
Hyperactive Akt1 Signaling Increases Tumor Progression and DNA Repair in Embryonal Rhabdomyosarcoma RD Line and Confers Susceptibility to Glycolysis and Mevalonate Pathway Inhibitors

 , , ,
, , , 

 ,
,  , ,
, ,  ,
,  , , ,
, , ,  ,
,  ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Plasmids
2.3. Cell Cultures
2.4. Immunoblotting
2.5. Immunofluorescence Analysis
2.6. Crystal Violet Assay
2.7. Clonogenic Assay
2.8. Zymography Assay
2.9. Wound Healing Assay
2.10. Neutral Red Assay
2.11. Flow Cytometric Analyses
2.12. Zebrafish Xenograft assay
2.13. Statistical Analysis
3. Results
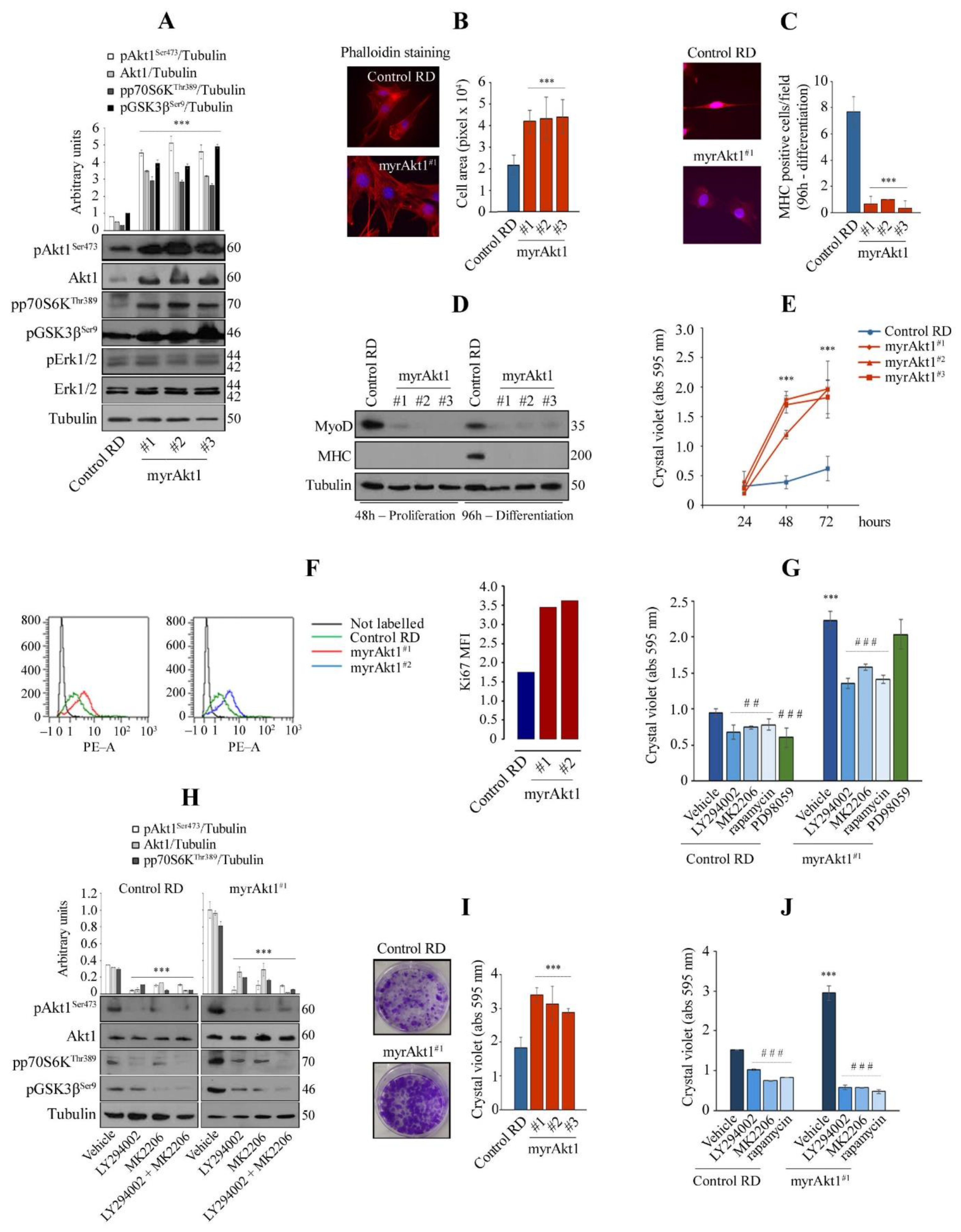
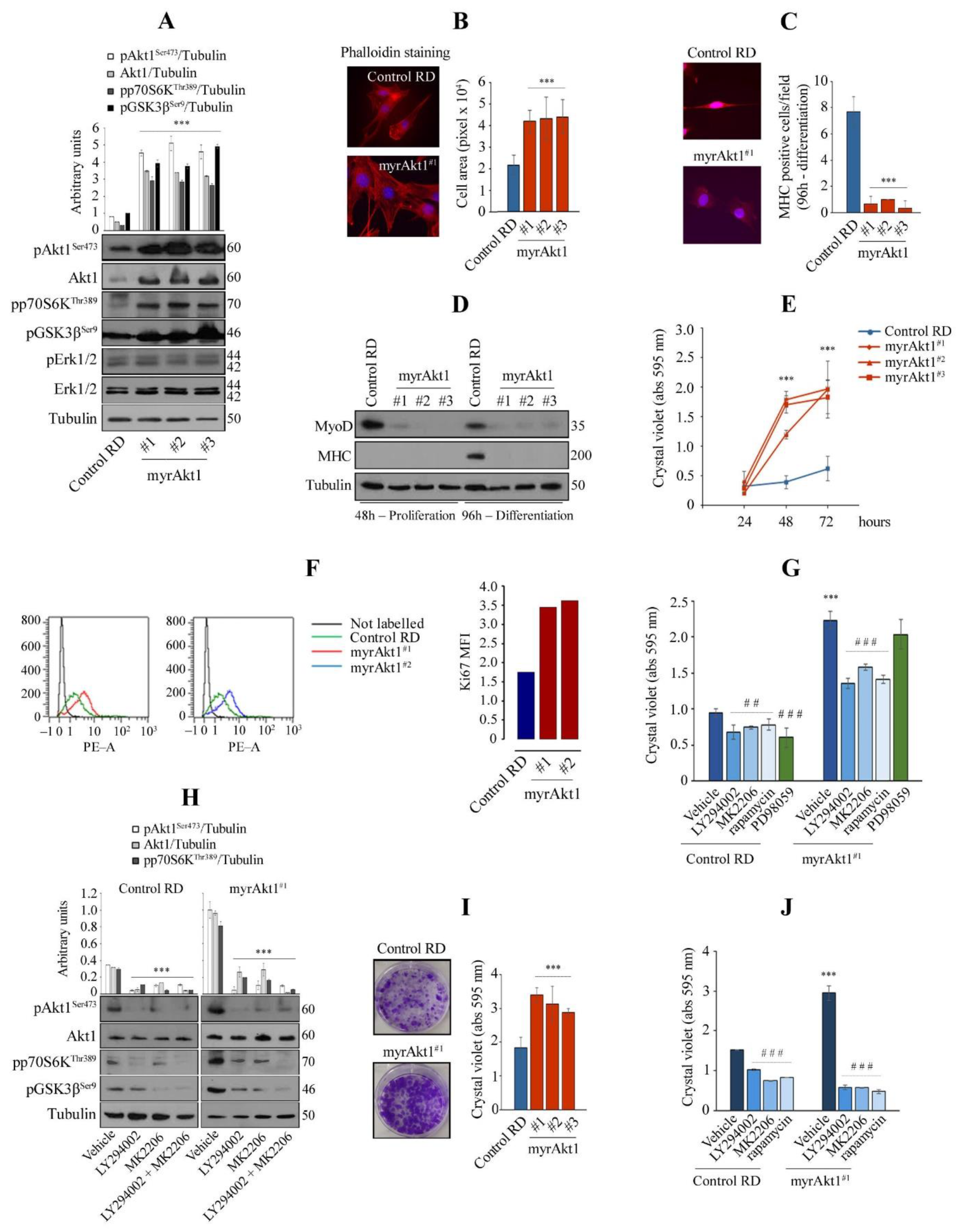
3.1. Hyperactive Akt1 Signaling Suppresses Myogenic Differentiation and Enhances Growth Rate and Clonogenicity in RD Line
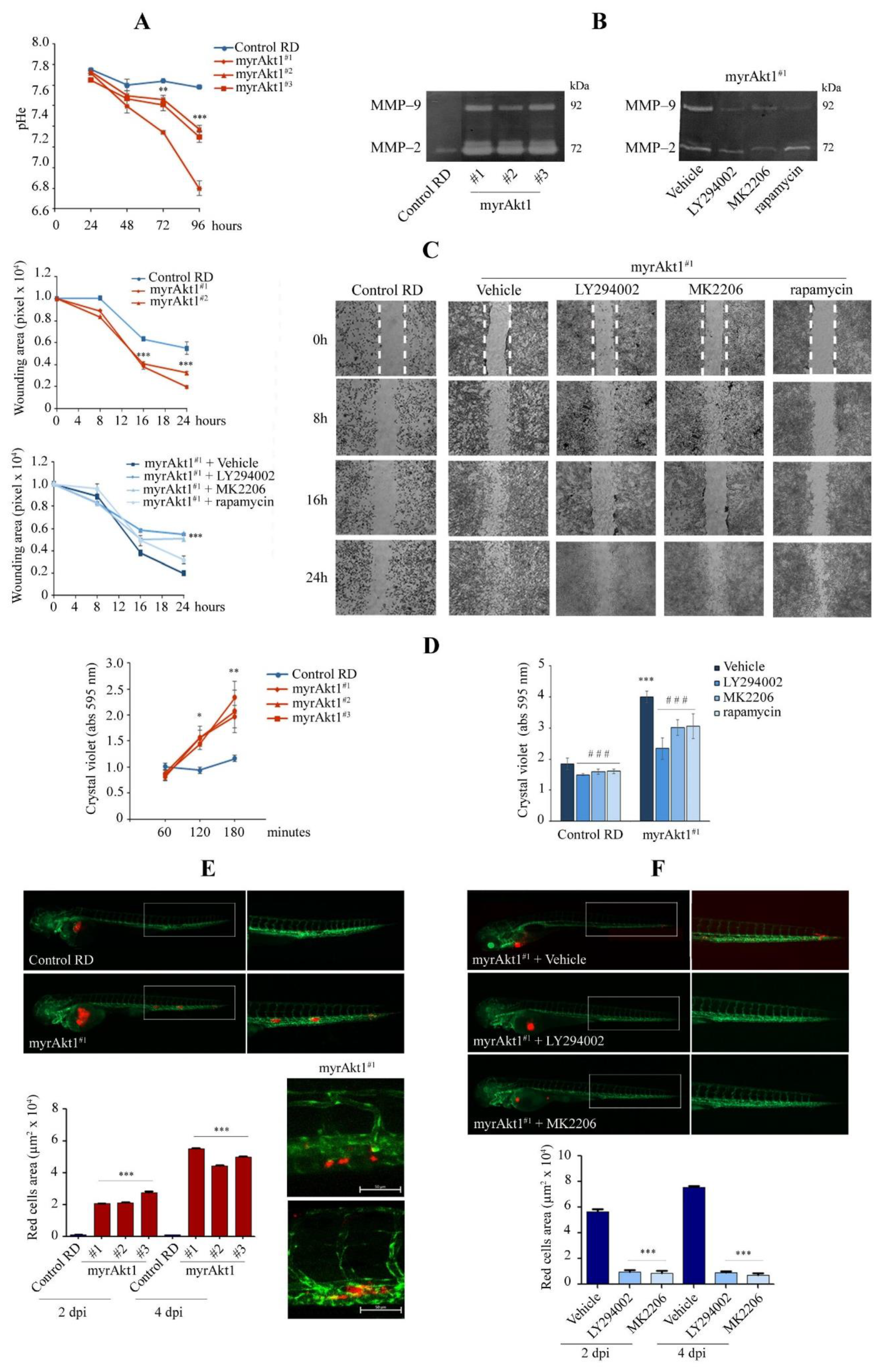
3.2. MyrAkt1 Enhances Tumor Cell Dissemination In Vitro and In Vivo
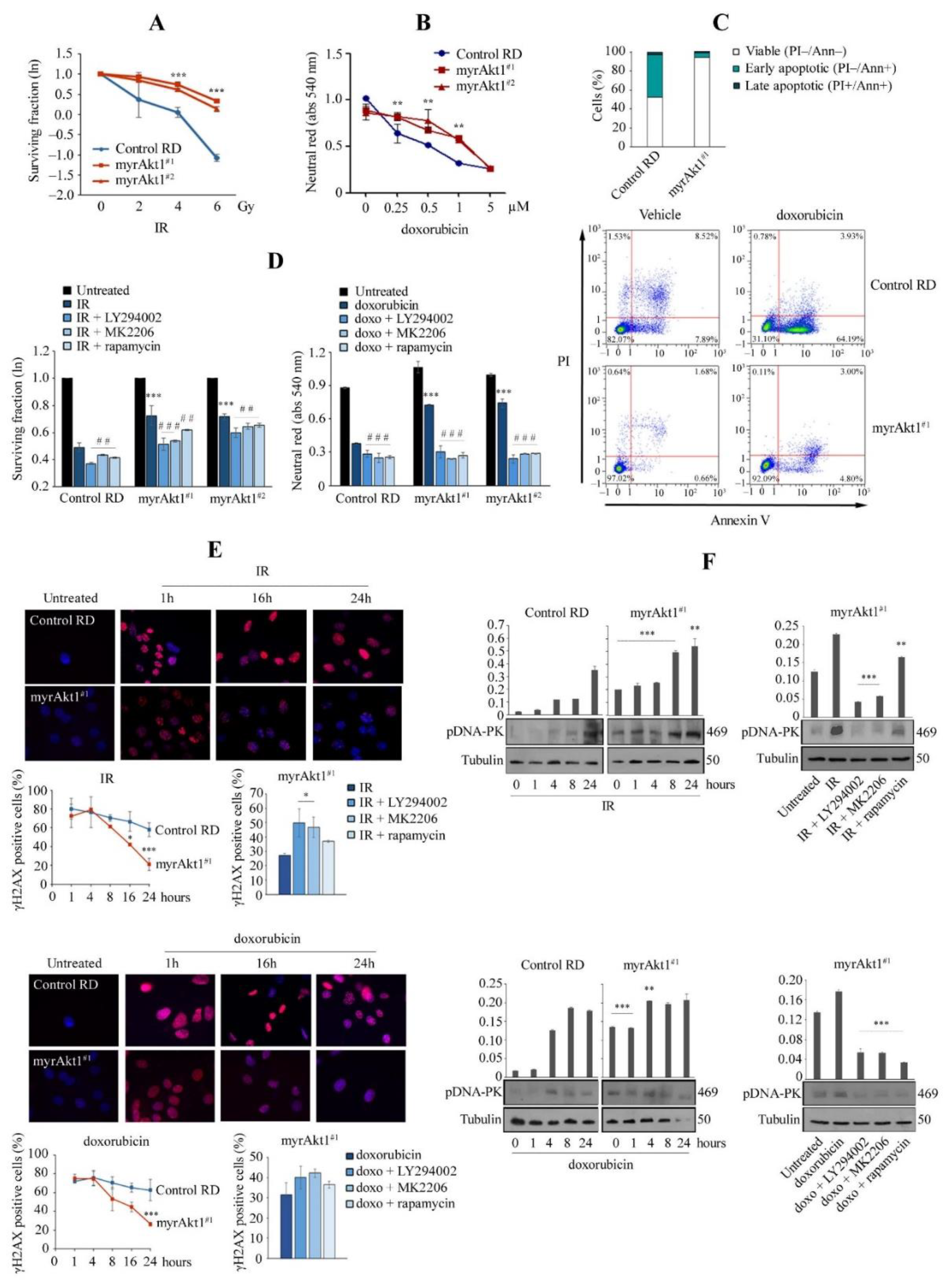
3.3. The Akt1/mTOR/DNA-PK Signaling Axis Correlates with Increase in DNA Repair
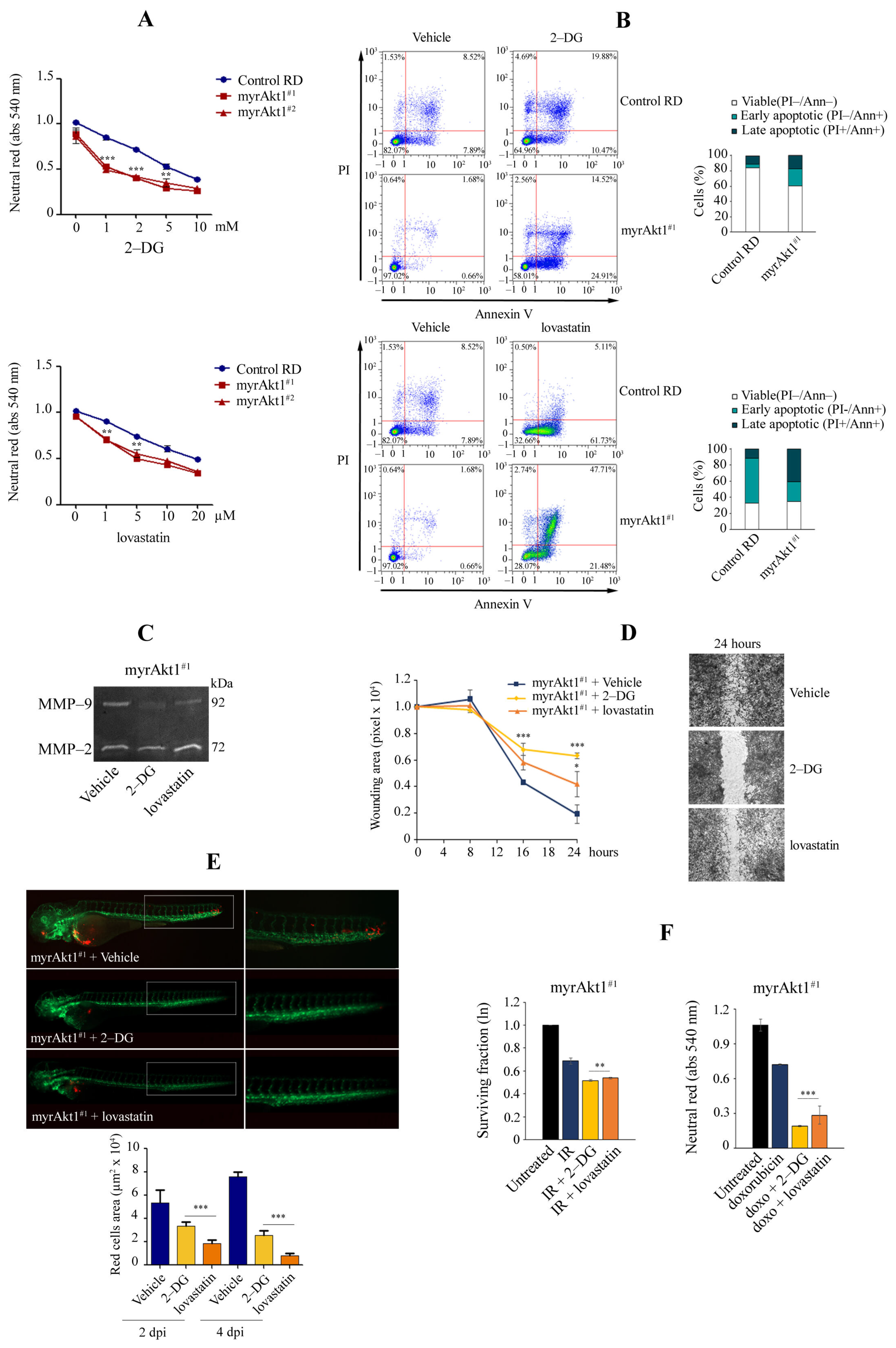
3.4. Treatments with 2-DG and Lovastatin, by Triggering Apoptosis, Inhibit myrAkt1-Driven Tumorigenicity
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Manning, B.D.; Toker, A. AKT/PKB Signaling: Navigating the Network. Cell 2017, 169, 381–405. [Google Scholar] [CrossRef] [PubMed]
- Song, M.; Bode, A.M.; Dong, Z.; Lee, M.H. AKT as a Therapeutic Target for Cancer. Cancer Res. 2019, 79, 1019–1031. [Google Scholar] [CrossRef] [PubMed]
- Xu, N.; Lao, Y.; Zhang, Y.; Gillespie, D.A. Akt: A double-edged sword in cell proliferation and genome stability. J. Oncol. 2012, 2012, 951724. [Google Scholar] [CrossRef] [PubMed]
- Hoxhaj, G.; Manning, B.D. The PI3K-AKT network at the interface of oncogenic signalling and cancer metabolism. Nat. Rev. Cancer 2020, 20, 74–88. [Google Scholar] [CrossRef]
- Cantor, J.R.; Sabatini, D.M. Cancer cell metabolism: One hallmark, many faces. Cancer Discov. 2012, 2, 881–898. [Google Scholar] [CrossRef]
- Liu, R.; Chen, Y.; Liu, G.; Li, C.; Song, Y.; Cao, Z.; Li, W.; Hu, J.; Lu, C.; Liu, Y. PI3K/AKT pathway as a key link modulates the multidrug resistance of cancers. Cell Death Dis. 2020, 11, 797. [Google Scholar] [CrossRef]
- Skapek, S.X.; Ferrari, A.; Gupta, A.A.; Lupo, P.J.; Butler, E.; Shipley, J.; Barr, F.G.; Hawkins, D.S. Rhabdomyosarcoma. Nat. Rev. Dis. Primers 2019, 5, 1. [Google Scholar] [CrossRef]
- Tenente, I.M.; Hayes, M.N.; Ignatius, M.S.; McCarthy, K.; Yohe, M.; Sindiri, S.; Gryder, B.; Oliveira, M.L.; Ramakrishnan, A.; Tang, Q.; et al. Myogenic regulatory transcription factors regulate growth in rhabdomyosarcoma. Elife 2017, 6, e19214. [Google Scholar] [CrossRef]
- Keller, C.; Guttridge, D.C. Mechanisms of impaired differentiation in rhabdomyosarcoma. FEBS J. 2013, 280, 4323–4334. [Google Scholar] [CrossRef]
- Galili, N.; Davis, R.J.; Fredericks, W.J.; Mukhopadhyay, S.; Rauscher, F.J.; Emanuel, B.S.; Rovera, G.; Barr, F.G. Fusion of a fork head domain gene to PAX3 in the solid tumour alveolar rhabdomyosarcoma. Nat. Genet. 1993, 5, 230–235. [Google Scholar] [CrossRef]
- Shapiro, D.N.; Sublett, J.E.; Li, B.; Downing, J.R.; Naeve, C.W. Fusion of PAX3 to a member of the forkhead family of transcription factors in human alveolar rhabdomyosarcoma. Cancer Res. 1993, 53, 5108–5112. [Google Scholar]
- Davis, R.J.; D’Cruz, C.M.; Lovell, M.A.; Biegel, J.A.; Barr, F.G. Fusion of PAX7 to FKHR by the variant t(1;13)(p36;q14) translocation in alveolar rhabdomyosarcoma. Cancer Res. 1994, 54, 2869–2872. [Google Scholar]
- Stratton, M.R.; Fisher, C.; Gusterson, B.A.; Cooper, C.S. Detection of point mutations in N-ras and K-ras genes of human embryonal rhabdomyosarcomas using oligonucleotide probes and the polymerase chain reaction. Cancer Res. 1989, 49, 6324–6327. [Google Scholar]
- Taylor, J.G.; Cheuk, A.T.; Tsang, P.S.; Chung, J.Y.; Song, Y.K.; Desai, K.; Yu, Y.; Chen, Q.R.; Shah, K.; Youngblood, V.; et al. Identification of FGFR4-activating mutations in human rhabdomyosarcomas that promote metastasis in xenotransplanted models. J. Clin. Investig. 2009, 119, 3395–3407. [Google Scholar]
- Shukla, N.; Ameur, N.; Yilmaz, I.; Nafa, K.; Lau, C.Y.; Marchetti, A.; Borsu, L.; Barr, F.G.; Ladanyi, M. Oncogene mutation profiling of pediatric solid tumors reveals significant subsets of embryonal rhabdomyosarcoma and neuroblastoma with mutated genes in growth signaling pathways. Clin. Cancer Res. 2012, 18, 748–757. [Google Scholar] [CrossRef]
- Shern, J.F.; Chen, L.; Chmielecki, J.; Wei, J.S.; Patidar, R.; Rosenberg, M.; Ambrogio, L.; Auclair, D.; Wang, J.; Song, Y.K.; et al. Comprehensive genomic analysis of rhabdomyosarcoma reveals a landscape of alterations affecting a common genetic axis in fusion-positive and fusion-negative tumors. Cancer Discov. 2014, 4, 216–231. [Google Scholar] [CrossRef]
- McKinnon, T.; Venier, R.; Yohe, M.; Sindiri, S.; Gryder, B.E.; Shern, J.F.; Kabaroff, L.; Dickson, B.; Schleicher, K.; Chouinard-Pelletier, G.; et al. Functional screening of FGFR4-driven tumorigenesis identifies PI3K/mTOR inhibition as a therapeutic strategy in rhabdomyosarcoma. Oncogene 2018, 37, 2630–2644. [Google Scholar] [CrossRef]
- Langdon, C.G.; Gadek, K.E.; Garcia, M.R.; Evans, M.K.; Reed, K.B.; Bush, M.; Hanna, J.A.; Drummond, C.J.; Maguire, M.C.; Leavey, P.J.; et al. Synthetic essentiality between PTEN and core dependency factor PAX7 dictates rhabdomyosarcoma identity. Nat. Commun. 2021, 12, 5520. [Google Scholar] [CrossRef]
- Petricoin, E.F.; Espina, V.; Araujo, R.P.; Midura, B.; Yeung, C.; Wan, X.; Eichler, G.S.; Johann, D.J.; Qualman, S.; Tsokos, M.; et al. Phosphoprotein pathway mapping: Akt/mammalian target of rapamycin activation is negatively associated with childhood rhabdomyosarcoma survival. Cancer Res. 2007, 67, 3431–3440. [Google Scholar] [CrossRef]
- Cen, L.; Arnoczky, K.J.; Hsieh, F.C.; Lin, H.J.; Qualman, S.J.; Yu, S.; Xiang, H.; Lin, J. Phosphorylation profiles of protein kinases in alveolar and embryonal rhabdomyosarcoma. Mod. Pathol. 2007, 20, 936–946. [Google Scholar] [CrossRef]
- Kohsaka, S.; Shukla, N.; Ameur, N.; Ito, T.; Ng, C.K.; Wang, L.; Lim, D.; Marchetti, A.; Viale, A.; Pirun, M.; et al. A recurrent neomorphic mutation in MYOD1 defines a clinically aggressive subset of embryonal rhabdomyosarcoma associated with PI3K-AKT pathway mutations. Nat. Genet. 2014, 46, 595–600. [Google Scholar] [CrossRef]
- Ramadan, F.; Fahs, A.; Ghayad, S.E.; Saab, R. Signaling pathways in Rhabdomyosarcoma invasion and metastasis. Cancer Metastasis Rev. 2020, 39, 287–301. [Google Scholar] [CrossRef]
- Jung, C.H.; Ro, S.H.; Cao, J.; Otto, N.M.; Kim, D.H. mTOR regulation of autophagy. FEBS Lett. 2010, 584, 1287–1295. [Google Scholar] [CrossRef]
- Fingar, D.C.; Blenis, J. Target of rapamycin (TOR): An integrator of nutrient and growth factor signals and coordinator of cell growth and cell cycle progression. Oncogene 2004, 23, 3151–3171. [Google Scholar] [CrossRef] [Green Version]
- Dada, S.; Demartines, N.; Dormond, O. mTORC2 regulates PGE2-mediated endothelial cell survival and migration. Biochem. Biophys. Res. Commun. 2008, 372, 875–879. [Google Scholar] [CrossRef]
- Sarbassov, D.D.; Ali, S.M.; Kim, D.H.; Guertin, D.A.; Latek, R.R.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr. Biol. 2004, 14, 1296–1302. [Google Scholar] [CrossRef]
- Kim, E.K.; Yun, S.J.; Ha, J.M.; Kim, Y.W.; Jin, I.H.; Yun, J.; Shin, H.K.; Song, S.H.; Kim, J.H.; Lee, J.S.; et al. Selective activation of Akt1 by mammalian target of rapamycin complex 2 regulates cancer cell migration, invasion, and metastasis. Oncogene 2011, 30, 2954–2963. [Google Scholar] [CrossRef]
- Wan, X.; Shen, N.; Mendoza, A.; Khanna, C.; Helman, L.J. CCI-779 inhibits rhabdomyosarcoma xenograft growth by an antiangiogenic mechanism linked to the targeting of mTOR/Hif-1alpha/VEGF signaling. Neoplasia 2006, 8, 394–401. [Google Scholar] [CrossRef] [PubMed]
- Cen, L.; Hsieh, F.C.; Lin, H.J.; Chen, C.S.; Qualman, S.J.; Lin, J. PDK-1/AKT pathway as a novel therapeutic target in rhabdomyosarcoma cells using OSU-03012 compound. Br. J. Cancer 2007, 97, 785–791. [Google Scholar] [CrossRef]
- Kaylani, S.Z.; Xu, J.; Srivastava, R.K.; Kopelovich, L.; Pressey, J.G.; Athar, M. Rapamycin targeting mTOR and hedgehog signaling pathways blocks human rhabdomyosarcoma growth in xenograft murine model. Biochem. Biophys. Res. Commun. 2013, 435, 557–561. [Google Scholar] [CrossRef]
- Guenther, M.K.; Graab, U.; Fulda, S. Synthetic lethal interaction between PI3K/Akt/mTOR and Ras/MEK/ERK pathway inhibition in rhabdomyosarcoma. Cancer Lett. 2013, 337, 200–209. [Google Scholar] [CrossRef] [PubMed]
- Renshaw, J.; Taylor, K.R.; Bishop, R.; Valenti, M.; De Haven Brandon, A.; Gowan, S.; Eccles, S.A.; Ruddle, R.R.; Johnson, L.D.; Raynaud, F.I.; et al. Dual Blockade of the PI3K/AKT/mTOR (AZD8055) and RAS/MEK/ERK (AZD6244) Pathways Synergistically Inhibits Rhabdomyosarcoma Cell Growth In Vitro and In Vivo. Clin. Cancer Res. 2013, 19, 5940–5951. [Google Scholar] [CrossRef] [PubMed]
- Graab, U.; Hahn, H.; Fulda, S. Identification of a novel synthetic lethality of combined inhibition of hedgehog and PI3K signaling in rhabdomyosarcoma. Oncotarget 2015, 6, 8722–8735. [Google Scholar] [CrossRef]
- Fulda, S. Cell death pathways as therapeutic targets in rhabdomyosarcoma. Sarcoma 2012, 2012, 326210. [Google Scholar] [CrossRef] [Green Version]
- Borinstein, S.C.; Steppan, D.; Hayashi, M.; Loeb, D.M.; Isakoff, M.S.; Binitie, O.; Brohl, A.S.; Bridge, J.A.; Stavas, M.; Shinohara, E.T.; et al. Consensus and controversies regarding the treatment of rhabdomyosarcoma. Pediatr. Blood Cancer 2018, 65, e26809. [Google Scholar] [CrossRef]
- Pacenta, H.L.; Allen-Rhoades, W.; Langenau, D.; Houghton, P.J.; Keller, C.; Heske, C.M.; Deel, M.D.; Linardic, C.M.; Shern, J.F.; Stewart, E.; et al. Prioritization of Novel Agents for Patients with Rhabdomyosarcoma: A Report from the Children’s Oncology Group (COG) New Agents for Rhabdomyosarcoma Task Force. J. Clin. Med. 2021, 10, 1416. [Google Scholar] [CrossRef]
- Jin, S.W.; Beis, D.; Mitchell, T.; Chen, J.N.; Stainier, D.Y. Cellular and molecular analyses of vascular tube and lumen for-mation in zebrafish. Development 2005, 132, 5199–5209. [Google Scholar] [CrossRef]
- Sims, D.; Maranyane, H.M.; Damerell, V.; Govender, D.; Isaacs, A.W.; Peres, J.; Prince, S. The c-Myc/AKT1/TBX3 Axis Is Important to Target in the Treatment of Embryonal Rhabdomyosarcoma. Cancers 2020, 12, 501. [Google Scholar] [CrossRef]
- Hinson, A.R.; Jones, R.; Crose, L.E.; Belyea, B.C.; Barr, F.G.; Linardic, C.M. Human rhabdomyosarcoma cell lines for rhabdomyosarcoma research: Utility and pitfalls. Front. Oncol. 2013, 3, 183. [Google Scholar] [CrossRef]
- Grillo-Hill, B.K.; Choi, C.; Jimenez-Vidal, M.; Barber, D.L. Increased H⁺ efflux is sufficient to induce dysplasia and necessary for viability with oncogene expression. Elife 2015, 4, e03270. [Google Scholar] [CrossRef]
- Hill, D.; Chen, L.; Snaar-Jagalska, E.; Chaudhry, B. Embryonic zebrafish xenograft assay of human cancer metastasis. F1000Res. 2018, 7, 1682. [Google Scholar] [CrossRef]
- Thorn, C.F.; Oshiro, C.; Marsh, S.; Hernandez-Boussard, T.; McLeod, H.; Klein, T.E.; Altman, R.B. Doxorubicin pathways: Pharmacodynamics and adverse effects. Pharmacogenet. Genom. 2011, 21, 440–446. [Google Scholar] [CrossRef] [PubMed]
- Kuo, L.J.; Yang, L.X. Gamma-H2AX—A novel biomarker for DNA double-strand breaks. In Vivo 2008, 22, 305–309. [Google Scholar] [PubMed]
- Bozulic, L.; Surucu, B.; Hynx, D.; Hemmings, B.A. PKBalpha/Akt1 acts downstream of DNA-PK in the DNA double-strand break response and promotes survival. Mol. Cell 2008, 30, 203–213. [Google Scholar] [CrossRef]
- Toulany, M.; Lee, K.J.; Fattah, K.R.; Lin, Y.F.; Fehrenbacher, B.; Schaller, M.; Chen, B.P.; Chen, D.J.; Rodemann, H.P. Akt promotes post-irradiation survival of human tumor cells through initiation, progression, and termination of DNA-PKcs-dependent DNA double-strand break repair. Mol. Cancer Res. 2012, 10, 945–957. [Google Scholar] [CrossRef]
- Elstrom, R.L.; Bauer, D.E.; Buzzai, M.; Karnauskas, R.; Harris, M.H.; Plas, D.R.; Zhuang, H.; Cinalli, R.M.; Alavi, A.; Rudin, C.M.; et al. Akt stimulates aerobic glycolysis in cancer cells. Cancer Res. 2004, 64, 3892–3899. [Google Scholar] [CrossRef]
- Porstmann, T.; Griffiths, B.; Chung, Y.L.; Delpuech, O.; Griffiths, J.R.; Downward, J.; Schulze, A. PKB/Akt induces transcription of enzymes involved in cholesterol and fatty acid biosynthesis via activation of SREBP. Oncogene 2005, 24, 6465–6481. [Google Scholar] [CrossRef]
- Manzella, G.; Schreck, L.D.; Breunis, W.B.; Molenaar, J.; Merks, H.; Barr, F.G.; Sun, W.; Römmele, M.; Zhang, L.; Tchinda, J.; et al. Phenotypic profiling with a living biobank of primary rhabdomyosarcoma unravels disease heterogeneity and AKT sensitivity. Nat. Commun. 2020, 11, 4629. [Google Scholar] [CrossRef]
- Maqoud, F.; Curci, A.; Scala, R.; Pannunzio, A.; Campanella, F.; Coluccia, M.; Passantino, G.; Zizzo, N.; Tricarico, D. Cell Cycle Regulation by Ca 2+-Activated K⁺ (BK) Channels Modulators in SH-SY5Y Neuroblastoma Cells. Int. J. Mol. Sci. 2018, 19, 2442. [Google Scholar] [CrossRef]
- Glass, D.J. PI3 kinase regulation of skeletal muscle hypertrophy and atrophy. Curr. Top. Microbiol. Immunol. 2010, 346, 267–278. [Google Scholar] [CrossRef]
- Knight, J.D.; Kothary, R. The myogenic kinome: Protein kinases critical to mammalian skeletal myogenesis. Skelet. Muscle 2011, 1, 29. [Google Scholar] [CrossRef] [PubMed]
- Gardner, S.; Anguiano, M.; Rotwein, P. Defining Akt actions in muscle differentiation. Am. J. Physiol. Cell Physiol. 2012, 303, C1292–C1300. [Google Scholar] [CrossRef] [PubMed]
- Fanzani, A.; Conraads, V.M.; Penna, F.; Martinet, W. Molecular and cellular mechanisms of skeletal muscle atrophy: An update. J. Cachexia Sarcopenia Muscle 2012, 3, 163–179. [Google Scholar] [CrossRef] [PubMed]
- Cetrone, M.; Mele, A.; Tricarico, D. Effects of the antidiabetic drugs on the age-related atrophy and sarcopenia associated with diabetes type II. Curr. Diabetes Rev. 2014, 10, 231–237. [Google Scholar] [CrossRef]
- Granados, V.A.; Avirneni-Vadlamudi, U.; Dalal, P.; Scarborough, S.R.; Galindo, K.A.; Mahajan, P.; Galindo, R.L. Selective Targeting of Myoblast Fusogenic Signaling and Differentiation-Arrest Antagonizes Rhabdomyosarcoma Cells. Cancer Res. 2019, 79, 4585–4591. [Google Scholar] [CrossRef]
- Davicioni, E.; Anderson, M.J.; Finckenstein, F.G.; Lynch, J.C.; Qualman, S.J.; Shimada, H.; Schofield, D.E.; Buckley, J.D.; Meyer, W.H.; Sorensen, P.H.; et al. Molecular classification of rhabdomyosarcoma--genotypic and phenotypic determinants of diagnosis: A report from the Children’s Oncology Group. Am. J. Pathol. 2009, 174, 550–564. [Google Scholar] [CrossRef]
- Tapscott, S.J.; Thayer, M.J.; Weintraub, H. Deficiency in rhabdomyosarcomas of a factor required for MyoD activity and myogenesis. Science 1993, 259, 1450–1453. [Google Scholar] [CrossRef]
- Yang, Z.; MacQuarrie, K.L.; Analau, E.; Tyler, A.E.; Dilworth, F.J.; Cao, Y.; Diede, S.J.; Tapscott, S.J. MyoD and E-protein heterodimers switch rhabdomyosarcoma cells from an arrested myoblast phase to a differentiated state. Genes. Dev. 2009, 23, 694–707. [Google Scholar] [CrossRef]
- Di Rocco, A.; Camero, S.; Benedetti, A.; Lozanoska-Ochser, B.; Megiorni, F.; Marchese, C.; Stramucci, L.; Ciccarelli, C.; Bouché, M.; Bossi, G.; et al. Anti-oncogenic and pro-myogenic action of the MKK6/p38/AKT axis induced by targeting MEK/ERK in embryonal rhabdomyosarcoma. Oncol. Rep. 2022, 48, 1–15. [Google Scholar] [CrossRef]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 169, 361–371. [Google Scholar] [CrossRef]
- Zhou, H.; Huang, S. Role of mTOR signaling in tumor cell motility, invasion and metastasis. Curr. Protein Pept. Sci. 2011, 12, 30–42. [Google Scholar] [CrossRef]
- Felkai, L.; Krencz, I.; Kiss, D.J.; Nagy, N.; Petővári, G.; Dankó, T.; Micsík, T.; Khoor, A.; Tornóczky, T.; Sápi, Z.; et al. Characterization of mTOR Activity and Metabolic Profile in Pediatric Rhabdomyosarcoma. Cancers 2020, 12, 1947. [Google Scholar] [CrossRef]
- Geoerger, B.; Kieran, M.W.; Grupp, S.; Perek, D.; Clancy, J.; Krygowski, M.; Ananthakrishnan, R.; Boni, J.P.; Berkenblit, A.; Spunt, S.L. Phase II trial of temsirolimus in children with high-grade glioma, neuroblastoma and rhabdomyosarcoma. Eur. J. Cancer 2012, 48, 253–262. [Google Scholar] [CrossRef]
- Balogh, P.; Bánusz, R.; Csóka, M.; Váradi, Z.; Varga, E.; Sápi, Z. Primary alveolar rhabdomyosarcoma of the bone: Two cases and review of the literature. Diagn. Pathol. 2016, 11, 99. [Google Scholar] [CrossRef] [Green Version]
- Codenotti, S.; Marampon, F.; Triggiani, L.; Bonù, M.L.; Magrini, S.M.; Ceccaroli, P.; Guescini, M.; Gastaldello, S.; Tombolini, V.; Poliani, P.L.; et al. Caveolin-1 promotes radioresistance in rhabdomyosarcoma through increased oxidative stress protection and DNA repair. Cancer Lett. 2021, 505, 1–12. [Google Scholar] [CrossRef]
- Petragnano, F.; Pietrantoni, I.; Camero, S.; Codenotti, S.; Milazzo, L.; Vulcano, F.; Macioce, G.; Giordani, I.; Tini, P.; Cheleschi, S.; et al. Clinically relevant radioresistant rhabdomyosarcoma cell lines: Functional, molecular and immune-related characterization. J. Biomed. Sci. 2020, 27, 90. [Google Scholar] [CrossRef]
- Boichuk, S.; Bikinieva, F.; Nurgatina, I.; Dunaev, P.; Valeeva, E.; Aukhadieva, A.; Sabirov, A.; Galembikova, A. Inhibition of AKT-Signaling Sensitizes Soft Tissue Sarcomas (STS) and Gastrointestinal Stromal Tumors (GIST) to Doxorubicin via Targeting of Homology-Mediated DNA Repair. Int. J. Mol. Sci. 2020, 21, 8842. [Google Scholar] [CrossRef]
- Pannunzio, N.R.; Watanabe, G.; Lieber, M.R. Nonhomologous DNA end-joining for repair of DNA double-strand breaks. J. Biol. Chem. 2018, 293, 10512–10523. [Google Scholar] [CrossRef]
- Fok, J.H.L.; Ramos-Montoya, A.; Vazquez-Chantada, M.; Wijnhoven, P.W.G.; Follia, V.; James, N.; Farrington, P.M.; Karmokar, A.; Willis, S.E.; Cairns, J.; et al. AZD7648 is a potent and selective DNA-PK inhibitor that enhances radiation, chemotherapy and olaparib activity. Nat. Commun. 2019, 10, 5065. [Google Scholar] [CrossRef]
- Carr, M.I.; Zimmermann, A.; Chiu, L.Y.; Zenke, F.T.; Blaukat, A.; Vassilev, L.T. DNA-PK Inhibitor, M3814, as a New Combination Partner of Mylotarg in the Treatment of Acute Myeloid Leukemia. Front. Oncol. 2020, 10, 127. [Google Scholar] [CrossRef]
- Bürkel, F.; Jost, T.; Hecht, M.; Heinzerling, L.; Fietkau, R.; Distel, L. Dual mTOR/DNA-PK Inhibitor CC-115 Induces Cell Death in Melanoma Cells and Has Radiosensitizing Potential. Int. J. Mol. Sci. 2020, 21, 9321. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef]
- Kurtoglu, M.; Gao, N.; Shang, J.; Maher, J.C.; Lehrman, M.A.; Wangpaichitr, M.; Savaraj, N.; Lane, A.N.; Lampidis, T.J. Under normoxia, 2-deoxy-D-glucose elicits cell death in select tumor types not by inhibition of glycolysis but by interfering with N-linked glycosylation. Mol. Cancer Ther. 2007, 6, 3049–3058. [Google Scholar] [CrossRef]
- Ramírez-Peinado, S.; Alcázar-Limones, F.; Lagares-Tena, L.; El Mjiyad, N.; Caro-Maldonado, A.; Tirado, O.M.; Muñoz-Pinedo, C. 2-deoxyglucose induces Noxa-dependent apoptosis in alveolar rhabdomyosarcoma. Cancer Res. 2011, 71, 6796–6806. [Google Scholar] [CrossRef] [Green Version]
- Clendening, J.W.; Pandyra, A.; Boutros, P.C.; El Ghamrasni, S.; Khosravi, F.; Trentin, G.A.; Martirosyan, A.; Hakem, A.; Hakem, R.; Jurisica, I.; et al. Dysregulation of the mevalonate pathway promotes transformation. Proc. Natl. Acad. Sci. USA 2010, 107, 15051–15056. [Google Scholar] [CrossRef] [PubMed]
- Kuzu, O.F.; Noory, M.A.; Robertson, G.P. The Role of Cholesterol in Cancer. Cancer Res. 2016, 76, 2063–2070. [Google Scholar] [CrossRef] [PubMed]
- Di Bello, E.; Zwergel, C.; Mai, A.; Valente, S. The Innovative Potential of Statins in Cancer: New Targets for New Therapies. Front. Chem. 2020, 8, 516. [Google Scholar] [CrossRef] [PubMed]
- Dimitroulakos, J.; Ye, L.Y.; Benzaquen, M.; Moore, M.J.; Kamel-Reid, S.; Freedman, M.H.; Yeger, H.; Penn, L.Z. Differential sensitivity of various pediatric cancers and squamous cell carcinomas to lovastatin-induced apoptosis: Therapeutic implications. Clin. Cancer Res. 2001, 7, 158–167. [Google Scholar]
- Mullen, P.J.; Yu, R.; Longo, J.; Archer, M.C.; Penn, L.Z. The interplay between cell signalling and the mevalonate pathway in cancer. Nat. Rev. Cancer 2016, 16, 718–731. [Google Scholar] [CrossRef]
- Hart, T.; Chandrashekhar, M.; Aregger, M.; Steinhart, Z.; Brown, K.R.; MacLeod, G.; Mis, M.; Zimmermann, M.; Fradet-Turcotte, A.; Sun, S.; et al. High-Resolution CRISPR Screens Reveal Fitness Genes and Genotype-Specific Cancer Liabilities. Cell 2015, 163, 1515–1526. [Google Scholar] [CrossRef]
- Wong, W.W.; Clendening, J.W.; Martirosyan, A.; Boutros, P.C.; Bros, C.; Khosravi, F.; Jurisica, I.; Stewart, A.K.; Bergsagel, P.L.; Penn, L.Z. Determinants of sensitivity to lovastatin-induced apoptosis in multiple myeloma. Mol. Cancer Ther. 2007, 6, 1886–1897. [Google Scholar] [CrossRef]
- De Vita, A.; Ferrari, A.; Miserocchi, G.; Vanni, S.; Domizio, C.; Fonzi, E.; Fausti, V.; Recine, F.; Bassi, M.; Campobassi, A.; et al. Identification of a novel RAB3IP-HMGA2 fusion transcript in an adult head and neck rhabdomyosarcoma. Oral Dis. 2021, in press. [Google Scholar] [CrossRef]
- Summer, H.; Li, O.; Bao, Q.; Zhan, L.; Peter, S.; Sathiyanathan, P.; Henderson, D.; Klonisch, T.; Goodman, S.D.; Dröge, P. HMGA2 exhibits dRP/AP site cleavage activity and protects cancer cells from DNA-damage-induced cytotoxicity during chemotherapy. Nucleic Acids Res. 2009, 37, 4371–4384. [Google Scholar] [CrossRef]
- Wu, Y.; Wang, X.; Xu, F.; Zhang, L.; Wang, T.; Fu, X.; Jin, T.; Zhang, W.; Ye, L. The regulation of acetylation and stability of HMGA2 via the HBXIP-activated Akt-PCAF pathway in promotion of esophageal squamous cell carcinoma growth. Nucleic Acids Res. 2020, 48, 4858–4876. [Google Scholar] [CrossRef]
- Thuault, S.; Tan, E.J.; Peinado, H.; Cano, A.; Heldin, C.H.; Moustakas, A. HMGA2 and Smads co-regulate SNAIL1 expression during induction of epithelial-to-mesenchymal transition. J. Biol. Chem. 2008, 283, 33437–33446. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Zhao, Z.; Xu, C.; Zhou, Z.; Zhu, Z.; You, T. HMGA2 induces transcription factor Slug expression to promote epithelial-to-mesenchymal transition and contributes to colon cancer progression. Cancer Lett. 2014, 355, 130–140. [Google Scholar] [CrossRef]
- De Vita, A.; Vanni, S.; Fausti, V.; Cocchi, C.; Recine, F.; Miserocchi, G.; Liverani, C.; Spadazzi, C.; Bassi, M.; Gessaroli, M.; et al. Deciphering the Genomic Landscape and Pharmacological Profile of Uncommon Entities of Adult Rhabdomyosarcomas. Int. J. Mol. Sci. 2021, 22, 11564. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Codenotti, S.; Zizioli, D.; Mignani, L.; Rezzola, S.; Tabellini, G.; Parolini, S.; Giacomini, A.; Asperti, M.; Poli, M.; Mandracchia, D.; et al. Hyperactive Akt1 Signaling Increases Tumor Progression and DNA Repair in Embryonal Rhabdomyosarcoma RD Line and Confers Susceptibility to Glycolysis and Mevalonate Pathway Inhibitors. Cells 2022, 11, 2859. https://doi.org/10.3390/cells11182859
Codenotti S, Zizioli D, Mignani L, Rezzola S, Tabellini G, Parolini S, Giacomini A, Asperti M, Poli M, Mandracchia D, et al. Hyperactive Akt1 Signaling Increases Tumor Progression and DNA Repair in Embryonal Rhabdomyosarcoma RD Line and Confers Susceptibility to Glycolysis and Mevalonate Pathway Inhibitors. Cells. 2022; 11(18):2859. https://doi.org/10.3390/cells11182859
Chicago/Turabian StyleCodenotti, Silvia, Daniela Zizioli, Luca Mignani, Sara Rezzola, Giovanna Tabellini, Silvia Parolini, Arianna Giacomini, Michela Asperti, Maura Poli, Delia Mandracchia, and et al. 2022. "Hyperactive Akt1 Signaling Increases Tumor Progression and DNA Repair in Embryonal Rhabdomyosarcoma RD Line and Confers Susceptibility to Glycolysis and Mevalonate Pathway Inhibitors" Cells 11, no. 18: 2859. https://doi.org/10.3390/cells11182859
APA StyleCodenotti, S., Zizioli, D., Mignani, L., Rezzola, S., Tabellini, G., Parolini, S., Giacomini, A., Asperti, M., Poli, M., Mandracchia, D., Vezzoli, M., Bernardi, S., Russo, D., Mitola, S., Monti, E., Triggiani, L., Tomasini, D., Gastaldello, S., Cassandri, M., ... Fanzani, A. (2022). Hyperactive Akt1 Signaling Increases Tumor Progression and DNA Repair in Embryonal Rhabdomyosarcoma RD Line and Confers Susceptibility to Glycolysis and Mevalonate Pathway Inhibitors. Cells, 11(18), 2859. https://doi.org/10.3390/cells11182859