

A Charcot-Marie-Tooth-Causing Mutation in HSPB1 Decreases Cell Adaptation to Repeated Stress by Disrupting Autophagic Clearance of Misfolded Proteins
 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells Lines and Primary Antibodies
2.2. Plasmid Construction and Cell Transfection
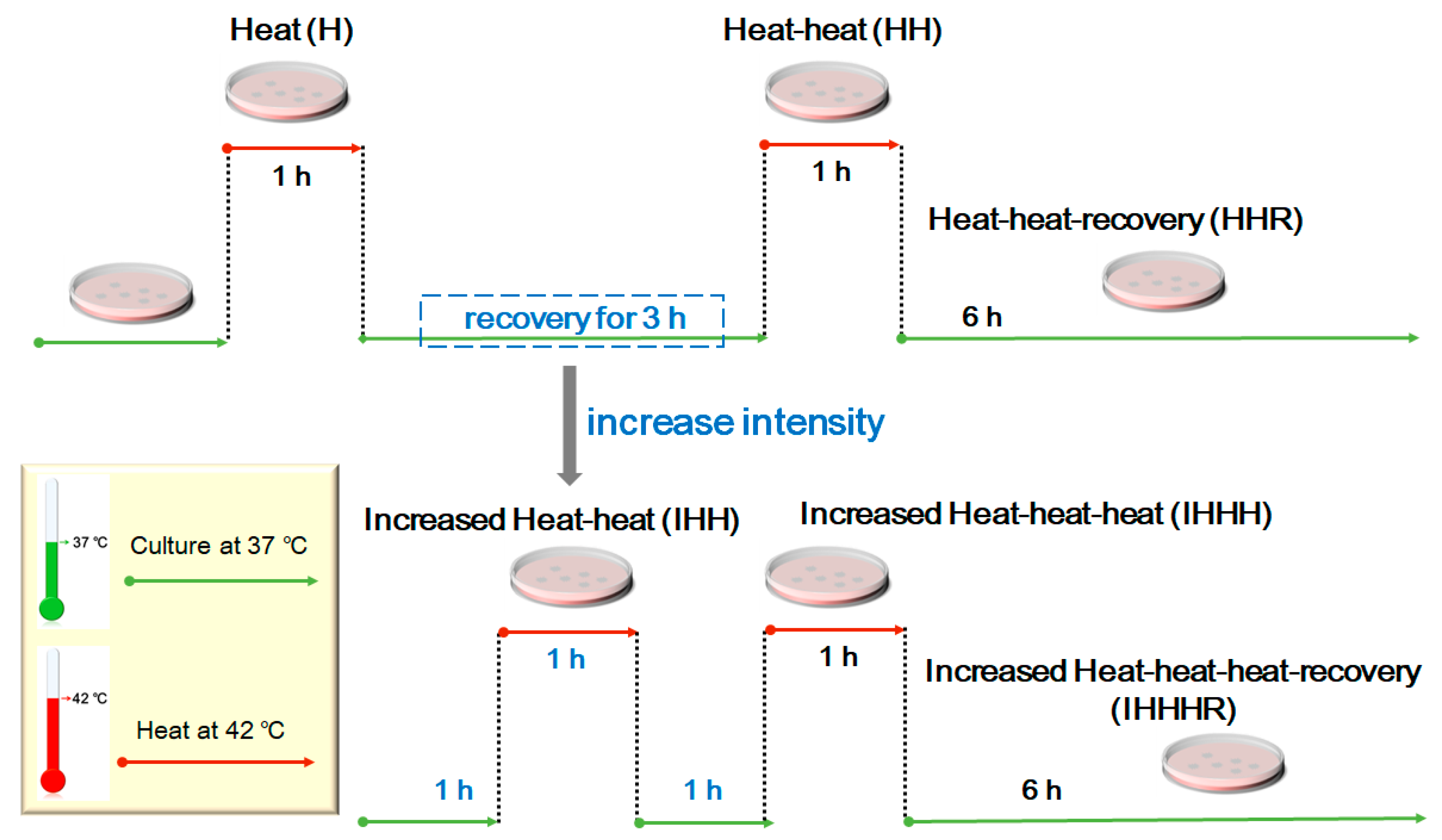
2.3. Cell Culture and Repeated Thermal Stimulation
2.4. Western Blotting
2.5. Co-Immunoprecipitation Assay
2.6. Immunofluorescence Assay
2.7. Statistical Analysis
3. Results
3.1. HSPB1S135F Binds Both to α-Tubulin and to Acetylated α-Tubulin
3.2. HSPB1S135F Blocks the Formation of Perinuclear Aggresomes under Repeated Heat Shock
3.3. HSPB1S135F Inhibits the Degradation of Autophagosome under Repeated Heat Shock
3.4. HSPB1S135F Prevents the Clearance of Misfolded Proteins
3.5. HSPB1S135F Decreases Cell Recovery from Repeated Heat Shock
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lupski, J.R.; Deocaluna, R.M.; Slaugenhaupt, S.; Pentao, L.; Guzzetta, V.; Trask, B.J.; Saucedocardenas, O.; Barker, D.F.; Killian, J.M.; Garcia, C.A.; et al. DNA Duplication Associated with Charcot-Marie-Tooth Disease Type-1a. Cell 1991, 66, 219–232. [Google Scholar] [CrossRef]
- Pyromali, I.; Benslimane, N.; Favreau, F.; Goizet, C.; Lazaro, L.; Vitry, M.; Derouault, P.; Sturtz, F.; Magdelaine, C.; Lia, A.S. From Negative to Positive Diagnosis: Structural Variation Could Be the Second Mutation You Are Looking for in a Recessive Autosomal Gene. J. Pers. Med. 2022, 12, 212. [Google Scholar] [CrossRef] [PubMed]
- Shen, S.D.; Benoy, V.; Bergman, J.K.; Kalin, J.H.; Frojuello, M.; Vistoli, G.; Haeck, W.; Van Den Bosch, L.; Kozikowski, A.P. Bicyclic-Capped Histone Deacetylase 6 Inhibitors with Improved Activity in a Model of Axonal Charcot-Marie-Tooth Disease. ACS Chem. Neurosci. 2016, 7, 240–258. [Google Scholar] [CrossRef]
- Evgrafov, O.V.; Mersiyanova, I.; Irobi, J.; Van Den Bosch, L.; Dierick, I.; Leung, C.L.; Schagina, O.; Verpoorten, N.; Van Impe, K.; Fedotov, V.; et al. Mutant small heat-shock protein 27 causes axonal Charcot-Marie-Tooth disease and distal hereditary motor neuropathy. Nat. Genet. 2004, 36, 602–606. [Google Scholar] [CrossRef] [PubMed]
- Dorsch, L.M.; Schuldt, M.; dos Remedios, C.G.; Schinkel, A.F.L.; de Jong, P.L.; Michels, M.; Kuster, D.W.D.; Brundel, B.J.J.M.; van der Velden, J. Protein Quality Control Activation and Microtubule Remodeling in Hypertrophic Cardiomyopathy. Cells 2019, 8, 741. [Google Scholar] [CrossRef]
- Gomes, V.M.; Wailemann, R.A.M.; Arini, G.S.; Oliveira, T.C.; Almeida, D.R.Q.; dos Santos, A.F.; Terra, L.F.; Lortz, S.; Labriola, L. HSPB1 Is Essential for Inducing Resistance to Proteotoxic Stress in Beta-Cells. Cells 2021, 10, 2178. [Google Scholar] [CrossRef] [PubMed]
- Ousman, S.S.; Frederick, A.; Lim, E.-M.F. Chaperone Proteins in the Central Nervous System and Peripheral Nervous System after Nerve Injury. Front. Neurosci. 2017, 11, 79. [Google Scholar] [CrossRef] [PubMed]
- Taga, A.; Cornblath, D.R. A novel HSPB1 mutation associated with a late onset CMT2 phenotype: Case presentation and systematic review of the literature. J. Peripher. Nerv. Syst. 2020, 25, 223–229. [Google Scholar] [CrossRef]
- Holguin, B.A.; Hildenbrand, Z.L.; Bernal, R.A. Insights Into the Role of Heat Shock Protein 27 in the Development of Neurodegeneration. Front. Mol. Neurosci. 2022, 15, 868089. [Google Scholar] [CrossRef]
- Vendredy, L.; Adriaenssens, E.; Timmerman, V. Small heat shock proteins in neurodegenerative diseases. Cell Stress Chaperones 2020, 25, 679–699. [Google Scholar] [CrossRef] [Green Version]
- Perlson, E.; Maday, S.; Fu, M.M.; Moughamian, A.J.; Holzbaur, E.L. Retrograde axonal transport: Pathways to cell death? Trends Neurosci. 2010, 33, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Kalmar, B.; Innes, A.; Wanisch, K.; Kolaszynska, A.K.; Pandraud, A.; Kelly, G.; Abramov, A.Y.; Reilly, M.M.; Schiavo, G.; Greensmith, L. Mitochondrial deficits and abnormal mitochondrial retrograde axonal transport play a role in the pathogenesis of mutant Hsp27-induced Charcot Marie Tooth Disease. Hum. Mol. Genet. 2017, 26, 3313–3326. [Google Scholar] [CrossRef] [PubMed]
- Almeida-Souza, L.; Goethals, S.; de Winter, V.; Dierick, I.; Gallardo, R.; Van Durme, J.; Irobi, J.; Gettemans, J.; Rousseau, F.; Schymkowitz, J.; et al. Increased Monomerization of Mutant HSPB1 Leads to Protein Hyperactivity in Charcot-Marie-Tooth Neuropathy. J. Biol. Chem. 2010, 285, 12778–12786. [Google Scholar] [CrossRef] [PubMed]
- d’Ydewalle, C.; Krishnan, J.; Chiheb, D.M.; Van Damme, P.; Irobi, J.; Kozikowski, A.P.; Vanden Berghe, P.; Timmerman, V.; Robberecht, W.; Van Den Bosch, L. HDAC6 inhibitors reverse axonal loss in a mouse model of mutant HSPB1-induced Charcot-Marie-Tooth disease. Nat. Med. 2011, 17, 968–974. [Google Scholar] [CrossRef] [PubMed]
- Almeida-Souza, L.; Asselbergh, B.; d’Ydewalle, C.; Moonens, K.; Goethals, S.; de Winter, V.; Azmi, A.; Irobi, J.; Timmermans, J.P.; Gevaert, K.; et al. Small heat-shock protein HSPB1 mutants stabilize microtubules in Charcot-Marie-Tooth neuropathy. J. Neurosci. 2011, 31, 15320–15328. [Google Scholar] [CrossRef] [PubMed]
- Almeida-Souza, L.; Timmerman, V.; Janssens, S. Microtubule dynamics in the peripheral nervous system: A matter of balance. Bioarchitecture 2011, 1, 267–270. [Google Scholar] [CrossRef]
- Prior, R.; Van Helleputte, L.; Benoy, V.; Van Den Bosch, L. Defective axonal transport: A common pathological mechanism in inherited and acquired peripheral neuropathies. Neurobiol. Dis. 2017, 105, 300–320. [Google Scholar] [CrossRef]
- Sun, Y.; MacRae, T.H. The small heat shock proteins and their role in human disease. FEBS J. 2005, 272, 2613–2627. [Google Scholar] [CrossRef] [PubMed]
- Chin, L.S.; Olzmann, J.A.; Li, L. Parkin-mediated ubiquitin signalling in aggresome formation and autophagy. Biochem. Soc. Trans. 2010, 38, 144–149. [Google Scholar] [CrossRef]
- Howard, J.; Hyman, A.A. Dynamics and mechanics of the microtubule plus end. Nature 2003, 422, 753–758. [Google Scholar] [CrossRef]
- Kapitein, L.C.; Hoogenraad, C.C. Building the Neuronal Microtubule Cytoskeleton. Neuron 2015, 87, 492–506. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, Y.; Kovacs, J.J.; McLaurin, A.; Vance, J.M.; Ito, A.; Yao, T.P. The deacetylase HDAC6 regulates aggresome formation and cell viability in response to misfolded protein stress. Cell 2003, 115, 727–738. [Google Scholar] [CrossRef]
- Weeks, S.D.; Muranova, L.K.; Heirbaut, M.; Beelen, S.; Gusev, N.B. Characterization of human small heat shock protein HSPB1 α-crystallin domain localized mutants associated with hereditary motor neuron diseases. Sci. Rep. 2018, 8, 688. [Google Scholar] [CrossRef] [PubMed]
- Xing, J.; Li, Y.; Lin, M.P.; Wang, J.F.; Wu, J.C.; Ma, Y.F.; Wang, Y.L.; Yang, L.; Luo, Y.F. Surface chemistry modulates osteoblasts sensitivity to low fluid shear stress. J. Biomed. Mater. Res. A 2014, 102, 4151–4160. [Google Scholar] [CrossRef] [PubMed]
- Bartelt-Kirbach, B.; Golenhofen, N. Reaction of small heat-shock proteins to different kinds of cellular stress in cultured rat hippocampal neurons. Cell Stress Chaperones 2014, 19, 145–153. [Google Scholar] [CrossRef] [PubMed]
- Hyttinen, J.M.T.; Amadio, M.; Viiri, J.; Pascale, A.; Salminen, A.; Kaarniranta, K. Clearance of misfolded and aggregated proteins by aggrephagy and implications for aggregation diseases. Ageing Res. Rev. 2014, 18, 16–28. [Google Scholar] [CrossRef]
- Pandey, U.B.; Nie, Z.P.; Batlevi, Y.; McCray, B.A.; Ritson, G.P.; Nedelsky, N.B.; Schwartz, S.L.; DiProspero, N.A.; Knight, M.A.; Schuldiner, O.; et al. HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature 2007, 447, 859–863. [Google Scholar] [CrossRef]
- Rossaert, E.; Van Den Bosch, L. HDAC6 inhibitors: Translating genetic and molecular insights into a therapy for axonal CMT. Brain Res. 2020, 1733, 146692. [Google Scholar] [CrossRef]
- Gal, J.; Bang, Y.; Choi, H.J. SIRT2 interferes with autophagy-mediated degradation of protein aggregates in neuronal cells under proteasome inhibition. Neurochem. Int. 2012, 61, 992–1000. [Google Scholar] [CrossRef]
- Mackeh, R.; Perdiz, D.; Lorin, S.; Codogno, P.; Pous, C. Autophagy and microtubules-new story, old players. J. Cell Sci. 2013, 126, 1071–1080. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.L.; Liu, X.G. The dual functions of alpha-tubulin acetylation in cellular apoptosis and autophage induced by tanespimycin in lung cancer cells. Cancer Cell Int. 2020, 20, 369. [Google Scholar] [CrossRef] [PubMed]
- Bucci, C.; Bakke, O.; Progida, C. Charcot-Marie-Tooth disease and intracellular traffic. Prog. Neurobiol. 2012, 99, 191–225. [Google Scholar] [CrossRef] [PubMed]
- Williams, K.L.; Rahimtula, M.; Mearow, K.M. Hsp27 and axonal growth in adult sensory neurons in vitro. BMC Neurosci. 2005, 6, 24–40. [Google Scholar] [CrossRef] [PubMed]
- Ylikallio, E.; Konovalova, S.; Dhungana, Y.; Hilander, T.; Junna, N.; Partanen, J.V.; Toppila, J.P.; Auranen, M.; Tyynismaa, H. Truncated HSPB1 causes axonal neuropathy and impairs tolerance to unfolded protein stress. BBA Clin. 2015, 3, 233–242. [Google Scholar] [CrossRef]
- Schmidt, M.F.; Gan, Z.Y.; Komander, D.; Dewson, G. Ubiquitin signalling in neurodegeneration: Mechanisms and therapeutic opportunities. Cell Death Differ. 2021, 28, 570–590. [Google Scholar] [CrossRef]
- Webster, J.M.; Darling, A.L.; Uversky, V.N.; Blair, L.J. Small Heat Shock Proteins, Big Impact on Protein Aggregation in Neurodegenerative Disease. Front. Pharmacol. 2019, 10, 1047. [Google Scholar] [CrossRef]
- Benn, S.C.; Perrelet, D.; Kato, A.C.; Scholz, J.; Decosterd, I.; Mannion, R.J.; Bakowska, J.C.; Woolf, C.J. Hsp27 upregulation and phosphorylation is required for injured sensory and motor neuron survival. Neuron 2002, 36, 45–56. [Google Scholar] [CrossRef]
- Alderson, T.R.; Adriaenssens, E.; Asselbergh, B.; Pritisanac, I.; Van Lent, J.; Gastall, H.Y.; Walti, M.A.; Louis, J.M.; Timmerman, V.; Baldwin, A.J.; et al. A weakened interface in the P182L variant of HSP27 associated with severe Charcot-Marie-Tooth neuropathy causes aberrant binding to interacting proteins. Embo J. 2021, 40, e103811. [Google Scholar] [CrossRef]
- Ackerley, S.; James, P.A.; Kalli, A.; French, S.; Davies, K.E.; Talbot, K. A mutation in the small heat-shock protein HSPB1 leading to distal hereditary motor neuronopathy disrupts neurofilament assembly and the axonal transport of specific cellular cargoes. Hum. Mol. Genet. 2006, 15, 347–354. [Google Scholar] [CrossRef]
- Folger, A.; Wang, Y.C. The Cytotoxicity and Clearance of Mutant Huntingtin and Other Misfolded Proteins. Cells 2021, 10, 2835. [Google Scholar] [CrossRef]
- Buchan, J.R.; Kolaitis, R.M.; Taylor, J.P.; Parker, R. Eukaryotic Stress Granules Are Cleared by Autophagy and Cdc48/VCP Function. Cell 2013, 153, 1461–1474. [Google Scholar] [CrossRef] [PubMed]
- Fortun, J.; Dunn, W.A.; Joy, S.; Li, J.; Notterpek, L. Emerging role for autophagy in the removal of aggresomes in Schwann cells. J. Neurosci. 2003, 23, 10672–10680. [Google Scholar] [CrossRef] [PubMed]
- Boyault, C.; Zhang, Y.; Fritah, S.; Caron, C.; Gilquin, B.; Kwon, S.H.; Garrido, C.; Yao, T.P.; Vourc’h, C.; Matthias, P.; et al. HDAC6 controls major cell response pathways to cytotoxic accumulation of protein aggregates. Genes Dev. 2007, 21, 2172–2181. [Google Scholar] [CrossRef] [PubMed]
- Wen, H.X.; Zhan, L.X.; Chen, S.Y.; Long, L.; Xu, E. Rab7 May be a Novel Therapeutic Target for Neurologic Diseases as a Key Regulator in Autophagy. J. Neurosci. Res. 2017, 95, 1993–2004. [Google Scholar] [CrossRef] [PubMed]
- Aplin, A.; Jasionowski, T.; Tuttle, D.L.; Lenk, S.E.; Dunn, W.A., Jr. Cytoskeletal elements are required for the formation and maturation of autophagic vacuoles. J. Cell Physiol. 1992, 152, 458–466. [Google Scholar] [CrossRef]
- Kochl, R.; Hu, X.W.; Chan, E.Y.; Tooze, S.A. Microtubules facilitate autophagosome formation and fusion of autophagosomes with endosomes. Traffic 2006, 7, 129–145. [Google Scholar] [CrossRef]
- Webb, J.L.; Ravikumar, B.; Rubinsztein, D.C. Microtubule disruption inhibits autophagosome-lysosome fusion: Implications for studying the roles of aggresomes in polyglutamine diseases. Int. J. Biochem. Cell Biol. 2004, 36, 2541–2550. [Google Scholar] [CrossRef]
- Taylor, J.P.; Tanaka, F.; Robitschek, J.; Sandoval, C.M.; Taye, A.; Markovic-Plese, S.; Fischbeck, K.H. Aggresomes protect cells by enhancing the degradation of toxic polyglutamine-containing protein. Hum. Mol. Genet. 2003, 12, 749–757. [Google Scholar] [CrossRef]
- Sleigh, J.N.; Rossor, A.M.; Fellows, A.D.; Tosolini, A.P.; Schiavo, G. Axonal transport and neurological disease. Nat. Rev. Neurol. 2019, 15, 691–703. [Google Scholar] [CrossRef]
- Gibbs, K.L.; Greensmith, L.; Schiavo, G. Regulation of Axonal Transport by Protein Kinases. Trends Biochem. Sci. 2015, 40, 597–610. [Google Scholar] [CrossRef] [Green Version]
- Kevenaar, J.T.; Hoogenraad, C.C. The axonal cytoskeleton: From organization to function. Front. Mol. Neurosci. 2015, 8, 44. [Google Scholar] [CrossRef] [PubMed]
- Gerdes, J.M.; Katsanis, N. Microtubule transport defects in neurological and ciliary disease. Cell Mol. Life Sci. 2005, 62, 1556–1570. [Google Scholar] [CrossRef]
- Wang, Y.; Song, M.X.; Song, F.Y. Neuronal autophagy and axon degeneration. Cell Mol. Life Sci. 2018, 75, 2389–2406. [Google Scholar] [CrossRef] [PubMed]
- Portran, D.; Schaedel, L.; Xu, Z.J.; Thery, M.; Nachury, M.V. Tubulin acetylation protects long-lived microtubules against mechanical ageing. Nat. Cell Biol. 2017, 19, 391–398. [Google Scholar] [CrossRef] [PubMed]
- Madeo, F.; Eisenberg, T.; Kroemer, G. Autophagy for the avoidance of neurodegeneration. Gene Dev. 2009, 23, 2253–2259. [Google Scholar] [CrossRef] [PubMed]
- Haidar, M.; Asselbergh, B.; Adriaenssens, E.; De Winter, V.; Timmermans, J.P.; Auer-Grumbach, M.; Juneja, M.; Timmerman, V. Neuropathy-causing mutations in HSPB1 impair autophagy by disturbing the formation of SQSTM1/p62 bodies. Autophagy 2019, 15, 1051–1068. [Google Scholar] [CrossRef]
- Ko, S.H.; Gonzalez, G.; Liu, Z.J.; Chen, L.Z. Axon Injury-Induced Autophagy Activation Is Impaired in a C. elegans Model of Tauopathy. Int. J. Mol. Sci. 2020, 21, 8559. [Google Scholar] [CrossRef]
- Saxton, W.M.; Hollenbeck, P.J. The axonal transport of mitochondria. J. Cell Sci. 2012, 125, 2095–2104. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, W.; Li, Z.; Hao, S.; Wang, B. A novel perspective on neuron study: Damaging and promoting effects in different neurons induced by mechanical stress. Biomech. Model. Mechanobiol. 2015, 15, 1–9. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, X.; Qiao, Y.; Han, R.; Gao, Y.; Yang, X.; Zhang, Y.; Wan, Y.; Yu, W.; Pan, X.; Xing, J. A Charcot-Marie-Tooth-Causing Mutation in HSPB1 Decreases Cell Adaptation to Repeated Stress by Disrupting Autophagic Clearance of Misfolded Proteins. Cells 2022, 11, 2886. https://doi.org/10.3390/cells11182886
Zhang X, Qiao Y, Han R, Gao Y, Yang X, Zhang Y, Wan Y, Yu W, Pan X, Xing J. A Charcot-Marie-Tooth-Causing Mutation in HSPB1 Decreases Cell Adaptation to Repeated Stress by Disrupting Autophagic Clearance of Misfolded Proteins. Cells. 2022; 11(18):2886. https://doi.org/10.3390/cells11182886
Chicago/Turabian StyleZhang, Xuelian, Yaru Qiao, Ronglin Han, Yingjie Gao, Xun Yang, Ying Zhang, Ying Wan, Wei Yu, Xianchao Pan, and Juan Xing. 2022. "A Charcot-Marie-Tooth-Causing Mutation in HSPB1 Decreases Cell Adaptation to Repeated Stress by Disrupting Autophagic Clearance of Misfolded Proteins" Cells 11, no. 18: 2886. https://doi.org/10.3390/cells11182886