

Identification of Cardiovascular Disease-Related Genes Based on the Co-Expression Network Analysis of Genome-Wide Blood Transcriptome

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
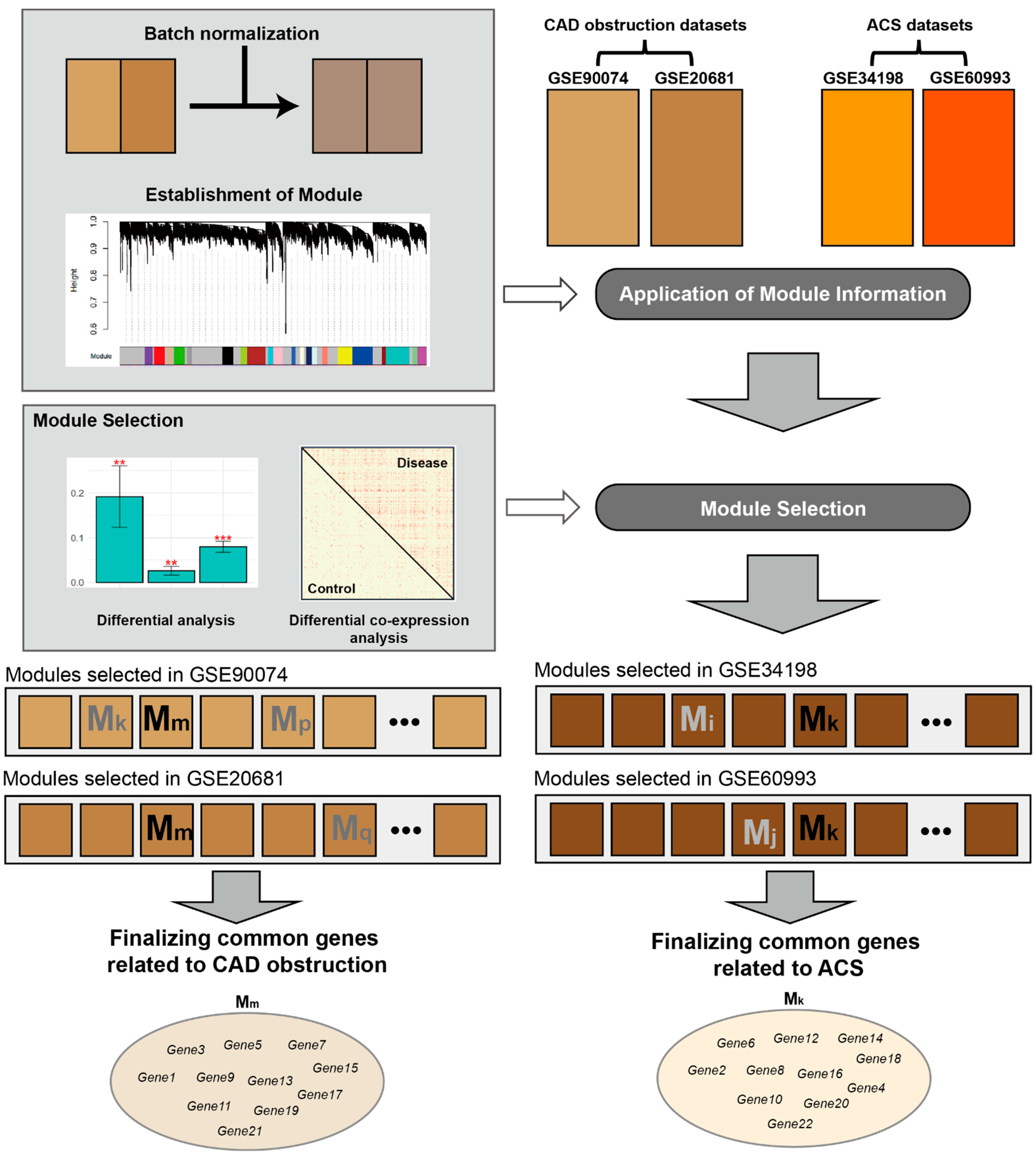
2.1. Datasets
2.2. Preprocessing
2.3. DE Analysis
2.4. Construction of Co-Expression Network and Modules
2.5. Selection of CVD-Related Modules
2.6. Identification of CVD-Related Genes
2.7. Pathway Analysis
2.8. Validation of the Candidate Genes
3. Results
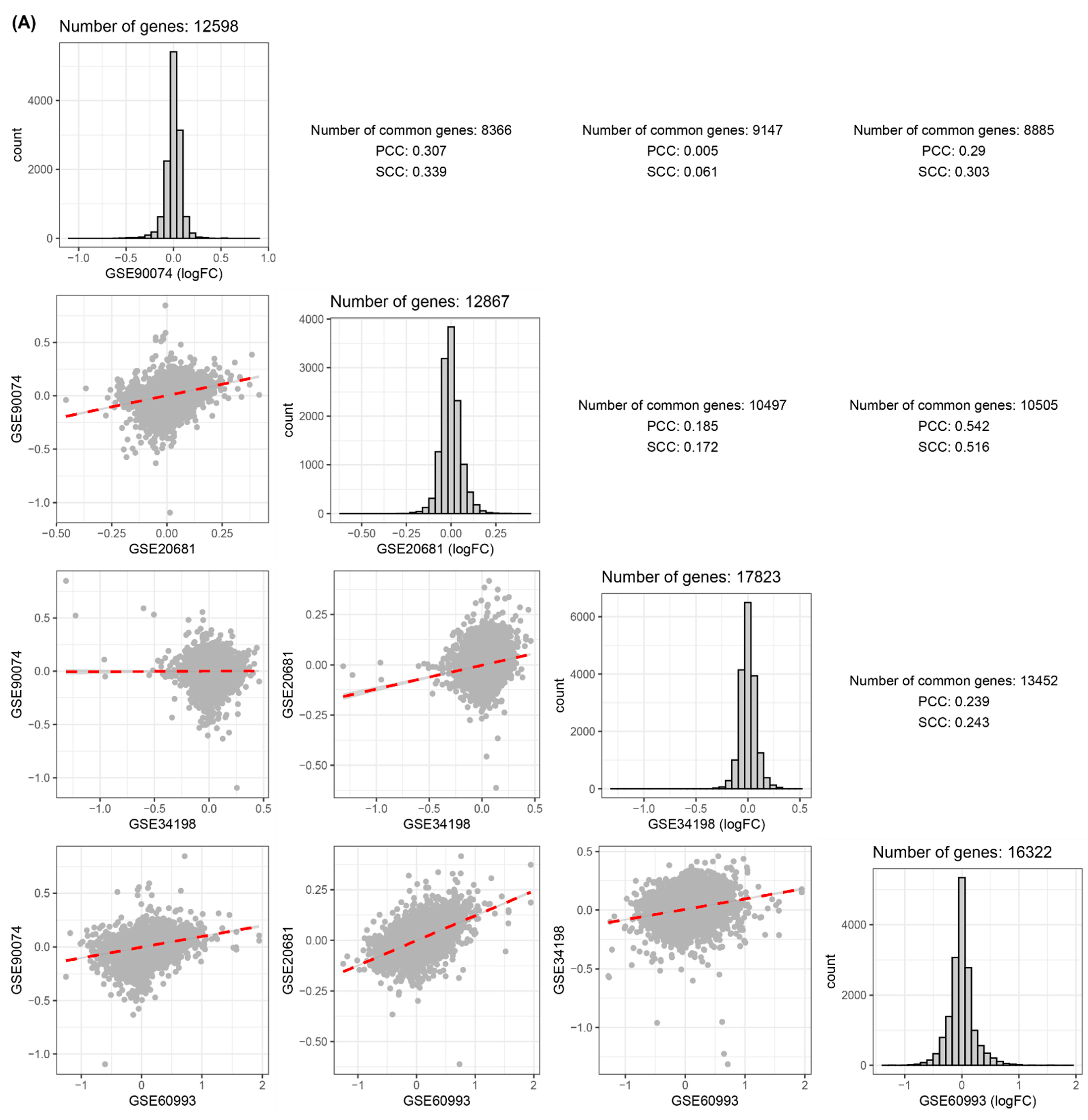
3.1. Comparisons of Disease-Related Signatures among the Four Blood CVD Datasets
3.2. Establishment of Modules
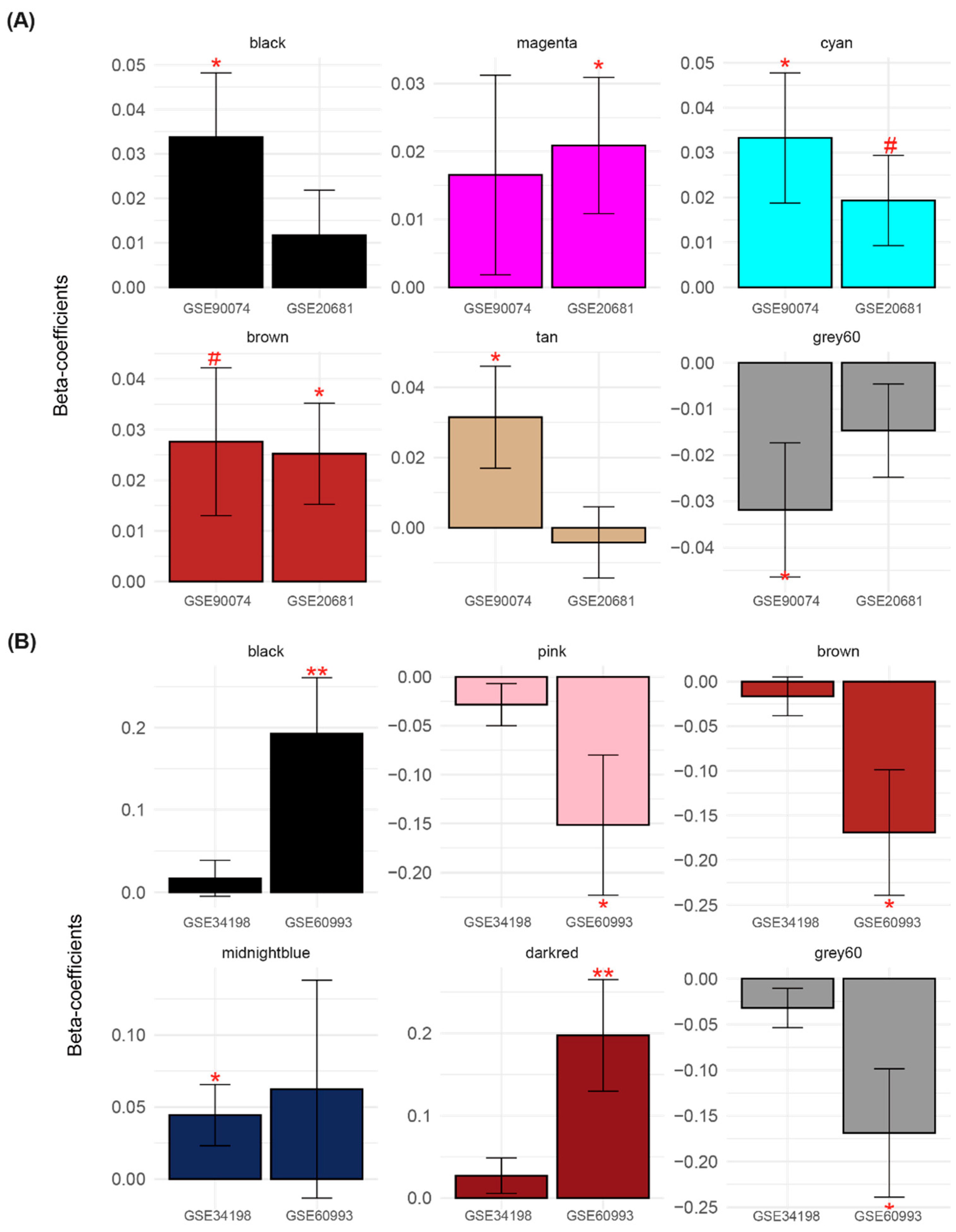
3.3. Identification of CVD-Related Module
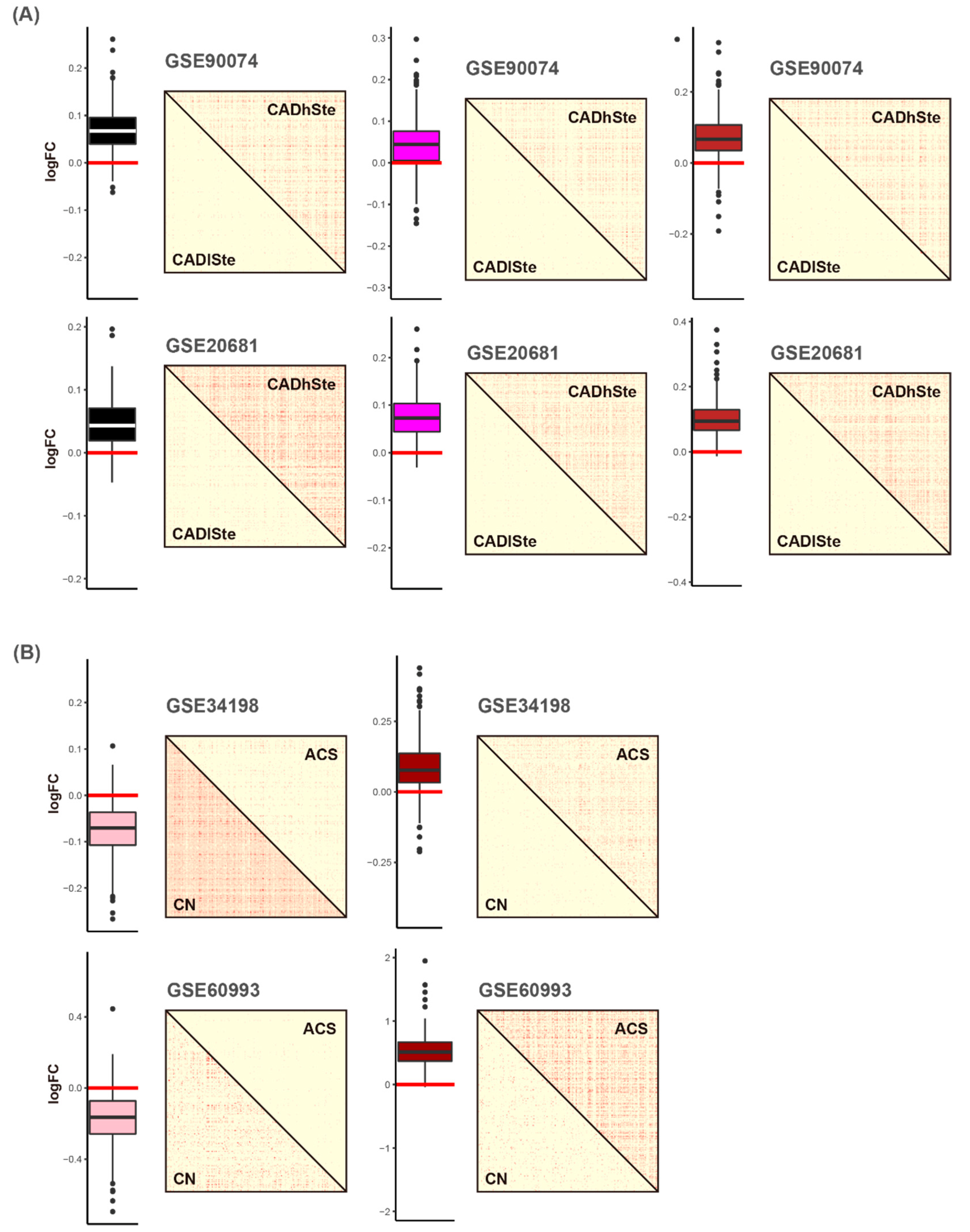
3.4. Validation for the Candidate Genes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Roth, G.A.; Mensah, G.A.; Johnson, C.O.; Addolorato, G.; Ammirati, E.; Baddour, L.M.; Barengo, N.C.; Beaton, A.Z.; Benjamin, E.J.; Benziger, C.P.; et al. Global Burden of Cardiovascular Diseases and Risk Factors, 1990–2019: Update From the GBD 2019 Study. J. Am. Coll. Cardiol. 2020, 76, 2982–3021. [Google Scholar] [CrossRef] [PubMed]
- Visseren, F.L.J.; Mach, F.; Smulders, Y.M.; Carballo, D.; Koskinas, K.C.; Bäck, M.; Benetos, A.; Biffi, A.; Boavida, J.M.; Capodanno, D.; et al. 2021 ESC Guidelines on cardiovascular disease prevention in clinical practice. Eur. Heart J. 2021, 42, 3227–3337. [Google Scholar] [CrossRef] [PubMed]
- Barabási, A.-L.; Gulbahce, N.; Loscalzo, J. Network medicine: A network-based approach to human disease. Nat. Rev. Genet. 2011, 12, 56–68. [Google Scholar] [CrossRef] [PubMed]
- Civelek, M.; Lusis, A.J. Systems genetics approaches to understand complex traits. Nat. Rev. Genet. 2014, 15, 34–48. [Google Scholar] [CrossRef]
- Deloukas, P.; Kanoni, S.; Willenborg, C.; Farrall, M.; Assimes, T.L.; Thompson, J.R.; Ingelsson, E.; Saleheen, D.; Erdmann, J.; Goldstein, B.A.; et al. Large-scale association analysis identifies new risk loci for coronary artery disease. Nat. Genet. 2013, 45, 25–33. [Google Scholar] [CrossRef]
- Peden, J.F.; Farrall, M. Thirty-five common variants for coronary artery disease: The fruits of much collaborative labour. Hum. Mol. Genet. 2011, 20, R198–R205. [Google Scholar] [CrossRef]
- Hindorff, L.A.; Sethupathy, P.; Junkins, H.A.; Ramos, E.M.; Mehta, J.P.; Collins, F.S.; Manolio, T.A. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc. Natl. Acad. Sci. USA 2009, 106, 9362–9367. [Google Scholar] [CrossRef]
- Tam, V.; Patel, N.; Turcotte, M.; Bossé, Y.; Paré, G.; Meyre, D. Benefits and limitations of genome-wide association studies. Nat. Rev. Genet. 2019, 20, 467–484. [Google Scholar] [CrossRef]
- Hägg, S.; Skogsberg, J.; Lundström, J.; Noori, P.; Nilsson, R.; Zhong, H.; Maleki, S.; Shang, M.M.; Brinne, B.; Bradshaw, M.; et al. Multi-organ expression profiling uncovers a gene module in coronary artery disease involving transendothelial migration of leukocytes and LIM domain binding 2: The Stockholm Atherosclerosis Gene Expression (STAGE) study. PLoS Genet. 2009, 5, e1000754. [Google Scholar] [CrossRef]
- Talukdar, H.A.; Foroughi Asl, H.; Jain, R.K.; Ermel, R.; Ruusalepp, A.; Franzén, O.; Kidd, B.A.; Readhead, B.; Giannarelli, C.; Kovacic, J.C.; et al. Cross-Tissue Regulatory Gene Networks in Coronary Artery Disease. Cell Syst. 2016, 2, 196–208. [Google Scholar] [CrossRef] [Green Version]
- Palou-Márquez, G.; Subirana, I.; Nonell, L.; Fernández-Sanlés, A.; Elosua, R. DNA methylation and gene expression integration in cardiovascular disease. Clin. Epigenetics 2021, 13, 75. [Google Scholar] [CrossRef]
- Fernández-Sanlés, A.; Sayols-Baixeras, S.; Subirana, I.; Sentí, M.; Pérez-Fernández, S.; de Castro Moura, M.; Esteller, M.; Marrugat, J.; Elosua, R. DNA methylation biomarkers of myocardial infarction and cardiovascular disease. Clin. Epigenetics 2021, 13, 86. [Google Scholar] [CrossRef]
- Zeng, L.; Talukdar, H.A.; Koplev, S.; Giannarelli, C.; Ivert, T.; Gan, L.M.; Ruusalepp, A.; Schadt, E.E.; Kovacic, J.C.; Lusis, A.J.; et al. Contribution of Gene Regulatory Networks to Heritability of Coronary Artery Disease. J. Am. Coll. Cardiol. 2019, 73, 2946–2957. [Google Scholar] [CrossRef]
- Halloran, J.W.; Zhu, D.; Qian, D.C.; Byun, J.; Gorlova, O.Y.; Amos, C.I.; Gorlov, I.P. Prediction of the gene expression in normal lung tissue by the gene expression in blood. BMC Med. Genom. 2015, 8, 77. [Google Scholar] [CrossRef]
- Basu, M.; Wang, K.; Ruppin, E.; Hannenhalli, S. Predicting tissue-specific gene expression from whole blood transcriptome. Sci. Adv. 2021, 7, eabd6991. [Google Scholar] [CrossRef]
- Lee, T.; Lee, H.; The Alzheimer’s Disease Neuroimaging Initiative. Identification of Disease-Related Genes That Are Common between Alzheimer’s and Cardiovascular Disease Using Blood Genome-Wide Transcriptome Analysis. Biomedicines 2021, 9, 1525. [Google Scholar] [CrossRef]
- Zhang, B.; Gaiteri, C.; Bodea, L.G.; Wang, Z.; McElwee, J.; Podtelezhnikov, A.A.; Zhang, C.; Xie, T.; Tran, L.; Dobrin, R.; et al. Integrated systems approach identifies genetic nodes and networks in late-onset Alzheimer’s disease. Cell 2013, 153, 707–720. [Google Scholar] [CrossRef]
- Lee, T.; Lee, H. Shared Blood Transcriptomic Signatures between Alzheimer’s Disease and Diabetes Mellitus. Biomedicines 2021, 9, 34. [Google Scholar] [CrossRef]
- Clough, E.; Barrett, T. The Gene Expression Omnibus Database. Methods Mol. Biol. 2016, 1418, 93–110. [Google Scholar] [CrossRef]
- Kang, D.D.; Sibille, E.; Kaminski, N.; Tseng, G.C. MetaQC: Objective quality control and inclusion/exclusion criteria for genomic meta-analysis. Nucleic Acids Res. 2012, 40, e15. [Google Scholar] [CrossRef] [Green Version]
- Ravi, S.; Schuck, R.N.; Hilliard, E.; Lee, C.R.; Dai, X.; Lenhart, K.; Willis, M.S.; Jensen, B.C.; Stouffer, G.A.; Patterson, C.; et al. Clinical Evidence Supports a Protective Role for CXCL5 in Coronary Artery Disease. Am. J. Pathol. 2017, 187, 2895–2911. [Google Scholar] [CrossRef]
- Beineke, P.; Fitch, K.; Tao, H.; Elashoff, M.R.; Rosenberg, S.; Kraus, W.E.; Wingrove, J.A. A whole blood gene expression-based signature for smoking status. BMC Med. Genom. 2012, 5, 58. [Google Scholar] [CrossRef]
- Elashoff, M.R.; Wingrove, J.A.; Beineke, P.; Daniels, S.E.; Tingley, W.G.; Rosenberg, S.; Voros, S.; Kraus, W.E.; Ginsburg, G.S.; Schwartz, R.S.; et al. Development of a blood-based gene expression algorithm for assessment of obstructive coronary artery disease in non-diabetic patients. BMC Med. Genom. 2011, 4, 26. [Google Scholar] [CrossRef]
- Valenta, Z.; Mazura, I.; Kolár, M.; Grünfeldová, H.; Feglarová, P.; Peleška, J.; Tomecková, M.; Kalina, J.; Slovák, D.; Zvárová, J. Determinants of excess genetic risk of acute myocardial infarction-a matched case-control study. Eur. J. Biomed. Inform. 2012, 8, 34–43. [Google Scholar] [CrossRef]
- Park, H.J.; Noh, J.H.; Eun, J.W.; Koh, Y.S.; Seo, S.M.; Park, W.S.; Lee, J.Y.; Chang, K.; Seung, K.B.; Kim, P.J.; et al. Assessment and diagnostic relevance of novel serum biomarkers for early decision of ST-elevation myocardial infarction. Oncotarget 2015, 6, 12970–12983. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef]
- Smyth, G.K. Limma: Linear models for microarray data. In Bioinformatics and Computational Biology Solutions Using R and Bioconductor; Springer: Berlin/Heidelberg, Germany, 2005; pp. 397–420. [Google Scholar]
- Leek, J.T.; Johnson, W.E.; Parker, H.S.; Jaffe, A.E.; Storey, J.D. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics 2012, 28, 882–883. [Google Scholar] [CrossRef]
- Johnson, W.E.; Li, C.; Rabinovic, A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics 2007, 8, 118–127. [Google Scholar] [CrossRef]
- Gandal, M.J.; Haney, J.R.; Parikshak, N.N.; Leppa, V.; Ramaswami, G.; Hartl, C.; Schork, A.J.; Appadurai, V.; Buil, A.; Werge, T.M.; et al. Shared molecular neuropathology across major psychiatric disorders parallels polygenic overlap. Science 2018, 359, 693–697. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- The Gene Ontology Resource: 20 years and still GOing strong. Nucleic Acids Res. 2019, 47, D330–D338. [CrossRef] [PubMed]
- Liberzon, A.; Birger, C.; Thorvaldsdóttir, H.; Ghandi, M.; Mesirov, J.P.; Tamayo, P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015, 1, 417–425. [Google Scholar] [CrossRef] [PubMed]
- Soh, J.; Cho, H.; Choi, C.-H.; Lee, H. Identification and Characterization of MicroRNAs Associated with Somatic Copy Number Alterations in Cancer. Cancers 2018, 10, 475. [Google Scholar] [CrossRef] [PubMed]
- Oh, E.; Kim, J.-H.; Um, J.; Jung, D.-W.; Williams, D.R.; Lee, H. Genome-Wide Transcriptomic Analysis of Non-Tumorigenic Tissues Reveals Aging-Related Prognostic Markers and Drug Targets in Renal Cell Carcinoma. Cancers 2021, 13, 3045. [Google Scholar] [CrossRef]
- Joehanes, R.; Ying, S.; Huan, T.; Johnson, A.D.; Raghavachari, N.; Wang, R.; Liu, P.; Woodhouse, K.A.; Sen, S.K.; Tanriverdi, K.; et al. Gene expression signatures of coronary heart disease. Arter. Thromb. Vasc. Biol. 2013, 33, 1418–1426. [Google Scholar] [CrossRef]
- Xu, M.; Zhang, D.F.; Luo, R.; Wu, Y.; Zhou, H.; Kong, L.L.; Bi, R.; Yao, Y.G. A systematic integrated analysis of brain expression profiles reveals YAP1 and other prioritized hub genes as important upstream regulators in Alzheimer's disease. Alzheimers Dement 2018, 14, 215–229. [Google Scholar] [CrossRef]
- Vogt, G.; El Choubassi, N.; Herczegfalvi, Á.; Kölbel, H.; Lekaj, A.; Schara, U.; Holtgrewe, M.; Krause, S.; Horvath, R.; Schuelke, M.; et al. Expanding the clinical and molecular spectrum of ATP6V1A related metabolic cutis laxa. J. Inherit. Metab. Dis. 2021, 44, 972–986. [Google Scholar] [CrossRef]
- Duan, X.; Yang, S.; Zhang, L.; Yang, T. V-ATPases and osteoclasts: Ambiguous future of V-ATPases inhibitors in osteoporosis. Theranostics 2018, 8, 5379–5399. [Google Scholar] [CrossRef]
- Suda, M.; Shimizu, I.; Katsuumi, G.; Hsiao, C.L.; Yoshida, Y.; Matsumoto, N.; Yoshida, Y.; Katayama, A.; Wada, J.; Seki, M.; et al. Glycoprotein nonmetastatic melanoma protein B regulates lysosomal integrity and lifespan of senescent cells. Sci. Rep. 2022, 12, 6522. [Google Scholar] [CrossRef]
- Santiago, F.S.; Li, Y.; Zhong, L.; Raftery, M.J.; Lins, L.; Khachigian, L.M. Truncated YY1 interacts with BASP1 through a 339KLK341 motif in YY1 and suppresses vascular smooth muscle cell growth and intimal hyperplasia after vascular injury. Cardiovasc. Res. 2021, 117, 2395–2406. [Google Scholar] [CrossRef]
- Tian, Z.; Sun, Y.; Sun, X.; Wang, J.; Jiang, T. LINC00473 inhibits vascular smooth muscle cell viability to promote aneurysm formation via miR-212-5p/BASP1 axis. Eur. J. Pharmacol. 2020, 873, 172935. [Google Scholar] [CrossRef]
- Khajavi, M.; Zhou, Y.; Schiffer, A.J.; Bazinet, L.; Birsner, A.E.; Zon, L.; D’Amato, R.J. Identification of Basp1 as a novel angiogenesis-regulating gene by multi-model system studies. FASEB J. 2021, 35, e21404. [Google Scholar] [CrossRef]
- Qi, F.; Zhang, W.; Huang, J.; Fu, L.; Zhao, J. Single-Cell RNA Sequencing Analysis of the Immunometabolic Rewiring and Immunopathogenesis of Coronavirus Disease 2019. Front. Immunol. 2021, 12, 651656. [Google Scholar] [CrossRef]
- Wang, Z.; Qiu, Z.; Hua, S.; Yang, W.; Chen, Y.; Huang, F.; Fan, Y.; Tong, L.; Xu, T.; Tong, X.; et al. Nuclear Tkt promotes ischemic heart failure via the cleaved Parp1/Aif axis. Basic Res. Cardiol. 2022, 117, 18. [Google Scholar] [CrossRef]
- Choublier, N.; Taghi, M.; Menet, M.-C.; Le Gall, M.; Bruce, J.; Chafey, P.; Guillonneau, F.; Moreau, A.; Denizot, C.; Parmentier, Y.; et al. Exposure of human cerebral microvascular endothelial cells hCMEC/D3 to laminar shear stress induces vascular protective responses. Fluids Barriers CNS 2022, 19, 41. [Google Scholar] [CrossRef]
- Tomas, L.; Edsfeldt, A.; Mollet, I.G.; Perisic Matic, L.; Prehn, C.; Adamski, J.; Paulsson-Berne, G.; Hedin, U.; Nilsson, J.; Bengtsson, E.; et al. Altered metabolism distinguishes high-risk from stable carotid atherosclerotic plaques. Eur. Heart J. 2018, 39, 2301–2310. [Google Scholar] [CrossRef]
- Lambert, S.A.; Jolma, A.; Campitelli, L.F.; Das, P.K.; Yin, Y.; Albu, M.; Chen, X.; Taipale, J.; Hughes, T.R.; Weirauch, M.T. The Human Transcription Factors. Cell 2018, 172, 650–665. [Google Scholar] [CrossRef]
- Fidalgo, M.; Shekar, P.C.; Ang, Y.S.; Fujiwara, Y.; Orkin, S.H.; Wang, J. Zfp281 functions as a transcriptional repressor for pluripotency of mouse embryonic stem cells. Stem Cells 2011, 29, 1705–1716. [Google Scholar] [CrossRef]
- Zhou, H.; Morales, M.G.; Hashimoto, H.; Dickson, M.E.; Song, K.; Ye, W.; Kim, M.S.; Niederstrasser, H.; Wang, Z.; Chen, B.; et al. ZNF281 enhances cardiac reprogramming by modulating cardiac and inflammatory gene expression. Genes Dev. 2017, 31, 1770–1783. [Google Scholar] [CrossRef]
- Sebastian, T.; Johnson, P.F. Stop and go: Anti-proliferative and mitogenic functions of the transcription factor C/EBPbeta. Cell Cycle 2006, 5, 953–957. [Google Scholar] [CrossRef] [Green Version]
- Boström, P.; Mann, N.; Wu, J.; Quintero, P.A.; Plovie, E.R.; Panáková, D.; Gupta, R.K.; Xiao, C.; MacRae, C.A.; Rosenzweig, A.; et al. C/EBPβ controls exercise-induced cardiac growth and protects against pathological cardiac remodeling. Cell 2010, 143, 1072–1083. [Google Scholar] [CrossRef]
- Lee, T.; Lee, H. Prediction of Alzheimer’s disease using blood gene expression data. Sci. Rep. 2020, 10, 3485. [Google Scholar] [CrossRef]
- Lee, J.W.; Lee, S.H.; Youn, Y.J.; Ahn, M.S.; Kim, J.Y.; Yoo, B.S.; Yoon, J.; Kwon, W.; Hong, I.S.; Lee, K.; et al. A randomized, open-label, multicenter trial for the safety and efficacy of adult mesenchymal stem cells after acute myocardial infarction. J. Korean Med. Sci. 2014, 29, 23–31. [Google Scholar] [CrossRef] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, T.; Hwang, S.; Seo, D.M.; Shin, H.C.; Kim, H.S.; Kim, J.-Y.; Uh, Y. Identification of Cardiovascular Disease-Related Genes Based on the Co-Expression Network Analysis of Genome-Wide Blood Transcriptome. Cells 2022, 11, 2867. https://doi.org/10.3390/cells11182867
Lee T, Hwang S, Seo DM, Shin HC, Kim HS, Kim J-Y, Uh Y. Identification of Cardiovascular Disease-Related Genes Based on the Co-Expression Network Analysis of Genome-Wide Blood Transcriptome. Cells. 2022; 11(18):2867. https://doi.org/10.3390/cells11182867
Chicago/Turabian StyleLee, Taesic, Sangwon Hwang, Dong Min Seo, Ha Chul Shin, Hyun Soo Kim, Jang-Young Kim, and Young Uh. 2022. "Identification of Cardiovascular Disease-Related Genes Based on the Co-Expression Network Analysis of Genome-Wide Blood Transcriptome" Cells 11, no. 18: 2867. https://doi.org/10.3390/cells11182867
APA StyleLee, T., Hwang, S., Seo, D. M., Shin, H. C., Kim, H. S., Kim, J.-Y., & Uh, Y. (2022). Identification of Cardiovascular Disease-Related Genes Based on the Co-Expression Network Analysis of Genome-Wide Blood Transcriptome. Cells, 11(18), 2867. https://doi.org/10.3390/cells11182867