
Pancreatic Ductal Adenocarcinoma: Molecular Pathology and Predictive Biomarkers
Abstract
:1. Introduction
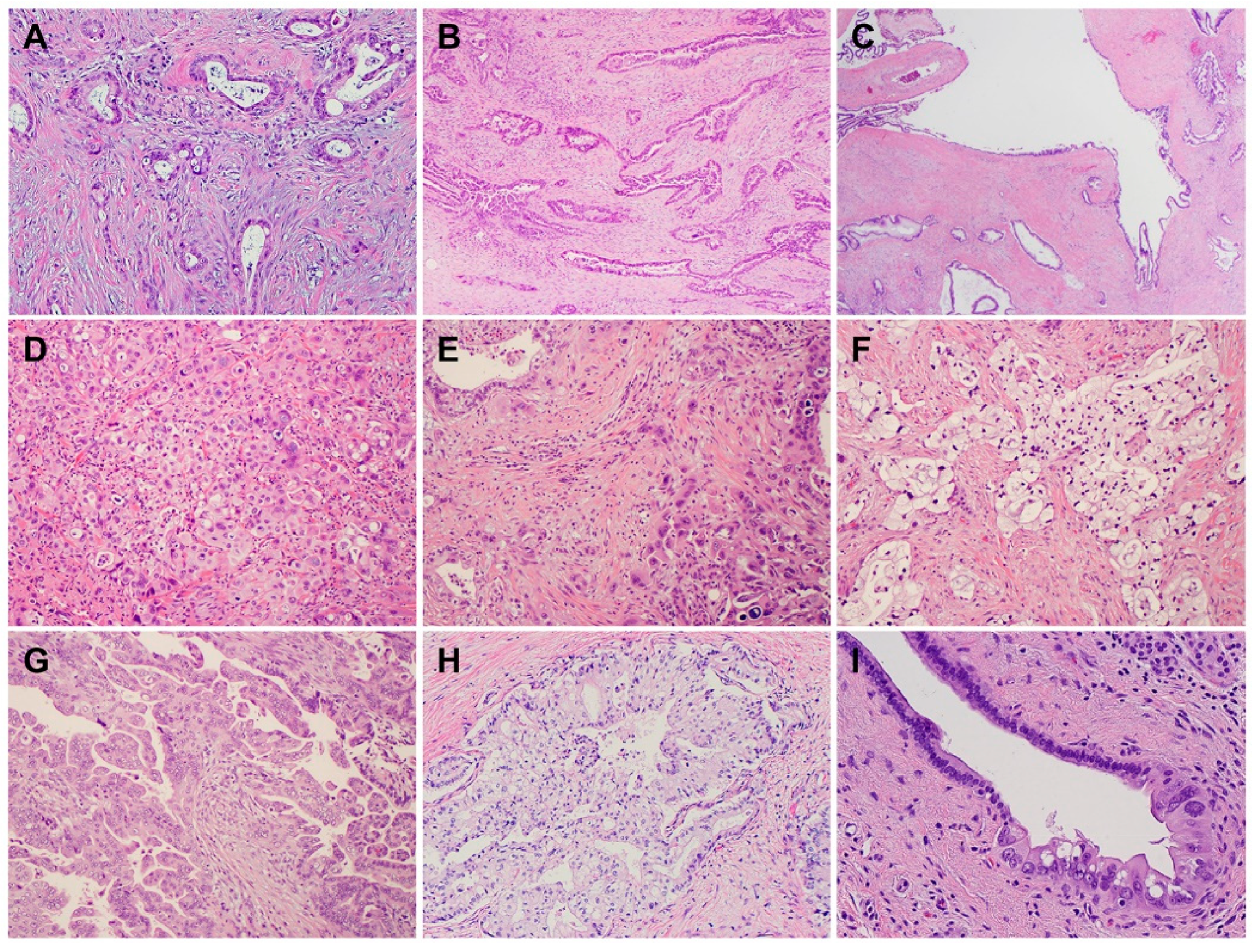
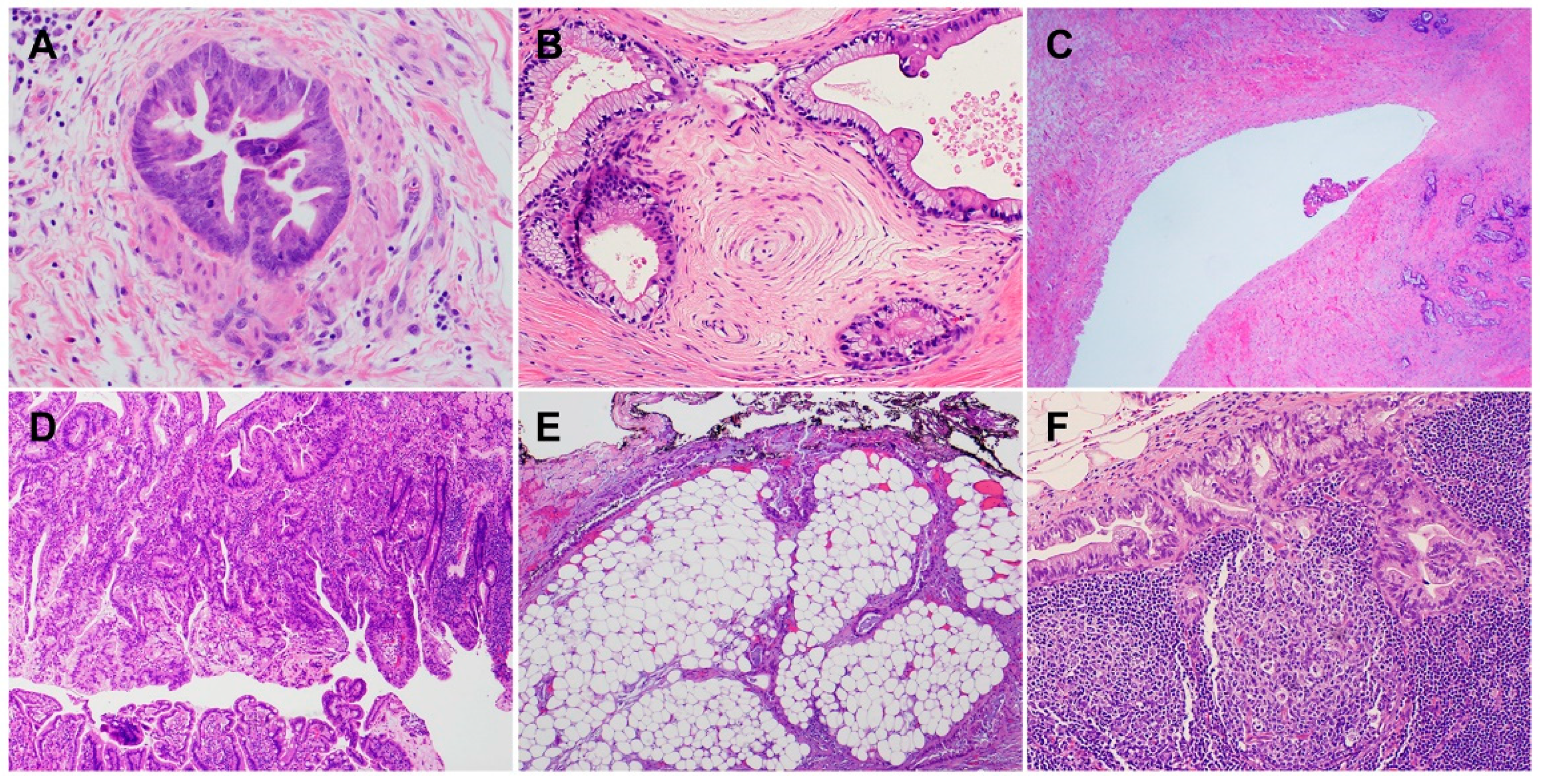
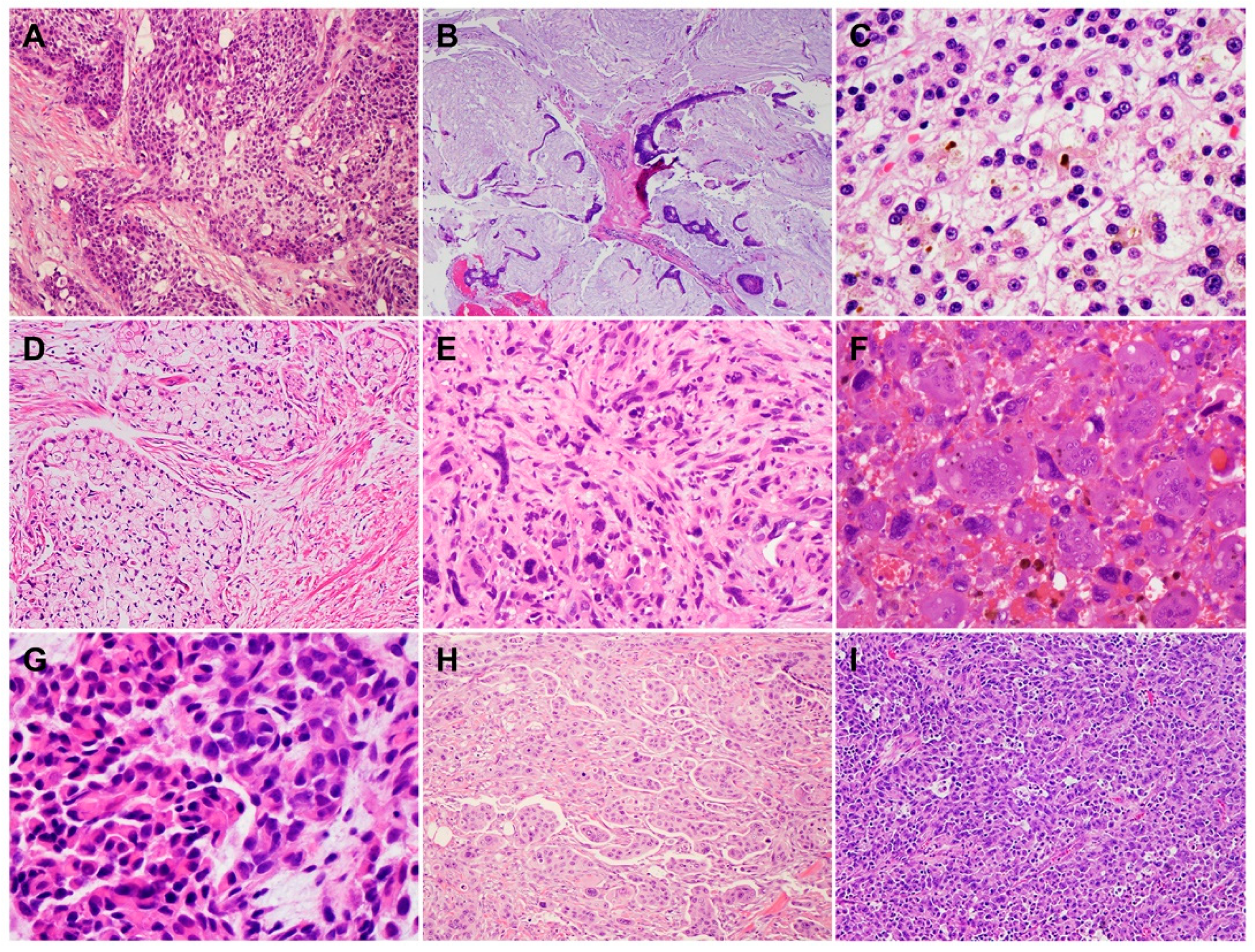
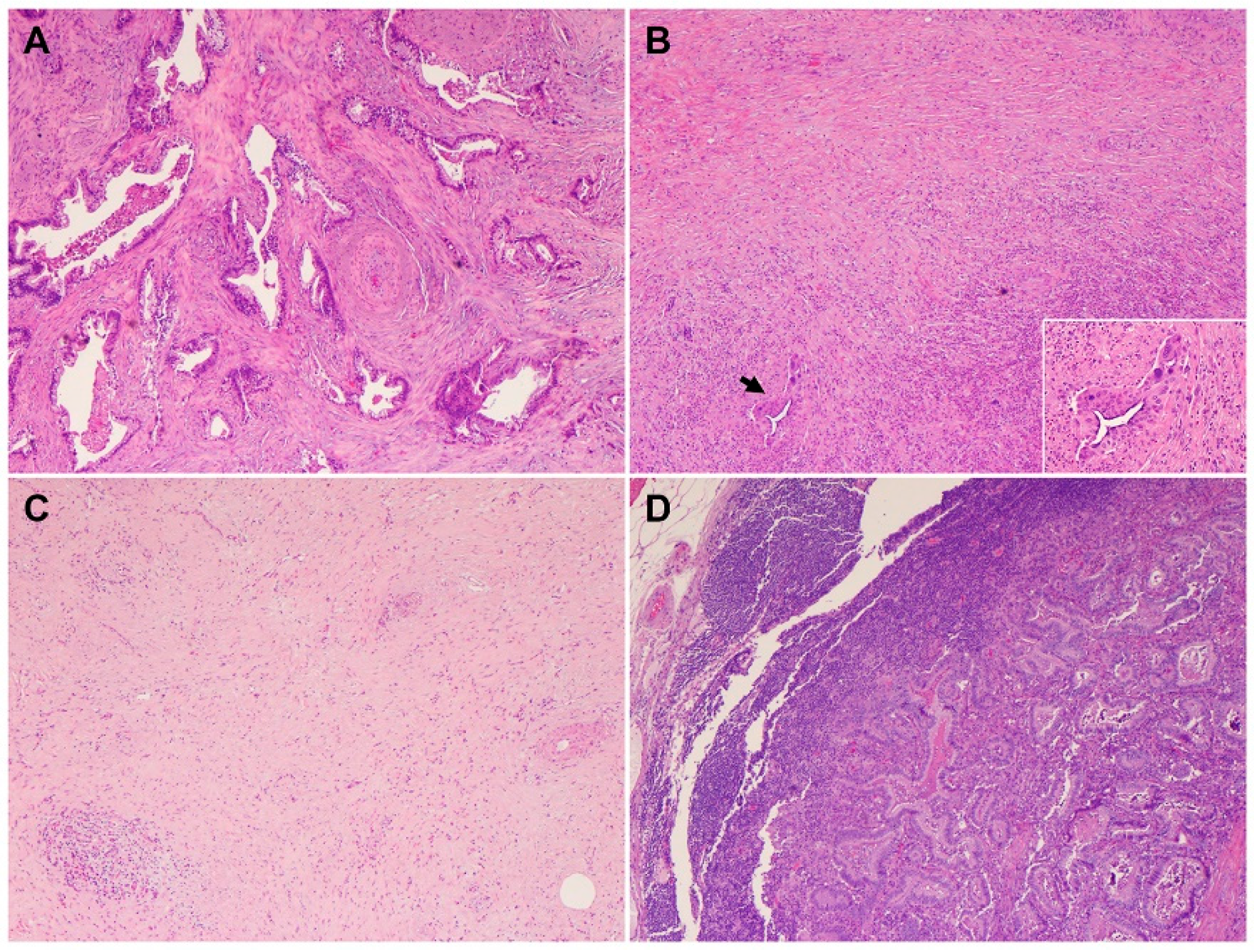
2. Histology and Morphologic Heterogeneity of PDAC
3. Genetic Alterations and Molecular Subtypes of PDAC
4. Heterogeneous Response of PDAC to Neoadjuvant Therapy
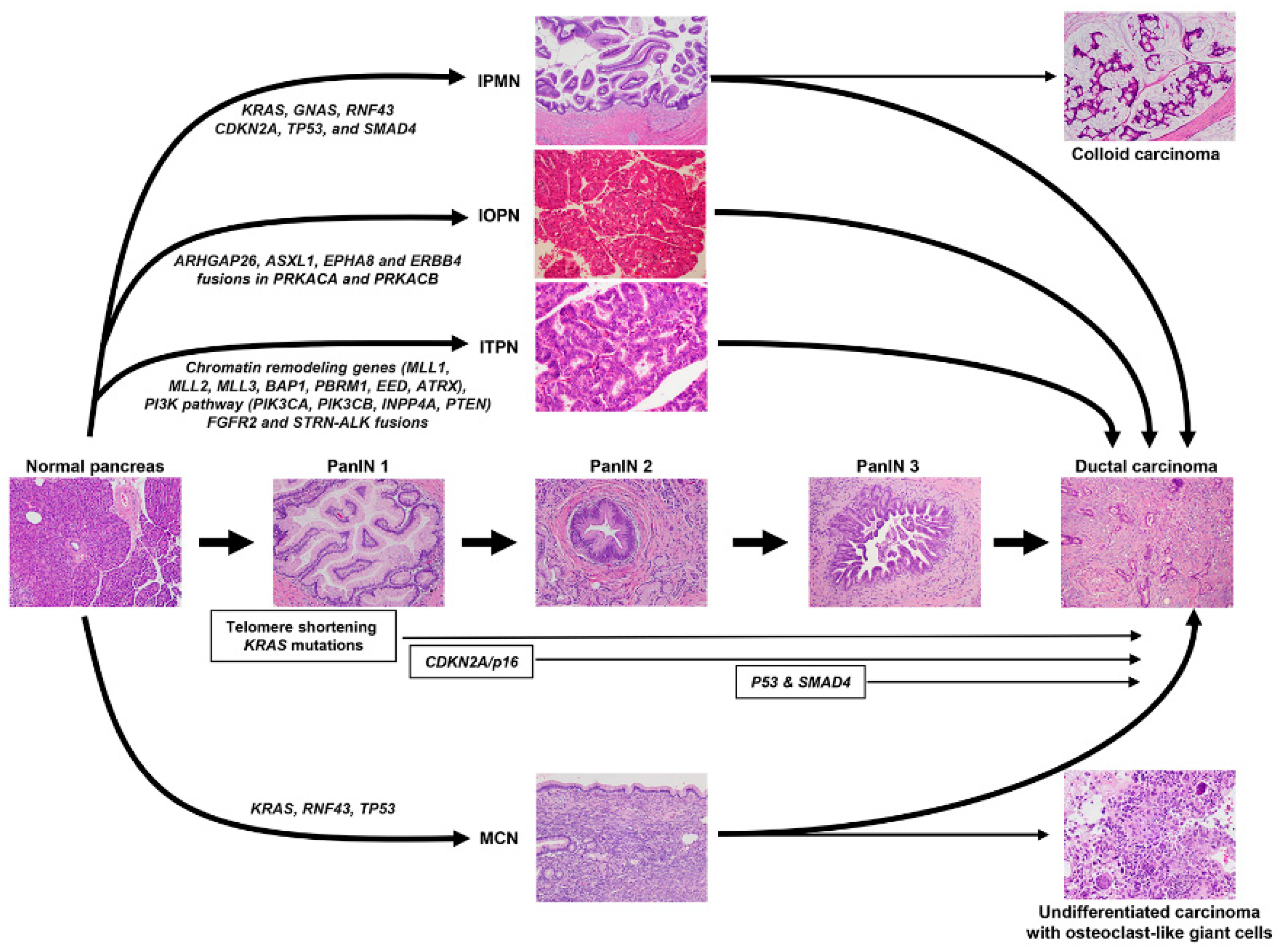
5. Precursor Lesions of PDAC
6. Emerging Predictive Markers and Targeted Therapies for PDAC Patients
6.1. Markers for the Defective DNA Damage Responses
6.2. KRASG12C Mutation
6.3. Neurotrophic Tropomyosin Receptor Kinase (NTRK) Fusions
6.4. Microsatellite Instability (MSI)/Defective Mismatch Repair (dMMR)
7. The Tumor Microenvironment of PDAC
8. Future Perspectives and Summary
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Rahib, L.; Wehner, M.R.; Matrisian, L.M.; Nead, K.T. Estimated Projection of US Cancer Incidence and Death to 2040. JAMA Netw. Open 2021, 4, e214708. [Google Scholar] [CrossRef] [PubMed]
- National Cancer Institute SRP, Cancer Statistics Branch. Surveillance Epidemiology and End Results (SEER) (1975–2018). 2021. Available online: http://seer.cancer.gov (accessed on 7 August 2022).
- Kamisawa, T.; Wood, L.D.; Itoi, T.; Takaori, K. Pancreatic cancer. Lancet 2016, 388, 73–85. [Google Scholar] [CrossRef]
- Maitra, A.; Hruban, R.H. Pancreatic cancer. Annu. Rev. Pathol. 2008, 3, 157–188. [Google Scholar] [CrossRef] [PubMed]
- Garcea, G.; Dennison, A.R.; Pattenden, C.J.; Neal, C.P.; Sutton, C.D.; Berry, D.P. Survival following curative resection for pancreatic ductal adenocarcinoma. A systematic review of the literature. JOP 2008, 9, 99–132. [Google Scholar] [PubMed]
- Hong, S.M.; Park, J.Y.; Hruban, R.H.; Goggins, M. Molecular signatures of pancreatic cancer. Arch. Pathol. Lab. Med. 2011, 135, 716–727. [Google Scholar] [CrossRef]
- Campbell, P.J.; Yachida, S.; Mudie, L.J.; Stephens, P.J.; Pleasance, E.D.; Stebbings, L.A.; Morsberger, L.A.; Latimer, C.; McLaren, S.; Lin, M.L.; et al. The patterns and dynamics of genomic instability in metastatic pancreatic cancer. Nature 2010, 467, 1109–1113. [Google Scholar] [CrossRef]
- Chatterjee, D.; Katz, M.H.; Rashid, A.; Wang, H.; Iuga, A.C.; Varadhachary, G.R.; Wolff, R.A.; Lee, J.E.; Pisters, P.W.; Crane, C.H.; et al. Perineural and intraneural invasion in posttherapy pancreaticoduodenectomy specimens predicts poor prognosis in patients with pancreatic ductal adenocarcinoma. Am. J. Surg. Pathol. 2012, 36, 409–417. [Google Scholar] [CrossRef]
- Chatterjee, D.; Rashid, A.; Wang, H.; Katz, M.H.; Wolff, R.A.; Varadhachary, G.R.; Lee, J.E.; Pisters, P.W.; Gomez, H.F.; Abbruzzese, J.L.; et al. Tumor invasion of muscular vessels predicts poor prognosis in patients with pancreatic ductal adenocarcinoma who have received neoadjuvant therapy and pancreaticoduodenectomy. Am. J. Surg. Pathol. 2012, 36, 552–559. [Google Scholar] [CrossRef]
- Fischer, L.K.; Katz, M.H.; Lee, S.M.; Liu, L.; Wang, H.; Varadhachary, G.R.; Wolff, R.A.; Lee, J.E.; Maitra, A.; Roland, C.L.; et al. The number and ratio of positive lymph nodes affect pancreatic cancer patient survival after neoadjuvant therapy and pancreaticoduodenectomy. Histopathology 2016, 68, 210–220. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Katz, M.H.; Lee, S.M.; Fischer, L.K.; Prakash, L.; Parker, N.; Wang, H.; Varadhachary, G.R.; Wolff, R.A.; Lee, J.E.; et al. Superior Mesenteric Artery Margin of Posttherapy Pancreaticoduodenectomy and Prognosis in Patients with Pancreatic Ductal Adenocarcinoma. Am. J. Surg. Pathol. 2015, 39, 1395–1403. [Google Scholar] [CrossRef] [PubMed]
- International Agency for Research on Cancer. WHO 2019 Classification of Tumors, Digestive System Tumours, 5th ed.; International Agency for Research on Cancer: Lyon, France, 2019; Volume 1, pp. 295–332. [Google Scholar]
- Adsay, N.V.; Merati, K.; Nassar, H.; Shia, J.; Sarkar, F.; Pierson, C.R.; Cheng, J.D.; Visscher, D.W.; Hruban, R.H.; Klimstra, D.S. Pathogenesis of colloid (pure mucinous) carcinoma of exocrine organs: Coupling of gel-forming mucin (MUC2) production with altered cell polarity and abnormal cell-stroma interaction may be the key factor in the morphogenesis and indolent behavior of colloid carcinoma in the breast and pancreas. Am. J. Surg. Pathol. 2003, 27, 571–578. [Google Scholar] [CrossRef] [PubMed]
- Adsay, N.V.; Pierson, C.; Sarkar, F.; Abrams, J.; Weaver, D.; Conlon, K.C.; Brennan, M.F.; Klimstra, D.S. Colloid (mucinous noncystic) carcinoma of the pancreas. Am. J. Surg. Pathol. 2001, 25, 26–42. [Google Scholar] [CrossRef] [PubMed]
- Poultsides, G.A.; Reddy, S.; Cameron, J.L.; Hruban, R.H.; Pawlik, T.M.; Ahuja, N.; Jain, A.; Edil, B.H.; Iacobuzio-Donahue, C.A.; Schulick, R.D.; et al. Histopathologic basis for the favorable survival after resection of intraductal papillary mucinous neoplasm-associated invasive adenocarcinoma of the pancreas. Ann. Surg. 2010, 251, 470–476. [Google Scholar] [CrossRef] [PubMed]
- Seidel, G.; Zahurak, M.; Iacobuzio-Donahue, C.; Sohn, T.A.; Adsay, N.V.; Yeo, C.J.; Lillemoe, K.D.; Cameron, J.L.; Hruban, R.H.; Wilentz, R.E. Almost all infiltrating colloid carcinomas of the pancreas and periampullary region arise from in situ papillary neoplasms: A study of 39 cases. Am. J. Surg. Pathol. 2002, 26, 56–63. [Google Scholar] [CrossRef]
- Brody, J.R.; Costantino, C.L.; Potoczek, M.; Cozzitorto, J.; McCue, P.; Yeo, C.J.; Hruban, R.H.; Witkiewicz, A.K. Adenosquamous carcinoma of the pancreas harbors KRAS2, DPC4 and TP53 molecular alterations similar to pancreatic ductal adenocarcinoma. Mod. Pathol. 2009, 22, 651–659. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef]
- Wilentz, R.E.; Goggins, M.; Redston, M.; Marcus, V.A.; Adsay, N.V.; Sohn, T.A.; Kadkol, S.S.; Yeo, C.J.; Choti, M.; Zahurak, M.; et al. Genetic, immunohistochemical, and clinical features of medullary carcinoma of the pancreas: A newly described and characterized entity. Am. J. Pathol. 2000, 156, 1641–1651. [Google Scholar] [CrossRef]
- Calhoun, E.S.; Jones, J.B.; Ashfaq, R.; Adsay, V.; Baker, S.J.; Valentine, V.; Hempen, P.M.; Hilgers, W.; Yeo, C.J.; Hruban, R.H.; et al. BRAF and FBXW7 (CDC4, FBW7, AGO, SEL10) mutations in distinct subsets of pancreatic cancer: Potential therapeutic targets. Am. J. Pathol. 2003, 163, 1255–1260. [Google Scholar] [CrossRef]
- Goggins, M.; Offerhaus, G.J.; Hilgers, W.; Griffin, C.A.; Shekher, M.; Tang, D.; Sohn, T.A.; Yeo, C.J.; Kern, S.E.; Hruban, R.H. Pancreatic adenocarcinomas with DNA replication errors (RER+) are associated with wild-type K-ras and characteristic histopathology. Poor differentiation, a syncytial growth pattern, and pushing borders suggest RER+. Am. J. Pathol. 1998, 152, 1501–1507. [Google Scholar]
- Yamamoto, H.; Itoh, F.; Nakamura, H.; Fukushima, H.; Sasaki, S.; Perucho, M.; Imai, K. Genetic and clinical features of human pancreatic ductal adenocarcinomas with widespread microsatellite instability. Cancer Res. 2001, 61, 3139–3144. [Google Scholar] [PubMed]
- Jones, S.; Zhang, X.; Parsons, D.W.; Lin, J.C.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Kamiyama, H.; Jimeno, A.; et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science 2008, 321, 1801–1806. [Google Scholar] [CrossRef] [PubMed]
- Mehlen, P.; Delloye-Bourgeois, C.; Chedotal, A. Novel roles for Slits and netrins: Axon guidance cues as anticancer targets? Nat. Rev. Cancer 2011, 11, 188–197. [Google Scholar] [CrossRef] [PubMed]
- Sabatier, C.; Plump, A.S.; Le, M.; Brose, K.; Tamada, A.; Murakami, F.; Lee, E.Y.; Tessier-Lavigne, M. The divergent Robo family protein rig-1/Robo3 is a negative regulator of slit responsiveness required for midline crossing by commissural axons. Cell 2004, 117, 157–169. [Google Scholar] [CrossRef]
- Trusolino, L.; Comoglio, P.M. Scatter-factor and semaphorin receptors: Cell signalling for invasive growth. Nat. Rev. Cancer 2002, 2, 289–300. [Google Scholar] [CrossRef] [PubMed]
- Samuel, N.; Hudson, T.J. The molecular and cellular heterogeneity of pancreatic ductal adenocarcinoma. Nat. Rev. Gastroenterol. Hepatol. 2011, 9, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Biankin, A.V.; Waddell, N.; Kassahn, K.S.; Gingras, M.C.; Muthuswamy, L.B.; Johns, A.L.; Miller, D.K.; Wilson, P.J.; Patch, A.M.; Wu, J.; et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature 2012, 491, 399–405. [Google Scholar] [CrossRef]
- Connor, A.A.; Denroche, R.E.; Jang, G.H.; Timms, L.; Kalimuthu, S.N.; Selander, I.; McPherson, T.; Wilson, G.W.; Chan-Seng-Yue, M.A.; Borozan, I.; et al. Association of Distinct Mutational Signatures with Correlates of Increased Immune Activity in Pancreatic Ductal Adenocarcinoma. JAMA Oncol. 2017, 3, 774–783. [Google Scholar] [CrossRef]
- Lowery, M.A.; Wong, W.; Jordan, E.J.; Lee, J.W.; Kemel, Y.; Vijai, J.; Mandelker, D.; Zehir, A.; Capanu, M.; Salo-Mullen, E.; et al. Prospective Evaluation of Germline Alterations in Patients With Exocrine Pancreatic Neoplasms. J. Natl. Cancer Inst. 2018, 110, 1067–1074. [Google Scholar] [CrossRef]
- Waddell, N.; Pajic, M.; Patch, A.M.; Chang, D.K.; Kassahn, K.S.; Bailey, P.; Johns, A.L.; Miller, D.; Nones, K.; Quek, K.; et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature 2015, 518, 495–501. [Google Scholar] [CrossRef]
- Perkhofer, L.; Golan, T.; Cuyle, P.J.; Matysiak-Budnik, T.; Van Laethem, J.L.; Macarulla, T.; Cauchin, E.; Kleger, A.; Beutel, A.K.; Gout, J.; et al. Targeting DNA Damage Repair Mechanisms in Pancreas Cancer. Cancers 2021, 13, 4259. [Google Scholar] [CrossRef] [PubMed]
- Sahin, I.H.; Lowery, M.A.; Stadler, Z.K.; Salo-Mullen, E.; Iacobuzio-Donahue, C.A.; Kelsen, D.P.; O’Reilly, E.M. Genomic instability in pancreatic adenocarcinoma: A new step towards precision medicine and novel therapeutic approaches. Expert Rev. Gastroenterol. Hepatol. 2016, 10, 893–905. [Google Scholar] [CrossRef] [PubMed]
- Singhi, A.D.; George, B.; Greenbowe, J.R.; Chung, J.; Suh, J.; Maitra, A.; Klempner, S.J.; Hendifar, A.; Milind, J.M.; Golan, T.; et al. Real-Time Targeted Genome Profile Analysis of Pancreatic Ductal Adenocarcinomas Identifies Genetic Alterations That Might Be Targeted With Existing Drugs or Used as Biomarkers. Gastroenterology 2019, 156, 2242–2253 e2244. [Google Scholar] [CrossRef]
- Bailey, P.; Chang, D.K.; Nones, K.; Johns, A.L.; Patch, A.M.; Gingras, M.C.; Miller, D.K.; Christ, A.N.; Bruxner, T.J.; Quinn, M.C.; et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016, 531, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network. Electronic address, a.a.d.h.e.; Cancer Genome Atlas Research, N. Integrated Genomic Characterization of Pancreatic Ductal Adenocarcinoma. Cancer Cell 2017, 32, 185–203 e113. [Google Scholar] [CrossRef] [PubMed]
- Collisson, E.A.; Bailey, P.; Chang, D.K.; Biankin, A.V. Molecular subtypes of pancreatic cancer. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 207–220. [Google Scholar] [CrossRef]
- Moffitt, R.A.; Marayati, R.; Flate, E.L.; Volmar, K.E.; Loeza, S.G.; Hoadley, K.A.; Rashid, N.U.; Williams, L.A.; Eaton, S.C.; Chung, A.H.; et al. Virtual microdissection identifies distinct tumor- and stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nat. Genet. 2015, 47, 1168–1178. [Google Scholar] [CrossRef]
- Cohen, S.J.; Alpaugh, R.K.; Palazzo, I.; Meropol, N.J.; Rogatko, A.; Xu, Z.; Hoffman, J.P.; Weiner, L.M.; Cheng, J.D. Fibroblast activation protein and its relationship to clinical outcome in pancreatic adenocarcinoma. Pancreas 2008, 37, 154–158. [Google Scholar] [CrossRef]
- Puleo, F.; Nicolle, R.; Blum, Y.; Cros, J.; Marisa, L.; Demetter, P.; Quertinmont, E.; Svrcek, M.; Elarouci, N.; Iovanna, J.; et al. Stratification of Pancreatic Ductal Adenocarcinomas Based on Tumor and Microenvironment Features. Gastroenterology 2018, 155, 1999–2013 e1993. [Google Scholar] [CrossRef]
- Torres, C.; Grippo, P.J. Pancreatic cancer subtypes: A roadmap for precision medicine. Ann. Med. 2018, 50, 277–287. [Google Scholar] [CrossRef]
- Chan-Seng-Yue, M.; Kim, J.C.; Wilson, G.W.; Ng, K.; Figueroa, E.F.; O’Kane, G.M.; Connor, A.A.; Denroche, R.E.; Grant, R.C.; McLeod, J.; et al. Transcription phenotypes of pancreatic cancer are driven by genomic events during tumor evolution. Nat. Genet. 2020, 52, 231–240. [Google Scholar] [CrossRef] [PubMed]
- Hwang, W.L.; Jagadeesh, K.A.; Guo, J.A.; Hoffman, H.I.; Yadollahpour, P.; Reeves, J.W.; Mohan, R.; Drokhlyansky, E.; Van Wittenberghe, N.; Ashenberg, O.; et al. Single-nucleus and spatial transcriptome profiling of pancreatic cancer identifies multicellular dynamics associated with neoadjuvant treatment. Nat. Genet. 2022, 54, 1178–1191. [Google Scholar] [CrossRef] [PubMed]
- Tempero, M.A.; Malafa, M.P.; Al-Hawary, M.; Behrman, S.W.; Berson III, A.B.; Cardin, D.B.; Cha, C.; Chiorean, E.G.; Chung, V.; Czito, B.; et al. NCCN Clinical Practice Guidelines in Oncology, Pancreatic Adenocarcinoma (Version 1.2021). Available online: https://www.nccn.org/professionals/physician_gls/PDF/pancreatic.pdf (accessed on 25 January 2021).
- Chatterjee, D.; Katz, M.H.; Rashid, A.; Varadhachary, G.R.; Wolff, R.A.; Wang, H.; Lee, J.E.; Pisters, P.W.; Vauthey, J.N.; Crane, C.; et al. Histologic grading of the extent of residual carcinoma following neoadjuvant chemoradiation in pancreatic ductal adenocarcinoma: A predictor for patient outcome. Cancer 2012, 118, 3182–3190. [Google Scholar] [CrossRef] [PubMed]
- Chou, A.; Ahadi, M.; Arena, J.; Sioson, L.; Sheen, A.; Fuchs, T.L.; Pavlakis, N.; Clarke, S.; Kneebone, A.; Hruby, G.; et al. A Critical Assessment of Postneoadjuvant Therapy Pancreatic Cancer Regression Grading Schemes with a Proposal for a Novel Approach. Am. J. Surg. Pathol. 2021, 45, 394–404. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.M.; Katz, M.H.; Liu, L.; Sundar, M.; Wang, H.; Varadhachary, G.R.; Wolff, R.A.; Lee, J.E.; Maitra, A.; Fleming, J.B.; et al. Validation of a Proposed Tumor Regression Grading Scheme for Pancreatic Ductal Adenocarcinoma After Neoadjuvant Therapy as a Prognostic Indicator for Survival. Am. J. Surg. Pathol. 2016, 40, 1653–1660. [Google Scholar] [CrossRef]
- Nagaria, T.S.; Wang, H.; Chatterjee, D.; Wang, H. Pathology of Treated Pancreatic Ductal Adenocarcinoma and Its Clinical Implications. Arch. Pathol. Lab. Med. 2020, 144, 838–845. [Google Scholar] [CrossRef]
- Hruban, R.H.; Takaori, K.; Klimstra, D.S.; Adsay, N.V.; Albores-Saavedra, J.; Biankin, A.V.; Biankin, S.A.; Compton, C.; Fukushima, N.; Furukawa, T.; et al. An illustrated consensus on the classification of pancreatic intraepithelial neoplasia and intraductal papillary mucinous neoplasms. Am. J. Surg. Pathol. 2004, 28, 977–987. [Google Scholar] [CrossRef]
- Hruban, R.H.; Adsay, N.V.; Albores-Saavedra, J.; Compton, C.; Garrett, E.S.; Goodman, S.N.; Kern, S.E.; Klimstra, D.S.; Kloppel, G.; Longnecker, D.S.; et al. Pancreatic intraepithelial neoplasia: A new nomenclature and classification system for pancreatic duct lesions. Am. J. Surg. Pathol. 2001, 25, 579–586. [Google Scholar] [CrossRef]
- Kanda, M.; Matthaei, H.; Wu, J.; Hong, S.M.; Yu, J.; Borges, M.; Hruban, R.H.; Maitra, A.; Kinzler, K.; Vogelstein, B.; et al. Presence of somatic mutations in most early-stage pancreatic intraepithelial neoplasia. Gastroenterology 2012, 142, 730–733 e739. [Google Scholar] [CrossRef]
- Hosoda, W.; Chianchiano, P.; Griffin, J.F.; Pittman, M.E.; Brosens, L.A.; Noe, M.; Yu, J.; Shindo, K.; Suenaga, M.; Rezaee, N.; et al. Genetic analyses of isolated high-grade pancreatic intraepithelial neoplasia (HG-PanIN) reveal paucity of alterations in TP53 and SMAD4. J. Pathol. 2017, 242, 16–23. [Google Scholar] [CrossRef]
- Hosoda, W.; Wood, L.D. Molecular Genetics of Pancreatic Neoplasms. Surg. Pathol. Clin. 2016, 9, 685–703. [Google Scholar] [CrossRef] [PubMed]
- Feldmann, G.; Beaty, R.; Hruban, R.H.; Maitra, A. Molecular genetics of pancreatic intraepithelial neoplasia. J. Hepatobiliary Pancreat. Surg. 2007, 14, 224–232. [Google Scholar] [CrossRef] [PubMed]
- Hata, T.; Suenaga, M.; Marchionni, L.; Macgregor-Das, A.; Yu, J.; Shindo, K.; Tamura, K.; Hruban, R.H.; Goggins, M. Genome-Wide Somatic Copy Number Alterations and Mutations in High-Grade Pancreatic Intraepithelial Neoplasia. Am. J. Pathol. 2018, 188, 1723–1733. [Google Scholar] [CrossRef] [PubMed]
- Mori, Y.; Ohtsuka, T.; Kono, H.; Ideno, N.; Aso, T.; Nagayoshi, Y.; Takahata, S.; Nakamura, M.; Ishigami, K.; Aishima, S.; et al. Management strategy for multifocal branch duct intraductal papillary mucinous neoplasms of the pancreas. Pancreas 2012, 41, 1008–1012. [Google Scholar] [CrossRef] [PubMed]
- Reid, M.D.; Lewis, M.M.; Willingham, F.F.; Adsay, N.V. The Evolving Role of Pathology in New Developments, Classification, Terminology, and Diagnosis of Pancreatobiliary Neoplasms. Arch. Pathol. Lab. Med. 2017, 141, 366–380. [Google Scholar] [CrossRef]
- Kloppel, G.; Basturk, O.; Schlitter, A.M.; Konukiewitz, B.; Esposito, I. Intraductal neoplasms of the pancreas. Semin. Diagn. Pathol. 2014, 31, 452–466. [Google Scholar] [CrossRef]
- Furukawa, T.; Kloppel, G.; Volkan Adsay, N.; Albores-Saavedra, J.; Fukushima, N.; Horii, A.; Hruban, R.H.; Kato, Y.; Klimstra, D.S.; Longnecker, D.S.; et al. Classification of types of intraductal papillary-mucinous neoplasm of the pancreas: A consensus study. Virchows Arch. 2005, 447, 794–799. [Google Scholar] [CrossRef]
- Adsay, N.V.; Merati, K.; Basturk, O.; Iacobuzio-Donahue, C.; Levi, E.; Cheng, J.D.; Sarkar, F.H.; Hruban, R.H.; Klimstra, D.S. Pathologically and biologically distinct types of epithelium in intraductal papillary mucinous neoplasms: Delineation of an “intestinal” pathway of carcinogenesis in the pancreas. Am. J. Surg. Pathol. 2004, 28, 839–848. [Google Scholar] [CrossRef]
- Fischer, C.G.; Wood, L.D. From somatic mutation to early detection: Insights from molecular characterization of pancreatic cancer precursor lesions. J. Pathol. 2018, 246, 395–404. [Google Scholar] [CrossRef]
- Kuboki, Y.; Shimizu, K.; Hatori, T.; Yamamoto, M.; Shibata, N.; Shiratori, K.; Furukawa, T. Molecular biomarkers for progression of intraductal papillary mucinous neoplasm of the pancreas. Pancreas 2015, 44, 227–235. [Google Scholar] [CrossRef]
- Abe, K.; Suda, K.; Arakawa, A.; Yamasaki, S.; Sonoue, H.; Mitani, K.; Nobukawa, B. Different patterns of p16INK4A and p53 protein expressions in intraductal papillary-mucinous neoplasms and pancreatic intraepithelial neoplasia. Pancreas 2007, 34, 85–91. [Google Scholar] [CrossRef]
- Furukawa, T.; Fujisaki, R.; Yoshida, Y.; Kanai, N.; Sunamura, M.; Abe, T.; Takeda, K.; Matsuno, S.; Horii, A. Distinct progression pathways involving the dysfunction of DUSP6/MKP-3 in pancreatic intraepithelial neoplasia and intraductal papillary-mucinous neoplasms of the pancreas. Mod. Pathol. 2005, 18, 1034–1042. [Google Scholar] [CrossRef] [PubMed]
- Biankin, A.V.; Biankin, S.A.; Kench, J.G.; Morey, A.L.; Lee, C.S.; Head, D.R.; Eckstein, R.P.; Hugh, T.B.; Henshall, S.M.; Sutherland, R.L. Aberrant p16(INK4A) and DPC4/Smad4 expression in intraductal papillary mucinous tumours of the pancreas is associated with invasive ductal adenocarcinoma. Gut 2002, 50, 861–868. [Google Scholar] [CrossRef] [PubMed]
- Inoue, H.; Furukawa, T.; Sunamura, M.; Takeda, K.; Matsuno, S.; Horii, A. Exclusion of SMAD4 mutation as an early genetic change in human pancreatic ductal tumorigenesis. Genes Chromosomes Cancer 2001, 31, 295–299. [Google Scholar] [CrossRef]
- Iacobuzio-Donahue, C.A.; Klimstra, D.S.; Adsay, N.V.; Wilentz, R.E.; Argani, P.; Sohn, T.A.; Yeo, C.J.; Cameron, J.L.; Kern, S.E.; Hruban, R.H. Dpc-4 protein is expressed in virtually all human intraductal papillary mucinous neoplasms of the pancreas: Comparison with conventional ductal adenocarcinomas. Am. J. Pathol. 2000, 157, 755–761. [Google Scholar] [CrossRef]
- Basturk, O.; Hong, S.M.; Wood, L.D.; Adsay, N.V.; Albores-Saavedra, J.; Biankin, A.V.; Brosens, L.A.; Fukushima, N.; Goggins, M.; Hruban, R.H.; et al. A Revised Classification System and Recommendations From the Baltimore Consensus Meeting for Neoplastic Precursor Lesions in the Pancreas. Am. J. Surg. Pathol. 2015, 39, 1730–1741. [Google Scholar] [CrossRef]
- Wang, T.; Askan, G.; Adsay, V.; Allen, P.; Jarnagin, W.R.; Memis, B.; Sigel, C.; Seven, I.E.; Klimstra, D.S.; Basturk, O. Intraductal Oncocytic Papillary Neoplasms: Clinical-Pathologic Characterization of 24 Cases, With An Emphasis on Associated Invasive Carcinomas. Am. J. Surg. Pathol. 2019, 43, 656–661. [Google Scholar] [CrossRef]
- Basturk, O.; Tan, M.; Bhanot, U.; Allen, P.; Adsay, V.; Scott, S.N.; Shah, R.; Berger, M.F.; Askan, G.; Dikoglu, E.; et al. The oncocytic subtype is genetically distinct from other pancreatic intraductal papillary mucinous neoplasm subtypes. Mod. Pathol. 2016, 29, 1058–1069. [Google Scholar] [CrossRef]
- Singhi, A.D.; Wood, L.D.; Parks, E.; Torbenson, M.S.; Felsenstein, M.; Hruban, R.H.; Nikiforova, M.N.; Wald, A.I.; Kaya, C.; Nikiforov, Y.E.; et al. Recurrent Rearrangements in PRKACA and PRKACB in Intraductal Oncocytic Papillary Neoplasms of the Pancreas and Bile Duct. Gastroenterology 2020, 158, 573–582.e2. [Google Scholar] [CrossRef]
- Basturk, O.; Berger, M.F.; Yamaguchi, H.; Adsay, V.; Askan, G.; Bhanot, U.K.; Zehir, A.; Carneiro, F.; Hong, S.M.; Zamboni, G.; et al. Pancreatic intraductal tubulopapillary neoplasm is genetically distinct from intraductal papillary mucinous neoplasm and ductal adenocarcinoma. Mod. Pathol. 2017, 30, 1760–1772. [Google Scholar] [CrossRef]
- Crippa, S.; Fernandez-Del Castillo, C.; Salvia, R.; Finkelstein, D.; Bassi, C.; Dominguez, I.; Muzikansky, A.; Thayer, S.P.; Falconi, M.; Mino-Kenudson, M.; et al. Mucin-producing neoplasms of the pancreas: An analysis of distinguishing clinical and epidemiologic characteristics. Clin. Gastroenterol. Hepatol. 2010, 8, 213–219. [Google Scholar] [CrossRef] [Green Version]
- Springer, S.; Wang, Y.; Dal Molin, M.; Masica, D.L.; Jiao, Y.; Kinde, I.; Blackford, A.; Raman, S.P.; Wolfgang, C.L.; Tomita, T.; et al. A combination of molecular markers and clinical features improve the classification of pancreatic cysts. Gastroenterology 2015, 149, 1501–1510. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Jiao, Y.; Dal Molin, M.; Maitra, A.; de Wilde, R.F.; Wood, L.D.; Eshleman, J.R.; Goggins, M.G.; Wolfgang, C.L.; Canto, M.I.; et al. Whole-exome sequencing of neoplastic cysts of the pancreas reveals recurrent mutations in components of ubiquitin-dependent pathways. Proc. Natl. Acad. Sci. USA 2011, 108, 21188–21193. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Matthaei, H.; Maitra, A.; Dal Molin, M.; Wood, L.D.; Eshleman, J.R.; Goggins, M.; Canto, M.I.; Schulick, R.D.; Edil, B.H.; et al. Recurrent GNAS mutations define an unexpected pathway for pancreatic cyst development. Sci. Transl. Med. 2011, 3, 92ra66. [Google Scholar] [CrossRef] [PubMed]
- Krasinskas, A.M.; Oakley, G.J.; Bagci, P.; Jang, K.T.; Kuan, S.F.; Reid, M.D.; Erbarut, I.; Adsay, V. “Simple Mucinous Cyst” of the Pancreas: A Clinicopathologic Analysis of 39 Examples of a Diagnostically Challenging Entity Distinct From Intraductal Papillary Mucinous Neoplasms and Mucinous Cystic Neoplasms. Am. J. Surg. Pathol. 2017, 41, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Milanetto, A.C.; Tonello, A.S.; Valotto, G.; Munari, G.; Luchini, C.; Fassan, M.; Pasquali, C. Simple mucinous cyst: Another potential cancer precursor in the pancreas? Case report with molecular characterization and systematic review of the literature. Virchows Arch. 2021, 479, 179–189. [Google Scholar] [CrossRef]
- Neoptolemos, J.P.; Palmer, D.H.; Ghaneh, P.; Psarelli, E.E.; Valle, J.W.; Halloran, C.M.; Faluyi, O.; O’Reilly, D.A.; Cunningham, D.; Wadsley, J.; et al. Comparison of adjuvant gemcitabine and capecitabine with gemcitabine monotherapy in patients with resected pancreatic cancer (ESPAC-4): A multicentre, open-label, randomised, phase 3 trial. Lancet 2017, 389, 1011–1024. [Google Scholar] [CrossRef]
- Conroy, T.; Hammel, P.; Hebbar, M.; Ben Abdelghani, M.; Wei, A.C.; Raoul, J.L.; Chone, L.; Francois, E.; Artru, P.; Biagi, J.J.; et al. FOLFIRINOX or Gemcitabine as Adjuvant Therapy for Pancreatic Cancer. N. Engl. J. Med. 2018, 379, 2395–2406. [Google Scholar] [CrossRef] [PubMed]
- Perkhofer, L.; Gout, J.; Roger, E.; Kude de Almeida, F.; Baptista Simoes, C.; Wiesmuller, L.; Seufferlein, T.; Kleger, A. DNA damage repair as a target in pancreatic cancer: State-of-the-art and future perspectives. Gut 2021, 70, 606–617. [Google Scholar] [CrossRef] [PubMed]
- Dreyer, S.B.; Upstill-Goddard, R.; Paulus-Hock, V.; Paris, C.; Lampraki, E.M.; Dray, E.; Serrels, B.; Caligiuri, G.; Rebus, S.; Plenker, D.; et al. Targeting DNA Damage Response and Replication Stress in Pancreatic Cancer. Gastroenterology 2021, 160, 362–377 e313. [Google Scholar] [CrossRef]
- Golan, T.; Hammel, P.; Reni, M.; Van Cutsem, E.; Macarulla, T.; Hall, M.J.; Park, J.O.; Hochhauser, D.; Arnold, D.; Oh, D.Y.; et al. Maintenance Olaparib for Germline BRCA-Mutated Metastatic Pancreatic Cancer. N. Engl. J. Med. 2019, 381, 317–327. [Google Scholar] [CrossRef] [PubMed]
- Weber, A.M.; Ryan, A.J. ATM and ATR as therapeutic targets in cancer. Pharmacol. Ther. 2015, 149, 124–138. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, S.A.; Schultz, C.W.; Azimi-Sadjadi, A.; Brody, J.R.; Pishvaian, M.J. ATM Dysfunction in Pancreatic Adenocarcinoma and Associated Therapeutic Implications. Mol. Cancer Ther. 2019, 18, 1899–1908. [Google Scholar] [CrossRef] [PubMed]
- Riches, L.C.; Trinidad, A.G.; Hughes, G.; Jones, G.N.; Hughes, A.M.; Thomason, A.G.; Gavine, P.; Cui, A.; Ling, S.; Stott, J.; et al. Pharmacology of the ATM Inhibitor AZD0156: Potentiation of Irradiation and Olaparib Responses Preclinically. Mol. Cancer Ther. 2020, 19, 13–25. [Google Scholar] [CrossRef]
- Durant, S.T.; Zheng, L.; Wang, Y.; Chen, K.; Zhang, L.; Zhang, T.; Yang, Z.; Riches, L.; Trinidad, A.G.; Fok, J.H.L.; et al. The brain-penetrant clinical ATM inhibitor AZD1390 radiosensitizes and improves survival of preclinical brain tumor models. Sci. Adv. 2018, 4, eaat1719. [Google Scholar] [CrossRef]
- Eso, Y.; Shimizu, T.; Takeda, H.; Takai, A.; Marusawa, H. Microsatellite instability and immune checkpoint inhibitors: Toward precision medicine against gastrointestinal and hepatobiliary cancers. J. Gastroenterol. 2020, 55, 15–26. [Google Scholar] [CrossRef]
- Hofmann, M.H.; Gerlach, D.; Misale, S.; Petronczki, M.; Kraut, N. Expanding the Reach of Precision Oncology by Drugging All KRAS Mutants. Cancer Discov. 2022, 12, 924–937. [Google Scholar] [CrossRef]
- Hong, D.S.; Fakih, M.G.; Strickler, J.H.; Desai, J.; Durm, G.A.; Shapiro, G.I.; Falchook, G.S.; Price, T.J.; Sacher, A.; Denlinger, C.S.; et al. KRAS(G12C) Inhibition with Sotorasib in Advanced Solid Tumors. N. Engl. J. Med. 2020, 383, 1207–1217. [Google Scholar] [CrossRef]
- Sotorasib Tackles KRASG12C-Mutated Pancreatic Cancer. Cancer Discov. 2022, 12, 878–879. [CrossRef]
- Drilon, A.; Laetsch, T.W.; Kummar, S.; DuBois, S.G.; Lassen, U.N.; Demetri, G.D.; Nathenson, M.; Doebele, R.C.; Farago, A.F.; Pappo, A.S.; et al. Efficacy of Larotrectinib in TRK Fusion-Positive Cancers in Adults and Children. N. Engl. J. Med. 2018, 378, 731–739. [Google Scholar] [CrossRef]
- Doebele, R.C.; Drilon, A.; Paz-Ares, L.; Siena, S.; Shaw, A.T.; Farago, A.F.; Blakely, C.M.; Seto, T.; Cho, B.C.; Tosi, D.; et al. Entrectinib in patients with advanced or metastatic NTRK fusion-positive solid tumours: Integrated analysis of three phase 1-2 trials. Lancet Oncol. 2020, 21, 271–282. [Google Scholar] [CrossRef]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef] [PubMed]
- Overman, M.J.; McDermott, R.; Leach, J.L.; Lonardi, S.; Lenz, H.J.; Morse, M.A.; Desai, J.; Hill, A.; Axelson, M.; Moss, R.A.; et al. Nivolumab in patients with metastatic DNA mismatch repair-deficient or microsatellite instability-high colorectal cancer (CheckMate 142): An open-label, multicentre, phase 2 study. Lancet Oncol. 2017, 18, 1182–1191. [Google Scholar] [CrossRef]
- Overman, M.J.; Lonardi, S.; Wong, K.Y.M.; Lenz, H.J.; Gelsomino, F.; Aglietta, M.; Morse, M.A.; Van Cutsem, E.; McDermott, R.; Hill, A.; et al. Durable Clinical Benefit With Nivolumab Plus Ipilimumab in DNA Mismatch Repair-Deficient/Microsatellite Instability-High Metastatic Colorectal Cancer. J. Clin. Oncol. 2018, 36, 773–779. [Google Scholar] [CrossRef] [PubMed]
- Luchini, C.; Brosens, L.A.A.; Wood, L.D.; Chatterjee, D.; Shin, J.I.; Sciammarella, C.; Fiadone, G.; Malleo, G.; Salvia, R.; Kryklyva, V.; et al. Comprehensive characterisation of pancreatic ductal adenocarcinoma with microsatellite instability: Histology, molecular pathology and clinical implications. Gut 2021, 70, 148–156. [Google Scholar] [CrossRef]
- Lupinacci, R.M.; Goloudina, A.; Buhard, O.; Bachet, J.B.; Marechal, R.; Demetter, P.; Cros, J.; Bardier-Dupas, A.; Collura, A.; Cervera, P.; et al. Prevalence of Microsatellite Instability in Intraductal Papillary Mucinous Neoplasms of the Pancreas. Gastroenterology 2018, 154, 1061–1065. [Google Scholar] [CrossRef]
- Luchini, C.; Mafficini, A.; Chatterjee, D.; Piredda, M.L.; Sciammarella, C.; Navale, P.; Malleo, G.; Mattiolo, P.; Marchegiani, G.; Pea, A.; et al. Histo-molecular characterization of pancreatic cancer with microsatellite instability: Intra-tumor heterogeneity, B2M inactivation, and the importance of metastatic sites. Virchows Arch. 2022, 480, 1261–1268. [Google Scholar] [CrossRef]
- Sohal, D.P.S.; Kennedy, E.B.; Cinar, P.; Conroy, T.; Copur, M.S.; Crane, C.H.; Garrido-Laguna, I.; Lau, M.W.; Johnson, T.; Krishnamurthi, S.; et al. Metastatic Pancreatic Cancer: ASCO Guideline Update. J. Clin. Oncol. 2020, 38, 3217–3230. [Google Scholar] [CrossRef]
- Boyd, L.N.C.; Andini, K.D.; Peters, G.J.; Kazemier, G.; Giovannetti, E. Heterogeneity and plasticity of cancer-associated fibroblasts in the pancreatic tumor microenvironment. Semin. Cancer Biol. 2022, 82, 184–196. [Google Scholar] [CrossRef]
- Herting, C.J.; Karpovsky, I.; Lesinski, G.B. The tumor microenvironment in pancreatic ductal adenocarcinoma: Current perspectives and future directions. Cancer Metastasis Rev. 2021, 40, 675–689. [Google Scholar] [CrossRef]
- Petersen, O.H.; Gerasimenko, J.V.; Gerasimenko, O.V.; Gryshchenko, O.; Peng, S. The roles of calcium and ATP in the physiology and pathology of the exocrine pancreas. Physiol. Rev. 2021, 101, 1691–1744. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Zhang, C.; Jiang, K.; Werner, J.; Bazhin, A.V.; D’Haese, J.G. The Role of Stellate Cells in Pancreatic Ductal Adenocarcinoma: Targeting Perspectives. Front. Oncol. 2020, 10, 621937. [Google Scholar] [CrossRef] [PubMed]
- Knudsen, E.S.; Vail, P.; Balaji, U.; Ngo, H.; Botros, I.W.; Makarov, V.; Riaz, N.; Balachandran, V.; Leach, S.; Thompson, D.M.; et al. Stratification of Pancreatic Ductal Adenocarcinoma: Combinatorial Genetic, Stromal, and Immunologic Markers. Clin. Cancer Res. 2017, 23, 4429–4440. [Google Scholar] [CrossRef] [PubMed]
- Danilova, L.; Ho, W.J.; Zhu, Q.; Vithayathil, T.; De Jesus-Acosta, A.; Azad, N.S.; Laheru, D.A.; Fertig, E.J.; Anders, R.; Jaffee, E.M.; et al. Programmed Cell Death Ligand-1 (PD-L1) and CD8 Expression Profiling Identify an Immunologic Subtype of Pancreatic Ductal Adenocarcinomas with Favorable Survival. Cancer Immunol. Res. 2019, 7, 886–895. [Google Scholar] [CrossRef]
- Karamitopoulou, E. Tumour microenvironment of pancreatic cancer: Immune landscape is dictated by molecular and histopathological features. Br. J. Cancer 2019, 121, 5–14. [Google Scholar] [CrossRef]
- Karamitopoulou, E. The Tumor Microenvironment of Pancreatic Cancer. Cancers 2020, 12, 3076. [Google Scholar] [CrossRef]
- Blando, J.; Sharma, A.; Higa, M.G.; Zhao, H.; Vence, L.; Yadav, S.S.; Kim, J.; Sepulveda, A.M.; Sharp, M.; Maitra, A.; et al. Comparison of immune infiltrates in melanoma and pancreatic cancer highlights VISTA as a potential target in pancreatic cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 1692–1697. [Google Scholar] [CrossRef]
- Wandmacher, A.M.; Letsch, A.; Sebens, S. Challenges and Future Perspectives of Immunotherapy in Pancreatic Cancer. Cancers 2021, 13, 4235. [Google Scholar] [CrossRef]
- Carstens, J.L.; Correa de Sampaio, P.; Yang, D.; Barua, S.; Wang, H.; Rao, A.; Allison, J.P.; LeBleu, V.S.; Kalluri, R. Spatial computation of intratumoral T cells correlates with survival of patients with pancreatic cancer. Nat. Commun. 2017, 8, 15095. [Google Scholar] [CrossRef]
- Nejati, R.; Goldstein, J.B.; Halperin, D.M.; Wang, H.; Hejazi, N.; Rashid, A.; Katz, M.H.; Lee, J.E.; Fleming, J.B.; Rodriguez-Canales, J.; et al. Prognostic Significance of Tumor-Infiltrating Lymphocytes in Patients with Pancreatic Ductal Adenocarcinoma Treated with Neoadjuvant Chemotherapy. Pancreas 2017, 46, 1180–1187. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Subtypes | Frequencies | Diagnostic Criteria | Specific IHC Markers | Type-Specific Genetic Alterations |
|---|---|---|---|---|
| ASC/SCC | 1–4% | ≥30% of SCC | CK5/6, P63, and P40 | UPF1, KMT2C, KMT2D, SMARCA4 (BRG1), KDM6, and KDM3 |
| Colloid carcinoma (CC) | ≥80% of CC | CK20, CDX2, and MUC2 | GNAS, ATM, and MSI/defective MMR | |
| Medullary carcinoma | <1% | NA | NA | MSI/defective MMR, POLE |
| Hepatoid carcinoma (HC) | <1% | ≥50% of HC | HepPar-1, Glypican 3, Arginase, and Albumin by FISH | BAP1 and Notch1 |
| Micropapillary carcinoma (MPC) | <5% | ≥50% of MPC | NA | KRAS, TP53, and SMAD4 |
| Signet ring cell carcinoma (SRC) | <1% | ≥80% of SRC | NA | PI3K and MEK |
| Undifferentiated carcinoma (UC) | 1–7% | NA | NA | CDH1 |
| UC with osteoclast-like giant cells | NA | NA | SERPINA3, MAGEB4, GLI3, MEGF8, TTN, and BRCA2 | |
| UC with rhabdoid cells | <1% | ≥50% | Loss nuclear expression of SMARCB1 (INI1) | SMARCB1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Taherian, M.; Wang, H.; Wang, H. Pancreatic Ductal Adenocarcinoma: Molecular Pathology and Predictive Biomarkers. Cells 2022, 11, 3068. https://doi.org/10.3390/cells11193068
Taherian M, Wang H, Wang H. Pancreatic Ductal Adenocarcinoma: Molecular Pathology and Predictive Biomarkers. Cells. 2022; 11(19):3068. https://doi.org/10.3390/cells11193068
Chicago/Turabian StyleTaherian, Mehran, Hua Wang, and Huamin Wang. 2022. "Pancreatic Ductal Adenocarcinoma: Molecular Pathology and Predictive Biomarkers" Cells 11, no. 19: 3068. https://doi.org/10.3390/cells11193068