
Krüppel-Like Factor 1: A Pivotal Gene Regulator in Erythropoiesis

Abstract
:1. Introduction

{kind=link}
{kind=link}
| Gene Symbol | Name/Function | References |
|---|---|---|
| CELL-CYCLE REGULATORS | ||
| E2F2 | E2F Transcription Factor 2 | [12,13,14] |
| P18 (CDKN2C) | Cyclin Dependent Kinase Inhibitor 2C | [14,15] |
| P21(CDKN1A) | Cyclin Dependent Kinase Inhibitor 1A | [14,15] |
| P27 (CDKN1B) | Cyclin Dependent Kinase Inhibitor 1B | [14,15] |
| HAEMOGLOBIN REGULATION | ||
| HBB | Human adult haemoglobin subunit β | [8,9,16] |
| Hbb-b1/Hbb-b2 | Murine adult haemoglobin subunit β | [1,8,9,16] |
| HBG | Human foetal haemoglobin subunit γ | [16,17] |
| Hbb-y | Murine embryonic haemoglobin subunit εy | [2,18,19] |
| Hbb-bh1 | Murine embryonic haemoglobin subunit βh1 | [2,18,19] |
| Hba-x | Murine embryonic haemoglobin subunit ζ | [2,18,19] |
| BCL11A | B cell CLL/lymphoma 11A | [20,21,22,23,24,25] |
| ZBTB7A | Zinc finger and BTB domain containing 7A | [26] |
| ADHESION MOLECULES/ANTIGENS | ||
| BCAM | Basal cell adhesion molecule (Lutheran blood group) | [27] |
| VCAM | Vascular cell adhesion molecule | [28] |
| CD44 | CD44 molecule (Indian blood group) | [29,30,31] |
| P1PK | alpha 1,4-galactosyltransferase (P blood group) | [2,32,33] |
| LW (ICAM4) | Intercellular adhesion molecule 4 (Landsteiner–Wiener blood group) | [2,32,33] |
| KNOPS | Complement C3b/C4b receptor 1 (Knops blood group) | [2,32,33] |
| OK | Basigin (Ok blood group) | [2,32,33] |
| RAPH | CD151 molecule (Raph blood group) | [2,32,33] |
| ERMAP/SCIANNA | Erythroblast membrane-associated protein (Scianna blood group) | [2,32,33] |
| AQP1 | Aquaporin 1 (Colton blood group) | [29] |
| HEME SYNTHESIS/IRON PROCESSING | ||
| ALAS2 | 5’-aminolevulinate synthase 2 | [34] |
| ALAD | Aminolevulinate dehydratase | [34] |
| HMBS | Hydroxymethylbilane synthase | [34] |
| SLC25A37 | Solute carrier family 25 member 37 | [34,35] |
| STEAP3 | STEAP3 metalloreductase | [34,35] |
| ABCG2 | ATP binding cassette subfamily G member 2 (Junior blood group) | [34,35] |
| ABCB10 | ATP binding cassette subfamily B member 1 | [34,35] |
| TFR2 | Transferrin receptor 2 | [34,35] |
| OTHER CATEGORIES/FUNCTIONS | ||
| PKLR | Pyruvate kinase L/R | [36] |
| DNASEII-ALPHA | Deoxyribonuclease 2, lysosomal | [37] |
| TER119 | Lymphocyte antigen 76 | [2] |
| AHSP | Alpha haemoglobin stabilizing protein | [2,38,39] |
| CD9 | CD9 molecule | [2,18] |
| CD24 | CD24 antigen (small cell lung carcinoma cluster 4 antigen) | [2,18] |
| DEMATIN | Erythrocyte membrane protein Band 4.9 | [2,40] |
| MGST3 | Microsomal glutathione S-transferase 3 | [2,18] |
| ACP3 | Acid phosphatase 3 | [2,18] |
| BZRP (TSPO) | Translocator protein | [2,18] |
| RH-CDE | Rhesus CDe complex | [2,18] |
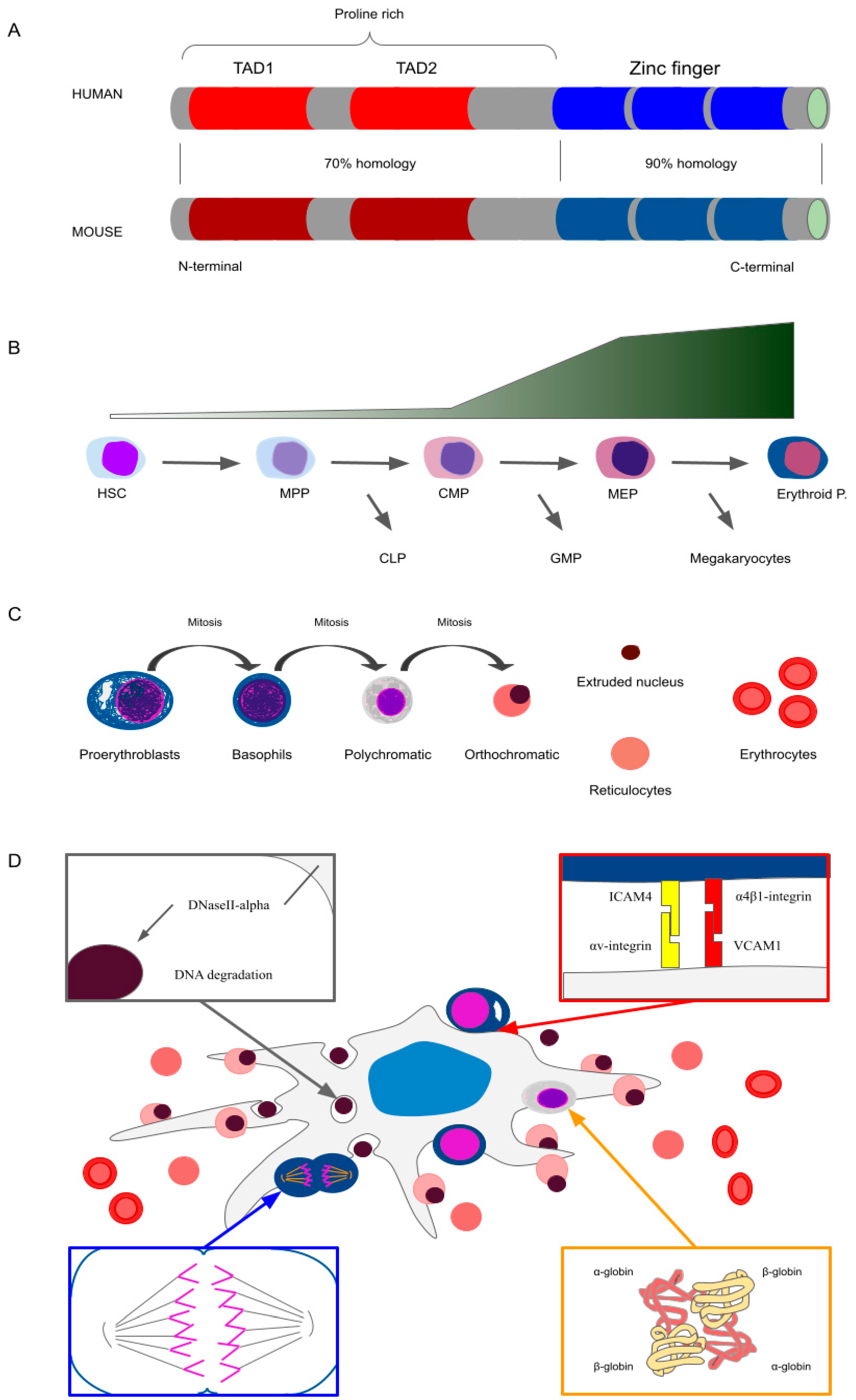
2. Erythropoiesis
2.1. Role of Klf1 in Primitive Erythropoiesis
2.2. Role of Klf1 in Definitive Erythropoiesis
2.2.1. Role of Klf1 in Cell-Cycle Regulation
2.2.2. Role of Klf1 in Central Macrophages of Erythroblastic Islands (CMEI) functions
2.2.3. Role of KLF1 in Erythroid Commitment
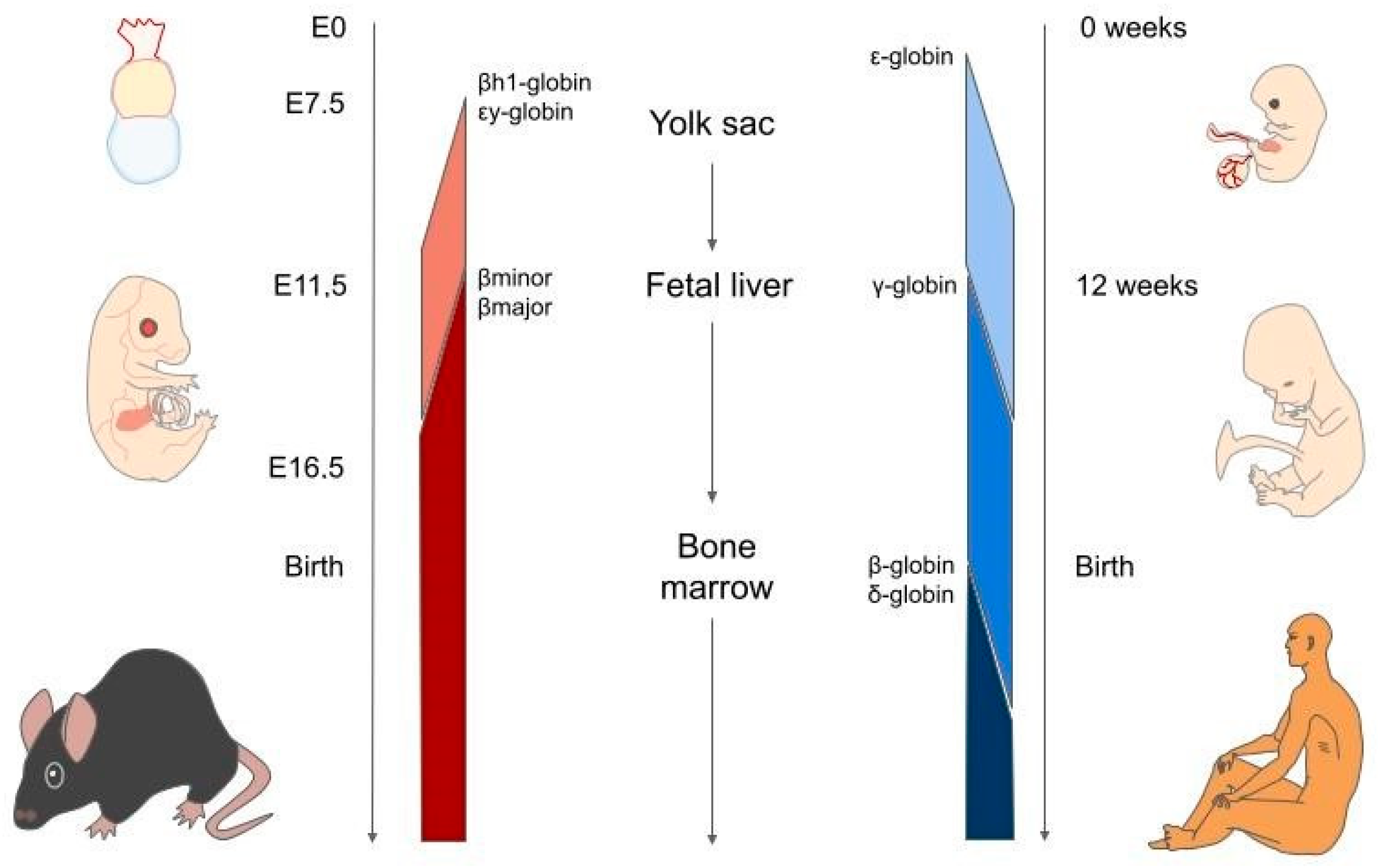
3. Haemoglobin Switching
4. Regulation of KLF1 Expression by Non-Coding RNA
5. Human KLF1 Variants and Phenotypes
5.1. In(Lu) Phenotype
5.2. Globin-Expression Dysregulation
5.3. Congenital Dyserythropoietic Anaemia (CDA)
5.4. Hydrops Foetalis
5.5. Non-Spherocytic Haemolytic Anaemia (NSHA)
5.6. Red Cell Protoporphyrin
5.7. Pyruvate Kinase Deficiency
6. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Miller, I.J.; Bieker, J.J. A Novel, Erythroid Cell-Specific Murine Transcription Factor That Binds to the CACCC Element and Is Related to the Krüppel Family of Nuclear Proteins. Mol. Cell. Biol. 1993, 13, 2776–2786. [Google Scholar] [CrossRef] [PubMed]
- Hodge, D.; Coghill, E.; Keys, J.; Maguire, T.; Hartmann, B.; McDowall, A.; Weiss, M.; Grimmond, S.; Perkins, A. A Global Role for EKLF in Definitive and Primitive Erythropoiesis. Blood 2006, 107, 3359–3370. [Google Scholar] [CrossRef] [PubMed]
- Tallack, M.R.; Magor, G.W.; Dartigues, B.; Sun, L.; Huang, S.; Fittock, J.M.; Fry, S.V.; Glazov, E.A.; Bailey, T.L.; Perkins, A.C. Novel Roles for KLF1 in Erythropoiesis Revealed by MRNA-Seq. Genome Res. 2012, 22, 2385–2398. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, L.; Chen, X.; Bieker, J.J. Regulation of Erythroid Krüppel-like Factor (EKLF) Transcriptional Activity by Phosphorylation of a Protein Kinase Casein Kinase II Site within Its Interaction Domain. J. Biol. Chem. 1998, 273, 23019–23025. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Kadam, S.; Emerson, B.M.; Bieker, J.J. Site-Specific Acetylation by P300 or CREB Binding Protein Regulates Erythroid Krüppel-like Factor Transcriptional Activity via Its Interaction with the SWI-SNF Complex. Mol. Cell. Biol. 2001, 21, 2413–2422. [Google Scholar] [CrossRef] [PubMed]
- Siatecka, M.; Xue, L.; Bieker, J.J. Sumoylation of EKLF Promotes Transcriptional Repression and Is Involved in Inhibition of Megakaryopoiesis. Mol. Cell. Biol. 2007, 27, 8547–8560. [Google Scholar] [CrossRef]
- Siatecka, M.; Soni, S.; Planutis, A.; Bieker, J.J. Transcriptional Activity of Erythroid Kruppel-like Factor (EKLF/KLF1) Modulated by PIAS3 (Protein Inhibitor of Activated STAT3). J. Biol. Chem. 2015, 290, 9929–9940. [Google Scholar] [CrossRef]
- Nuez, B.; Michalovich, D.; Bygrave, A.; Ploemacher, R.; Grosveld, F. Defective Haematopoiesis in Fetal Liver Resulting from Inactivation of the EKLF Gene. Nature 1995, 375, 316–318. [Google Scholar] [CrossRef]
- Perkins, A.C.; Sharpe, A.H.; Orkin, S.H. Lethal β-Thalassaemia in Mice Lacking the Erythroid CACCC-Transcription Factor EKLF. Nature 1995, 375, 318–322. [Google Scholar] [CrossRef]
- Perkins, A.C.; Peterson, K.R.; Stamatoyannopoulos, G.; Witkowska, H.E.; Orkin, S.H. Fetal Expression of a Human Agamma Globin Transgene Rescues Globin Chain Imbalance but Not Hemolysis in EKLF Null Mouse Embryos. Blood 2000, 95, 1827–1833. [Google Scholar] [CrossRef]
- Grech, L. Control of Globin Gene Expression by Kruppel-like Factors. XJENZA Line 2014, 2, 64–71. [Google Scholar] [CrossRef]
- Pilon, A.M.; Arcasoy, M.O.; Dressman, H.K.; Vayda, S.E.; Maksimova, Y.D.; Sangerman, J.I.; Gallagher, P.G.; Bodine, D.M. Failure of Terminal Erythroid Differentiation in EKLF-Deficient Mice Is Associated with Cell Cycle Perturbation and Reduced Expression of E2F2. Mol. Cell. Biol. 2008, 28, 7394–7401. [Google Scholar] [CrossRef] [PubMed]
- Tallack, M.R.; Keys, J.R.; Humbert, P.O.; Perkins, A.C. EKLF/KLF1 Controls Cell Cycle Entry via Direct Regulation of E2f2. J. Biol. Chem. 2009, 284, 20966–20974. [Google Scholar] [CrossRef] [PubMed]
- Tallack, M.R.; Keys, J.R.; Perkins, A.C. Erythroid Kruppel-like Factor Regulates the G1 Cyclin Dependent Kinase Inhibitor P18INK4c. J. Mol. Biol. 2007, 369, 313–321. [Google Scholar] [CrossRef] [PubMed]
- Gnanapragasam, M.N.; McGrath, K.E.; Catherman, S.; Xue, L.; Palis, J.; Bieker, J.J. EKLF/KLF1-Regulated Cell Cycle Exit Is Essential for Erythroblast Enucleation. Blood 2016, 128, 1631–1641. [Google Scholar] [CrossRef]
- Donze, D.; Townes, T.M.; Bieker, J.J. Role of Erythroid Kruppel-like Factor in Human γ- to β-Globin Gene Switching. J. Biol. Chem. 1995, 270, 1955–1959. [Google Scholar] [CrossRef]
- Wijgerde, M.; Gribnau, J.; Trimborn, T.; Nuez, B.; Philipsen, S.; Grosveld, F.; Fraser, P. The Role of EKLF in Human Beta-Globin Gene Competition. Genes Dev. 1996, 10, 2894–2902. [Google Scholar] [CrossRef]
- Isern, J.; Fraser, S.T.; He, Z.; Zhang, H.; Baron, M.H. Dose-Dependent Regulation of Primitive Erythroid Maturation and Identity by the Transcription Factor Eklf. Blood 2010, 116, 3972–3980. [Google Scholar] [CrossRef]
- Basu, P.; Lung, T.K.; Lemsaddek, W.; Sargent, T.G.; Williams, D.C.; Basu, M.; Redmond, L.C.; Lingrel, J.B.; Haar, J.L.; Lloyd, J.A. EKLF and KLF2 Have Compensatory Roles in Embryonic β-Globin Gene Expression and Primitive Erythropoiesis. Blood 2007, 110, 3417–3425. [Google Scholar] [CrossRef]
- Menzel, S.; Garner, C.; Gut, I.; Matsuda, F.; Yamaguchi, M.; Heath, S.; Foglio, M.; Zelenika, D.; Boland, A.; Rooks, H.; et al. A QTL Influencing F Cell Production Maps to a Gene Encoding a Zinc-Finger Protein on Chromosome 2p15. Nat. Genet. 2007, 39, 1197–1199. [Google Scholar] [CrossRef]
- Uda, M.; Galanello, R.; Sanna, S.; Lettre, G.; Sankaran, V.G.; Chen, W.; Usala, G.; Busonero, F.; Maschio, A.; Albai, G.; et al. Genome-Wide Association Study Shows BCL11A Associated with Persistent Fetal Hemoglobin and Amelioration of the Phenotype of Beta-Thalassemia. Proc. Natl. Acad. Sci. USA 2008, 105, 1620–1625. [Google Scholar] [CrossRef] [PubMed]
- Sedgewick, A.E.; Timofeev, N.; Sebastiani, P.; So, J.C.C.; Ma, E.S.K.; Chan, L.C.; Fucharoen, G.; Fucharoen, S.; Barbosa, C.G.; Vardarajan, B.N.; et al. BCL11A Is a Major HbF Quantitative Trait Locus in Three Different Populations with Beta-Hemoglobinopathies. Blood Cells. Mol. Dis. 2008, 41, 255–258. [Google Scholar] [CrossRef] [PubMed]
- Lettre, G.; Sankaran, V.G.; Bezerra, M.A.C.; Araújo, A.S.; Uda, M.; Sanna, S.; Cao, A.; Schlessinger, D.; Costa, F.F.; Hirschhorn, J.N.; et al. DNA Polymorphisms at the BCL11A, HBS1L-MYB, and Beta-Globin Loci Associate with Fetal Hemoglobin Levels and Pain Crises in Sickle Cell Disease. Proc. Natl. Acad. Sci. USA 2008, 105, 11869–11874. [Google Scholar] [CrossRef] [PubMed]
- Sankaran, V.G.; Xu, J.; Ragoczy, T.; Ippolito, G.C.; Walkley, C.R.; Maika, S.D.; Fujiwara, Y.; Ito, M.; Groudine, M.; Bender, M.A.; et al. Developmental and Species-Divergent Globin Switching Are Driven by BCL11A. Nature 2009, 460, 1093–1097. [Google Scholar] [CrossRef]
- Esteghamat, F.; Gillemans, N.; Bilic, I.; van den Akker, E.; Cantù, I.; van Gent, T.; Klingmüller, U.; van Lom, K.; von Lindern, M.; Grosveld, F.; et al. Erythropoiesis and Globin Switching in Compound Klf1::Bcl11a Mutant Mice. Blood 2013, 121, 2553–2562. [Google Scholar] [CrossRef]
- Norton, L.J.; Funnell, A.P.W.; Burdach, J.; Wienert, B.; Kurita, R.; Nakamura, Y.; Philipsen, S.; Pearson, R.C.M.; Quinlan, K.G.R.; Crossley, M. KLF1 Directly Activates Expression of the Novel Fetal Globin Repressor ZBTB7A/LRF in Erythroid Cells. Blood Adv. 2017, 1, 685–692. [Google Scholar] [CrossRef]
- Singleton, B.K.; Burton, N.M.; Green, C.; Brady, R.L.; Anstee, D.J. Mutations in EKLF/KLF1 Form the Molecular Basis of the Rare Blood Group In(Lu) Phenotype. Blood 2008, 112, 2081–2088. [Google Scholar] [CrossRef]
- Xue, L.; Galdass, M.; Gnanapragasam, M.N.; Manwani, D.; Bieker, J.J. Extrinsic and Intrinsic Control by EKLF (KLF1) within a Specialized Erythroid Niche. Dev. Camb. Engl. 2014, 141, 2245–2254. [Google Scholar] [CrossRef]
- Arnaud, L.; Saison, C.; Helias, V.; Lucien, N.; Steschenko, D.; Giarratana, M.-C.; Prehu, C.; Foliguet, B.; Montout, L.; de Brevern, A.G.; et al. A Dominant Mutation in the Gene Encoding the Erythroid Transcription Factor KLF1 Causes a Congenital Dyserythropoietic Anemia. Am. J. Hum. Genet. 2010, 87, 721–727. [Google Scholar] [CrossRef]
- Hariharan, P.; Colah, R.; Ghosh, K.; Nadkarni, A. Differential Role of Kruppel like Factor 1 (KLF1) Gene in Red Blood Cell Disorders. Genomics 2019, 111, 1771–1776. [Google Scholar] [CrossRef]
- Ortolano, R.; Forouhar, M.; Warwick, A.; Harper, D. A Case of Congenital Dyserythropoeitic Anemia Type IV Caused by E325K Mutation in Erythroid Transcription Factor KLF1. J. Pediatr. Hematol. Oncol. 2018, 40, e389–e391. [Google Scholar] [CrossRef] [PubMed]
- Fraser, N.S.; Knauth, C.M.; Schoeman, E.M.; Moussa, A.; Perkins, A.C.; Walsh, T.; Millard, G.M.; Dean, M.M.; Hyland, C.A.; Flower, R.L. Investigation of the Variable In(Lu) Phenotype Caused by KLF1 Variants. Transfusion (Paris) 2018, 58, 2414–2420. [Google Scholar] [CrossRef] [PubMed]
- Fraser, N.S.; Knauth, C.M.; Moussa, A.; Dean, M.M.; Hyland, C.A.; Perkins, A.C.; Flower, R.L.; Schoeman, E.M. Genetic Variants Within the Erythroid Transcription Factor, KLF1, and Reduction of the Expression of Lutheran and Other Blood Group Antigens: Review of the In(Lu) Phenotype. Transfus. Med. Rev. 2019, 33, 111–117. [Google Scholar] [CrossRef]
- Magor, G.W.; Tallack, M.R.; Gillinder, K.R.; Bell, C.C.; McCallum, N.; Williams, B.; Perkins, A.C. KLF1-Null Neonates Display Hydrops Fetalis and a Deranged Erythroid Transcriptome. Blood 2015, 125, 2405–2417. [Google Scholar] [CrossRef]
- Tallack, M.R.; Whitington, T.; Yuen, W.S.; Wainwright, E.N.; Keys, J.R.; Gardiner, B.B.; Nourbakhsh, E.; Cloonan, N.; Grimmond, S.M.; Bailey, T.L.; et al. A Global Role for KLF1 in Erythropoiesis Revealed by ChIP-Seq in Primary Erythroid Cells. Genome Res. 2010, 20, 1052–1063. [Google Scholar] [CrossRef] [PubMed]
- Su, M.Y.; Steiner, L.A.; Bogardus, H.; Mishra, T.; Schulz, V.P.; Hardison, R.C.; Gallagher, P.G. Identification of Biologically Relevant Enhancers in Human Erythroid Cells. J. Biol. Chem. 2013, 288, 8433–8444. [Google Scholar] [CrossRef] [PubMed]
- Porcu, S.; Manchinu, M.F.; Marongiu, M.F.; Sogos, V.; Poddie, D.; Asunis, I.; Porcu, L.; Marini, M.G.; Moi, P.; Cao, A.; et al. Klf1 Affects DNase II-Alpha Expression in the Central Macrophage of a Fetal Liver Erythroblastic Island: A Non-Cell-Autonomous Role in Definitive Erythropoiesis. Mol. Cell. Biol. 2011, 31, 4144–4154. [Google Scholar] [CrossRef]
- Yu, X.; Kong, Y.; Dore, L.C.; Abdulmalik, O.; Katein, A.M.; Zhou, S.; Choi, J.K.; Gell, D.; Mackay, J.P.; Gow, A.J.; et al. An Erythroid Chaperone That Facilitates Folding of Alpha-Globin Subunits for Hemoglobin Synthesis. J. Clin. Investig. 2007, 117, 1856–1865. [Google Scholar] [CrossRef]
- Keys, J.R.; Tallack, M.R.; Hodge, D.J.; Cridland, S.O.; David, R.; Perkins, A.C. Genomic Organisation and Regulation of Murine Alpha Haemoglobin Stabilising Protein by Erythroid Kruppel-like Factor. Br. J. Haematol. 2007, 136, 150–157. [Google Scholar] [CrossRef]
- Khanna, R.; Chang, S.H.; Andrabi, S.; Azam, M.; Kim, A.; Rivera, A.; Brugnara, C.; Low, P.S.; Liu, S.-C.; Chishti, A.H. Headpiece Domain of Dematin Is Required for the Stability of the Erythrocyte Membrane. Proc. Natl. Acad. Sci. USA 2002, 99, 6637–6642. [Google Scholar] [CrossRef] [Green Version]
- Hung, C.-H.; Wang, K.-Y.; Liou, Y.-H.; Wang, J.-P.; Huang, A.Y.-S.; Lee, T.-L.; Jiang, S.-T.; Liao, N.-S.; Shyu, Y.-C.; Shen, C.-K.J. Negative Regulation of the Differentiation of Flk2− CD34− LSK Hematopoietic Stem Cells by EKLF/KLF1. Int. J. Mol. Sci. 2020, 21, 8448. [Google Scholar] [CrossRef] [PubMed]
- Frontelo, P.; Manwani, D.; Galdass, M.; Karsunky, H.; Lohmann, F.; Gallagher, P.G.; Bieker, J.J. Novel Role for EKLF in Megakaryocyte Lineage Commitment. Blood 2007, 110, 3871–3880. [Google Scholar] [CrossRef] [PubMed]
- Crossley, M.; Tsang, A.P.; Bieker, J.J.; Orkin, S.H. Regulation of the Erythroid Kruppel-like Factor (EKLF) Gene Promoter by the Erythroid Transcription Factor GATA-1. J. Biol. Chem. 1994, 269, 15440–15444. [Google Scholar] [CrossRef]
- Wong, P.M.; Chung, S.W.; Chui, D.H.; Eaves, C.J. Properties of the Earliest Clonogenic Hemopoietic Precursors to Appear in the Developing Murine Yolk Sac. Proc. Natl. Acad. Sci. USA 1986, 83, 3851–3854. [Google Scholar] [CrossRef]
- Palis, J.; Robertson, S.; Kennedy, M.; Wall, C.; Keller, G. Development of Erythroid and Myeloid Progenitors in the Yolk Sac and Embryo Proper of the Mouse. Development 1999, 126, 5073–5084. [Google Scholar] [CrossRef]
- Palis, J. Primitive and Definitive Erythropoiesis in Mammals. Front. Physiol. 2014, 5. [Google Scholar] [CrossRef]
- Allen, T.D.; Dexter, T.M. Ultrastructural Aspects of Erythropoietic Differentiation in Long-Term Bone Marrow Culture. Differentiation 1982, 21, 86–94. [Google Scholar] [CrossRef]
- Manwani, D.; Bieker, J.J. Chapter 2 The Erythroblastic Island. In Current Topics in Developmental Biology; Elsevier: Amsterdam, The Netherlands, 2008; Volume 82, pp. 23–53. ISBN 978-0-12-374366-4. [Google Scholar]
- Southwood, C.M.; Downs, K.M.; Bieker, J.J. Erythroid Krüppel-like Factor Exhibits an Early and Sequentially Localized Pattern of Expression during Mammalian Erythroid Ontogeny. Dev. Dyn. 1996, 206, 248–259. [Google Scholar] [CrossRef]
- Basu, P.; Morris, P.E.; Haar, J.L.; Wani, M.A.; Lingrel, J.B.; Gaensler, K.M.L.; Lloyd, J.A. KLF2 Is Essential for Primitive Erythropoiesis and Regulates the Human and Murine Embryonic β-like Globin Genes in Vivo. Blood 2005, 106, 2566–2571. [Google Scholar] [CrossRef]
- Kuo, C.T.; Veselits, M.L.; Barton, K.P.; Lu, M.M.; Clendenin, C.; Leiden, J.M. The LKLF Transcription Factor Is Required for Normal Tunica Media Formation and Blood Vessel Stabilization during Murine Embryogenesis. Genes Dev. 1997, 11, 2996–3006. [Google Scholar] [CrossRef] [Green Version]
- Wani, M.A.; Means, R.T.; Lingrel, J.B. Loss of LKLF Function Results in Embryonic Lethality in Mice. Transgenic Res. 1998, 7, 229–238. [Google Scholar] [CrossRef] [PubMed]
- Pang, C.J.; Lemsaddek, W.; Alhashem, Y.N.; Bondzi, C.; Redmond, L.C.; Ah-Son, N.; Dumur, C.I.; Archer, K.J.; Haar, J.L.; Lloyd, J.A.; et al. Krüppel-Like Factor 1 (KLF1), KLF2, and Myc Control a Regulatory Network Essential for Embryonic Erythropoiesis. Mol. Cell. Biol. 2012, 32, 2628–2644. [Google Scholar] [CrossRef] [PubMed]
- Kina, T.; Ikuta, K.; Takayama, E.; Wada, K.; Majumdar, A.S.; Weissman, I.L.; Katsura, Y. The Monoclonal Antibody TER-119 Recognizes a Molecule Associated with Glycophorin A and Specifically Marks the Late Stages of Murine Erythroid Lineage: Characterization of the Monoclonal Antibody TER-119. Br. J. Haematol. 2000, 109, 280–287. [Google Scholar] [CrossRef] [PubMed]
- Koulnis, M.; Pop, R.; Porpiglia, E.; Shearstone, J.R.; Hidalgo, D.; Socolovsky, M. Identification and Analysis of Mouse Erythroid Progenitors Using the CD71/TER119 Flow-Cytometric Assay. J. Vis. Exp. 2011, e2809. [Google Scholar] [CrossRef]
- Drissen, R.; von Lindern, M.; Kolbus, A.; Driegen, S.; Steinlein, P.; Beug, H.; Grosveld, F.; Philipsen, S. The Erythroid Phenotype of EKLF-Null Mice: Defects in Hemoglobin Metabolism and Membrane Stability. Mol. Cell. Biol. 2005, 25, 5205–5214. [Google Scholar] [CrossRef]
- Fischer, M.; Schade, A.E.; Branigan, T.B.; Müller, G.A.; DeCaprio, J.A. Coordinating Gene Expression during the Cell Cycle. Trends Biochem. Sci. 2022, in press. [Google Scholar] [CrossRef]
- Manchinu, M.F.; Brancia, C.; Caria, C.A.; Musu, E.; Porcu, S.; Simbula, M.; Asunis, I.; Perseu, L.; Ristaldi, M.S. Deficiency in Interferon Type 1 Receptor Improves Definitive Erythropoiesis in Klf1 Null Mice. Cell Death Differ. 2018, 25, 589–599. [Google Scholar] [CrossRef]
- Luo, Q.; Ma, X.; Wahl, S.M.; Bieker, J.J.; Crossley, M.; Montaner, L.J. Activation and Repression of Interleukin-12 P40 Transcription by Erythroid Kruppel-like Factor in Macrophages. J. Biol. Chem. 2004, 279, 18451–18456. [Google Scholar] [CrossRef]
- Mukherjee, K.; Xue, L.; Planutis, A.; Gnanapragasam, M.N.; Chess, A.; Bieker, J.J. EKLF/KLF1 Expression Defines a Unique Macrophage Subset during Mouse Erythropoiesis. eLife 2021, 10, e61070. [Google Scholar] [CrossRef]
- Li, W.; Wang, Y.; Zhao, H.; Zhang, H.; Xu, Y.; Wang, S.; Guo, X.; Huang, Y.; Zhang, S.; Han, Y.; et al. Identification and Transcriptome Analysis of Erythroblastic Island Macrophages. Blood 2019, 134, 480–491. [Google Scholar] [CrossRef]
- Lopez-Yrigoyen, M.; Yang, C.-T.; Fidanza, A.; Cassetta, L.; Taylor, A.H.; McCahill, A.; Sellink, E.; von Lindern, M.; van den Akker, E.; Mountford, J.C.; et al. Genetic Programming of Macrophages Generates an in Vitro Model for the Human Erythroid Island Niche. Nat. Commun. 2019, 10, 881. [Google Scholar] [CrossRef] [PubMed]
- Nagata, S. DNA Degradation in Development and Programmed Cell Death. Annu. Rev. Immunol. 2005, 23, 853–875. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H.; Okabe, Y.; Kawane, K.; Fukuyama, H.; Nagata, S. Lethal Anemia Caused by Interferon-Beta Produced in Mouse Embryos Carrying Undigested DNA. Nat. Immunol. 2005, 6, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Tewari, R. Erythroid Kruppel-like Factor (EKLF) Is Active in Primitive and Definitive Erythroid Cells and Is Required for the Function of 5′HS3 of the Beta -Globin Locus Control Region. EMBO J. 1998, 17, 2334–2341. [Google Scholar] [CrossRef]
- Tallack, M.R.; Perkins, A.C. Megakaryocyte-Erythroid Lineage Promiscuity in EKLF Null Mouse Blood. Haematologica 2010, 95, 144–147. [Google Scholar] [CrossRef]
- Starck, J.; Cohet, N.; Gonnet, C.; Sarrazin, S.; Doubeikovskaia, Z.; Doubeikovski, A.; Verger, A.; Duterque-Coquillaud, M.; Morle, F. Functional Cross-Antagonism between Transcription Factors FLI-1 and EKLF. Mol. Cell. Biol. 2003, 23, 1390–1402. [Google Scholar] [CrossRef]
- Bouilloux, F.; Juban, G.; Cohet, N.; Buet, D.; Guyot, B.; Vainchenker, W.; Louache, F.; Morlé, F. EKLF Restricts Megakaryocytic Differentiation at the Benefit of Erythrocytic Differentiation. Blood 2008, 112, 576–584. [Google Scholar] [CrossRef]
- Kuvardina, O.N.; Herglotz, J.; Kolodziej, S.; Kohrs, N.; Herkt, S.; Wojcik, B.; Oellerich, T.; Corso, J.; Behrens, K.; Kumar, A.; et al. RUNX1 Represses the Erythroid Gene Expression Program during Megakaryocytic Differentiation. Blood 2015, 125, 3570–3579. [Google Scholar] [CrossRef]
- Sankaran, V.G.; Xu, J.; Orkin, S.H. Advances in the Understanding of Haemoglobin Switching. Br. J. Haematol. 2010, 149, 181–194. [Google Scholar] [CrossRef]
- Peschle, C.; Mavilio, F.; Carè, A.; Migliaccio, G.; Migliaccio, A.R.; Salvo, G.; Samoggia, P.; Petti, S.; Guerriero, R.; Marinucci, M. Haemoglobin Switching in Human Embryos: Asynchrony of Zeta—Alpha and Epsilon—Gamma-Globin Switches in Primitive and Definite Erythropoietic Lineage. Nature 1985, 313, 235–238. [Google Scholar] [CrossRef]
- Whitelaw, E.; Lamb, P.; Hogben, P.; Proudfoot, N.J. The Globin Switch at the Level of MRNA in the Developing Mouse. Prog. Clin. Biol. Res. 1989, 316A, 323–333. [Google Scholar] [PubMed]
- Kingsley, P.D.; Malik, J.; Emerson, R.L.; Bushnell, T.P.; McGrath, K.E.; Bloedorn, L.A.; Bulger, M.; Palis, J. “Maturational” Globin Switching in Primary Primitive Erythroid Cells. Blood 2006, 107, 1665–1672. [Google Scholar] [CrossRef] [PubMed]
- Sankaran, V.G.; Orkin, S.H. The Switch from Fetal to Adult Hemoglobin. Cold Spring Harb. Perspect. Med. 2013, 3, a011643. [Google Scholar] [CrossRef] [PubMed]
- McColl, B.; Kao, B.R.; Lourthai, P.; Chan, K.; Wardan, H.; Roosjen, M.; Delagneau, O.; Gearing, L.J.; Blewitt, M.E.; Svasti, S.; et al. An in Vivo Model for Analysis of Developmental Erythropoiesis and Globin Gene Regulation. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2014, 28, 2306–2317. [Google Scholar] [CrossRef]
- Perkins, A.C.; Gaensler, K.M.; Orkin, S.H. Silencing of Human Fetal Globin Expression Is Impaired in the Absence of the Adult Beta-Globin Gene Activator Protein EKLF. Proc. Natl. Acad. Sci. USA 1996, 93, 12267–12271. [Google Scholar] [CrossRef]
- Bank, A. Regulation of Human Fetal Hemoglobin: New Players, New Complexities. Blood 2006, 107, 435–443. [Google Scholar] [CrossRef]
- Kim, Y.W.; Yun, W.J.; Kim, A. Erythroid Activator NF-E2, TAL1 and KLF1 Play Roles in Forming the LCR HSs in the Human Adult β-Globin Locus. Int. J. Biochem. Cell Biol. 2016, 75, 45–52. [Google Scholar] [CrossRef]
- Topfer, S.K.; Feng, R.; Huang, P.; Ly, L.C.; Martyn, G.E.; Blobel, G.A.; Weiss, M.J.; Quinlan, K.G.R.; Crossley, M. Disrupting the Adult Globin Promoter Alleviates Promoter Competition and Reactivates Fetal Globin Gene Expression. Blood 2022, 139, 2107–2118. [Google Scholar] [CrossRef]
- Starlard-Davenport, A.; Gu, Q.; Pace, B.S. Targeting Genetic Modifiers of HBG Gene Expression in Sickle Cell Disease: The MiRNA Option. Mol. Diagn. Ther. 2022, 26, 497–509. [Google Scholar] [CrossRef]
- Li, Y.; Liu, D.; Zhang, X.; Li, Z.; Ye, Y.; Liu, Q.; Shen, J.; Chen, Z.; Huang, H.; Liang, Y.; et al. MiR-326 Regulates HbF Synthesis by Targeting EKLF in Human Erythroid Cells. Exp. Hematol. 2018, 63, 33–40.e2. [Google Scholar] [CrossRef]
- Ward, C.M.; Li, B.; Pace, B.S. Original Research: Stable Expression of MiR-34a Mediates Fetal Hemoglobin Induction in K562 Cells. Exp. Biol. Med. 2016, 241, 719–729. [Google Scholar] [CrossRef] [PubMed]
- Obeidi, N.; Pourfathollah, A.A.; Soleimani, M.; Nikougoftar Zarif, M.; Kouhkan, F. The Effect of Mir-451 Upregulation on Erythroid Lineage Differentiation of Murine Embryonic Stem Cells. Cell J. 2016, 18, 165–178. [Google Scholar] [CrossRef] [PubMed]
- Rankine-Mullings, A.E.; Nevitt, S.J. Hydroxyurea (Hydroxycarbamide) for Sickle Cell Disease. Cochrane Database Syst. Rev. 2022, 9, CD002202. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Sun, G.; Wu, P.; Chen, C.; Kuang, Y.; Liu, L.; Zheng, Z.; He, Y.; Gu, Q.; Lu, T.; et al. WDR82-Binding Long Noncoding RNA LncEry Controls Mouse Erythroid Differentiation and Maturation. J. Exp. Med. 2022, 219, e20211688. [Google Scholar] [CrossRef]
- Perkins, A.; Xu, X.; Higgs, D.R.; Patrinos, G.P.; Arnaud, L.; Bieker, J.J.; Philipsen, S. KLF1 Consensus Workgroup Krüppeling Erythropoiesis: An Unexpected Broad Spectrum of Human Red Blood Cell Disorders Due to KLF1 Variants. Blood 2016, 127, 1856–1862. [Google Scholar] [CrossRef]
- Jaffray, J.A.; Mitchell, W.B.; Gnanapragasam, M.N.; Seshan, S.V.; Guo, X.; Westhoff, C.M.; Bieker, J.J.; Manwani, D. Erythroid Transcription Factor EKLF/KLF1 Mutation Causing Congenital Dyserythropoietic Anemia Type IV in a Patient of Taiwanese Origin: Review of All Reported Cases and Development of a Clinical Diagnostic Paradigm. Blood Cells. Mol. Dis. 2013, 51, 71–75. [Google Scholar] [CrossRef]
- Borg, J.; Papadopoulos, P.; Georgitsi, M.; Gutiérrez, L.; Grech, G.; Fanis, P.; Phylactides, M.; Verkerk, A.J.M.H.; van der Spek, P.J.; Scerri, C.A.; et al. Haploinsufficiency for the Erythroid Transcription Factor KLF1 Causes Hereditary Persistence of Fetal Hemoglobin. Nat. Genet. 2010, 42, 801–805. [Google Scholar] [CrossRef]
- Perseu, L.; Satta, S.; Moi, P.; Demartis, F.R.; Manunza, L.; Sollaino, M.C.; Barella, S.; Cao, A.; Galanello, R. KLF1 Gene Mutations Cause Borderline HbA(2). Blood 2011, 118, 4454–4458. [Google Scholar] [CrossRef]
- Satta, S.; Perseu, L.; Moi, P.; Asunis, I.; Cabriolu, A.; Maccioni, L.; Demartis, F.R.; Manunza, L.; Cao, A.; Galanello, R. Compound Heterozygosity for KLF1 Mutations Associated with Remarkable Increase of Fetal Hemoglobin and Red Cell Protoporphyrin. Haematologica 2011, 96, 767–770. [Google Scholar] [CrossRef]
- Xu, L.; Zhu, D.; Zhang, Y.; Liang, G.; Liang, M.; Wei, X.; Feng, X.; Wu, X.; Shang, X. Compound Heterozygosity for KLF1 Mutations Causing Hemolytic Anemia in Children: A Case Report and Literature Review. Front. Genet. 2021, 12, 691461. [Google Scholar] [CrossRef]
- Huang, J.; Zhang, X.; Liu, D.; Wei, X.; Shang, X.; Xiong, F.; Yu, L.; Yin, X.; Xu, X. Compound Heterozygosity for KLF1 Mutations Is Associated with Microcytic Hypochromic Anemia and Increased Fetal Hemoglobin. Eur. J. Hum. Genet. 2015, 23, 1341–1348. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Jiang, F.; Li, J.; Tang, F.; Li, D.-Z. Congenital Nonspherocytic Hemolytic Anemia Caused by Krüppel-Like Factor 1 Gene Variants: Another Case Report. Hemoglobin 2019, 43, 292–295. [Google Scholar] [CrossRef] [PubMed]
- Tangsricharoen, T.; Natesirinilkul, R.; Phusua, A.; Fanhchaksai, K.; Ittiwut, C.; Chetruengchai, W.; Juntharaniyom, M.; Charoenkwan, P.; Viprakasit, V.; Phokaew, C.; et al. Severe Neonatal Haemolytic Anaemia Caused by Compound Heterozygous KLF1 Mutations: Report of Four Families and Literature Review. Br. J. Haematol. 2021, 194, 626–634. [Google Scholar] [CrossRef] [PubMed]
- Viprakasit, V.; Ekwattanakit, S.; Riolueang, S.; Chalaow, N.; Fisher, C.; Lower, K.; Kanno, H.; Tachavanich, K.; Bejrachandra, S.; Saipin, J.; et al. Mutations in Kruppel-like Factor 1 Cause Transfusion-Dependent Hemolytic Anemia and Persistence of Embryonic Globin Gene Expression. Blood 2014, 123, 1586–1595. [Google Scholar] [CrossRef]
- Helias, V.; Saison, C.; Peyrard, T.; Vera, E.; Prehu, C.; Cartron, J.-P.; Arnaud, L. Molecular Analysis of the Rare In(Lu) Blood Type: Toward Decoding the Phenotypic Outcome of Haploinsufficiency for the Transcription Factor KLF1. Hum. Mutat. 2013, 34, 221–228. [Google Scholar] [CrossRef]
- Keller, J.; Vege, S.; Horn, T.; Keller, M.A.; Leger, R.M.; Aeschlimann, J.; Lomas-Francis, C.; Westhoff, C.M. Novel Mutations in KLF1 Encoding the In(Lu) Phenotype Reflect a Diversity of Clinical Presentations. Transfusion (Paris) 2018, 58, 196–199. [Google Scholar] [CrossRef]
- Funnell, A.P.W.; Maloney, C.A.; Thompson, L.J.; Keys, J.; Tallack, M.; Perkins, A.C.; Crossley, M. Erythroid Krüppel-Like Factor Directly Activates the Basic Krüppel-Like Factor Gene in Erythroid Cells. Mol. Cell. Biol. 2007, 27, 2777–2790. [Google Scholar] [CrossRef]
- Nilson, D.G.; Sabatino, D.E.; Bodine, D.M.; Gallagher, P.G. Major Erythrocyte Membrane Protein Genes in EKLF-Deficient Mice. Exp. Hematol. 2006, 34, 705–712. [Google Scholar] [CrossRef]
- Singleton, B.K.; Lau, W.; Fairweather, V.S.S.; Burton, N.M.; Wilson, M.C.; Parsons, S.F.; Richardson, B.M.; Trakarnsanga, K.; Brady, R.L.; Anstee, D.J.; et al. Mutations in the Second Zinc Finger of Human EKLF Reduce Promoter Affinity but Give Rise to Benign and Disease Phenotypes. Blood 2011, 118, 3137–3145. [Google Scholar] [CrossRef]
- Feng, W.C.; Southwood, C.M.; Bieker, J.J. Analyses of Beta-Thalassemia Mutant DNA Interactions with Erythroid Krüppel-like Factor (EKLF), an Erythroid Cell-Specific Transcription Factor. J. Biol. Chem. 1994, 269, 1493–1500. [Google Scholar] [CrossRef]
- Huisman, T.H.J. Levels of Hb A2 in Heterozygotes and Homozygotes for Beta-Thalassemia Mutations: Influence of Mutations in the CACCC and ATAAA Motifs of the Beta-Globin Gene Promoter. Acta Haematol. 1997, 98, 187–194. [Google Scholar] [CrossRef] [PubMed]
- Wienert, B.; Martyn, G.E.; Kurita, R.; Nakamura, Y.; Quinlan, K.G.R.; Crossley, M. KLF1 Drives the Expression of Fetal Hemoglobin in British HPFH. Blood 2017, 130, 803–807. [Google Scholar] [CrossRef] [PubMed]
- Iolascon, A.; Russo, R.; Delaunay, J. Congenital Dyserythropoietic Anemias. Curr. Opin. Hematol. 2011, 18, 146–151. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, P.; Fermo, E.; Vercellati, C.; Boschetti, C.; Barcellini, W.; Iurlo, A.; Marcello, A.P.; Righetti, P.G.; Zanella, A. Congenital Dyserythropoietic Anemia Type II (CDAII) Is Caused by Mutations in the SEC23B Gene. Hum. Mutat. 2009, 30, 1292–1298. [Google Scholar] [CrossRef] [PubMed]
- Renella, R.; Roberts, N.A.; Brown, J.M.; De Gobbi, M.; Bird, L.E.; Hassanali, T.; Sharpe, J.A.; Sloane-Stanley, J.; Ferguson, D.J.P.; Cordell, J.; et al. Codanin-1 Mutations in Congenital Dyserythropoietic Anemia Type 1 Affect HP1{alpha} Localization in Erythroblasts. Blood 2011, 117, 6928–6938. [Google Scholar] [CrossRef]
- Liljeholm, M.; Irvine, A.F.; Vikberg, A.-L.; Norberg, A.; Month, S.; Sandström, H.; Wahlin, A.; Mishima, M.; Golovleva, I. Congenital Dyserythropoietic Anemia Type III (CDA III) Is Caused by a Mutation in Kinesin Family Member, KIF23. Blood 2013, 121, 4791–4799. [Google Scholar] [CrossRef]
- Ravindranath, Y.; Johnson, R.M.; Goyette, G.; Buck, S.; Gadgeel, M.; Gallagher, P.G. KLF1 E325K-Associated Congenital Dyserythropoietic Anemia Type IV: Insights Into the Variable Clinical Severity. J. Pediatr. Hematol. Oncol. 2018, 40, e405–e409. [Google Scholar] [CrossRef]
- Heruth, D.P.; Hawkins, T.; Logsdon, D.P.; Gibson, M.I.; Sokolovsky, I.V.; Nsumu, N.N.; Major, S.L.; Fegley, B.; Woods, G.M.; Lewing, K.B.; et al. Mutation in Erythroid Specific Transcription Factor KLF1 Causes Hereditary Spherocytosis in the Nan Hemolytic Anemia Mouse Model. Genomics 2010, 96, 303–307. [Google Scholar] [CrossRef]
- Siatecka, M.; Sahr, K.E.; Andersen, S.G.; Mezei, M.; Bieker, J.J.; Peters, L.L. Severe Anemia in the Nan Mutant Mouse Caused by Sequence-Selective Disruption of Erythroid Kruppel-like Factor. Proc. Natl. Acad. Sci. USA 2010, 107, 15151–15156. [Google Scholar] [CrossRef]
- Planutis, A.; Xue, L.; Trainor, C.D.; Dangeti, M.; Gillinder, K.; Siatecka, M.; Nebor, D.; Peters, L.L.; Perkins, A.C.; Bieker, J.J. Neomorphic Effects of the Neonatal Anemia (Nan-Eklf) Mutation Contribute to Deficits throughout Development. Dev. Camb. Engl. 2017, 144, 430–440. [Google Scholar] [CrossRef] [Green Version]
- Nébor, D.; Graber, J.H.; Ciciotte, S.L.; Robledo, R.F.; Papoin, J.; Hartman, E.; Gillinder, K.R.; Perkins, A.C.; Bieker, J.J.; Blanc, L.; et al. Mutant KLF1 in Adult Anemic Nan Mice Leads to Profound Transcriptome Changes and Disordered Erythropoiesis. Sci. Rep. 2018, 8, 12793. [Google Scholar] [CrossRef] [PubMed]
- Gillinder, K.R.; Ilsley, M.D.; Nébor, D.; Sachidanandam, R.; Lajoie, M.; Magor, G.W.; Tallack, M.R.; Bailey, T.; Landsberg, M.J.; Mackay, J.P.; et al. Promiscuous DNA-Binding of a Mutant Zinc Finger Protein Corrupts the Transcriptome and Diminishes Cell Viability. Nucleic Acids Res. 2017, 45, 1130–1143. [Google Scholar] [CrossRef] [PubMed]
- Ilsley, M.D.; Huang, S.; Magor, G.W.; Landsberg, M.J.; Gillinder, K.R.; Perkins, A.C. Corrupted DNA-Binding Specificity and Ectopic Transcription Underpin Dominant Neomorphic Mutations in KLF/SP Transcription Factors. BMC Genomics 2019, 20, 417. [Google Scholar] [CrossRef] [PubMed]
- Kulczynska-Figurny, K.; Bieker, J.J.; Siatecka, M. Severe Anemia Caused by Dominant Mutations in Krüppel-like Factor 1 (KLF1). Mutat. Res. Rev. Mutat. Res. 2020, 786, 108336. [Google Scholar] [CrossRef]
- Pilon, A.M.; Ajay, S.S.; Kumar, S.A.; Steiner, L.A.; Cherukuri, P.F.; Wincovitch, S.; Anderson, S.M.; Mullikin, J.C.; Gallagher, P.G.; NISC Comparative Sequencing Center; et al. Genome-Wide ChIP-Seq Reveals a Dramatic Shift in the Binding of the Transcription Factor Erythroid Kruppel-like Factor during Erythrocyte Differentiation. Blood 2011, 118, e139–e148. [Google Scholar] [CrossRef]
- dos Santos, C.O.; Costa, F.F. AHSP and Beta-Thalassemia: A Possible Genetic Modifier. Hematol. Amst. Neth. 2005, 10, 157–161. [Google Scholar] [CrossRef]
- Che Yaacob, N.S.; Islam, M.A.; Alsaleh, H.; Ibrahim, I.K.; Hassan, R. Alpha-Hemoglobin-Stabilizing Protein (AHSP): A Modulatory Factor in β-Thalassemia. Int. J. Hematol. 2020, 111, 352–359. [Google Scholar] [CrossRef]
- Lee, H.H.L.; Mak, A.S.L.; Kou, K.O.; Poon, C.F.; Wong, W.S.; Chiu, K.H.; Au, P.K.C.; Chan, K.Y.K.; Kan, A.S.Y.; Tang, M.H.Y.; et al. An Unusual Hydrops Fetalis Associated with Compound Heterozygosity for Krüppel-like Factor 1 Mutations. Hemoglobin 2016, 40, 431–434. [Google Scholar] [CrossRef]
- Desgardin, A.D.; Abramova, T.; Rosanwo, T.O.; Kartha, S.; Shim, E.-H.; Jane, S.M.; Cunningham, J.M. Regulation of Delta-Aminolevulinic Acid Dehydratase by Krüppel-Like Factor 1. PLoS ONE 2012, 7, e46482. [Google Scholar] [CrossRef]
- White, R.A.; Sokolovsky, I.V.; Britt, M.I.; Nsumu, N.N.; Logsdon, D.P.; McNulty, S.G.; Wilmes, L.A.; Brewer, B.P.; Wirtz, E.; Joyce, H.R.; et al. Hematologic Characterization and Chromosomal Localization of the Novel Dominantly Inherited Mouse Hemolytic Anemia, Neonatal Anemia (Nan). Blood Cells. Mol. Dis. 2009, 43, 141–148. [Google Scholar] [CrossRef]
- Wienert, B.; Funnell, A.P.W.; Norton, L.J.; Pearson, R.C.M.; Wilkinson-White, L.E.; Lester, K.; Vadolas, J.; Porteus, M.H.; Matthews, J.M.; Quinlan, K.G.R.; et al. Editing the Genome to Introduce a Beneficial Naturally Occurring Mutation Associated with Increased Fetal Globin. Nat. Commun. 2015, 6, 7085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
| Mutation Type | Nucleotide Change | Predicted Amino Acid Change |
|---|---|---|
| Regulatory | -154C > T | |
| Regulatory | -124T > C | |
| Nonsense | c.89G > A | Trp30Term |
| Nonsense | c.172C > T | Gln58Term |
| Nonsense | c.380T > A | Leu127Term |
| Missense | c.544T > C | Phe182Leu |
| Small insertion | c.526_527insCGGCGCC | Gly176AlafsX179 |
| Small insertion | c.519_525dupCGGCGCC | Gly176ArgfsX179 |
| Small deletion | c.569delC | Pro190LeufsX47 |
| Nonsense | c.809C > A | Ser270Term |
| Nonsense | c.862A > T | Lys288Term |
| Nonsense | c.874A > T | Lys292Term |
| Missense | c.892G > C | Ala298Pro |
| Missense | c.895C > T | His299Tyr |
| Small insertion | c.954dupG | Arg319GlufsX34 |
| Missense | c.902G > A | Arg301His |
| Missense | c.973G > A | Glu325Lys |
| Missense | c.983G > T | Arg328Leu |
| Missense | c.983G > A | Arg328His |
| Missense | c.991C > T | Arg331Gly |
| Missense | c.991C > T | Arg331Trp |
| Missense | c.994A > C | Lys332Gln |
| Missense | c.1003G > A | Gly335Arg |
| Missense | c.1012C > T | Pro338Ser |
| Missense | c.1012C > A | Pro338Thr |
| Monoallelic mutations genotypes-phenotypes | ||
| Genotypes | Phenotypes | References |
| Lys292Term/WT | Blood group variant In(Lu) | [27] |
| Arg319GlufsX34/WT | Blood group variant In(Lu) | [27] |
| Pro190LeufsX47/WT | Blood group variant In(Lu) | [27] |
| His299Tyr/WT | Blood group variant In(Lu) | [27] |
| Arg328Leu/WT | Blood group variant In(Lu) | [27] |
| Arg328His/WT | Blood group variant In(Lu) | [27] |
| Arg331Gly/WT | Blood group variant In(Lu) | [27] |
| Leu127Term/WT | Blood group variant In(Lu) | [27] |
| -124T>C/WT | Blood group variant In(Lu) | [27] |
| Lys288Ter/WT | HPFH | [88] |
| Ser270Term/WT | Increased HbA2 levels | [89] |
| Glu325Lys/WT | Type IV CDA | [29] |
| Compound heterozygous mutations genotypes-phenotypes | ||
| Genotypes | Phenotypes | References |
| p.S270X/p.K332Q | HPFH and red cell protoporphyrin | [90] |
| Trp30Term/Arg319GlufsX34 | Hydrops fetalis | [34] |
| Ala298Pro/Gly176AlafsX179 | NSHA | [91] |
| Pro338Ser/Gly176ArgfsX179 | NSHA | [92] |
| Pro338Thr/Gly176ArgfsX179 | NSHA | [93] |
| Gly335Arg/Gly176ArgfsX179 | NSHA | [94] |
| Arg331Trp/Gly335Arg | NSHA | [95] |
| Arg301His/Gly176ArgfsX179 | NSHA | [94] |
| -154 C > T/Ala298Pro | NSHA | [95] |
| Gln58Ter/Ala298Pro | NSHA | [95] |
| Ala298Pro/Gly176ArgfsX179 | NSHA and pyruvate kinase deficiency | [95] |
| Homozygous mutation genotype-phenotype | ||
| Genotype | Phenotype | References |
| Phe182Leu/Phe182Leu | Increased HbA2 levels | [30] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Caria, C.A.; Faà, V.; Ristaldi, M.S. Krüppel-Like Factor 1: A Pivotal Gene Regulator in Erythropoiesis. Cells 2022, 11, 3069. https://doi.org/10.3390/cells11193069
Caria CA, Faà V, Ristaldi MS. Krüppel-Like Factor 1: A Pivotal Gene Regulator in Erythropoiesis. Cells. 2022; 11(19):3069. https://doi.org/10.3390/cells11193069
Chicago/Turabian StyleCaria, Cristian Antonio, Valeria Faà, and Maria Serafina Ristaldi. 2022. "Krüppel-Like Factor 1: A Pivotal Gene Regulator in Erythropoiesis" Cells 11, no. 19: 3069. https://doi.org/10.3390/cells11193069