
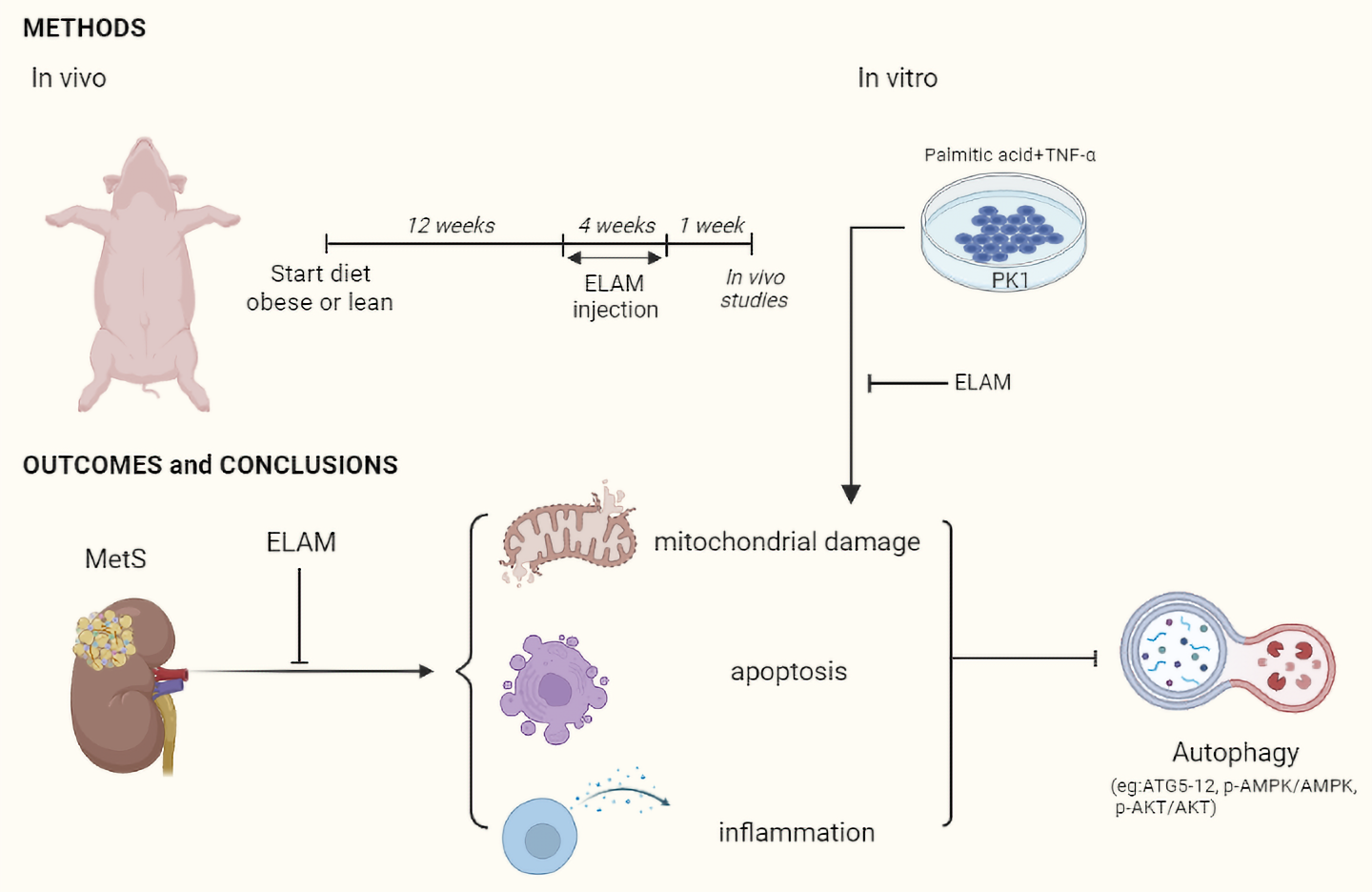
Effects of Elamipretide on Autophagy in Renal Cells of Pigs with Metabolic Syndrome
 ,
,  , and
, and
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Animal Experiments
2.2. Autophagy Markers
2.3. Mitochondrial Energy Production and Cellular Apoptosis
2.4. Fibrosis and Lipid Deposits
2.5. Inflammatory Biomarkers Levels
2.6. Cell Culture
2.7. TMRE, MitoSOX and Western Blot
2.8. Statistical Analysis
3. Results
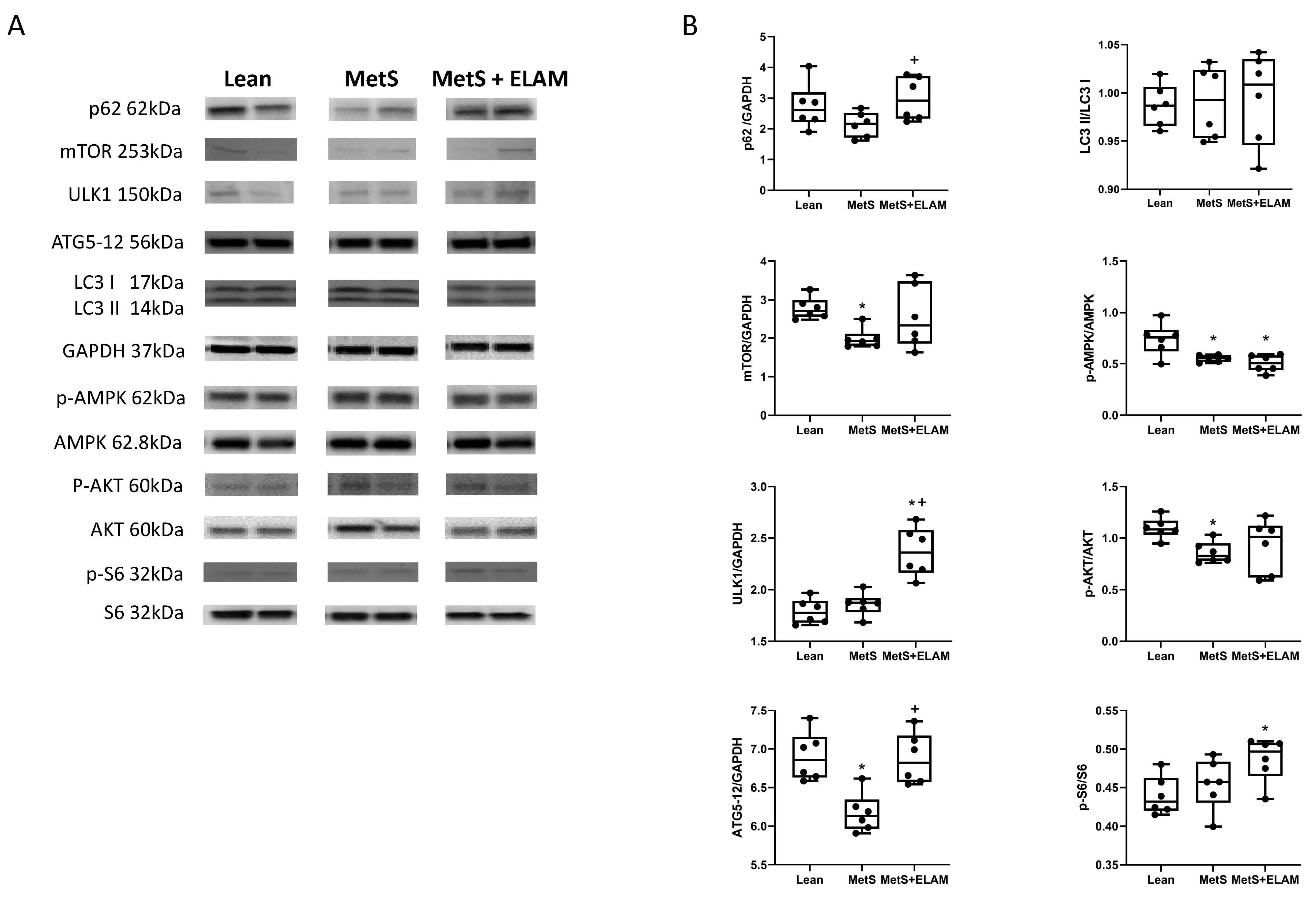
3.1. MetS Affects Autophagy
3.2. Elamipretide Ameliorates MetS-Induced Cellular Mitochondrial Damage and Apoptosis
3.3. Elamipretide Attenuates Renal Fibrosis and Injury
3.4. Elamipretide Attenuates MetS-Induced Renal Inflammation
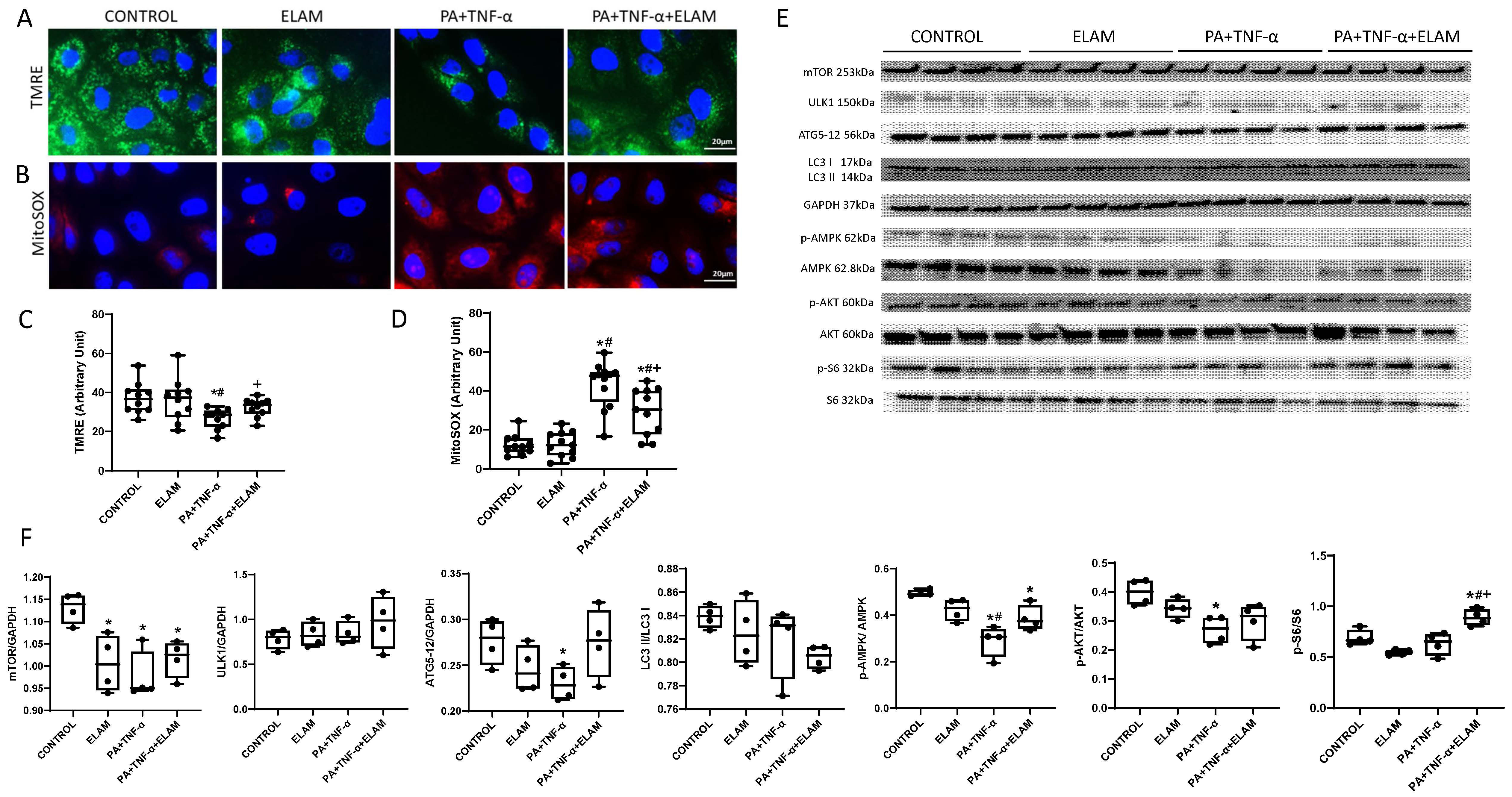
3.5. ELAM Attenuates Mitochondrial Dysfunction in Injured PK1 Cells
3.6. Autophagy in PA+TNF-α-Injured PK1 Cells
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bhurosy, T.; Jeewon, R. Overweight and obesity epidemic in developing countries: A problem with diet, physical activity, or socioeconomic status? Sci. World J. 2014, 2014, 964236. [Google Scholar] [CrossRef]
- Romieu, I.; Dossus, L.; Barquera, S.; Blottière, H.M.; Franks, P.W.; Gunter, M.; Hwalla, N.; Hursting, S.D.; Leitzmann, M.; Margetts, B.; et al. Energy balance and obesity: What are the main drivers? Cancer Causes Control 2017, 28, 247–258. [Google Scholar] [CrossRef] [PubMed]
- Ezquerro, S.; Mocha, F.; Frühbeck, G.; Guzmán-Ruiz, R.; Valentí, V.; Mugueta, C.; Becerril, S.; Catalán, V.; Gómez-Ambrosi, J.; Silva, C.; et al. Ghrelin Reduces TNF-α-Induced Human Hepatocyte Apoptosis, Autophagy, and Pyroptosis: Role in Obesity-Associated NAFLD. J. Clin. Endocrinol. Metab. 2019, 104, 21–37. [Google Scholar] [CrossRef]
- Liu, H.; Javaheri, A.; Godar, R.J.; Murphy, J.; Ma, X.; Rohatgi, N.; Mahadevan, J.; Hyrc, K.; Saftig, P.; Marshall, C.; et al. Intermittent fasting preserves beta-cell mass in obesity-induced diabetes via the autophagy-lysosome pathway. Autophagy 2017, 13, 1952–1968. [Google Scholar] [CrossRef] [PubMed]
- Ghandriz, R.; Lerman, L.O. Renal Cellular Autophagy in Obesity: Boon or Bane? Semin. Nephrol. 2021, 41, 349–357. [Google Scholar] [CrossRef]
- Mizushima, N.; Levine, B. Autophagy in Human Diseases. N. Engl. J. Med. 2020, 383, 1564–1576. [Google Scholar] [CrossRef]
- Klionsky, D.J. Autophagy: From phenomenology to molecular understanding in less than a decade. Nat. Rev. Mol. Cell Biol. 2007, 8, 931–937. [Google Scholar] [CrossRef]
- Uttenweiler, A.; Schwarz, H.; Mayer, A. Microautophagic vacuole invagination requires calmodulin in a Ca2+-independent function. J. Biol. Chem. 2005, 280, 33289–33297. [Google Scholar] [CrossRef]
- Mortimore, G.E.; Hutson, N.J.; Surmacz, C.A. Quantitative correlation between proteolysis and macro- and microautophagy in mouse hepatocytes during starvation and refeeding. Proc. Natl. Acad. Sci. USA 1983, 80, 2179–2183. [Google Scholar] [CrossRef]
- Senft, D.; Ronai, Z.A. UPR, autophagy, and mitochondria crosstalk underlies the ER stress response. Trends Biochem. Sci. 2015, 40, 141–148. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Kitada, M.; Ogura, Y.; Koya, D. Relationship between Autophagy and Metabolic Syndrome Characteristics in the Pathogenesis of Atherosclerosis. Front. Cell Dev. Biol. 2021, 9, 641852. [Google Scholar] [CrossRef] [PubMed]
- Fetterman, J.L.; Holbrook, M.; Flint, N.; Feng, B.; Bretón-Romero, R.; Linder, E.A.; Berk, B.D.; Duess, M.A.; Farb, M.G.; Gokce, N.; et al. Restoration of autophagy in endothelial cells from patients with diabetes mellitus improves nitric oxide signaling. Atherosclerosis 2016, 247, 207–217. [Google Scholar] [CrossRef] [PubMed]
- LaRocca, T.J.; Gioscia-Ryan, R.A.; Hearon, C.M., Jr.; Seals, D.R. The autophagy enhancer spermidine reverses arterial aging. Mech. Ageing Dev. 2013, 134, 314–320. [Google Scholar] [CrossRef] [PubMed]
- Mahajan, N.; Hoover, B.; Rajendram, M.; Shi, H.Y.; Kawasaki, K.; Weibel, D.B.; Zhang, M. Maspin binds to cardiolipin in mitochondria and triggers apoptosis. FASEB J. 2019, 33, 6354–6364. [Google Scholar] [CrossRef]
- Giorgio, M.; Migliaccio, E.; Orsini, F.; Paolucci, D.; Moroni, M.; Contursi, C.; Pelliccia, G.; Luzi, L.; Minucci, S.; Marcaccio, M.; et al. Electron transfer between cytochrome c and p66Shc generates reactive oxygen species that trigger mitochondrial apoptosis. Cell 2005, 122, 221–233. [Google Scholar] [CrossRef]
- Soubannier, V.; McLelland, G.L.; Zunino, R.; Braschi, E.; Rippstein, P.; Fon, E.A.; McBride, H.M. A vesicular transport pathway shuttles cargo from mitochondria to lysosomes. Curr. Biol. 2012, 22, 135–141. [Google Scholar] [CrossRef]
- Filomeni, G.; De Zio, D.; Cecconi, F. Oxidative stress and autophagy: The clash between damage and metabolic needs. Cell Death Differ. 2015, 22, 377–388. [Google Scholar] [CrossRef]
- Jounai, N.; Kobiyama, K.; Shiina, M.; Ogata, K.; Ishii, K.J.; Takeshita, F. NLRP4 negatively regulates autophagic processes through an association with beclin1. J. Immunol. 2011, 186, 1646–1655. [Google Scholar] [CrossRef]
- Abate, M.; Festa, A.; Falco, M.; Lombardi, A.; Luce, A.; Grimaldi, A.; Zappavigna, S.; Sperlongano, P.; Irace, C.; Caraglia, M.; et al. Mitochondria as playmakers of apoptosis, autophagy and senescence. Semin. Cell Dev. Biol. 2020, 98, 139–153. [Google Scholar] [CrossRef]
- Luo, Y.; Wu, M.Y.; Deng, B.Q.; Huang, J.; Hwang, S.H.; Li, M.Y.; Zhou, C.Y.; Zhang, Q.Y.; Yu, H.B.; Zhao, D.K.; et al. Inhibition of soluble epoxide hydrolase attenuates a high-fat diet-mediated renal injury by activating PAX2 and AMPK. Proc. Natl. Acad. Sci. USA 2019, 116, 5154–5159. [Google Scholar] [CrossRef] [Green Version]
- Mattson, M.P.; Longo, V.D.; Harvie, M. Impact of intermittent fasting on health and disease processes. Ageing Res. Rev. 2017, 39, 46–58. [Google Scholar] [CrossRef] [PubMed]
- Pfaller, W.; Seppi, T.; Ohno, A.; Giebisch, G.; Beck, F.X. Quantitative morphology of renal cortical structures during compensatory hypertrophy. Exp. Nephrol. 1998, 6, 308–319. [Google Scholar] [CrossRef] [PubMed]
- Pallet, N. Autophagy in the kidney. Med. Sci. 2017, 33, 275–282. [Google Scholar]
- Youle, R.J.; van der Bliek, A.M. Mitochondrial fission, fusion, and stress. Science 2012, 337, 1062–1065. [Google Scholar] [CrossRef] [PubMed]
- Nargesi, A.A.; Zhang, L.; Tang, H.; Jordan, K.L.; Saadiq, I.M.; Textor, S.C.; Lerman, L.O.; Eirin, A. Coexisting renal artery stenosis and metabolic syndrome magnifies mitochondrial damage, aggravating poststenotic kidney injury in pigs. J. Hypertens. 2019, 37, 2061–2073. [Google Scholar] [CrossRef]
- Xue, H.; Li, P.; Luo, Y.; Wu, C.; Liu, Y.; Qin, X.; Huang, X.; Sun, C. Salidroside stimulates the Sirt1/PGC-1α axis and ameliorates diabetic nephropathy in mice. Phytomedicine 2019, 54, 240–247. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.M.; Sung, J.; Lee, K. Longitudinal relationships of metabolic syndrome and obesity with kidney function: Healthy Twin Study. Clin. Exp. Nephrol. 2015, 19, 887–894. [Google Scholar] [CrossRef]
- Tsuboi, N.; Okabayashi, Y.; Shimizu, A.; Yokoo, T. The Renal Pathology of Obesity. Kidney Int. Rep. 2017, 2, 251–260. [Google Scholar] [CrossRef]
- Coughlan, M.T.; Nguyen, T.V.; Penfold, S.A.; Higgins, G.C.; Thallas-Bonke, V.; Tan, S.M.; Van Bergen, N.J.; Sourris, K.C.; Harcourt, B.E.; Thorburn, D.R.; et al. Mapping time-course mitochondrial adaptations in the kidney in experimental diabetes. Clin. Sci. 2016, 130, 711–720. [Google Scholar] [CrossRef]
- Higgins, G.C.; Coughlan, M.T. Mitochondrial dysfunction and mitophagy: The beginning and end to diabetic nephropathy? Br. J. Pharmacol. 2014, 171, 1917–1942. [Google Scholar] [CrossRef]
- De Cavanagh, E.M.; Inserra, F.; Ferder, M.; Ferder, L. From mitochondria to disease: Role of the renin-angiotensin system. Am. J. Nephrol. 2007, 27, 545–553. [Google Scholar] [CrossRef] [PubMed]
- Eirin, A.; Hedayat, A.F.; Ferguson, C.M.; Textor, S.C.; Lerman, A.; Lerman, L.O. Mitoprotection preserves the renal vasculature in porcine metabolic syndrome. Exp. Physiol. 2018, 103, 1020–1029. [Google Scholar] [CrossRef] [PubMed]
- Eirin, A.; Woollard, J.R.; Ferguson, C.M.; Jordan, K.L.; Tang, H.; Textor, S.C.; Lerman, A.; Lerman, L.O. The metabolic syndrome induces early changes in the swine renal medullary mitochondria. Transl. Res. 2017, 184, 45–56.e9. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.H.; Zhu, X.Y.; Eirin, A.; Nargesi, A.A.; Woollard, J.R.; Santelli, A.; Sun, I.O.; Textor, S.C.; Lerman, L.O. Early podocyte injury and elevated levels of urinary podocyte-derived extracellular vesicles in swine with metabolic syndrome: Role of podocyte mitochondria. Am. J. Physiol. Renal. Physiol. 2019, 317, F12–F22. [Google Scholar] [CrossRef] [PubMed]
- Iverson, S.L.; Orrenius, S. The cardiolipin-cytochrome c interaction and the mitochondrial regulation of apoptosis. Arch. Biochem. Biophys. 2004, 423, 37–46. [Google Scholar] [CrossRef]
- Szeto, H.H. Stealth Peptides Target Cellular Powerhouses to Fight Rare and Common Age-Related Diseases. Protein Pept. Lett. 2018, 25, 1108–1123. [Google Scholar] [CrossRef]
- Pawar, A.S.; Zhu, X.Y.; Eirin, A.; Tang, H.; Jordan, K.L.; Woollard, J.R.; Lerman, A.; Lerman, L.O. Adipose tissue remodeling in a novel domestic porcine model of diet-induced obesity. Obesity 2015, 23, 399–407. [Google Scholar] [CrossRef]
- Eirin, A.; Ebrahimi, B.; Kwon, S.H.; Fiala, J.A.; Williams, B.J.; Woollard, J.R.; He, Q.; Gupta, R.C.; Sabbah, H.N.; Prakash, Y.S.; et al. Restoration of Mitochondrial Cardiolipin Attenuates Cardiac Damage in Swine Renovascular Hypertension. J. Am. Heart Assoc. 2016, 5, e003118. [Google Scholar] [CrossRef]
- Eirin, A.; Ebrahimi, B.; Zhang, X.; Zhu, X.Y.; Woollard, J.R.; He, Q.; Textor, S.C.; Lerman, A.; Lerman, L.O. Mitochondrial protection restores renal function in swine atherosclerotic renovascular disease. Cardiovasc. Res. 2014, 103, 461–472. [Google Scholar] [CrossRef]
- Li, Z.L.; Woollard, J.R.; Ebrahimi, B.; Crane, J.A.; Jordan, K.L.; Lerman, A.; Wang, S.M.; Lerman, L.O. Transition from obesity to metabolic syndrome is associated with altered myocardial autophagy and apoptosis. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 1132–1141. [Google Scholar] [CrossRef]
- Eirin, A.; Zhu, X.Y.; Krier, J.D.; Tang, H.; Jordan, K.L.; Grande, J.P.; Lerman, A.; Textor, S.C.; Lerman, L.O. Adipose tissue-derived mesenchymal stem cells improve revascularization outcomes to restore renal function in swine atherosclerotic renal artery stenosis. Stem Cells 2012, 30, 1030–1041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gómez-Sánchez, R.; Pizarro-Estrella, E.; Yakhine-Diop, S.M.; Rodríguez-Arribas, M.; Bravo-San Pedro, J.M.; Fuentes, J.M.; González-Polo, R.A. Routine Western blot to check autophagic flux: Cautions and recommendations. Anal. Biochem. 2015, 477, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Manders, E.M.M.; Verbeek, F.J.; Aten, J.A. Measurement of co-localization of objects in dual-colour confocal images. J. Microsc. 1993, 169, 375–382. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Li, Z.L.; Crane, J.A.; Jordan, K.L.; Pawar, A.S.; Textor, S.C.; Lerman, A.; Lerman, L.O. Valsartan regulates myocardial autophagy and mitochondrial turnover in experimental hypertension. Hypertension 2014, 64, 87–93. [Google Scholar] [CrossRef]
- Eirin, A.; Zhu, X.Y.; Urbieta-Caceres, V.H.; Grande, J.P.; Lerman, A.; Textor, S.C.; Lerman, L.O. Persistent kidney dysfunction in swine renal artery stenosis correlates with outer cortical microvascular remodeling. Am. J. Physiol. Renal. Physiol. 2011, 300, F1394–F1401. [Google Scholar] [CrossRef]
- Urbieta-Caceres, V.H.; Zhu, X.Y.; Jordan, K.L.; Tang, H.; Textor, K.; Lerman, A.; Lerman, L.O. Selective improvement in renal function preserved remote myocardial microvascular integrity and architecture in experimental renovascular disease. Atherosclerosis 2012, 221, 350–358. [Google Scholar] [CrossRef] [PubMed]
- Eirin, A.; Gloviczki, M.L.; Tang, H.; Gössl, M.; Jordan, K.L.; Woollard, J.R.; Lerman, A.; Grande, J.P.; Textor, S.C.; Lerman, L.O. Inflammatory and injury signals released from the post-stenotic human kidney. Eur. Heart J. 2013, 34, 540–548a. [Google Scholar] [CrossRef]
- Zhu, X.Y.; Urbieta-Caceres, V.; Krier, J.D.; Textor, S.C.; Lerman, A.; Lerman, L.O. Mesenchymal stem cells and endothelial progenitor cells decrease renal injury in experimental swine renal artery stenosis through different mechanisms. Stem Cells 2013, 31, 117–125. [Google Scholar] [CrossRef]
- Liang, X.; Chen, Y.; Zhang, L.; Jiang, F.; Wang, W.; Ye, Z.; Liu, S.; Yu, C.; Shi, W. Necroptosis, a novel form of caspase-independent cell death, contributes to renal epithelial cell damage in an ATP-depleted renal ischemia model. Mol. Med. Rep. 2014, 10, 719–724. [Google Scholar] [CrossRef]
- Zhang, L.; Jiang, F.; Chen, Y.; Luo, J.; Liu, S.; Zhang, B.; Ye, Z.; Wang, W.; Liang, X.; Shi, W. Necrostatin-1 attenuates ischemia injury induced cell death in rat tubular cell line NRK-52E through decreased Drp1 expression. Int. J. Mol. Sci. 2013, 14, 24742–24754. [Google Scholar] [CrossRef]
- Yamamoto, T.; Takabatake, Y.; Minami, S.; Sakai, S.; Fujimura, R.; Takahashi, A.; Namba-Hamano, T.; Matsuda, J.; Kimura, T.; Matsui, I.; et al. Eicosapentaenoic acid attenuates renal lipotoxicity by restoring autophagic flux. Autophagy 2021, 17, 1700–1713. [Google Scholar] [CrossRef] [PubMed]
- Mori, Y.; Ajay, A.K.; Chang, J.H.; Mou, S.; Zhao, H.; Kishi, S.; Li, J.; Brooks, C.R.; Xiao, S.; Woo, H.M.; et al. KIM-1 mediates fatty acid uptake by renal tubular cells to promote progressive diabetic kidney disease. Cell Metab. 2021, 33, 1042–1061.e7. [Google Scholar] [CrossRef] [PubMed]
- Reddy, P.H. Mitochondrial medicine for aging and neurodegenerative diseases. Neuromol. Med. 2008, 10, 291–315. [Google Scholar] [CrossRef] [PubMed]
- Farrelly, E.; Amaral, M.C.; Marshall, L.; Huang, S.G. A high-throughput assay for mitochondrial membrane potential in permeabilized yeast cells. Anal. Biochem. 2001, 293, 269–276. [Google Scholar] [CrossRef]
- Foster, M.C.; Hwang, S.J.; Porter, S.A.; Massaro, J.M.; Hoffmann, U.; Fox, C.S. Fatty kidney, hypertension, and chronic kidney disease: The Framingham Heart Study. Hypertension 2011, 58, 784–790. [Google Scholar] [CrossRef]
- De Vries, A.P.; Ruggenenti, P.; Ruan, X.Z.; Praga, M.; Cruzado, J.M.; Bajema, I.M.; D’Agati, V.D.; Lamb, H.J.; Pongrac Barlovic, D.; Hojs, R.; et al. Fatty kidney: Emerging role of ectopic lipid in obesity-related renal disease. Lancet Diabetes Endocrinol. 2014, 2, 417–426. [Google Scholar] [CrossRef]
- Chagnac, A.; Zingerman, B.; Rozen-Zvi, B.; Herman-Edelstein, M. Consequences of Glomerular Hyperfiltration: The Role of Physical Forces in the Pathogenesis of Chronic Kidney Disease in Diabetes and Obesity. Nephron 2019, 143, 38–42. [Google Scholar] [CrossRef]
- Tai, H.; Wang, Z.; Gong, H.; Han, X.; Zhou, J.; Wang, X.; Wei, X.; Ding, Y.; Huang, N.; Qin, J.; et al. Autophagy impairment with lysosomal and mitochondrial dysfunction is an important characteristic of oxidative stress-induced senescence. Autophagy 2017, 13, 99–113. [Google Scholar] [CrossRef]
- Liu, S.; Hartleben, B.; Kretz, O.; Wiech, T.; Igarashi, P.; Mizushima, N.; Walz, G.; Huber, T.B. Autophagy plays a critical role in kidney tubule maintenance, aging and ischemia-reperfusion injury. Autophagy 2012, 8, 826–837. [Google Scholar] [CrossRef]
- Jia, J.; Abudu, Y.P.; Claude-Taupin, A.; Gu, Y.; Kumar, S.; Choi, S.W.; Peters, R.; Mudd, M.H.; Allers, L.; Salemi, M.; et al. Galectins control MTOR and AMPK in response to lysosomal damage to induce autophagy. Autophagy 2019, 15, 169–171. [Google Scholar] [CrossRef]
- Dikic, I.; Elazar, Z. Mechanism and medical implications of mammalian autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 349–364. [Google Scholar] [CrossRef] [PubMed]
- Minami, S.; Yamamoto, T.; Takabatake, Y.; Takahashi, A.; Namba, T.; Matsuda, J.; Kimura, T.; Kaimori, J.Y.; Matsui, I.; Hamano, T.; et al. Lipophagy maintains energy homeostasis in the kidney proximal tubule during prolonged starvation. Autophagy 2017, 13, 1629–1647. [Google Scholar] [CrossRef]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Egan, D.F.; Shackelford, D.B.; Mihaylova, M.M.; Gelino, S.; Kohnz, R.A.; Mair, W.; Vasquez, D.S.; Joshi, A.; Gwinn, D.M.; Taylor, R.; et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science 2011, 331, 456–461. [Google Scholar] [CrossRef]
- Pattingre, S.; Tassa, A.; Qu, X.; Garuti, R.; Liang, X.H.; Mizushima, N.; Packer, M.; Schneider, M.D.; Levine, B. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 2005, 122, 927–939. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.C.; Guan, K.L. mTOR: A pharmacologic target for autophagy regulation. J. Clin. Investig. 2015, 125, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Sahani, M.H.; Itakura, E.; Mizushima, N. Expression of the autophagy substrate SQSTM1/p62 is restored during prolonged starvation depending on transcriptional upregulation and autophagy-derived amino acids. Autophagy 2014, 10, 431–441. [Google Scholar] [CrossRef]
- Runwal, G.; Stamatakou, E.; Siddiqi, F.H.; Puri, C.; Zhu, Y.; Rubinsztein, D.C. LC3-positive structures are prominent in autophagy-deficient cells. Sci. Rep. 2019, 9, 10147. [Google Scholar] [CrossRef]
- Lee, S.H.; Cho, W.J.; Najy, A.J.; Saliganan, A.D.; Pham, T.; Rakowski, J.; Loughery, B.; Ji, C.H.; Sakr, W.; Kim, S.; et al. p62/SQSTM1-induced caspase-8 aggresomes are essential for ionizing radiation-mediated apoptosis. Cell Death Dis. 2021, 12, 997. [Google Scholar] [CrossRef]
- Matsuda, N.; Sato, S.; Shiba, K.; Okatsu, K.; Saisho, K.; Gautier, C.A.; Sou, Y.S.; Saiki, S.; Kawajiri, S.; Sato, F.; et al. PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J. Cell Biol. 2010, 189, 211–221. [Google Scholar] [CrossRef]
- Twig, G.; Elorza, A.; Molina, A.J.; Mohamed, H.; Wikstrom, J.D.; Walzer, G.; Stiles, L.; Haigh, S.E.; Katz, S.; Las, G.; et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008, 27, 433–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, H.; Wang, Y.; Li, W.; Chen, H.; Du, L.; Liu, D.; Wang, X.; Xu, T.; Liu, L.; Chen, Q. Deficiency of mitophagy receptor FUNDC1 impairs mitochondrial quality and aggravates dietary-induced obesity and metabolic syndrome. Autophagy 2019, 15, 1882–1898. [Google Scholar] [CrossRef] [PubMed]
- Rambold, A.S.; Lippincott-Schwartz, J. Mechanisms of mitochondria and autophagy crosstalk. Cell Cycle 2011, 10, 4032–4038. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.S.; Nitta, T.; Mohuczy, D.; O’Malley, K.A.; Moldawer, L.L.; Dunn, W.A., Jr.; Behrns, K.E. Impaired autophagy: A mechanism of mitochondrial dysfunction in anoxic rat hepatocytes. Hepatology 2008, 47, 1725–1736. [Google Scholar] [CrossRef]
- Florens, N.; Calzada, C.; Lyasko, E.; Juillard, L.; Soulage, C.O. Modified Lipids and Lipoproteins in Chronic Kidney Disease: A New Class of Uremic Toxins. Toxins 2016, 8, 376. [Google Scholar] [CrossRef] [PubMed]
- Haunerland, N.H.; Spener, F. Fatty acid-binding proteins—Insights from genetic manipulations. Prog. Lipid Res. 2004, 43, 328–349. [Google Scholar] [CrossRef]
- Bremer, J. Carnitine—Metabolism and functions. Physiol. Rev. 1983, 63, 1420–1480. [Google Scholar] [CrossRef]
- Brivet, M.; Boutron, A.; Slama, A.; Costa, C.; Thuillier, L.; Demaugre, F.; Rabier, D.; Saudubray, J.M.; Bonnefont, J.P. Defects in activation and transport of fatty acids. J. Inherit. Metab. Dis. 1999, 22, 428–441. [Google Scholar] [CrossRef]
- Nsiah-Sefaa, A.; McKenzie, M. Combined defects in oxidative phosphorylation and fatty acid β-oxidation in mitochondrial disease. Biosci. Rep. 2016, 36, e00313. [Google Scholar] [CrossRef]
- Pietrocola, F.; Galluzzi, L.; Bravo-San Pedro, J.M.; Madeo, F.; Kroemer, G. Acetyl coenzyme A: A central metabolite and second messenger. Cell Metab. 2015, 21, 805–821. [Google Scholar] [CrossRef]
- Shi, L.; Tu, B.P. Acetyl-CoA and the regulation of metabolism: Mechanisms and consequences. Curr. Opin. Cell. Biol. 2015, 33, 125–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, H.L.; Sui, Y.; Guan, J.; He, L.; Zhu, X.; Fan, R.R.; Xu, G.; Kong, A.P.; Ho, C.S.; Lai, F.M.; et al. Fat redistribution and adipocyte transformation in uninephrectomized rats. Kidney Int. 2008, 74, 467–477. [Google Scholar] [CrossRef] [PubMed]
- Szeto, H.H.; Liu, S.; Soong, Y.; Alam, N.; Prusky, G.T.; Seshan, S.V. Protection of mitochondria prevents high-fat diet-induced glomerulopathy and proximal tubular injury. Kidney Int. 2016, 90, 997–1011. [Google Scholar] [CrossRef]
- Herman-Edelstein, M.; Scherzer, P.; Tobar, A.; Levi, M.; Gafter, U. Altered renal lipid metabolism and renal lipid accumulation in human diabetic nephropathy. J. Lipid Res. 2014, 55, 561–572. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, S.; Zaritsky, J.J.; Fornoni, A.; Smoyer, W.E. Dyslipidaemia in nephrotic syndrome: Mechanisms and treatment. Nat. Rev. Nephrol. 2018, 14, 57–70, Erratum in Nat. Rev. Nephrol. 2018, 14, 70. [Google Scholar] [CrossRef]
- Du, X.G.; Ruan, X.Z. Lipid Metabolism Disorder and Renal Fibrosis. Adv. Exp. Med. Biol. 2019, 1165, 525–541. [Google Scholar]
- Saad, A.; Herrmann, S.M.S.; Eirin, A.; Ferguson, C.M.; Glockner, J.F.; Bjarnason, H.; McKusick, M.A.; Misra, S.; Lerman, L.O.; Textor, S.C. Phase 2a Clinical Trial of Mitochondrial Protection (Elamipretide) During Stent Revascularization in Patients with Atherosclerotic Renal Artery Stenosis. Circ. Cardiovasc. Interv. 2017, 10, e005487. [Google Scholar] [CrossRef]
- Allen, M.E.; Pennington, E.R.; Perry, J.B.; Dadoo, S.; Makrecka-Kuka, M.; Dambrova, M.; Moukdar, F.; Patel, H.D.; Han, X.; Kidd, G.K.; et al. The cardiolipin-binding peptide elamipretide mitigates fragmentation of cristae networks following cardiac ischemia reperfusion in rats. Commun. Biol. 2020, 3, 389. [Google Scholar] [CrossRef]
- Levine, B.; Kroemer, G. Autophagy in the pathogenesis of disease. Cell 2008, 132, 27–42. [Google Scholar] [CrossRef]
- Ducasa, G.M.; Mitrofanova, A.; Mallela, S.K.; Liu, X.; Molina, J.; Sloan, A.; Pedigo, C.E.; Ge, M.; Santos, J.V.; Hernandez, Y.; et al. ATP-binding cassette A1 deficiency causes cardiolipin-driven mitochondrial dysfunction in podocytes. J. Clin. Investig. 2019, 129, 3387–3400. [Google Scholar] [CrossRef]
- Chu, C.T.; Ji, J.; Dagda, R.K.; Jiang, J.F.; Tyurina, Y.Y.; Kapralov, A.A.; Tyurin, V.A.; Yanamala, N.; Shrivastava, I.H.; Mohammadyani, D.; et al. Cardiolipin externalization to the outer mitochondrial membrane acts as an elimination signal for mitophagy in neuronal cells. Nat. Cell Biol. 2013, 15, 1197–1205. [Google Scholar] [CrossRef] [PubMed]
- Petcherski, A.; Trudeau, K.M.; Wolf, D.M.; Segawa, M.; Lee, J.; Taddeo, E.P.; Deeney, J.T.; Liesa, M. Elamipretide Promotes Mitophagosome Formation and Prevents Its Reduction Induced by Nutrient Excess in INS1 β-cells. J. Mol. Biol. 2018, 430, 4823–4833. [Google Scholar] [CrossRef] [PubMed]
- Settembre, C.; Fraldi, A.; Jahreiss, L.; Spampanato, C.; Venturi, C.; Medina, D.; de Pablo, R.; Tacchetti, C.; Rubinsztein, D.C.; Ballabio, A. A block of autophagy in lysosomal storage disorders. Hum. Mol. Genet. 2008, 17, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Lei, Y.; Devarapu, S.K.; Motrapu, M.; Cohen, C.D.; Lindenmeyer, M.T.; Moll, S.; Kumar, S.V.; Anders, H.J. Interleukin-1β Inhibition for Chronic Kidney Disease in Obese Mice with Type 2 Diabetes. Front. Immunol. 2019, 10, 1223. [Google Scholar] [CrossRef] [PubMed]
- Bandach, I.; Segev, Y.; Landau, D. Experimental modulation of Interleukin 1 shows its key role in chronic kidney disease progression and anemia. Sci. Rep. 2021, 11, 6288. [Google Scholar] [CrossRef]
- Tagawa, A.; Yasuda, M.; Kume, S.; Yamahara, K.; Nakazawa, J.; Chin-Kanasaki, M.; Araki, H.; Araki, S.; Koya, D.; Asanuma, K.; et al. Impaired Podocyte Autophagy Exacerbates Proteinuria in Diabetic Nephropathy. Diabetes 2016, 65, 755–767. [Google Scholar] [CrossRef]
- Shi, C.S.; Shenderov, K.; Huang, N.N.; Kabat, J.; Abu-Asab, M.; Fitzgerald, K.A.; Sher, A.; Kehrl, J.H. Activation of autophagy by inflammatory signals limits IL-1β production by targeting ubiquitinated inflammasomes for destruction. Nat. Immunol. 2012, 13, 255–263. [Google Scholar] [CrossRef]
- Paul, S.; Kashyap, A.K.; Jia, W.; He, Y.W.; Schaefer, B.C. Selective autophagy of the adaptor protein Bcl10 modulates T cell receptor activation of NF-κB. Immunity 2012, 36, 947–958. [Google Scholar] [CrossRef]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011, 469, 221–225. [Google Scholar] [CrossRef]
- Nakahira, K.; Haspel, J.A.; Rathinam, V.A.; Lee, S.J.; Dolinay, T.; Lam, H.C.; Englert, J.A.; Rabinovitch, M.; Cernadas, M.; Kim, H.P.; et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 2011, 12, 222–230. [Google Scholar] [CrossRef]
- Bonet-Ponce, L.; Saez-Atienzar, S.; da Casa, C.; Flores-Bellver, M.; Barcia, J.M.; Sancho-Pelluz, J.; Romero, F.J.; Jordan, J.; Galindo, M.F. On the mechanism underlying ethanol-induced mitochondrial dynamic disruption and autophagy response. Biochim. Biophys. Acta 2015, 1852, 1400–1409. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Kang, R.; Livesey, K.M.; Cheh, C.W.; Farkas, A.; Loughran, P.; Hoppe, G.; Bianchi, M.E.; Tracey, K.J.; Zeh, H.J., 3rd; et al. Endogenous HMGB1 regulates autophagy. J. Cell Biol. 2010, 190, 881–892. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Lean | Mets | Mets + ELAM |
|---|---|---|---|
| Body weight (Kg) | 68 ± 6.0 | 92.3 ± 4.3 * | 93.1 ± 3.4 * |
| Glucose (mg/dL) | 111 ± 3.5 | 108.1 ± 26.5 | 109.3 ± 24.3 |
| Fasting Insulin (µU/mL) | 0.4 ± 0.008 | 0.9 ± 0.2 * | 0.7 ± 0.1 * |
| HOMA-IR score | 0.6 ± 0.1 | 1.3 ± 0.2 * | 1.2 ± 0.1 * |
| Total cholesterol (mg/dL) | 72.3 ± 6.9 | 564.2 ± 68.3 * | 603.0 ± 62.3 * |
| HDL cholesterol (mg/dL) | 42.9 ± 1.4 | 164.8 ± 28.1 * | 130.3 ± 9.8 * |
| LDL cholesterol (mg/dL) | 29.1 ± 2.7 | 389.1 ± 48.9 * | 390.3 ± 50.1 * |
| Triglycerides (mg/dL) | 7.4 ± 0.6 | 12.5 ± 2.1 * | 13.2 ± 1.8 * |
| Creatinine (mg/dL) | 1.67 ± 0.24 | 1.45 ± 0.20 | 1.62 ± 0.40 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hong, S.; Ghandriz, R.; Siddiqi, S.; Zhu, X.-Y.; Saadiq, I.M.; Jordan, K.L.; Tang, H.; Ali, K.A.; Lerman, A.; Eirin, A.; et al. Effects of Elamipretide on Autophagy in Renal Cells of Pigs with Metabolic Syndrome. Cells 2022, 11, 2891. https://doi.org/10.3390/cells11182891
Hong S, Ghandriz R, Siddiqi S, Zhu X-Y, Saadiq IM, Jordan KL, Tang H, Ali KA, Lerman A, Eirin A, et al. Effects of Elamipretide on Autophagy in Renal Cells of Pigs with Metabolic Syndrome. Cells. 2022; 11(18):2891. https://doi.org/10.3390/cells11182891
Chicago/Turabian StyleHong, Siting, Ramyar Ghandriz, Sarosh Siddiqi, Xiang-Yang Zhu, Ishran M. Saadiq, Kyra L. Jordan, Hui Tang, Khaled A. Ali, Amir Lerman, Alfonso Eirin, and et al. 2022. "Effects of Elamipretide on Autophagy in Renal Cells of Pigs with Metabolic Syndrome" Cells 11, no. 18: 2891. https://doi.org/10.3390/cells11182891