling, T.; de Oliveira Mann, C.C.; Ablasser, A.; Moldt, M.; Witte, G.; Hornung, V.; Hopfner, K.P. Structural mechanism of cytosolic DNA sensing by cGAS. Nature 2013, 498, 332–337. [Google Scholar] [CrossRef] [PubMed]
- Gao, D.; Wu, J.; Wu, Y.T.; Du, F.; Aroh, C.; Yan, N.; Sun, L.; Chen, Z.J. Cyclic GMP-AMP synthase is an innate immune sensor of HIV and other retroviruses. Science 2013, 341, 903–906. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Chu, H.; Li, C.; Wong, B.H.-Y.; Cheng, Z.-S.; Poon, V.K.-M.; Sun, T.; Lau, C.C.-Y.; Wong, K.K.-Y.; Chan, J.Y.-W.; et al. Active replication of Middle East respiratory syndrome coronavirus and aberrant induction of inflammatory cytokines and chemokines in human macrophages: Implications for pathogenesis. J. Infect. Dis. 2014, 209, 1331–1342. [Google Scholar] [CrossRef]
- de Oliveira Mann, C.C.; Hopfner, K.P. Nuclear cGAS: Guard or prisoner? EMBO J. 2021, 40, e108293. [Google Scholar] [CrossRef]
- Lahaye, X.; Gentili, M.; Silvin, A.; Conrad, C.; Picard, L.; Jouve, M.; Zueva, E.; Maurin, M.; Nadalin, F.; Knott, G.J.; et al. NONO Detects the Nuclear HIV Capsid to Promote cGAS-Mediated Innate Immune Activation. Cell 2018, 175, 488–501.e422. [Google Scholar] [CrossRef]
- Mathavarajah, S.; Salsman, J.; Dellaire, G. An emerging role for calcium signalling in innate and autoimmunity via the cGAS-STING axis. Cytokine Growth Factor Rev. 2019, 50, 43–51. [Google Scholar] [CrossRef]
- Ablasser, A.; Schmid-Burgk, J.L.; Hemmerling, I.; Horvath, G.L.; Schmidt, T.; Latz, E.; Hornung, V. Cell intrinsic immunity spreads to bystander cells via the intercellular transfer of cGAMP. Nature 2013, 503, 530–534. [Google Scholar] [CrossRef]
- Wu, J.; Sun, L.; Chen, X.; Du, F.; Shi, H.; Chen, C.; Chen, Z.J. Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science 2013, 339, 826–830. [Google Scholar] [CrossRef] [Green Version]
- Lund, J.; Sato, A.; Akira, S.; Medzhitov, R.; Iwasaki, A. Toll-like receptor 9-mediated recognition of Herpes simplex virus-2 by plasmacytoid dendritic cells. J. Exp. Med. 2003, 198, 513–520. [Google Scholar] [CrossRef] [PubMed]
- Horan, K.A.; Hansen, K.; Jakobsen, M.R.; Holm, C.K.; Søby, S.; Unterholzner, L.; Thompson, M.; West, J.A.; Iversen, M.B.; Rasmussen, S.B.; et al. Proteasomal degradation of herpes simplex virus capsids in macrophages releases DNA to the cytosol for recognition by DNA sensors. J. Immunol. 2013, 190, 2311–2319. [Google Scholar] [CrossRef] [PubMed]
- Tan, X.; Sun, L.; Chen, J.; Chen, Z.J. Detection of Microbial Infections Through Innate Immune Sensing of Nucleic Acids. Annu. Rev. Microbiol. 2018, 72, 447–478. [Google Scholar] [CrossRef] [PubMed]
- Unterholzner, L.; Keating, S.E.; Baran, M.; Horan, K.A.; Jensen, S.B.; Sharma, S.; Sirois, C.M.; Jin, T.; Latz, E.; Xiao, T.S.; et al. IFI16 is an innate immune sensor for intracellular DNA. Nat. Immunol. 2010, 11, 997–1004. [Google Scholar] [CrossRef]
- Kerur, N.; Veettil, M.V.; Sharma-Walia, N.; Bottero, V.; Sadagopan, S.; Otageri, P.; Chandran, B. IFI16 acts as a nuclear pathogen sensor to induce the inflammasome in response to Kaposi Sarcoma-associated herpesvirus infection. Cell Host Microbe. 2011, 9, 363–375. [Google Scholar] [CrossRef]
- Zhang, Z.; Yuan, B.; Bao, M.; Lu, N.; Kim, T.; Liu, Y.J. The helicase DDX41 senses intracellular DNA mediated by the adaptor STING in dendritic cells. Nat. Immunol. 2011, 12, 959–965. [Google Scholar] [CrossRef]
- Chiu, Y.H.; Macmillan, J.B.; Chen, Z.J. RNA polymerase III detects cytosolic DNA and induces type I interferons through the RIG-I pathway. Cell 2009, 138, 576–591. [Google Scholar] [CrossRef]
- Takaoka, A.; Wang, Z.; Choi, M.K.; Yanai, H.; Negishi, H.; Ban, T.; Lu, Y.; Miyagishi, M.; Kodama, T.; Honda, K.; et al. DAI (DLM-1/ZBP1) is a cytosolic DNA sensor and an activator of innate immune response. Nature 2007, 448, 501–505. [Google Scholar] [CrossRef]
- Keating, S.E.; Baran, M.; Bowie, A.G. Cytosolic DNA sensors regulating type I interferon induction. Trends Immunol. 2011, 32, 574–581. [Google Scholar] [CrossRef]
- Yu, L.; Liu, P. Cytosolic DNA sensing by cGAS: Regulation, function, and human diseases. Signal Transduct Target 2021, 6, 170. [Google Scholar] [CrossRef]
- Karayel, E.; Burckstummer, T.; Bilban, M.; Durnberger, G.; Weitzer, S.; Martinez, J.; Superti-Furga, G. The TLR-independent DNA recognition pathway in murine macrophages: Ligand features and molecular sigNature. Eur. J. Immunol. 2009, 39, 1929–1936. [Google Scholar] [CrossRef] [PubMed]
- Luecke, S.; Holleufer, A.; Christensen, M.H.; Jonsson, K.L.; Boni, G.A.; Sorensen, L.K.; Johannsen, M.; Jakobsen, M.R.; Hartmann, R.; Paludan, S.R. cGAS is activated by DNA in a length-dependent manner. EMBO Rep. 2017, 18, 1707–1715. [Google Scholar] [CrossRef] [PubMed]
- Stetson, D.B.; Medzhitov, R. Recognition of cytosolic DNA activates an IRF3-dependent innate immune response. Immunity 2006, 24, 93–103. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Meng, Q.; Qin, Y.; Liang, P.; Tan, P.; He, L.; Zhou, Y.; Chen, Y.; Huang, J.; Wang, R.F.; et al. TRIM14 Inhibits cGAS Degradation Mediated by Selective Autophagy Receptor p62 to Promote Innate Immune Responses. Mol. Cell 2016, 64, 105–119. [Google Scholar] [CrossRef]
- Ishikawa, H.; Barber, G.N. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 2008, 455, 674–678. [Google Scholar] [CrossRef]
- Kranzusch, P.J.; Wilson, S.C.; Lee, A.S.; Berger, J.M.; Doudna, J.A.; Vance, R.E. Ancient Origin of cGAS-STING reveals Mechanism of Universal 2’, 3’ cGAMP Signaling. Mol. Cell 2015, 59, 891–903. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, H.; Li, W. DNA-binding mechanisms of human and mouse cGAS: A comparative MD and MM/GBSA study. Phys. Chem. Chem. Phys. 2020, 22, 26390–26401. [Google Scholar] [CrossRef]
- Kranzusch, P.J.; Vance, R.E. cGAS dimerization entangles DNA recognition. Immunity 2013, 39, 992–994. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Huang, T.; Du, M.; Chen, X.; Du, F.; Ren, J.; Chen, Z.J. Phosphorylation and chromatin tethering prevent cGAS activation during mitosis. Science 2021, 371, 1204. [Google Scholar] [CrossRef]
- Barnett, K.C.; Coronas-Serna, J.M.; Zhou, W.; Ernandes, M.J.; Cao, A.; Kranzusch, P.J.; Kagan, J.C. Phosphoinositide Interactions Position cGAS at the Plasma Membrane to Ensure Efficient Distinction between Self- and Viral DNA. Cell 2019, 176, 1432–1446.e1411. [Google Scholar] [CrossRef] [Green Version]
- Gentili, M.; Lahaye, X.; Nadalin, F.; Nader, G.P.F.; Puig Lombardi, E.; Herve, S.; De Silva, N.S.; Rookhuizen, D.C.; Zueva, E.; Goudot, C.; et al. The N-Terminal Domain of cGAS Determines Preferential Association with Centromeric DNA and Innate Immune Activation in the Nucleus. Cell Rep. 2019, 26, 2377–2393.e2313. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Whiteley, A.T.; de Oliveira Mann, C.C.; Morehouse, B.R.; Nowak, R.P.; Fischer, E.S.; Gray, N.S.; Mekalanos, J.J.; Kranzusch, P.J. Structure of the Human cGAS-DNA Complex reveals Enhanced Control of Immune Surveillance. Cell 2018, 174, 300–311.e311. [Google Scholar] [CrossRef] [PubMed]
- Kranzusch, P.J. cGAS and CD-NTase enzymes: Structure, mechanism, and evolution. Curr. Opin. Struct. Biol. 2019, 59, 178–187. [Google Scholar] [CrossRef]
- Hooy, R.M.; Sohn, J. The allosteric activation of cGAS underpins its dynamic signaling landscape. eLife 2018, 7, e39984. [Google Scholar] [CrossRef]
- Gekara, N.O.; Jiang, H. The innate immune DNA sensor cGAS: A membrane, cytosolic, or nuclear protein? Sci. Signal. 2019, 12, eaax3521. [Google Scholar] [CrossRef]
- Zierhut, C.; Yamaguchi, N.; Paredes, M.; Luo, J.-D.; Carroll, T.; Funabiki, H. The cytoplasmic DNA sensor cGAS promotes mitotic cell death. Cell 2019, 178, 302–315.e323. [Google Scholar] [CrossRef]
- Schoggins, J.W.; MacDuff, D.A.; Imanaka, N.; Gainey, M.D.; Shrestha, B.; Eitson, J.L.; Mar, K.B.; Richardson, R.B.; Ratushny, A.V.; Litvak, V. Pan-viral specificity of IFN-induced genes reveals new roles for cGAS in innate immunity. Nature 2014, 505, 691–695. [Google Scholar] [CrossRef]
- Mazaleuskaya, L.; Veltrop, R.; Ikpeze, N.; Martin-Garcia, J.; Navas-Martin, S. Protective role of Toll-like receptor 3-induced type I interferon in murine coronavirus infection of macrophages. Viruses 2012, 4, 901–923. [Google Scholar] [CrossRef]
- van der Made, C.I.; Hoischen, A.; Netea, M.G.; van de Veerdonk, F.L. Primary immunodeficiencies in cytosolic pattern-recognition receptor pathways: Toward host-directed treatment strategies. Immunol. Ogical. Rev. 2020, 297, 247–272. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Wang, J.; Hui, P.; Yarovinsky, T.O.; Badeti, S.; Pham, K.; Liu, C. Potential role of IFN-α in COVID-19 patients and its underlying treatment options. Appl. Microbiol. Biotechnol. 2021, 105, 4005–4015. [Google Scholar] [CrossRef]
- Karayel, E. Zhang, Q.; Tang, Z.; An, R.; Ye, L.; Zhong, B. USP29 maintains the stability of cGAS and promotes cellular antiviral responses and autoimmunity. Cell Res. 2020, 30, 914–927. [Google Scholar]
- Du, J.-M.; Qian, M.-J.; Yuan, T.; Chen, R.-H.; He, Q.-J.; Yang, B.; Ling, Q.; Zhu, H. cGAS and cancer therapy: A double-edged sword. Acta. Pharmacol. Sin. 2022, 43, 2202–2211. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Chen, Z.J. The cGAS-cGAMP-STING pathway connects DNA damage to inflammation, senescence, and cancer. J. Exp. Med. 2018, 215, 1287–1299. [Google Scholar] [CrossRef] [PubMed]
- Fenech, M.; Morley, A.A. Measurement of micronuclei in lymphocytes. Mutat. Res. Environ. Mutag. Relat. Subjects. 1985, 147, 29–36. [Google Scholar] [CrossRef]
- Nelson, B.C.; Dizdaroglu, M. Implications of DNA damage and DNA repair on human diseases. Mutagenesis 2020, 35, 1–3. [Google Scholar] [CrossRef]
- Du, M.; Chen, Z.J. DNA-induced liquid phase condensation of cGAS activates innate immune signaling. Science 2018, 361, 704–709. [Google Scholar] [CrossRef]
- Ma, Z.; Jacobs, S.R.; West, J.A.; Stopford, C.; Zhang, Z.; Davis, Z.; Barber, G.N.; Glaunsinger, B.A.; Dittmer, D.P.; Damania, B. Modulation of the cGAS-STING DNA sensing pathway by gammaherpesviruses. Proc. Natl. Acad. Sci. USA 2015, 112, E4306–E4315. [Google Scholar] [CrossRef]
- Su, Q.; Mehta, S.; Zhang, J. Liquid-liquid phase separation: Orchestrating cell signaling through time and space. Mol. Cell 2021, 81, 4137–4146. [Google Scholar] [CrossRef]
- Lu, Q.; Haragopal, H.; Slepchenko, K.G.; Stork, C.; Li, Y.V. Intracellular zinc distribution in mitochondria, ER and the Golgi apparatus. Int. J. Physiol. Pathophysiol. Pharmacol. 2016, 8, 35. [Google Scholar]
- Liu, H.; Zhang, H.; Wu, X.; Ma, D.; Wu, J.; Wang, L.; Jiang, Y.; Fei, Y.; Zhu, C.; Tan, R.; et al. Nuclear cGAS suppresses DNA repair and promotes tumorigenesis. Nature 2018, 563, 131–136. [Google Scholar] [CrossRef]
- Wu, J.; Chen, Y.J.; Dobbs, N.; Sakai, T.; Liou, J.; Miner, J.J.; Yan, N. STING-mediated disruption of calcium homeostasis chronically activates ER stress and primes T cell death. J. Exp. Med. 2019, 216, 867–883. [Google Scholar] [CrossRef]
- Ablasser, A.; Chen, Z.J. cGAS in action: Expanding roles in immunity and inflammation. Science 2019, 363, eaat8657. [Google Scholar] [CrossRef] [PubMed]
- Liang, Q.; Seo, G.J.; Choi, Y.J.; Kwak, M.-J.; Ge, J.; Rodgers, M.A.; Shi, M.; Leslie, B.J.; Hopfner, K.-P.; Ha, T. Crosstalk between the cGAS DNA sensor and Beclin-1 autophagy protein shapes innate antimicrobial immune responses. Cell Host Microbe. 2014, 15, 228–238. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.-Y.; Pang, X.-Y.; Xu, Y.-Y.; Zhou, G.-P.; Xu, H.-G. Transcriptional regulation of human cyclic GMP-AMP synthase gene. Cell. Signal. 2019, 62, 109355. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.-Z.; Cheng, W.-C.; Chen, S.-F.; Nieh, S.; O’Connor, C.; Liu, C.-L.; Tsai, W.-W.; Wu, C.-J.; Martin, L.; Lin, Y.-S. miR-25/93 mediates hypoxia-induced immunosuppression by repressing cGAS. Nat. Cell Biol. 2017, 19, 1286–1296. [Google Scholar] [CrossRef] [PubMed]
- Song, B.; Greco, T.M.; Lum, K.K.; Taber, C.E.; Cristea, I.M. The DNA Sensor cGAS is Decorated by Acetylation and Phosphorylation Modifications in the Context of Immune Signaling. Mol. Cell Proteom. 2020, 19, 1193–1208. [Google Scholar] [CrossRef]
- Seo, G.J.; Yang, A.; Tan, B.; Kim, S.; Liang, Q.; Choi, Y.; Yuan, W.; Feng, P.; Park, H.S.; Jung, J.U. Akt Kinase-Mediated Checkpoint of cGAS DNA Sensing Pathway. Cell Rep. 2015, 13, 440–449. [Google Scholar] [CrossRef]
- Zhong, L.; Hu, M.M.; Bian, L.J.; Liu, Y.; Chen, Q.; Shu, H.B. Phosphorylation of cGAS by CDK1 impairs self-DNA sensing in mitosis. Cell Discov. 2020, 6, 26. [Google Scholar] [CrossRef]
- Li, M.; Shu, H.B. Dephosphorylation of cGAS by PPP6C impairs its substrate binding activity and innate antiviral response. Protein Cell 2020, 11, 584–599. [Google Scholar] [CrossRef]
- Seo, G.J.; Kim, C.; Shin, W.J.; Sklan, E.H.; Eoh, H.; Jung, J.U. TRIM56-mediated monoubiquitination of cGAS for cytosolic DNA sensing. Nat. Commun. 2018, 9, 613. [Google Scholar] [CrossRef]
- Liu, Z.S.; Zhang, Z.Y.; Cai, H.; Zhao, M.; Mao, J.; Dai, J.; Xia, T.; Zhang, X.M.; Li, T. RINCK-mediated monoubiquitination of cGAS promotes antiviral innate immune responses. Cell Biosci. 2018, 8, 35. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Huang, L.; Hong, Z.; Lv, Z.; Mao, Z.; Tang, Y.; Kong, X.; Li, S.; Cui, Y.; Liu, H.; et al. The E3 ubiquitin ligase RNF185 facilitates the cGAS-mediated innate immune response. PLoS Pathog. 2017, 13, e1006264. [Google Scholar] [CrossRef]
- Kato, Y.; Park, J.; Takamatsu, H.; Konaka, H.; Aoki, W.; Aburaya, S.; Ueda, M.; Nishide, M.; Koyama, S.; Hayama, Y.; et al. Apoptosis-derived membrane vesicles drive the cGAS-STING pathway and enhance type I IFN production in systemic lupus erythematosus. Ann. Rheum. Dis. 2018, 77, 1507–1515. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Jiang, F.; Kong, L.; Li, B.; Yang, Y.; Zhang, L.; Liu, B.; Zheng, Y.; Gao, C. Cutting Edge: USP27X DeubiquitiNat.es and Stabilizes the DNA Sensor cGAS to Regulate Cytosolic DNA-Mediated Signaling. J. Immunol. 2019, 203, 2049–2054. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.M.; Yang, Q.; Xie, X.Q.; Liao, C.Y.; Lin, H.; Liu, T.T.; Yin, L.; Shu, H.B. Sumoylation Promotes the Stability of the DNA Sensor cGAS and the Adaptor STING to Regulate the Kinetics of Response to DNA Virus. Immunity 2016, 45, 555–569. [Google Scholar] [CrossRef]
- Cui, Y.; Yu, H.; Zheng, X.; Peng, R.; Wang, Q.; Zhou, Y.; Wang, R.; Wang, J.; Qu, B.; Shen, N.; et al. SENP7 Potentiates cGAS Activation by Relieving SUMO-Mediated Inhibition of Cytosolic DNA Sensing. PLoS Pathog. 2017, 13, e1006156. [Google Scholar] [CrossRef]
- Narita, T.; Weinert, B.T.; Choudhary, C. Functions and mechanisms of non-histone protein acetylation. Nat. Rev. Mol. Cell Biol. 2019, 20, 156–174. [Google Scholar] [CrossRef]
- Dai, J.; Huang, Y.J.; He, X.; Zhao, M.; Wang, X.; Liu, Z.S.; Xue, W.; Cai, H.; Zhan, X.Y.; Huang, S.Y.; et al. Acetylation Blocks cGAS Activity and Inhibits Self-DNA-Induced Autoimmunity. Cell 2019, 176, 1447–1460.e1414. [Google Scholar] [CrossRef]
- Song, Z.M.; Lin, H.; Yi, X.M.; Guo, W.; Hu, M.M.; Shu, H.B. KAT5 acetylates cGAS to promote innate immune response to DNA virus. Proc. Natl Acad. Sci. USA 2020, 117, 21568–21575. [Google Scholar] [CrossRef]
- Barber, G.N. STING: Infection, inflammation and cancer. Nat. Rev. Immunol. 2015, 15, 760–770. [Google Scholar] [CrossRef]
- Place, D.E.; Kanneganti, T.-D. Cell death–mediated cytokine release and its therapeutic implications. J. Exp. Med. 2019, 216, 1474–1486. [Google Scholar] [CrossRef]
- Kuang, S.; Zheng, J.; Yang, H.; Li, S.; Duan, S.; Shen, Y.; Ji, C.; Gan, J.; Xu, X.-W.; Li, J. Structure insight of GSDMD reveals the basis of GSDMD autoinhibition in cell pyroptosis. Proc. Natl. Acad. Sci. USA 2017, 114, 10642–10647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Zhang, Z.; Ruan, J.; Pan, Y.; Magupalli, V.G.; Wu, H.; Lieberman, J. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature 2016, 535, 153–158. [Google Scholar] [CrossRef]
- Huang, L.S.; Hong, Z.; Wu, W.; Xiong, S.; Zhong, M.; Gao, X.; Rehman, J.; Malik, A.B. mtDNA activates cGAS signaling and suppresses the YAP-mediated endothelial cell proliferation program to promote inflammatory injury. Immunity 2020, 52, 475–486.e475. [Google Scholar] [CrossRef]
- Díaz-García, E.; García-Tovar, S.; Alfaro, E.; Jaureguizar, A.; Casitas, R.; Sánchez-Sánchez, B.; Zamarrón, E.; Fernández-Lahera, J.; López-Collazo, E.; Cubillos-Zapata, C. Inflammasome activation: A keystone of proinflammatory response in obstructive sleep apnea. Am. J. Respir. Crit. Care Med. 2022, 205, 1337–1348. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Zhou, Q.; Xu, W.; Cai, Y.; Yin, Z.; Gao, X.; Xiong, S. DNA-dependent activator of interferon-regulatory factors (DAI) promotes lupus nephritis by activating the calcium pathway. J. Biol. Chem. 2013, 288, 13534–13550. [Google Scholar] [CrossRef]
- Berridge, M.J.; Lipp, P.; Bootman, M.D. The versatility and universality of calcium signalling. Nat. Rev. Mol. Cell. Biol. 2000, 1, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Tassiulas, I.; Park-Min, K.H.; Reid, A.C.; Gil-Henn, H.; Schlessinger, J.; Baron, R.; Zhang, J.J.; Ivashkiv, L.B. “Tuning” of type I interferon-induced Jak-STAT1 signaling by calcium-dependent kinases in macrophages. Nat. Immunol. 2008, 9, 186–193. [Google Scholar] [CrossRef] [PubMed]
- Tano, J.Y.; Vazquez, G. Requirement for non-regulated, constitutive calcium influx in macrophage survival signaling. Biochem. Biophys. Res. Commun. 2011, 407, 432–437. [Google Scholar] [CrossRef]
- Srikanth, S.; Woo, J.S.; Wu, B.; El-Sherbiny, Y.M.; Leung, J.; Chupradit, K.; Rice, L.; Seo, G.J.; Calmettes, G.; Ramakrishna, C. The Ca2+ sensor STIM1 regulates the type I interferon response by retaining the signaling adaptor STING at the endoplasmic reticulum. Nat. Immunol. 2019, 20, 152–162. [Google Scholar] [CrossRef]
- Kyttaris, V.C.; Zhang, Z.; Kampagianni, O.; Tsokos, G.C. Calcium signaling in systemic lupus erythematosus T cells: A treatment target. Arthritis Rheumatism. 2011, 63, 2058–2066. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.; Jefferies, C. Role of DNA/RNA sensors and contribution to autoimmunity. Cytokine Growth Factor Rev. 2014, 25, 745–757. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, N.; Tauseef, M.; Amin, R.; Joshi, B.; Joshi, J.C.; Kini, V.; Klomp, J.; Li, W.; Knezevic, N.; Barbera, N.; et al. Noncanonical function of long myosin light chain kinase in increasing ER-PM junctions and augmentation of SOCE. FASEB J. 2020, 34, 12805–12819. [Google Scholar] [CrossRef] [PubMed]
- Yazbeck, P.; Tauseef, M.; Kruse, K.; Amin, M.R.; Sheikh, R.; Feske, S.; Komarova, Y.; Mehta, D. STIM1 Phosphorylation at Y361 Recruits Orai1 to STIM1 Puncta and Induces Ca(2+) Entry. Sci. Rep. 2017, 7, 42758. [Google Scholar] [CrossRef]
- Soni, D.; Regmi, S.C.; Wang, D.-M.; DebRoy, A.; Zhao, Y.-Y.; Vogel, S.M.; Malik, A.B.; Tiruppathi, C. Pyk2 phosphorylation of VE-PTP downstream of STIM1-induced Ca2+ entry regulates disassembly of adherens junctions. Am. J. Physiol. -Lung Cell. Mol. Physiol. 2017, 312, L1003–L1017. [Google Scholar] [CrossRef]
- Rayees, S.; Joshi, J.C.; Tauseef, M.; Anwar, M.; Baweja, S.; Rochford, I.; Joshi, B.; Hollenberg, M.D.; Reddy, S.P.; Mehta, D. PAR2-Mediated cAMP Generation Suppresses TRPV4-Dependent Ca(2+) Signaling in Alveolar Macrophages to Resolve TLR4-Induced Inflammation. Cell Rep. 2019, 27, 793–805.e794. [Google Scholar] [CrossRef]
- Hurley, R.L.; Anderson, K.A.; Franzone, J.M.; Kemp, B.E.; Means, A.R.; Witters, L.A. The Ca2+/calmodulin-dependent protein kinase kinases are AMP-activated protein kinase kinases. J. Biol. Chem. 2005, 280, 29060–29066. [Google Scholar] [CrossRef]
- Prantner, D.; Darville, T.; Nagarajan, U.M. Stimulator of IFN gene is critical for induction of IFN-beta during Chlamydia muridarum infection. J. Immunol. 2010, 184, 2551–2560. [Google Scholar] [CrossRef]
- Konno, H.; Konno, K.; Barber, G.N. Cyclic dinucleotides trigger ULK1 (ATG1) phosphorylation of STING to prevent sustained innate immune signaling. Cell 2013, 155, 688–698. [Google Scholar] [CrossRef]
- Kwon, D.; Sesaki, H.; Kang, S.J. Intracellular calcium is a rheostat for the STING signaling pathway. Biochem. Biophys. Res. Commun. 2018, 500, 497–503. [Google Scholar] [CrossRef]
- Son, A.; de Jesus, A.A.; Schwartz, D.M. STIM1 holds a STING in its (N-terminal) tail. Cell Calcium 2019, 80, 192–193. [Google Scholar] [CrossRef]
- Matthay, M.A.; Zemans, R.L. The acute respiratory distress syndrome: Pathogenesis and treatment. Annu. Rev. Pathol. 2011, 6, 147–163. [Google Scholar] [CrossRef] [Green Version]
- Joshi, J.C.; Joshi, B.; Rochford, I.; Rayees, S.; Akhter, M.Z.; Baweja, S.; Chava, K.R.; Tauseef, M.; Abdelkarim, H.; Natarajan, V.; et al. SPHK2-Generated S1P in CD11b(+) Macrophages Blocks STING to Suppress the Inflammatory Function of Alveolar Macrophages. Cell Rep. 2020, 30, 4096–4109.e4095. [Google Scholar] [CrossRef] [PubMed]
- Bellani, G.; Laffey, J.G.; Pham, T.; Fan, E.; Brochard, L.; Esteban, A.; Gattinoni, L.; van Haren, F.; Larsson, A.; McAuley, D.F.; et al. Epidemiology, Patterns of Care, and Mortality for Patients with Acute Respiratory Distress Syndrome in Intensive Care Units in 50 Countries. JAMA 2016, 315, 788–800. [Google Scholar] [CrossRef] [PubMed]
- Rochford, I.; Joshi, J.C.; Rayees, S.; Anwar, M.; Akhter, M.Z.; Yalagala, L.; Banerjee, S.; Mehta, D. Evidence for reprogramming of monocytes into reparative alveolar macrophages in vivo by targeting PDE4b. Am. J. Physiol. Lung Cell Mol. Physiol. 2021, 321, L686–L702. [Google Scholar] [CrossRef] [PubMed]
- Bachmaier, K.; Stuart, A.; Singh, A.; Mukhopadhyay, A.; Chakraborty, S.; Hong, Z.; Wang, L.; Tsukasaki, Y.; Maienschein-Cline, M.; Ganesh, B.B.; et al. Albumin Nanoparticle Endocytosing Subset of Neutrophils for Precision Therapeutic Targeting of Inflammatory Tissue Injury. ACS Nano 2022, 16, 4084–4101. [Google Scholar] [CrossRef]
- Leseigneur, C.; Le-Bury, P.; Pizarro-Cerda, J.; Dussurget, O. Emerging Evasion Mechanisms of Macrophage Defenses by Pathogenic Bacteria. Front. Cell Infect. Microbiol. 2020, 10, 577559. [Google Scholar] [CrossRef]
- Korns, D.; Frasch, S.C.; Fernandez-Boyanapalli, R.; Henson, P.M.; Bratton, D.L. Modulation of macrophage efferocytosis in inflammation. Front. Immunol. 2011, 2, 57. [Google Scholar] [CrossRef]
- Elliott, M.R.; Koster, K.M.; Murphy, P.S. Efferocytosis Signaling in the Regulation of Macrophage Inflammatory Responses. J. Immunol. 2017, 198, 1387–1394. [Google Scholar] [CrossRef]
- Funes, S.C.; Rios, M.; Escobar-Vera, J.; Kalergis, A.M. Implications of macrophage polarization in autoimmunity. Immunol. Ogy. 2018, 154, 186–195. [Google Scholar] [CrossRef]
- Mosser, D.M.; Edwards, J.P. Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 2008, 8, 958–969. [Google Scholar] [CrossRef]
- Galbraith, M.D.; Kinning, K.T.; Sullivan, K.D.; Araya, P.; Smith, K.P.; Granrath, R.E.; Shaw, J.R.; Baxter, R.; Jordan, K.R.; Russell, S.; et al. Specialized interferon action in COVID-19. Proc. Natl. Acad Sci. USA 2022, 119, e2116730119. [Google Scholar] [CrossRef] [PubMed]
- Decout, A.; Katz, J.D.; Venkatraman, S.; Ablasser, A. The cGAS-STING pathway as a therapeutic target in inflammatory diseases. Nat. Rev. Immunol. 2021, 21, 548–569. [Google Scholar] [CrossRef] [PubMed]
- Lucas, C.; Wong, P.; Klein, J.; Castro, T.B.R.; Silva, J.; Sundaram, M.; Ellingson, M.K.; Mao, T.; Oh, J.E.; Israelow, B.; et al. Longitudinal analyses reveal immunological misfiring in severe COVID-19. Nature 2020, 584, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Nienhold, R.; Ciani, Y.; Koelzer, V.H.; Tzankov, A.; Haslbauer, J.D.; Menter, T.; Schwab, N.; Henkel, M.; Frank, A.; Zsikla, V.; et al. Two distinct immunopathological profiles in autopsy lungs of COVID-19. Nat. Commun. 2020, 11, 5086. [Google Scholar] [CrossRef]
- Park, A.; Iwasaki, A. Type I and Type III Interferons—Induction, Signaling, Evasion, and Application to Combat COVID-19. Cell Host Microbe. 2020, 27, 870–878. [Google Scholar] [CrossRef]
- Hadjadj, J.; Yatim, N.; Barnabei, L.; Corneau, A.; Boussier, J.; Smith, N.; Pere, H.; Charbit, B.; Bondet, V.; Chenevier-Gobeaux, C.; et al. Impaired type I interferon activity and inflammatory responses in severe COVID-19 patients. Science 2020, 369, 718–724. [Google Scholar] [CrossRef]
- Schulte-Schrepping, J.; Reusch, N.; Paclik, D.; Bassler, K.; Schlickeiser, S.; Zhang, B.; Kramer, B.; Krammer, T.; Brumhard, S.; Bonaguro, L.; et al. Severe COVID-19 Is Marked by a Dysregulated Myeloid Cell Compartment. Cell 2020, 182, 1419–1440.e1423. [Google Scholar] [CrossRef]
- Rice, G.I.; Forte, G.M.; Szynkiewicz, M.; Chase, D.S.; Aeby, A.; Abdel-Hamid, M.S.; Ackroyd, S.; Allcock, R.; Bailey, K.M.; Balottin, U.; et al. Assessment of interferon-related biomarkers in Aicardi-Goutieres syndrome associated with mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, and ADAR: A case-control study. Lancet. Neurol. 2013, 12, 1159–1169. [Google Scholar] [CrossRef]
- Israelow, B.; Song, E.; Mao, T.; Lu, P.; Meir, A.; Liu, F.; Alfajaro, M.M.; Wei, J.; Dong, H.; Homer, R.J. Mouse model of SARS-CoV-2 Reveals inflammatory role of type I interferon signaling. J. Exp. Med. 2020, 217, e20201241. [Google Scholar] [CrossRef]
- Domizio, J.D.; Gulen, M.F.; Saidoune, F.; Thacker, V.V.; Yatim, A.; Sharma, K.; Nass, T.; Guenova, E.; Schaller, M.; Conrad, C. The cGAS–STING pathway drives type I IFN immunopathology in COVID-19. Nature 2022, 603, 145–151. [Google Scholar] [CrossRef] [PubMed]
- Heuberger, J.; Trimpert, J.; Vladimirova, D.; Goosmann, C.; Lin, M.; Schmuck, R.; Mollenkopf, H.J.; Brinkmann, V.; Tacke, F.; Osterrieder, N. Epithelial response to IFN-γ promotes SARS-CoV-2 infection. EMBO Mol. Med. 2021, 13, e13191. [Google Scholar] [CrossRef] [PubMed]
- Akamatsu, H.; Toi, Y.; Hayashi, H.; Fujimoto, D.; Tachihara, M.; Furuya, N.; Otani, S.; Shimizu, J.; Katakami, N.; Azuma, K. Efficacy of Osimertinib Plus Bevacizumab vs Osimertinib in Patients With EGFR T790M–Mutated Non–Small Cell Lung Cancer PRev..iously Treated With Epidermal Growth Factor Receptor–Tyrosine Kinase Inhibitor: West Japan Oncology Group 8715L Phase 2 Randomized Clinical Trial. JAMA Oncol. 2021, 7, 386–394. [Google Scholar] [PubMed]
- Bueno, M.; Zank, D.; Buendia-Roldan, I.; Fiedler, K.; Mays, B.G.; Alvarez, D.; Sembrat, J.; Kimball, B.; Bullock, J.K.; Martin, J.L.; et al. PINK1 attenuates mtDNA release in alveolar epithelial cells and TLR9 mediated profibrotic responses. PLoS ONE 2019, 14, e0218003. [Google Scholar] [CrossRef]
- Schuliga, M.; Pechkovsky, D.V.; Read, J.; Waters, D.W.; Blokland, K.E.C.; Reid, A.T.; Hogaboam, C.M.; Khalil, N.; Burgess, J.K.; Prele, C.M.; et al. Mitochondrial dysfunction contributes to the senescent phenotype of IPF lung fibroblasts. J. Cell. Mol. Med. 2018, 22, 5847–5861. [Google Scholar] [CrossRef]
- Schuliga, M.; Read, J.; Blokland, K.E.C.; Waters, D.W.; Burgess, J.; Prele, C.; Mutsaers, S.E.; Jaffar, J.; Westall, G.; Reid, A.; et al. Self DNA perpetuates IPF lung fibroblast senescence in a cGAS-dependent manner. Clin. Sci. (Lond.) 2020, 134, 889–905. [Google Scholar] [CrossRef]
- da Silva, A.L.; Bresciani, M.J.; Karnopp, T.E.; Weber, A.F.; Ellwanger, J.H.; Henriques, J.A.; Valim, A.R.; Possuelo, L.G. DNA damage and cellular abnormalities in tuberculosis, lung cancer and chronic obstructive pulmonary disease. Multidiscip. Respir. Med. 2015, 10, 38. [Google Scholar] [CrossRef]
- Maluf, S.W.; Mergener, M.; Dalcanale, L.; Costa, C.C.; Pollo, T.; Kayser, M.; da Silva, L.B.; Pra, D.; Teixeira, P.J.Z. DNA damage in peripheral blood of patients with chronic obstructive pulmonary disease (COPD). Mutat. Res. 2007, 626, 180–184. [Google Scholar] [CrossRef]
- Channappanavar, R.; Fehr, A.R.; Zheng, J.; Wohlford-Lenane, C.; Abrahante, J.E.; Mack, M.; Sompallae, R.; McCray, P.B.; Meyerholz, D.K.; Perlman, S. IFN-I response timing relative to virus replication determines MERS coronavirus infection outcomes. J. Clin. Investig. 2019, 129, 3625–3639. [Google Scholar] [CrossRef]
- Guerriero, J.L. Macrophages: Their Untold Story in T Cell Activation and Function. Int. Rev. Cell Mol. Biol. 2019, 342, 73–93. [Google Scholar]
- Bergsmedh, A.; Szeles, A.; Henriksson, M.; Bratt, A.; Folkman, M.J.; Spetz, A.L.; Holmgren, L. Horizontal transfer of oncogenes by uptake of apoptotic bodies. Proc. Natl. Acad. Sci. USA 2001, 98, 6407–6411. [Google Scholar] [CrossRef] [PubMed]
- Kwon, J.; Bakhoum, S.F. The Cytosolic DNA-Sensing cGAS-STING Pathway in Cancer. Cancer Discov. 2020, 10, 26–39. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, F.; Zhang, X. STING-associated vasculopathy with onset in infancy: A familial case series report and literature Review. Ann. Transl. Med. 2021, 9, 176. [Google Scholar] [CrossRef] [PubMed]
- Cuadrado, E.; Michailidou, I.; van Bodegraven, E.J.; Jansen, M.H.; Sluijs, J.A.; Geerts, D.; Couraud, P.O.; De Filippis, L.; Vescovi, A.L.; Kuijpers, T.W.; et al. Phenotypic variation in Aicardi-Goutieres syndrome explained by cell-specific IFN-stimulated gene response and cytokine release. J. Immunol. 2015, 194, 3623–3633. [Google Scholar] [CrossRef] [PubMed]
- Crow, Y.J.; Hayward, B.E.; Parmar, R.; Robins, P.; Leitch, A.; Ali, M.; Black, D.N.; van Bokhoven, H.; Brunner, H.G.; Hamel, B.C.; et al. Mutations in the gene encoding the 3’-5’ DNA exonuclease TREX1 cause Aicardi-Goutieres syndrome at the AGS1 locus. Nat. Genet. 2006, 38, 917–920. [Google Scholar] [CrossRef]
- Yan, N. Immune Diseases Associated with TREX1 and STING Dysfunction. J. Interferon Cytokine Res. 2017, 37, 198–206. [Google Scholar] [CrossRef]
- Stetson, D.B.; Ko, J.S.; Heidmann, T.; Medzhitov, R. Trex1 prevents cell-intrinsic initiation of autoimmunity. Cell 2008, 134, 587–598. [Google Scholar] [CrossRef]
- Lundgren, M.C.; Molitor, J.A.; Spilseth, B.; Adeyi, O. A Fatal Case of Diffuse Alveolar Hemorrhage in the Setting of Systemic Lupus Erythematosus: A Case Report and Review of Noninfectious Causes of Acute Pulmonary Hemorrhage in Adults. Case Rep. Rheumatol. 2021, 2021, 6620701. [Google Scholar] [CrossRef]
- Hepburn, A.L.; Narat, S.; Mason, J.C. The management of peripheral blood cytopenias in systemic lupus erythematosus. Rheumatology 2010, 49, 2243–2254. [Google Scholar] [CrossRef]
- Knight, J.S.; Subramanian, V.; O’Dell, A.A.; Yalavarthi, S.; Zhao, W.; Smith, C.K.; Hodgin, J.B.; Thompson, P.R.; Kaplan, M.J. Peptidylarginine deiminase inhibition disrupts NET formation and protects against kidney, skin and vascular disease in lupus-prone MRL/lpr mice. Ann. Rheum. Dis. 2015, 74, 2199–2206. [Google Scholar] [CrossRef]
- Zheng, J.; Mo, J.; Zhu, T.; Zhuo, W.; Yi, Y.; Hu, S.; Yin, J.; Zhang, W.; Zhou, H.; Liu, Z. Comprehensive elaboration of the cGAS-STING signaling axis in cancer development and immunotherapy. Mol. Cancer 2020, 19, 133. [Google Scholar] [CrossRef] [PubMed]
- Takashima, K.; Takeda, Y.; Oshiumi, H.; Shime, H.; Okabe, M.; Ikawa, M.; Matsumoto, M.; Seya, T. STING in tumor and host cells cooperatively work for NK cell-mediated tumor growth retardation. Biochem. Biophys. Res. Commun. 2016, 478, 1764–1771. [Google Scholar] [CrossRef] [PubMed]
- Ho, S.S.W.; Zhang, W.Y.L.; Tan, N.Y.J.; Khatoo, M.; Suter, M.A.; Tripathi, S.; Cheung, F.S.G.; Lim, W.K.; Tan, P.H.; Ngeow, J.; et al. The DNA Structure-Specific Endonuclease MUS81 Mediates DNA Sensor STING-Dependent Host Rejection of Prostate Cancer Cells. Immunity 2016, 44, 1177–1189. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.; Boire, A.; Jin, X.; Valiente, M.; Er, E.E.; Lopez-Soto, A.; Jacob, L.S.; Patwa, R.; Shah, H.; Xu, K.; et al. Carcinoma-astrocyte gap junctions promote brain metastasis by cGAMP transfer. Nature 2016, 533, 493–498. [Google Scholar] [CrossRef] [PubMed]
- Petrovic, M.; Borchard, G.; Jordan, O. Considerations for the delivery of STING ligands in cancer immunotherapy. J. Control. Release 2021, 339, 235–247. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Zhao, W.; Ju, Z.; Wang, L.; Peng, Y.; Labrie, M.; Yap, T.A.; Mills, G.B.; Peng, G. PARPi Triggers the STING-Dependent Immune Response and Enhances the Therapeutic Efficacy of Immune Checkpoint Blockade Independent of BRCAness. Cancer Res. 2019, 79, 311–319. [Google Scholar] [CrossRef]
- Grabosch, S.; Bulatovic, M.; Zeng, F.; Ma, T.; Zhang, L.; Ross, M.; Brozick, J.; Fang, Y.; Tseng, G.; Kim, E.; et al. Cisplatin-induced immune modulation in ovarian cancer mouse models with distinct inflammation profiles. Oncogene 2019, 38, 2380–2393. [Google Scholar] [CrossRef]
- Xie, W.; Lama, L.; Adura, C.; Tomita, D.; Glickman, J.F.; Tuschl, T.; Patel, D.J. Human cGAS catalytic domain has an additional DNA-binding interface that enhances enzymatic activity and liquid-phase condensation. Proc. Natl. Acad. Sci. USA 2019, 116, 11946–11955. [Google Scholar] [CrossRef]
- Marcus, A.; Mao, A.J.; Lensink-Vasan, M.; Wang, L.; Vance, R.E.; Raulet, D.H. Tumor-Derived cGAMP Triggers a STING-Mediated Interferon Response in Non-tumor Cells to Activate the NK Cell Response. Immunity 2018, 49, 754–763.e754. [Google Scholar] [CrossRef]
- Ohkuri, T.; Ghosh, A.; Kosaka, A.; Zhu, J.; Ikeura, M.; David, M.; Watkins, S.C.; Sarkar, S.N.; Okada, H. STING contributes to antiglioma immunity via triggering type I IFN signals in the tumor microenvironment. Cancer Immunol. Res. 2014, 2, 1199–1208. [Google Scholar] [CrossRef]
- Klarquist, J.; Hennies, C.M.; Lehn, M.A.; Reboulet, R.A.; Feau, S.; Janssen, E.M. STING-mediated DNA sensing promotes antitumor and autoimmune responses to dying cells. J. Immunol. 2014, 193, 6124–6134. [Google Scholar] [CrossRef] [PubMed]
- Demaria, O.; De Gassart, A.; Coso, S.; Gestermann, N.; Di Domizio, J.; Flatz, L.; Gaide, O.; Michielin, O.; Hwu, P.; Petrova, T.V.; et al. STING activation of tumor endothelial cells initiates spontaneous and therapeutic antitumor immunity. Proc. Natl. Acad. Sci. USA 2015, 112, 15408–15413. [Google Scholar] [CrossRef] [PubMed]
- Woo, S.R.; Fuertes, M.B.; Corrales, L.; Spranger, S.; Furdyna, M.J.; Leung, M.Y.; Duggan, R.; Wang, Y.; Barber, G.N.; Fitzgerald, K.A.; et al. STING-dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity 2014, 41, 830–842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crasta, K.; Ganem, N.J.; Dagher, R.; Lantermann, A.B.; Ivanova, E.V.; Pan, Y.; Nezi, L.; Protopopov, A.; Chowdhury, D.; Pellman, D. DNA breaks and chromosome pulverization from errors in mitosis. Nature 2012, 482, 53–58. [Google Scholar] [CrossRef]
- Harding, S.M.; Benci, J.L.; Irianto, J.; Discher, D.E.; Minn, A.J.; Greenberg, R.A. Mitotic progression following DNA damage enables pattern recognition within micronuclei. Nature 2017, 548, 466–470. [Google Scholar] [CrossRef]
- Mackenzie, K.J.; Carroll, P.; Martin, C.-A.; Murina, O.; Fluteau, A.; Simpson, D.J.; Olova, N.; Sutcliffe, H.; Rainger, J.K.; Leitch, A.; et al. cGAS surveillance of micronuclei links genome instability to innate immunity. Nature 2017, 548, 461–465. [Google Scholar] [CrossRef]
- Kitajima, S.; Ivanova, E.; Guo, S.; Yoshida, R.; Campisi, M.; Sundararaman, S.K.; Tange, S.; Mitsuishi, Y.; Thai, T.C.; Masuda, S.; et al. Suppression of STING Associated with LKB1 Loss in KRAS-Driven Lung Cancer. Cancer Discov. 2019, 9, 34–45. [Google Scholar] [CrossRef]
- Platnich, J.M.; Chung, H.; Lau, A.; Sandall, C.F.; Bondzi-Simpson, A.; Chen, H.M.; Komada, T.; Trotman-Grant, A.C.; Brandelli, J.R.; Chun, J.; et al. Shiga Toxin/Lipopolysaccharide Activates Caspase-4 and Gasdermin D to Trigger Mitochondrial Reactive Oxygen Species Upstream of the NLRP3 Inflammasome. Cell Rep. 2018, 25, 1525–1536.e1527. [Google Scholar] [CrossRef]
- Sansone, P.; Savini, C.; Kurelac, I.; Chang, Q.; Amato, L.B.; Strillacci, A.; Stepanova, A.; Iommarini, L.; Mastroleo, C.; Daly, L.; et al. Packaging and transfer of mitochondrial DNA via exosomes regulate escape from dormancy in hormonal therapy-resistant breast cancer. Proc. Natl Acad. Sci. USA 2017, 114, E9066–E9075. [Google Scholar] [CrossRef]
- Dou, Z.; Ghosh, K.; Vizioli, M.G.; Zhu, J.; Sen, P.; Wangensteen, K.J.; Simithy, J.; Lan, Y.; Lin, Y.; Zhou, Z.; et al. Cytoplasmic chromatin triggers inflammation in senescence and cancer. Nature 2017, 550, 402–406. [Google Scholar] [CrossRef]
- Yang, H.; Wang, H.; Ren, J.; Chen, Q.; Chen, Z.J. cGAS is essential for cellular senescence. Proc. Natl. Acad. Sci. USA 2017, 114, E4612–E4620. [Google Scholar] [CrossRef] [PubMed]
- Coppe, J.P.; Patil, C.K.; Rodier, F.; Sun, Y.; Munoz, D.P.; Goldstein, J.; Nelson, P.S.; Desprez, P.Y.; Campisi, J. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008, 6, 2853–2868. [Google Scholar] [CrossRef] [PubMed]
- Gluck, S.; Guey, B.; Gulen, M.F.; Wolter, K.; Kang, T.W.; Schmacke, N.A.; Bridgeman, A.; Rehwinkel, J.; Zender, L.; Ablasser, A. Innate immune sensing of cytosolic chromatin fragments through cGAS promotes senescence. Nat. Cell Biol. 2017, 19, 1061–1070. [Google Scholar] [CrossRef] [PubMed]
- Ranoa, D.R.E.; Widau, R.C.; Mallon, S.; Parekh, A.D.; Nicolae, C.M.; Huang, X.; Bolt, M.J.; Arina, A.; Parry, R.; Kron, S.J.; et al. STING Promotes Homeostasis via Regulation of Cell Proliferation and Chromosomal Stability. Cancer Res. 2019, 79, 1465–1479. [Google Scholar] [CrossRef]
- Sauter, B.; Albert, M.L.; Francisco, L.; Larsson, M.; Somersan, S.; Bhardwaj, N. Consequences of cell death: Exposure to necrotic tumor cells, but not primary tissue cells or apoptotic cells, induces the maturation of immunostimulatory dendritic cells. J. Exp. Med. 2000, 191, 423–434. [Google Scholar] [CrossRef]
- Smyth, M.J.; Godfrey, D.I.; Trapani, J.A. A fresh look at tumor immunosurveillance and immunotherapy. Nat. Immunol. 2001, 2, 293–299. [Google Scholar] [CrossRef]
- Deng, L.; Liang, H.; Xu, M.; Yang, X.; Burnette, B.; Arina, A.; Li, X.D.; Mauceri, H.; Beckett, M.; Darga, T.; et al. STING-Dependent Cytosolic DNA Sensing Promotes Radiation-Induced Type I Interferon-Dependent Antitumor Immunity in Immunogenic Tumors. Immunity 2014, 41, 843–852. [Google Scholar] [CrossRef]
- Diamond, M.S.; Kinder, M.; Matsushita, H.; Mashayekhi, M.; Dunn, G.P.; Archambault, J.M.; Lee, H.; Arthur, C.D.; White, J.M.; Kalinke, U.; et al. Type I interferon is selectively required by dendritic cells for immune rejection of tumors. J. Exp. Med. 2011, 208, 1989–2003. [Google Scholar] [CrossRef]
- Ritchie, C.; Cordova, A.F.; Hess, G.T.; Bassik, M.C.; Li, L. SLC19A1 Is an Importer of the Immunotransmitter cGAMP. Mol. Cell 2019, 75, 372–381.e375. [Google Scholar] [CrossRef]
- Nowarski, R.; Gagliani, N.; Huber, S.; Flavell, R.A. Innate immune cells in inflammation and cancer. Cancer Immunol. Res. 2013, 1, 77–84. [Google Scholar] [CrossRef]
- Bao, T.; Liu, J.; Leng, J.; Cai, L. The cGAS-STING pathway: More than fighting against viruses and cancer. Cell Biosci. 2021, 11, 209. [Google Scholar] [CrossRef] [PubMed]
- Fryer, A.L.; Abdullah, A.; Taylor, J.M.; Crack, P.J. The Complexity of the cGAS-STING Pathway in CNS Pathologies. Front. Neurosci. 2021, 15, 621501. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Rao, X.; Wang, X.; Luo, Z.; Wang, J.; Sheng, S.; Liu, Y.; Zhang, N.; Jin, S.; Chen, H.; et al. cGAS-STING Signaling Pathway and Liver Disease: From Basic Research to Clinical Practice. Front. Pharm. 2021, 12, 719644. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Du, J.; Zhu, H.; Ling, Q. The role of cGAS-STING signalling in liver diseases. JHEP Rep. 2021, 3, 100324. [Google Scholar] [CrossRef] [PubMed]
- Jauhari, A.; Baranov, S.V.; Suofu, Y.; Kim, J.; Singh, T.; Yablonska, S.; Li, F.; Wang, X.; Oberly, P.; Minnigh, M.B.; et al. Melatonin inhibits cytosolic mitochondrial DNA—induced neuroinflammatory signaling in accelerated aging and neurodegeneration. J. Clin. Invest. 2020, 130, 3124–3136. [Google Scholar] [CrossRef]
- Bai, J.; Liu, F. cGASSTING signaling and function in metabolism and kidney diseases. J. Mol. Cell Biol. 2021, 13, 728–738. [Google Scholar] [CrossRef]
- Ding, R.; Li, H.; Liu, Y.; Ou, W.; Zhang, X.; Chai, H.; Huang, X.; Yang, W.; Wang, Q. Activating cGAS-STING axis contributes to neuroinflammation in CVST mouse model and induces inflammasome activation and microglia pyroptosis. J. Neuroinflamm. 2022, 19, 137. [Google Scholar] [CrossRef]
- Skopelja-Gardner, S.; An, J.; Elkon, K.B. Role of the cGAS-STING pathway in systemic and organ-specific diseases. Nat. Rev. Nephrol. 2022, 18, 558–572. [Google Scholar] [CrossRef]
- Barrett, J.P.; Knoblach, S.M.; Bhattacharya, S.; Gordish-Dressman, H.; Stoica, B.A.; Loane, D.J. Traumatic Brain Injury Induces cGAS Activation and Type I Interferon Signaling in Aged Mice. Front. Immunol. 2021, 12, 710608. [Google Scholar] [CrossRef]
- Larrick, J.W.; Mendelsohn, A.R. Modulation of cGAS-STING Pathway by Nicotinamide Riboside in Alzheimer’s Disease. Rejuven. Res. 2021, 24, 397–402. [Google Scholar] [CrossRef]
- Li, F.; Wang, N.; Zheng, Y.; Luo, Y.; Zhang, Y. cGAS- Stimulator of Interferon Genes Signaling in Central Nervous System Disorders. Aging. Dis. 2021, 12, 1658–1674. [Google Scholar] [CrossRef] [PubMed]
- Jin, M.; Shiwaku, H.; Tanaka, H.; Obita, T.; Ohuchi, S.; Yoshioka, Y.; Jin, X.; Kondo, K.; Fujita, K.; Homma, H.; et al. activates microglia via the PQBP1-cGAS-STING pathway to promote brain inflammation. Nat. Commun. 2021, 12, 6565. [Google Scholar] [CrossRef] [PubMed]
- Paul, B.D.; Snyder, S.H.; Bohr, V.A. Signaling by cGAS-STING in Neurodegeneration, Neuroinflammation, and Aging. Trends Neurosci. 2021, 44, 83–96. [Google Scholar] [CrossRef]
- Xu, D.; Tian, Y.; Xia, Q.; Ke, B. The cGAS-STING Pathway: Novel Perspectives in Liver Diseases. Front. Immunol. 2021, 12, 682736. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Rao, H.; Zhao, J.; Wee, A.; Li, X.; Fei, R.; Huang, R.; Wu, C.; Liu, F.; Wei, L. STING expression in monocyte-derived macrophages is associated with the progression of liver inflammation and fibrosis in patients with nonalcoholic fatty liver disease. Lab. Invest. 2020, 100, 542–552. [Google Scholar] [CrossRef]
- Luther, J.; Khan, S.; Gala, M.K.; Kedrin, D.; Sridharan, G.; Goodman, R.P.; Garber, J.J.; Masia, R.; Diagacomo, E.; Adams, D.; et al. Hepatic gap junctions amplify alcohol liver injury by propagating cGAS-mediated IRF3 activation. Proc. Natl. Acad. Sci. USA 2020, 117, 11667–11673. [Google Scholar] [CrossRef]
- Li, H.; Hu, L.; Wang, L.; Wang, Y.; Shao, M.; Chen, Y.; Wu, W.; Wang, L. Iron Activates cGAS-STING Signaling and Promotes Hepatic Inflammation. J. Agric. Food Chem. 2022, 70, 2211–2220. [Google Scholar] [CrossRef] [PubMed]
- Chung, K.W.; Dhillon, P.; Huang, S.; Sheng, X.; Shrestha, R.; Qiu, C.; Kaufman, B.A.; Park, J.; Pei, L.; Baur, J.; et al. Mitochondrial Damage and Activation of the STING Pathway Lead to Renal Inflammation and Fibrosis. Cell Metab. 2019, 30, 784–799.e785. [Google Scholar] [CrossRef] [PubMed]
- Steinhagen, F.; Zillinger, T.; Peukert, K.; Fox, M.; Thudium, M.; Barchet, W.; Putensen, C.; Klinman, D.; Latz, E.; Bode, C. Suppressive oligodeoxynucleotides containing TTAGGG motifs inhibit cGAS activation in human monocytes. Eur. J. Immunol. 2018, 48, 605–611. [Google Scholar] [CrossRef]
- An, J.; Woodward, J.J.; Lai, W.; Minie, M.; Sun, X.; Tanaka, L.; Snyder, J.M.; Sasaki, T.; Elkon, K.B. Inhibition of Cyclic GMP-AMP Synthase Using a Novel Antimalarial Drug Derivative in Trex1-Deficient Mice. Arthritis Rheumatol. 2018, 70, 1807–1819. [Google Scholar] [CrossRef]
- Huang, Y.; Liang, W.; Li, K.; Liao, X.; Chen, J.; Qiu, X.; Liu, K.; Qiu, D.; Qin, Y. Sorafenib suppresses the activation of type I interferon pathway induced by RLR-MAVS and cGAS-STING signaling. Biochem. Biophys. Res. Commun. 2022, 623, 181–188. [Google Scholar] [CrossRef] [PubMed]
- Hall, J.; Brault, A.; Vincent, F.; Weng, S.; Wang, H.; Dumlao, D.; Aulabaugh, A.; Aivazian, D.; Castro, D.; Chen, M.; et al. Discovery of PF-06928215 as a high affinity inhibitor of cGAS enabled by a novel fluorescence polarization assay. PLoS ONE 2017, 12, e0184843. [Google Scholar]
- Hong, Z.; Mei, J.; Guo, H.; Zhu, J.; Wang, C. Intervention of cGASSTING signaling in sterile inflammatory diseases. J. Mol. Cell Biol. 2022, 14, mjac005. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Sooreshjani, M.A.; Mikek, C.; Opoku-Temeng, C.; Sintim, H.O. Suramin potently inhibits cGAMP synthase, cGAS, in THP1 cells to modulate IFN-beta levels. Future Med. Chem. 2018, 10, 1301–1317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vincent, J.; Adura, C.; Gao, P.; Luz, A.; Lama, L.; Asano, Y.; Okamoto, R.; Imaeda, T.; Aida, J.; Rothamel, K.; et al. Small molecule inhibition of cGAS reduces interferon expression in primary macrophages from autoimmune mice. Nat. Commun. 2017, 8, 750. [Google Scholar] [CrossRef]
- Lama, L.; Adura, C.; Xie, W.; Tomita, D.; Kamei, T.; Kuryavyi, V.; Gogakos, T.; Steinberg, J.I.; Miller, M.; Ramos-Espiritu, L.; et al. Development of human cGAS-specific small-molecule inhibitors for repression of dsDNA-triggered interferon expression. Nat. Commun. 2019, 10, 2261. [Google Scholar] [CrossRef]
- Chu, L.; Li, C.; Li, Y.; Yu, Q.; Yu, H.; Li, C.; Meng, W.; Zhu, J.; Wang, Q.; Wang, C.; et al. Perillaldehyde Inhibition of cGAS Reduces dsDNA-Induced Interferon Response. Front. Immunol. 2021, 12, 655637. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Joshi, B.; Joshi, J.C.; Mehta, D. Regulation of cGAS Activity and Downstream Signaling. Cells 2022, 11, 2812. https://doi.org/10.3390/cells11182812
Joshi B, Joshi JC, Mehta D. Regulation of cGAS Activity and Downstream Signaling. Cells. 2022; 11(18):2812. https://doi.org/10.3390/cells11182812
Chicago/Turabian StyleJoshi, Bhagwati, Jagdish Chandra Joshi, and Dolly Mehta. 2022. "Regulation of cGAS Activity and Downstream Signaling" Cells 11, no. 18: 2812. https://doi.org/10.3390/cells11182812
APA StyleJoshi, B., Joshi, J. C., & Mehta, D. (2022). Regulation of cGAS Activity and Downstream Signaling. Cells, 11(18), 2812. https://doi.org/10.3390/cells11182812