
Exosome Release by Glucose Deprivation Is Important for the Viability of TSC-Null Cells


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents and Antibodies
2.2. Cell Culture
2.3. Chemical Treatments
2.4. siRNA Treatment and Transfection
2.5. Immunoblot Analysis
2.6. Immunocytochemistry
2.7. Exosome Purification and Isolation
2.8. Transmission Electron Microscopy (TEM)
2.9. Nanoparticle Tracking Analysis
2.10. Cell Viability Assay
2.11. Statistical Analysis
3. Results
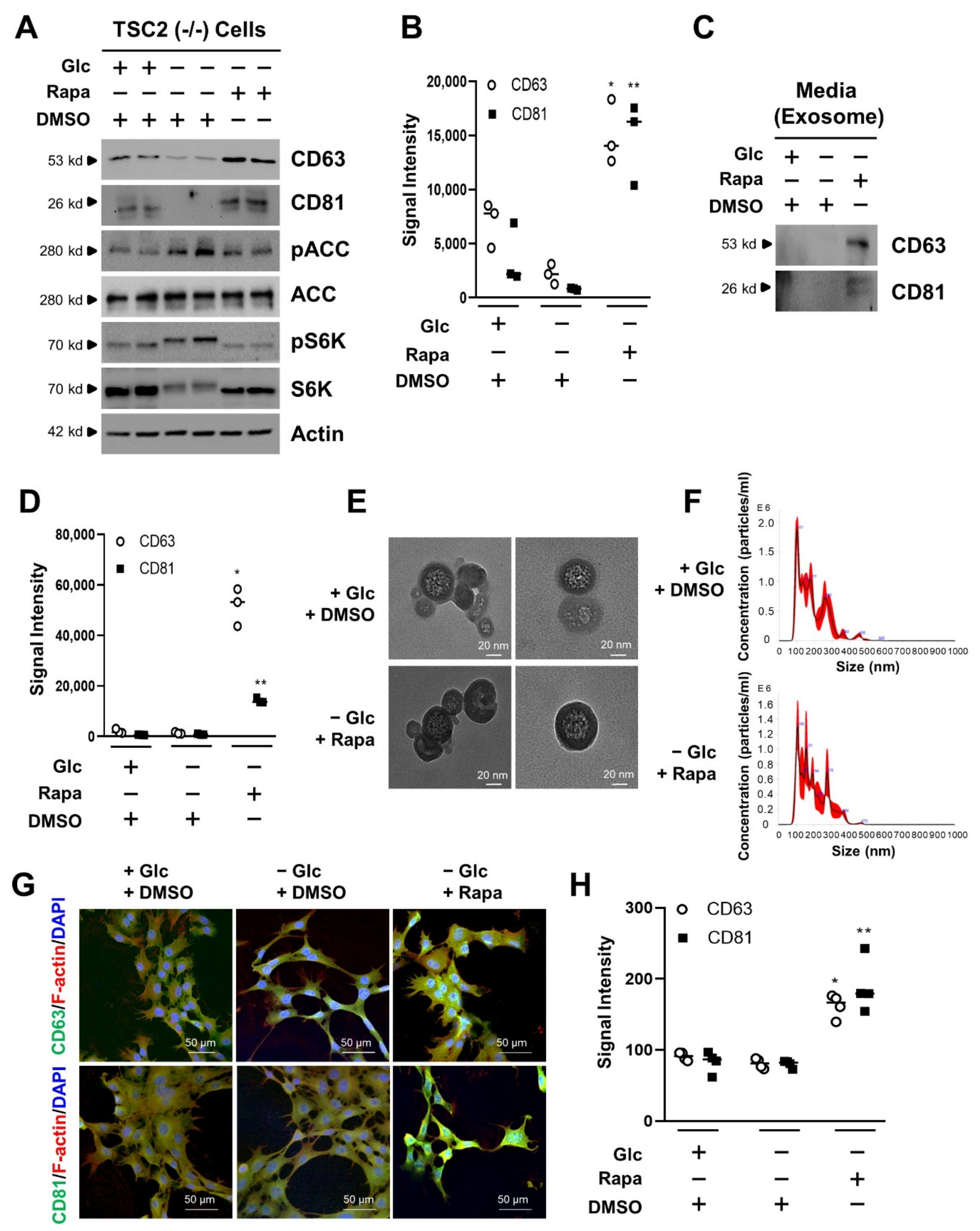
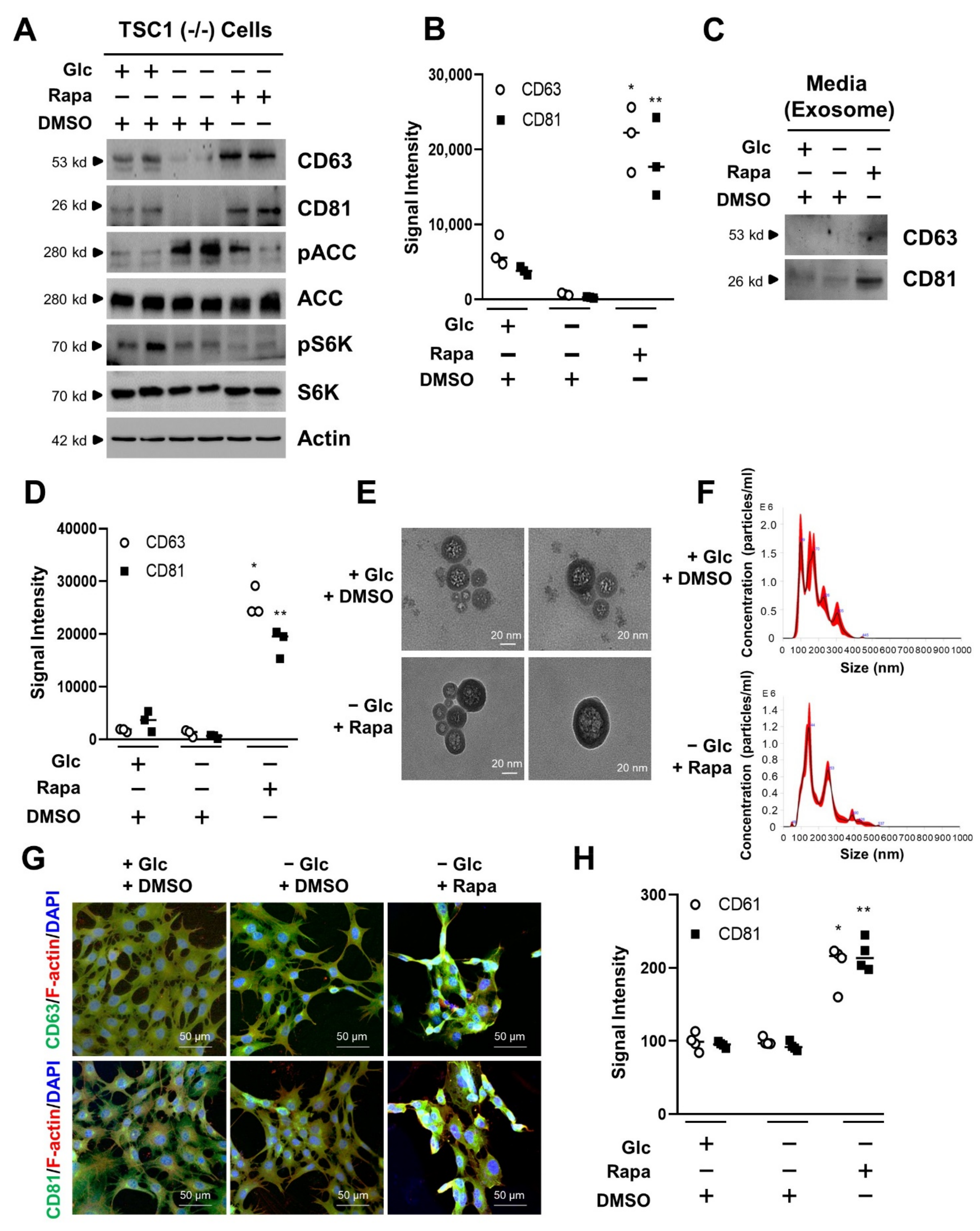
3.1. mTORC1 Inhibition Increases the Exosome Release of TSC2 (−/−) MEF Cells in Glucose Deprivation
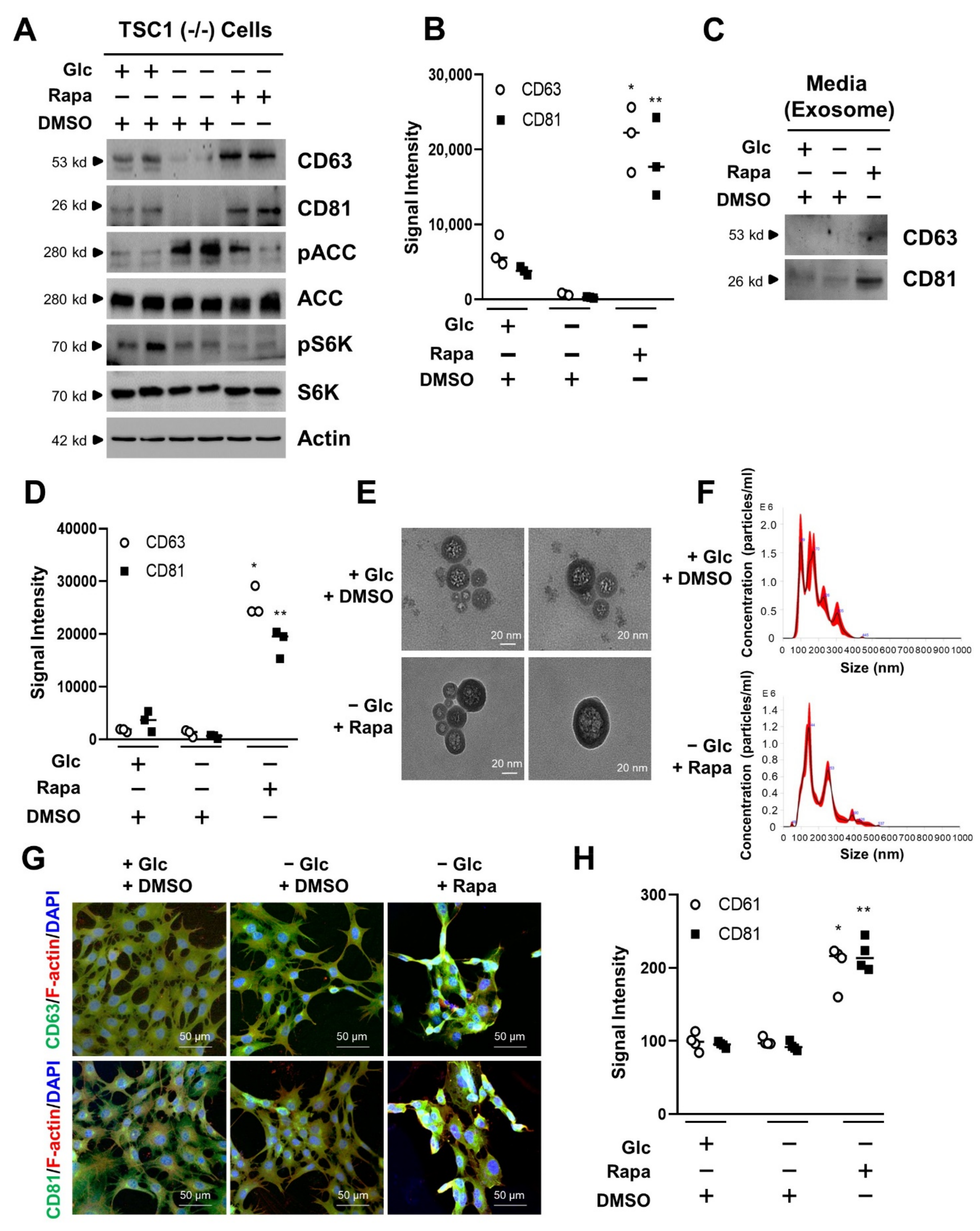
3.2. mTORC1 Inhibition Increases the Exosome Release of TSC1 (−/−) MEF Cells in Glucose Deprivation
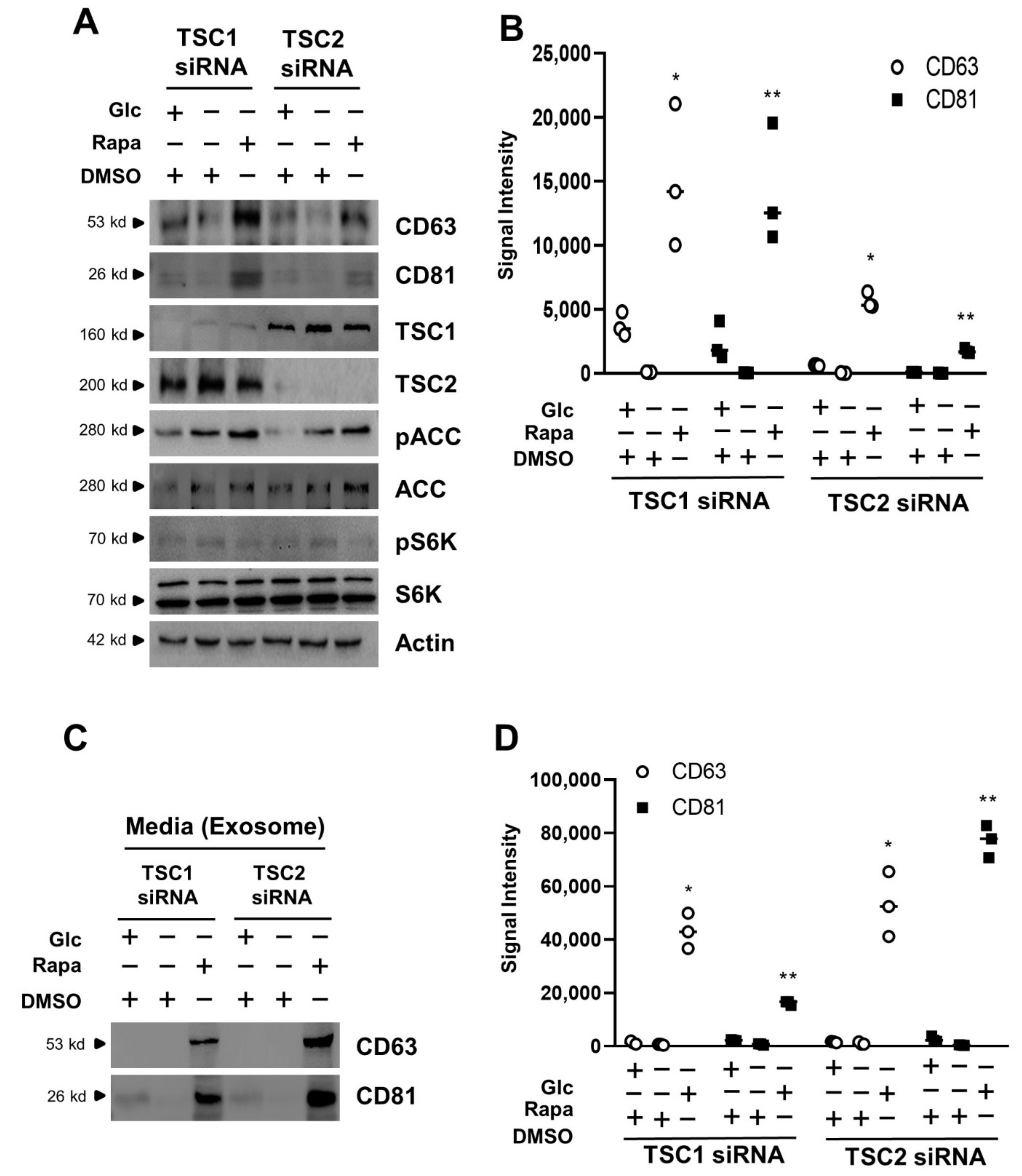
3.3. Exosome Release Is Increased by mTORC1 Inhibition in Transient Silencing of TSC1/2
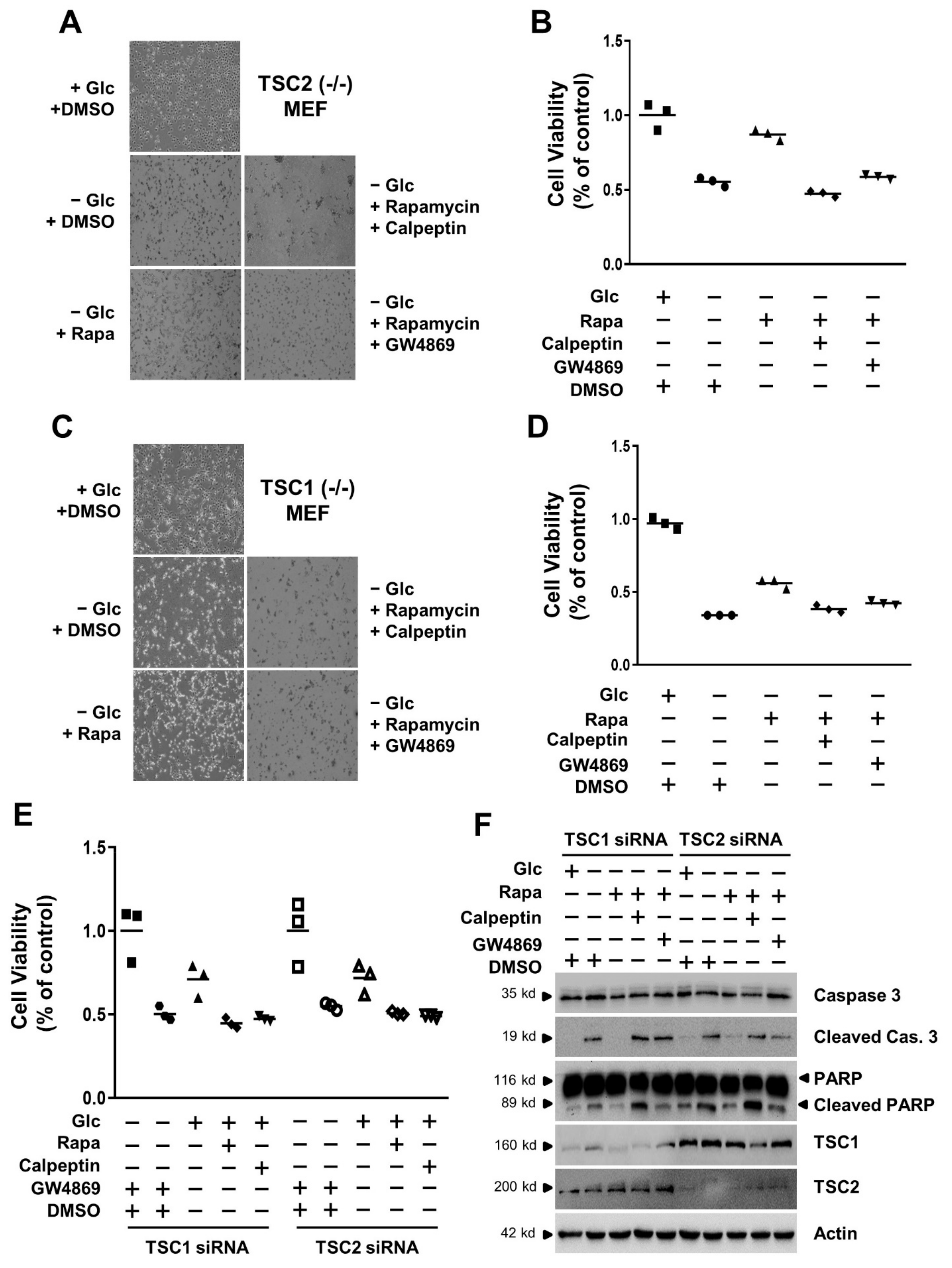
3.4. mTORC1 Inhibition Increases Cell Viability under Conditions of Glucose Deprivation
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sapir, L.; Tzlil, S. Talking over the extracellular matrix: How do cells communicate mechanically? Semin. Cell Dev. Biol. 2017, 71, 99–105. [Google Scholar] [CrossRef] [PubMed]
- Tkach, M.; Théry, C. Communication by Extracellular Vesicles: Where we are and where we need to go. Cell 2016, 164, 1226–1232. [Google Scholar] [CrossRef] [PubMed]
- Wagner, J.U.G.; Dimmeler, S. Cellular cross-talks in the diseased and aging heart. J. Mol. Cell Cardiol. 2020, 138, 136–146. [Google Scholar] [CrossRef] [PubMed]
- Trosko, J.E.; Ruch, R.J. Cell-cell communication in carcinogenesis. Front. Biosci. 1998, 3, d208–d236. [Google Scholar] [CrossRef]
- Dominiak, A.; Chełstowska, B.; Olejarz, W.; Nowicka, G. Communication in the cancer microenvironment as a target for therapeutic interventions. Cancers 2020, 12, 1232. [Google Scholar] [CrossRef] [PubMed]
- Paskeh, M.D.A.; Entezari, M.; Mirzaei, S.; Zabolian, A.; Saleki, H.; Naghdi, M.J.; Sabet, S.; Khoshbakht, M.A.; Hashemi, M.; Hushmandi, K.; et al. Emerging role of exosomes in cancer progression and tumor microenvironment remodeling. J. Hematol. Oncol. 2022, 15, 83. [Google Scholar] [CrossRef] [PubMed]
- LeBleu, V.S.; Kalluri, R. Exosomes as a multicomponent biomarker platform in cancer. Trends Cancer. 2020, 6, 767–774. [Google Scholar] [CrossRef]
- Kalluri, R. The biology and function of exosomes in cancer. J. Clin. Investig. 2016, 126, 1208–1215. [Google Scholar] [CrossRef]
- Kahlert, C.; Kalluri, R. Exosomes in tumor microenvironment influence cancer progression and metastasis. J. Mol. Med. 2013, 91, 431–437. [Google Scholar] [CrossRef]
- Trams, E.G.; Lauter, C.J.; Salem, N., Jr.; Heine, U. Exfoliation of membrane ecto-enzymes in the form of micro-vesicles. Biochim. Biophys. Acta 1981, 6, 63–70. [Google Scholar] [CrossRef]
- Johnstone, R.M.; Adam, M.; Hammond, J.R.; Orr, L.; Turbide, C. Vesicle formation during reticulocyte maturation. Association of plasma membrane activities with released vesicles (exosomes). J. Biol. Chem. 1987, 5, 9412–9420. [Google Scholar] [CrossRef]
- Van Niel, G.; D’Angelo, G.; Raposo, G. Shedding light on the cell biology of extracellular vesicles. Nat. Rev. Mol. Cell Biol. 2018, 19, 213–228. [Google Scholar] [CrossRef] [PubMed]
- McAndrews, K.M.; Kalluri, R. Mechanisms associated with biogenesis of exosomes in cancer. Mol. Cancer 2019, 18, 52. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; LeBleu, V.S. The biology, function, and biomedical applications of exosomes. Science 2020, 367, 640. [Google Scholar] [CrossRef]
- Kowal, J.; Tkach, M.; Théry, C. Biogenesis and secretion of exosomes. Curr. Opin. Cell Biol. 2014, 29, 116–125. [Google Scholar] [CrossRef]
- Halova, I.; Draber, P. Tetraspanins and transmembrane adaptor Proteins as plasma membrane organizers-mast cell case. Front. Cell Dev. Biol. 2016, 4, 43. [Google Scholar] [CrossRef]
- Mathieu, M.; Martin-Jaular, L.; Lavieu, G.; Théry, C. Specificities of secretion and uptake of exosomes and other extracellular vesicles for cell-to-cell communication. Nat. Cell Biol. 2019, 21, 9–17. [Google Scholar] [CrossRef]
- Bebelman, M.P.; Smit, M.J.; Pegtel, D.M.; Baglio, S.R. Biogenesis and function of extracellular vesicles in cancer. Pharmacol. Ther. 2018, 188, 1–11. [Google Scholar] [CrossRef]
- Choudhury, S.R.; Dutta, S.; Bhaduri, U.; Rao, M.R.S. LncRNA Hmrhl regulates expression of cancer related genes in chronic myelogenous leukemia through chromatin association. NAR Cancer 2021, 3, zcab042. [Google Scholar] [CrossRef]
- Yang, L.; Wu, X.H.; Wang, D.; Luo, C.L.; Chen, L.X. Bladder cancer cell-derived exosomes inhibit tumor cell apoptosis and induce cell proliferation in vitro. Mol. Med. Rep. 2013, 8, 1272–1278. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Shi, P.; Zhao, G.; Xu, J.; Peng, W.; Zhang, J.; Zhang, G.; Wang, X.; Dong, Z.; Chen, F.; et al. Targeting cancer stem cell pathways for cancer therapy. Signal Transduct. Target. Ther. 2020, 5, 8. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.; Lou, K.; Zou, X.; Zou, J.; Zhang, G. The potential role of exosomal proteins in prostate cancer. Front. Oncol. 2022, 12, 2683. [Google Scholar] [CrossRef] [PubMed]
- Gyukity-Sebestyén, E.; Harmati, M.; Dobra, G.; Németh, I.B.; Mihály, J.; Zvara, Á.; Hunyadi-Gulyás, É.; Katona, R.; Nagy, I.; Horváth, P.; et al. Melanoma-derived exosomes induce PD-1 overexpression and tumor progression via mesenchymal stem cell oncogenic reprogramming. Front. Immunol. 2019, 10, 2459. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Zhang, Y.; Arrazola, P.; Hino, O.; Kobayashi, T.; Yeung, R.S.; Ru, B.; Pan, D. Tsc tumour suppressor proteins antagonize amino-acid-TOR signalling. Nat. Cell Biol. 2002, 4, 699–704. [Google Scholar] [CrossRef]
- Tee, A.R.; Fingar, D.C.; Manning, B.D.; Kwiatkowski, D.J.; Cantley, C.L.; Blenis, J. Tuberous sclerosis complex-1 and -2 gene products function together to inhibit mammalian target of rapamycin (mTOR)-mediated downstream signaling. Proc. Natl. Acad. Sci. USA 2002, 99, 13571–13576. [Google Scholar] [CrossRef]
- Sancak, Y.; Bar-Peled, L.; Zoncu, R.; Markhard, A.L.; Nada, S.; Sabatini, D.M. Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell 2010, 141, 290–303. [Google Scholar] [CrossRef]
- Yoshida, S.; Pacitto, R.; Yao, Y.; Inoki, K.; Swanson, J.A. Growth factor signaling to mTORC1 by amino acid-laden macropinosomes. J. Cell Biol. 2015, 211, 159–172. [Google Scholar] [CrossRef]
- Choo, A.Y.; Kim, S.G.; Vander Heiden, M.G.; Mahoney, S.J.; Vu, H.; Yoon, S.O.; Cantley, L.C.; Blenis, J. Glucose addiction of TSC null cells is caused by failed mTORC1-dependent balancing of metabolic demand with supply. Mol. Cell 2010, 38, 487–499. [Google Scholar] [CrossRef]
- Marygold, S.J.; Leevers, S.J. Growth signaling: TSC takes its place. Curr. Biol. 2002, 12, R785–R787. [Google Scholar] [CrossRef]
- Hengstschläger, M.; Rodman, D.M.; Miloloza, A.; Hengstschläger-Ottnad, E.; Rosner, M.; Kubista, M. Tuberous sclerosis gene products in proliferation control. Mutat. Res. 2001, 488, 233–239. [Google Scholar] [CrossRef]
- Valianou, M.; Filippidou, N.; Johnson, D.L.; Vogel, P.; Zhang, E.Y.; Liu, X.; Lu, Y.; Yu, J.J.; Bisser, J.J.; Astrinidis, A. Rapalog resistance is associated with mesenchymal-type changes in Tsc2-null cells. Sci. Rep. 2019, 9, 3015. [Google Scholar] [CrossRef]
- Bissler, J.J.; McCormack, F.X.; Young, L.R.; Elwing, J.M.; Chuck, G.; Leonard, J.M.; Schmithorst, V.J.; Laor, T.; Brody, A.S.; Bean, J.; et al. Sirolimus for angiomyolipoma in tuberous sclerosis complex or lymphangioleiomyomatosis. N. Engl. J. Med. 2008, 358, 140–151. [Google Scholar] [CrossRef] [PubMed]
- McCormack, F.X.; Inoue, Y.; Moss, J.; Singer, L.G.; Strange, C.; Nakata, K.; Barker, A.F.; Chapman, J.T.; Brantly, M.L.; Stocks, J.M.; et al. Efficacy and safety of sirolimus in lymphangioleiomyomatosis. N. Engl. J. Med. 2011, 364, 1595–1606. [Google Scholar] [CrossRef] [PubMed]
- Lobb, R.J.; Becker, M.; Wen, S.W.; Wong, C.S.; Wiegmans, A.P.; Leimgruber, A.; Möller, A. Optimized exosome isolation protocol for cell culture supernatant and human plasma. J. Extracell. Vesicles 2015, 4, 27031. [Google Scholar] [CrossRef] [PubMed]
- Hewson, C.; Capraro, D.; Burdach, J.; Whitaker, N.; Morris, K.V. Extracellular vesicle associated long non-coding RNAs functionally enhance cell viability. Noncoding RNA Res. 2016, 1, 3–11. [Google Scholar] [CrossRef]
- Lee, S.S.; Won, J.H.; Lim, G.J.; Han, J.; Lee, J.Y.; Cho, K.O.; Bae, Y.K. A novel population of extracellular vesicles smaller than exosomes promotes cell proliferation. Cell Commun. Signal 2019, 17, 95. [Google Scholar] [CrossRef] [PubMed]
- Zou, W.; Lai, M.; Zhang, Y.; Zheng, L.; Xing, Z.; Li, T.; Zou, Z.; Song, Q.; Zhao, X.; Xia, L.; et al. Exosome release is regulated by mTORC1. Adv. Sci. 2018, 6, 1801313. [Google Scholar] [CrossRef]
- Patel, B.; Patel, J.; Cho, J.H.; Manne, S.; Bonala, S.; Henske, E.; Roegiers, F.; Markiewski, M.; Karbowniczek, M. Exosomes mediate the acquisition of the disease phenotypes by cells with normal genome in tuberous sclerosis complex. Oncogene 2016, 35, 3027–3036. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bae, J.-H.; Kim, J.H. Exosome Release by Glucose Deprivation Is Important for the Viability of TSC-Null Cells. Cells 2022, 11, 2862. https://doi.org/10.3390/cells11182862
Bae J-H, Kim JH. Exosome Release by Glucose Deprivation Is Important for the Viability of TSC-Null Cells. Cells. 2022; 11(18):2862. https://doi.org/10.3390/cells11182862
Chicago/Turabian StyleBae, Ji-Hyun, and Jong Hyun Kim. 2022. "Exosome Release by Glucose Deprivation Is Important for the Viability of TSC-Null Cells" Cells 11, no. 18: 2862. https://doi.org/10.3390/cells11182862
APA StyleBae, J.-H., & Kim, J. H. (2022). Exosome Release by Glucose Deprivation Is Important for the Viability of TSC-Null Cells. Cells, 11(18), 2862. https://doi.org/10.3390/cells11182862