Abstract
Background: Platelets can support cancer progression via the release of microparticles and microvesicles that enhance the migratory behaviour of recipient cancer cells. We recently showed that platelet-derived extracellular vesicles (PEVs) stimulate migration and invasiveness in highly metastatic MDA-MB-231 cells by stimulating the phosphorylation of p38 MAPK and the myosin light chain 2 (MLC2). Herein, we assessed whether the pro-migratory effect of PEVs involves the remodelling of the Ca2+ handling machinery, which drives MDA-MB-231 cell motility. Methods: PEVs were isolated from human blood platelets, and Fura-2/AM Ca2+ imaging, RT-qPCR, and immunoblotting were exploited to assess their effect on intracellular Ca2+ dynamics and Ca2+-dependent migratory processes in MDA-MB-231 cells. Results: Pretreating MDA-MB-231 cells with PEVs for 24 h caused an increase in Ca2+ release from the endoplasmic reticulum (ER) due to the up-regulation of SERCA2B and InsP3R1/InsP3R2 mRNAs and proteins. The consequent enhancement of ER Ca2+ depletion led to a significant increase in store-operated Ca2+ entry. The larger Ca2+ mobilization from the ER was required to potentiate serum-induced migration by recruiting p38 MAPK and MLC2. Conclusions: PEVs stimulate migration in the highly metastatic MDA-MB-231 breast cancer cell line by inducing a partial remodelling of the Ca2+ handling machinery.
1. Introduction
Breast cancer represents the most widespread cancer in women and accounted for about 685.000 deaths in 2020. This death toll has been forecasted to rise to 7 million by 2040 [1]. Triple negative breast cancer (TNBC) is featured by the absence of the human epidermal growth factor receptor 2 (HER2), estrogen receptor, and progesterone receptor. Therefore, TNBC is barely sensitive to anti-HER2 and hormonal therapies, with poor survival rates [2]. Accordingly, TNBC presents high aggressiveness and short median time to relapse due to its ability to leave the primary tumour site and spread to distant organs, thereby causing patient death [3]. Understanding the cellular and molecular mechanisms that stimulate TNBC cells to migrate and colonize their new niches is indispensable when it comes to designing alternative treatments for TNBC patients.
Platelet-derived extracellular vesicles (PEVs) are gaining growing interest as mediators of platelet function in different physiological and pathological contexts, from haemostasis to cardiovascular diseases [4,5]. In the last two decades, PEVs have also been recognized as critical players in the complex interplay occurring between blood platelets and cancer [6]. Exosomes and plasma membrane-derived vesicles are the two main classes of PEVs released by platelets into the bloodstream [4]. Platelet exosomes (also known as small PEVs) are stored in intracellular multivesicular bodies and released during cell secretion. Conversely, membrane-derived vesicles derive from the plasma membrane and are now typically defined as medium-large PEVs, but they were previously referred to as platelet-derived microparticles (PMPs) and platelet-derived microvesicles (PMVs) [7].
Medium-large PEVs (henceforth PEVs for brevity) have been widely studied in the frame of the platelet–cancer crosstalk. Their levels in the circulation are increased in oncological patients bearing diverse types of cancer, including cutaneous malignant melanoma [8], colorectal carcinoma [9], lung cancer [10], and breast cancer [11]. Moreover, PEVs were found to mediate intercellular communication by delivering bioactive compounds, thus initiating phenotypic and functional changes in recipient cells. These observations boosted research on the role played by these PEVs in cancer aiming to unravel their contribution in the progression of the disease but also to determine their potential use as early diagnostic markers and novel drug delivery tools [12,13]. PEVs are now known to directly regulate cancer cells as well as the cellular components of the tumour-microenvironment (TME). In this context, most studies have hinted at PEVs as crucial drivers of cancer progression, although a few investigations have discussed their anti-cancer functions [6,14,15].
We showed that thrombin-induced PEVs are internalized by different breast cancer cell lines, thereby exerting cell-specific reactions [16]. In particular, the TNBC MDA-MB-231 cell line efficiently internalizes PEVs, and this event potentiates cell migration and invasiveness. In agreement with these observations, long-term (up to 24 h) exposure to PEVs stimulates specific signalling pathways, such as p38 mitogen-activated protein kinase (p38 MAPK), myosin light chain-2 (MLC2), and Rho-associated protein kinase (ROCK) [16], that drive cancer cell motility and spreading [17,18,19,20]. Nevertheless, the molecular mechanism(s) whereby a prolonged exposure to PEVs promote(s) migration in MDA-MB-231 cells is still unclear. The remodelling of the Ca2+ handling machinery supports several cancer hallmarks, including tissue invasion and metastasis [21,22,23,24,25]. An increase in intracellular Ca2+ concentration ([Ca2+]i) in MDA-MB-231 cells can be elicited by inositol-1,4,5-trisphosphate (InsP3) [26], which gates three subtypes of InsP3 receptors (InsP3R1, InsP3R2, and InsP3R3) to release Ca2+ from the endoplasmic reticulum (ER) [22]. InsP3-induced Ca2+ mobilization, in turn, causes a strong reduction in ER Ca2+ concentration, which leads to the activation of a Ca2+ entry pathway on the plasma membrane, known as store-operated Ca2+ entry (SOCE) [27,28,29]. In MDA-MB-231 cells, SOCE is mediated by the physical association between Stromal Interaction Molecule 1 (STIM1), which functions as a sensor of ER Ca2+ concentration and is activated by a drop in intraluminal Ca2+ levels, and Orai1, which forms the Ca2+-permeable channel on the plasma membrane [24,27,30,31]. The interaction between InsP3-induced ER Ca2+ mobilization and SOCE finely shapes the intracellular Ca2+ signals that increase the migration capacity of this highly invasive breast cancer cell line [24,26,31,32]. An increase in the expression of genes encoding for several InsP3R isoforms can stimulate multiple cancer hallmarks, including proliferation, migration, invasion, and apoptosis resistance [33,34]. Similarly, SOCE up-regulation, because of the over-expression of STIM and/or Orai1 proteins, can result in the activation of many pro-oncogenic signalling pathways in neoplastic cells [22,29,35]. Furthermore, the overexpression of several members of the Transient Receptor Potential (TRP) superfamily of non-selective cation channels can also support neoplastic transformation in virtually all cancer cell types [22,36].
Intriguingly, the signalling pathways recruited downstream of PEV stimulation, e.g., p38 MAPK and MLC2, can be activated following an elevation in [Ca2+]i [16,18]. Herein, we thus sought to assess whether long-term exposure to PEVs stimulates MDA-MB-231 cell migration through the remodelling of the Ca2+ handling machinery. By using a variety of approaches, we showed that PEVs cause a remarkable elevation in InsP3-induced ER Ca2+ release by increasing Sarco-Endoplasmic Ca2+-ATPase 2B (SERCA2B) and InsP3R1/InsP3R2 transcript and protein expression. The larger depletion in ER Ca2+ content, in turn, leads to enhanced SOCE activation. In agreement with this observation, serum elicited larger intracellular Ca2+ signals and potentiated migration in MDA-MB-231 cells exposed to PEVs through the Ca2+-dependent recruitment of p38 MAPK and MLC2. Our evidence indicates that InsP3Rs rather than SOCE are required to drive MDA-MB-231 cell migration. These data shed novel light on the signalling pathways whereby PEVs can stimulate the metastatic spreading of TNBC cells and hint at the Ca2+ toolkit as a promising target to prevent the detrimental interaction between PEVs and these highly aggressive breast cancer cells.
2. Materials and Methods
2.1. Cancer Cell Culture
The triple negative breast cancer line, MDA-MB-231, was provided by Professor Livia Visai (Department of Molecular Medicine, University of Pavia). Cancer cells were periodically checked to verify the absence of bacterial contamination and were maintained in DMEM supplemented with 10% foetal bovine serum (FBS), 2 mM L-glutamine, 100 U/mL penicillin, and 100 μg/mL streptomycin, split every two days, and used for the experiments within 10 passages. The count of vital cells was determined by Trypan Blue staining and phase contrast microscopy analysis.
2.2. PEV Isolation
Human blood platelets were purified from healthy donors as recently described [16]. Upon the washing procedure, platelets were resuspended at a concentration of 3 × 108/mL in HEPES buffer (10 mM HEPES, 137 mM NaCl, 2.9 mM KCl, and 12 mM NaHCO3, pH 7.4) supplemented with 1 mM CaCl2, 0.5 mM MgCl2 and 5.5 mM glucose. To induce the release of PEVs, platelets were stimulated with the physiological agonist thrombin (0.2 U/mL) for 30 min at 37 °C under constant stirring. Platelets were pelleted by low-speed centrifugation (750× g, 20 min) and the supernatant was then centrifuged at 18,500× g for 90 min at 10 °C to collect medium-large PEVs, which were finally resuspended in HEPES buffer. The protein content of the different preparations of PEVs was determined by BCA assay.
2.3. [Ca2+]i Measurements
As described elsewhere, Ca2+ imaging was used to measure intracellular Ca2+ signals in PEV-treated cancer cells [16,37]. MDA-MB-231 cells were loaded with 4 μM Fura-2/AM (1 mM stock in DMSO) in physiological salt solution (PSS) (150 mM NaCl, 6 mM KCl, 1.5 mM CaCl2, 1 mM MgCl2, 10 mM Glucose, 10 mM HEPES, pH 7.4) for 30 min at 37 °C and 5% CO2. After washing in PSS, the coverslip was fixed to the bottom of a Petri dish and the cells were either left untreated or treated with 30 µg/mL thrombin-induced PEVs. The cells were observed by an upright epifluorescence Axiolab microscope (Carl Zeiss, Oberkochen, Germany) equipped with a Zeiss ×40 Achroplan objective (water-immersion, 2.0 mm working distance, 0.9 numerical aperture). The cells were excited alternately at 340 and 380 nm, and the emitted light was detected at 510 nm. Custom software, working in the LINUX environment, was used to drive the camera (Extended-ISIS Camera, Photonic Science, Millham, UK) and the filter wheel, and to measure and plot on-line the fluorescence from rectangular “regions of interest” (ROI) enclosing 20–30 single cells. [Ca2+]i was monitored by measuring, for each ROI, the ratio of the mean fluorescence emitted at 510 nm when exciting alternatively at 340 and 380 nm [ratio (F340/F380)]. An increase in [Ca2+]i causes an increase in the ratio [38,39]. Ratio measurements were performed and plotted on-line every 3 s. The experiments were performed at room temperature (22 °C).
The resting Ca2+ entry was evaluated by exploiting the Mn2+-quenching technique. Mn2+ has been shown to quench Fura-2/AM fluorescence. Since Mn2+ and Ca2+ share common entry pathways in the plasma membrane, Fura-2/AM quenching by Mn2+ is regarded as an index of divalent cation influx [31,40,41]. Experiments were carried out at the 360 nm wavelength, the isosbestic wavelength for Fura-2/AM, and in Ca2+-free medium supplemented with 0.5 mM EGTA, as previously described [40,42]. This avoids Ca2+ competition for Mn2+ entry and therefore enhances Mn2+ quenching. The basal [Ca2+]i was evaluated by using the Grynkiewicz equation, as shown elsewhere [35,43].
2.4. Cell Migration Assay
The effect of PEVs on the migration of MDA-MB-231 cells was evaluated using Falcon cell culture inserts (8 μm pore size) positioned in a 24-well plate, as shown in [16]. Cancer cells were serum starved for 6 h and then resuspended in DMEM with the addition of 0.5% FBS. Cells were either left untreated or treated with PEVs, supplemented or not with 2-Aminoethyl diphenyl borate (2-APB; 50 µM) or YM-58483 (also known as BTP-2; 20 µM), and then transferred inside the inserts. DMEM containing 10% FBS was added to the lower chamber. After 24 h, cells that moved through the porous membrane were stained with 0.5% crystal violet and counted at 10× microscope magnification with an Olympus BX51 microscope (Olympus Corporation, Japan).
2.5. Real-Time Quantitative Reverse Transcription PCR (qRT-PCR)
The total RNA was isolated from PEVs as well as PEV-treated and untreated MDA-MB-231 cells using Trizol reagent (Thermo Fisher Scientific, Waltham, MA, USA) according to the manufacturer’s instructions. For RNA extraction from PEVs, 20 µg of RNA grade glycogen (Thermo Fisher Scientific) was added in the precipitation step [44]. After DNAse treatment (Turbo DNA-free™ kit, Thermo Fisher Scientific), RNA was quantified with a BioPhotometer D30 (Eppendorf, Hamburg, Germany). cDNA was synthetised from 200 ng of MDA-MB-231 RNA and 100 ng of PEV RNA using the iScript cDNA Synthesis Kit (BioRad, Hercules, CA, USA). Gene expression analyses were performed in triplicate with specific primers (Table S1) and the SsoFast™ EvaGreen® Supermix (BioRad) using a CFX Connect Real-Time System (BioRad) using the following program: an initial step at 95 °C for 30 s, 40 cycles of 5 s at 95 °C, and 5 s at 58 °C. Fluorescence measurements were taken at the end of each elongation step. The PCR mixture consisted of 10 µL SsoFast™ EvaGreen® Supermix, 7 µL nuclease-free water, 1 µL cDNA, and 1 µL of each forward and reverse primer. Melting curve analysis was performed to ensure that single amplicons were obtained for each target gene after each qRT-PCR and primer efficiencies were determined using at least five different cDNA concentrations. Primers were designed on an exon-intron junction using the NCBI Primer tool [45]. Gene expression was evaluated using the ΔΔCt method [46,47]. The genes actin beta and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) were used as endogenous references for normalizing target mRNA. Data are presented as means ± standard error (SE) from three biological replicates. Results were analysed using the GraphPad Prism 5 software (version 5.01; GraphPad Software, San Diego, CA, USA) and the significance was determined using the unpaired Student’s t-test. p-values < 0.05 were considered significant.
2.6. SDS-PAGE and Immunoblotting
MDA-MB-231 cells were grown in 35 mm Petri dish, incubated with PEVs for 24 h, and lysed with Lysis Buffer (50 mM Tris–HCl, 150 mM NaCl, 1% Nonidet P-40, 1 mM EDTA, 0.25% deoxycholic acid, 0.1% SDS, pH 7.4, 1 mM PMSF, 5 μg/mL leupeptin, and 5 μg/mL aprotinin). Protein expression and phosphorylation were analysed by SDS-PAGE followed by immunoblotting, as previously described [48]. Membrane staining was performed using the different antibodies diluted 1:1000 in TBS (20 mM Tris, 500 mM NaCl, pH 7.5) containing 0.5% BSA and 0.1% Tween-20 in combination with the appropriate HRP-conjugated secondary antibodies (1:2000 in PBS plus 0.1% Tween-20). Quantification of the band intensity was performed by computer-assisted densitometric scanning using Quantity One–4.6.8 software (BioRad).
2.7. Statistical Analysis
All the reported figures are representative of three different experiments. As to the Ca2+ data, the amplitude of Ca2+ release in response to extracellular stimulation was measured as the difference between the ratio at the peak of intracellular Ca2+ mobilization and the mean ratio of 1 min baseline before the peak. The magnitude of SOCE evoked by extracellular stimulation upon Ca2+ restoration to the bath was measured as the difference between the ratio at the peak of extracellular Ca2+ entry and the mean ratio of 1 in baseline before Ca2+ re-addition. The rate of Mn2+ influx was evaluated by measuring the slope of the fluorescence intensity curve at 400 s after Mn2+ addition [40,42]. Pooled data are given as mean ± SEM, while the number of cells analysed is indicated above the corresponding histogram bars (number of responding cells/total number of analysed cells).
For immunoblotting and cell migration analyses, all reported figures are representative of at least three different experiments and the quantitative data are reported as mean ± SD. Data from immunoblotting scanning were normalised considering the intensity of the band of interest in the sample not incubated with PEVs as one arbitrary unit (A. U.). For cell migration analyses, data are expressed as number of migrated cells per field at 10X microscope magnification.
Comparisons between two groups were done using the Student’s t-test, whereas multiple comparisons were performed using One-Way Analysis of Variance (ANOVA) with the Bonferroni post-hoc test. p-values less than 0.05 were considered statistically significant. Data were analysed using the GraphPad Prism (Version 5.01) software.
3. Results
3.1. Long-Term Exposure to PEVs Increases ER Ca2+ Mobilization and SOCE Activation in MDA-MB-231 Cells
The dose-response relationship showed in a previous study from our group demonstrated that 30 µg/mL represents the PEV dose that is more efficiently internalized and is more effective at inducing migration in MDA-MB-231 cells [16,49]. The long-term effect of PEVs on intracellular Ca2+ dynamics was therefore evaluated in MDA-MB-231 cells maintained or not (control, Ctrl) in the presence of 30 µg/mL PEVs for 24 h and loaded with the Ca2+-sensitive fluorophore, Fura-2/AM. Preliminary recordings revealed that PEVs did not affect either the basal [Ca2+]i (Figure S1A) or the modest constitutive Ca2+ entry that has previously been reported in MDA-MB-231 cells (Figure S1B,C) [50]. Next, we exploited the Ca2+ add-back protocol to evaluate whether long-term exposure to PEVs influences ER Ca2+ mobilization and SOCE activation. As described elsewhere [27,28,30,37,50,51], this manoeuvre consists in stimulating the cells with cyclopiazonic acid (CPA), a selective inhibitor of SERCA, in the absence of extracellular Ca2+ (0Ca2+) to cause passive ER Ca2+ efflux through Ca2+-permeable leakage channels. Subsequently, 1.8 mM Ca2+ is restituted to external Ca2+ to monitor SOCE through Orai1 channels that have previously been activated by STIM1 upon the depletion of the ER Ca2+ store. Figure 1A,B show that CPA (10 µM) caused a transient increase in [Ca2+]i under 0Ca2+ conditions, which reflects ER Ca2+ releasing ability in the absence (Ctrl) and presence of PEVs. PEVs caused a significant (p < 0.05) elevation in CPA-evoked ER Ca2+ mobilization (Figure 1C). The following restitution of extracellular Ca2+ induced a second increase in [Ca2+]i (Figure 1A,B), which was entirely due to SOCE through Orai1 channels [27,28,30,50]. Statistical analysis revealed that long-term exposure to PEVs also increased SOCE in MDA-MB-231 cells (Figure 1D). Altogether, these findings indicate that PEVs induce a remarkable remodelling of the Ca2+ handling machinery in highly aggressive breast cancer MDA-MB-231 cells by enhancing both ER Ca2+ release and SOCE.
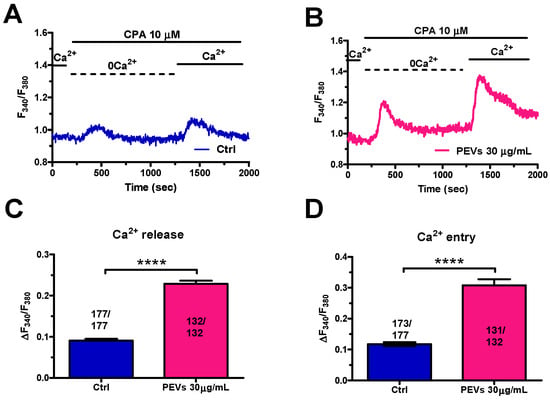
Figure 1.
Long-term exposure of MDA-MB-231 cells to PEVs increases ER Ca2+ mobilization and SOCE activation. (A). Control (Ctrl) cells were challenged with CPA (10 μM), a selective SERCA inhibitor, in the absence of extracellular Ca2+ (0Ca2+). This manoeuvre caused a transient increase in [Ca2+]i due to the depletion of the intracellular ER Ca2+ store. The restoration of extracellular Ca2+ induced a second increase in [Ca2+]i, which is representative of SOCE. (B). Cells treated for 24 h with PEVs (30 μg/mL) showed a higher Ca2+ transient when stimulated with CPA under 0Ca2+ conditions. Likewise, the restoration of extracellular Ca2+ induced an enhanced increase in [Ca2+]i that is indicative of a greater SOCE. (C). Mean ± SEM of the CPA-dependent intracellular Ca2+ mobilization in control and PEV-treated cells. **** indicate p < 0.0001. (D). Mean ± SEM of the CPA-dependent SOCE amplitude in control and PEV-treated cells. **** indicate p < 0.0001.
3.2. Long-Term Exposure to PEVs Increases InsP3-Induced ER Ca2+ Release and InsP3-Sensitive SOCE
The migratory mechanism in MDA-MB-231 cells exposed to chemotactic cues involves an initial phase of Ca2+ release from the ER through InsP3Rs followed by SOCE activation [26,30,31,52,53]. Adenosine triphosphate (ATP) represents an autocrine/paracrine messenger that has long been used to evaluate InsP3-dependent Ca2+ signals in multiple types of cancer cells [23,40,53,54], including MDA-MB-231 cells [55,56]. ATP binds to Gq-coupled P2Y receptors, thereby stimulating phospholipase Cβ (PLCβ) to cleave phosphatidylinositol-4,5-bisphosphate (PIP2) into diacylglycerol (DAG) and InsP3 [23,40,54,57]. Preliminary experiments confirmed that ATP-evoked intracellular Ca2+ release was mediated by InsP3Rs (Figure S2). The Ca2+ add-back protocol confirmed that both phases of the Ca2+ response to ATP (100 µM), i.e., intracellular Ca2+ release through InsP3Rs and SOCE, were significantly enhanced in MDA-MB-231 pre-treated with PEVs for 24 h when compared to untreated (Ctrl) cells (Figure 2A–C). As described elsewhere [37,40,57], the agonist was removed from the perfusate 100 s before the re-addition of extracellular Ca2+ to prevent the activation of second messengers-operated channels and ionotropic P2X receptors. To circumvent the Gq-protein-PLCβ-InsP3 signalling pathway, InsP3Rs were directly activated with a membrane-permeant esterified form of InsP3, known as InsP3-BM (1 µM) [58]. The Ca2+ add-back protocol showed that InsP3-BM-evoked ER Ca2+ mobilization and SOCE were significantly larger in MDA-MB-231 pre-treated with PEVs for 24 h when compared to untreated (Ctrl) cells. Overall, these findings provide further support to the notion that long-term exposure to PEVs potentiates InsP3-dependent Ca2+ signals in MDA-MB-231 cells.
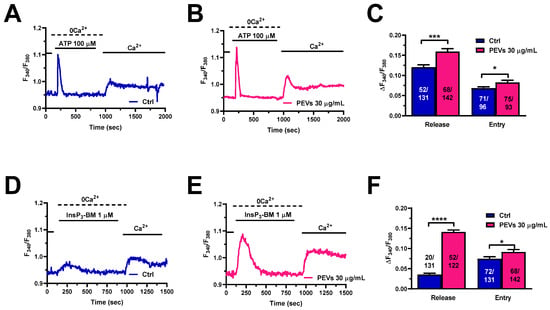
Figure 2.
Long-term exposure of MDA-MB-231 cells to PEVs increases InsP3-induced ER Ca2+ mobilization and SOCE. (A). ATP (100 µM) evoked a transient increase in [Ca2+]i under 0Ca2+ conditions in control (Ctrl) cells. ATP was then removed from the recording bath, while extracellular Ca2+ was restored to the solution to measure SOCE. (B). The treatment of MDA-MB-231 cells with PEVs (30 μg/mL, 24 h) increased ATP-induced endogenous Ca2+ mobilization and SOCE activation. (C). Mean ± SEM of ATP-induced intracellular Ca2+ release and SOCE in control (Ctrl) and PEV-treated cells. * indicates p < 0.05 and *** indicates p < 0.001. (D). InsP3-BM, a membrane-permeant esterified form of InsP3, induced a small increase in [Ca2+]i under 0Ca2+ conditions in control (Ctrl) cells. Restoration of extracellular Ca2+, in the absence of the agonist, caused a second bump in [Ca2+]i that was indicative of SOCE activation. (E). The treatment of MDA-MB-231 cells with PEVs (30 µg/mL, 24 h) caused an enhancement in intracellular Ca2+ mobilization and SOCE in MDA-MB-231 cells stimulated with InsP3-BM. (F). Mean ± SEM of InsP3-BM-induced endogenous Ca2+ mobilization and SOCE in control (Ctrl) and PEV-treated cells. **** indicates p < 0.0001 and * indicates p < 0.05.
3.3. Molecular Characterization of the Ca2+ Handling Machinery in MDA-MB-231 Cells Exposed to PEVs
In order to gain further insights into the molecular mechanisms whereby PEVs increase InsP3-induced ER Ca2+ mobilization and SOCE activity, we carried out a thorough RT-qPCR analysis of mRNAs isolated from MDA-MB-231 cells exposed or not (Ctrl) to PEVs (30 µg/mL; 24 h). Figure 3 shows a significant increase in the expression levels of the transcripts encoding for SERCA2B (Figure 3A), i.e., the major SERCA isoform in MDA-MB-231 cells [59], and for InsP3R1 (Figure 3B) and InsP3R2 (Figure 3C), which mediate ER Ca2+ release [26]. Conversely, there was no change in the expression of the transcripts encoding for InsP3R3 (Figure 3D) and for all the molecular components of the SOCE machinery in MDA-MB-231 cells, i.e., STIM1 (Figure 3E) and Orai1 (Figure 3F) [24,27,30,31]. Notably, RT-qPCR analysis of PEV transcripts did not reveal detectable levels of any of these mRNAs (data not shown), thereby showing that SERCA2B, InsP3R1, and InsP3R2 transcripts are not directly transferred to MDA-MB-231 cells from PEVs (mean Ct values ± SD of 37.5 ± 1, 38.5 ± 1, 38.2 ± 1.1, 34.9 ± 1.4, 34.8 ± 1, 37.3 ± 1.4, and 36.6 ± 1.3 for SERCA2B, InsP3R1, InsP3R3, Orai1, SERCA3, InsP3R2, and STIM1, respectively).
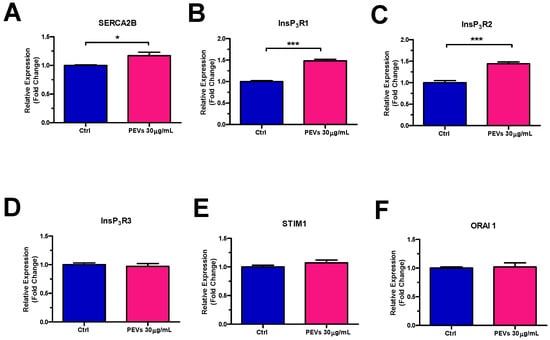
Figure 3.
Long-term exposure to PEVs enhances the expression of SERCA2B, InsP3R1 and InsP3R2 transcripts. RT-qPCR was performed on the transcripts encoding for SERCA2B (A), InsP3R1 (B), InsP3R2 (C), InsP3R3 (D), STIM1 (E), and Orai1 (F) and harvested from MDA-MB-231 cells treated with 30 µg/mL for 24 h as compared to untreated (Ctrl) cells. Results were normalized with the level of the GAPDH control. * indicates p < 0.05, *** indicate p < 0.001.
Next, western blot analysis of SERCA2B, InsP3Rs, STIM1, and Orai1 protein expression was performed by exploiting affinity-antibodies, as illustrated in [37,40,53]. Immunoblots revealed a major band of ≈115 kDa for SERCA2B (Figure 4A, left panel) and a large band over 250 kDa, deriving from the sum of the 313/260/250 kDa bands corresponding to InsP3R1, InsP3R2, and InsP3R3 (Figure 4B, left panel), as previously shown in [53]. Furthermore, two major bands of ≈86 and ≈35 kDa were detected for, respectively, STIM1 (Figure 4C, left panel) and Orai1 (Figure 4D, left panel). Densitometric analysis confirmed that SERCA2B (Figure 4A, right panel) and InsP3R (Figure 4B, right panel) proteins were significantly up-regulated in MDA-MB-231 cells exposed to PEVs (30 µg/mL; 24 h). Conversely, and in agreement with the qRT-PCR data, there was no significant difference in the expression level of STIM1 (Figure 4C, right panel) and Orai1 proteins (Figure 4D, right panel). Overall, these findings strongly suggest that the increase in the amount of ER Ca2+ that is releasable through InsP3Rs is due to the overexpression of SERCA2B and InsP3R proteins. Since there is no difference in the expression levels of its underlying constituents, i.e., STIM1 and Orai1, the increase in SOCE is likely to reflect the larger drop in ER Ca2+ concentration following agonist stimulation of MDA-MB-231 cells pre-exposed to PEVs [60,61,62].
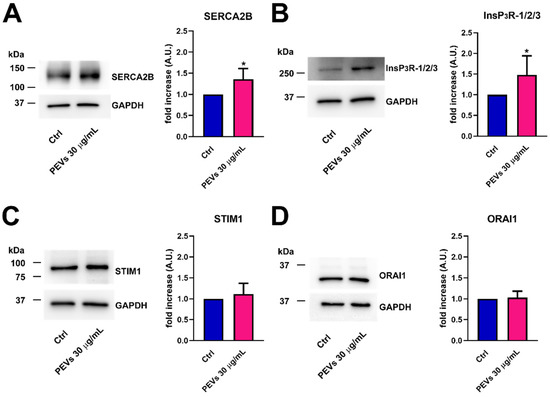
Figure 4.
The treatment of MDA-MB-231 cells with PEVs increases the protein expression of SERCA2B and InsP3Rs. Representative immunoblots of SERCA2B and InsP3Rs are reported in (A,B), respectively, where GAPDH is for equal loading control (left panel). Quantification of the results performed by densitometric scanning is reported in the right panels as fold increase (A.U.) of protein expression over basal (control, Ctrl). Results are the mean ± SD of three different experiments. * indicates p < 0.05. Conversely, long term exposure of MDA-MB-231 cells to PEVs do not cause differences in the expression level of STIM1 and Orai1. The left panels of (C,D) show the representative immunoblots of STIM1 and Orai1, respectively, while the right panels show band quantifications reported as fold increase (A.U.) of protein expression over basal (Ctrl). Results are the mean ± SD of three different experiments.
3.4. Serum-Induced Intracellular Ca2+ Signals Are Larger in MDA-MB-231 Cells Exposed to PEVs
It has long been known that FBS stimulates the migration in cancer cells through an increase in [Ca2+]i [26,30,31,52,53]. FBS consists of a mixture of growth factors that bind to specific tyrosine kinase receptors (TKRs) that are coupled to PLCγ and stimulate InsP3-production and InsP3-dependent Ca2+ signalling. A recent report from our group showed that 24 h treatment with PEVs potentiated serum-evoked migration in MDA-MB-231 cells [16]. Therefore, we reasoned that the increase in the chemotactic response to FBS could be associated to an elevation in the underlying increase in [Ca2+]i. The Ca2+ add-back protocol confirmed that, upon long-term exposure to PEVs (30 µg/mL; 24 h), MDA-MB-231 cells displayed a remarkable increase in serum-evoked intracellular Ca2+ release and SOCE activation when compared to non-treated (Ctrl) cells (Figure 5A–C). The increase in extracellular Ca2+ entry was further confirmed by measuring the area under the curve (AUC) corresponding to the increase in [Ca2+]i evoked by the re-addition of extracellular Ca2+ (Figure S3). Pharmacological characterization revealed for the first time that serum-evoked intracellular Ca2+ release was mediated by ER Ca2+ release through InsP3Rs in MDA-MB-231 cells. Indeed, the intracellular Ca2+ response to 20% FBS was dramatically reduced by each of the following manoeuvres [16,37,40,63]: (1) blockade of PLCγ activity with the specific aminosteroid, U73122 (10 µM); (2) specific inhibition of InsP3Rs with 2-APB (50 µM) (Figure 5D,F); and (3) depletion of the ER Ca2+ store with CPA (30 µM) (Figure 5E,F).
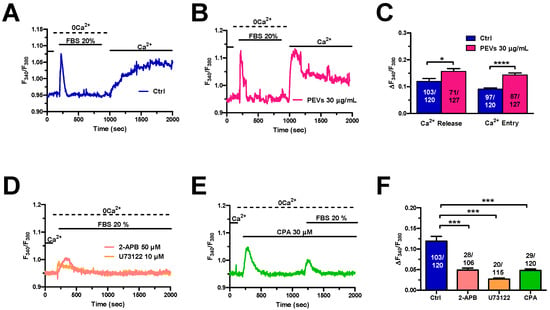
Figure 5.
Long-term exposure to PEVs enhances FBS-induced ER Ca2+ mobilization and SOCE. (A). A transient increase in [Ca2+]i was evoked by 20% FBS under 0Ca2+ conditions in control (Ctrl) cells. FBS was then removed from the recording bath, while extracellular Ca2+ was restored to the perfusate to measure SOCE. (B). Pre-treatment of MDA-MB-231 cells with PEVs (30 μg/mL, 24 h) increased the endogenous Ca2+ mobilization and the SOCE induced by 20% FBS. (C). Mean ± SEM of 20% FBS-induced intracellular Ca2+ release and SOCE in control and treated cells. **** indicate p < 0.0001 and * indicates p < 0.05. (D). The Ca2+ response to 20% FBS, under 0Ca2+ conditions, was inhibited by U73122 (10 µM, 30 min), a selective PLC blocker. Moreover, the Ca2+ signal was inhibited by blocking InsP3Rs with 2-APB (50 µM, 30 min). (E). Emptying the ER with CPA (30 µM, 20 min) prevented (not shown) or significantly reduced an FBS-induced increase in [Ca2+]i. (F). Mean ± SEM of the amplitude of 20% FBS-evoked Ca2+ peak under the designated treatments. *** indicate p < 0.001.
In agreement with previous reports on other cancer cell types [40,52], serum-evoked SOCE (Figure 6A) was dampened by blocking Orai1 channels with either Pyr6 (10 µM) (Figure 6B,D) or BTP-2 (20 µM) (Figure 6C,D) in MDA-MB-231 cells. Taken together, these findings show that long-term exposure to PEVs leads to an increase in InsP3-induced ER Ca2+ mobilization and SOCE that could potentiate the pro-migratory Ca2+ response to serum stimulation.
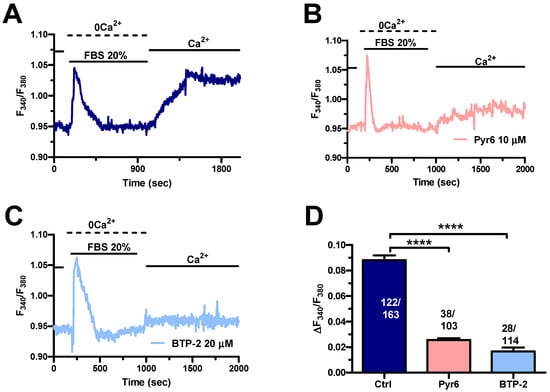
Figure 6.
The Ca2+ response to FBS is sustained by SOCE in control MDA-MB-231 cells. (A). An intracellular Ca2+ response was induced by 20% FBS under 0Ca2+ conditions. Restoration of extracellular Ca2+, in the absence of the agonist, induced a second increase in [Ca2+]i, which was indicative of SOCE. (B). Extracellular Ca2+ entry evoked by 20% FBS was suppressed or significantly reduced by blocking Orai1 with Pyr6 (10 µM, 10 min). (C). Extracellular Ca2+ entry evoked by 20% FBS was suppressed or significantly reduced by blocking Orai1 with BTP-2 (20 µM, 10 min). (D). Mean ± SEM of the amplitude of Ca2+ peak in cells under the designated treatments. **** indicate p < 0.0001.
3.5. PEVs Potentiate Serum-Dependent Migration through the Ca2+-Dependent Recruitment of p38 MAPK and MLC2 in MDA-MB-231 Cells
To assess whether the partial remodelling of the Ca2+ handling machinery potentiates serum-induced migration upon pretreatment with PEVs, we evaluated MDA-MB-231 motility in the absence (Ctrl) and presence of specific InsP3R and SOCE inhibitors. The pharmacological blockade of InsP3Rs with 2-APB (50 µM) significantly inhibited serum-migration in MDA-MB-231 cells pre-treated with PEVs (30 µg/mL, 24 h), whereas this process was not affected by SOCE inhibition with BTP-2 (20 µM) (Figure 7A,B). Next, we assessed whether a serum-evoked increase in [Ca2+]i is required to boost p38 MAPK and MLC2 activation in the presence of PEVs (30 µg/mL, 24 h). We confirmed that PEVs potentiated p38 MAPK and MLC2 phosphorylation (Figure 7C–F), while they did not hyper-activate the Ca2+-sensitive Pyk2 (Figure S4). We then examined the phosphorylation of p38 MAPK and MLC2 in the absence (Ctrl) and presence of 2-APB and BTP-2. Unlike migration, PEV-dependent increase in p38 MAPK and MLC2 phosphorylation was sensitive both to InsP3R inhibition with 2-APB (50 µM) and to SOCE blockade with BTP-2 (20 µM) (Figure 7C–F). These findings demonstrate that InsP3Rs, rather than SOCE, support PEV-dependent migration in MDA-MB-231, although they can both induce p38 MAPK and MLC2 phosphorylation.
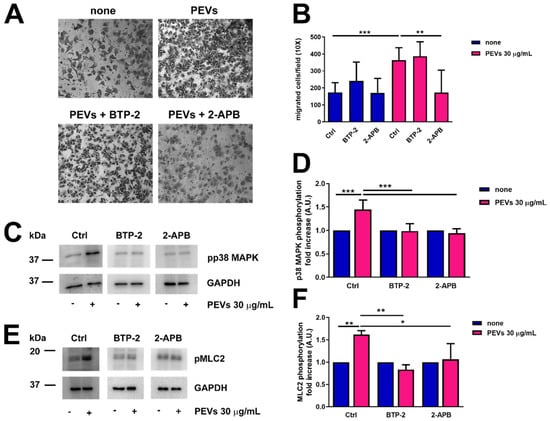
Figure 7.
Pharmacological blockade of InsP3Rs with 2-APB significantly inhibited migration in MDA-MB-231 cells treated with PEVs. (A). MDA-MB-231 cells were incubated with PEVs (30 µg/mL, 24 h) or left untreated (none) before being treated with 2-APB (50 µM) and BTP-2 (20 µM) and then transferred into cell culture inserts. The cells migrated through the porous membrane were stained and counted. Representative images of migrated stained cells are reported. The quantification of the results is shown in (B) as the mean ± SD of three experiments. ** p < 0.01 and *** p < 0.005. Phosphorylation of p38MAPK (C,D) and MLC2 (E,F) in MDA-MB-231 cells incubated with PEVs (30 µg/mL, 24 h) and then treated with 2-APB (50 µM) and BTP-2 (20 µM). Representative immunoblots are reported in (C,E) for phospho-p38MAPK and phospho-MLC2, respectively, where GAPDH staining is for equal loading control. The quantification of the results is shown in (D) for phospho-p38MAPK and in (F) for phospho-MLC2 as the mean ± SD of three experiments. * indicates p < 0.05, ** indicate p < 0.01 and *** indicate p < 0.005.
4. Discussion
Circulating platelets can contribute to cancer progression by releasing PEVs that facilitate (by shrouding disseminated tumour cells from recognition by NK cells) or stimulate cancer cell spreading from the primary site [6,13]. Multiple pieces of evidence support the notion that PEVs promote cancer cell invasiveness by mediating the transfer of platelet-derived instructive cues, consisting either in signalling proteins or genetic material, to the target cells [6,13,64]. The relationship between breast cancer and platelets is complex and is yet to be fully unravelled. Nevertheless, PEVs have also been shown to directly engage signal transduction pathways, such as Src, focal adhesion kinases (FAKs), p38 MAPK, and MLC2, which stimulate migration in the TNBC MDA-MB-231 cell line [16,65]. In accord, MDA-MB-231 cells can induce platelet activation and aggregation [49,66], thereby leading to the release of robust amounts of PEVs that, in turn, increase cancer cell migration and invasion [16]. Intriguingly, an increase in [Ca2+]i may occur upstream of p38 MAPK and MLC2 recruitment [16,18] and can modulate p38 MAPK and MLC2 phosphorylation [67,68,69]. It has long been known that a complex remodelling of Ca2+ handling machinery supports several cancer hallmarks, including migration and invasion [21,22,70]. Therefore, we sought to investigate whether the long-term exposure of the highly invasive MDA-MB-231 cells to PEVs potentiates migration by rewiring their Ca2+ transport system.
4.1. PEVs Induce Remodelling of the Ca2+ Handling Machinery in MDA-MB-231 Cells
A sustained increase in [Ca2+]i is required to trigger the release of PEVs from the plasma membrane of activated platelets [4]. Preliminary reports suggested that PEVs can, in turn, also influence intracellular Ca2+ dynamics in target cells. For instance, a recent investigation showed that PEVs elicit intracellular Ca2+ oscillations in aortic vascular smooth muscle cells, thereby promoting migration and neointimal hyperplasia in a rat model of vascular injury [71]. Furthermore, we documented that 30 µg/mL PEVs induced a robust increase in [Ca2+]i in MDA-MB-231 cells, with this being initiated by InsP3-induced ER Ca2+ release and maintained by SOCE activation [16]. Nevertheless, MDA-MB-231 cells displayed enhanced migration and invasiveness upon 24 h incubation with PEVs [16]. Therefore, the immediate Ca2+ response to PEVs is unlikely to engage the Ca2+-dependent molecular machinery that potentiates migration during the late stages of PEV stimulation. These pieces of evidence prompted us to assess whether long-term exposure to PEVs stimulates the p38 MAPK and MLC2 signalling pathways and potentiates MDA-MB-231 cell migration through remodelling the Ca2+ handling machinery.
4.1.1. PEV-Dependent Increase in InsP3-Induced ER Ca2+ Release Is Associated to SERCA2B and InsP3R Up-Regulation
Pre-treatment with PEVs did not change either the resting [Ca2+]i or the basal plasma membrane permeability to Ca2+, which is mediated by a yet-to-be-identified Ca2+-permeable route in MDA-MB-231 cells, while it is mediated by Orai1 channels in low-migrating MCF-7 cells [41]. Since PEVs did not affect resting Ca2+ influx in MDA-MB-231 cells, we did not further investigate its molecular structure.
The Ca2+ response of breast cancer cells to chemotactic cues is triggered by InsP3-induced Ca2+ release from the ER and sustained over time by SOCE [24,26,32]. The Ca2+ add-back protocol represents the most common strategy to evaluate both the amount of ER Ca2+ that can be released through InsP3Rs and the extent of SOCE activation in cancer cells, including MDA-MB-231 cells [27,28,30,37,50]. We found that, in the absence of extracellular Ca2+, CPA-evoked intracellular Ca2+ release was significantly increased in MDA-MB-231 cells exposed to PEVs when compared to untreated cells. The amount of Ca2+ introduced into the cytosol via ER Ca2+ leakage channels upon SERCA inhibition with CPA or thapsigargin is recognized as a reliable indicator of the releasable ER Ca2+ pool. In accord, this protocol has been widely exploited to evaluate the differences in ER Ca2+ content in primary vs. metastatic cancer cells [40] and in cancer cells exposed to different pharmacological or genetic treatments [26,30,54,57,72,73]. Molecular analysis showed that long-term exposure to PEVs caused an increase in the transcript and protein levels of SERCA2B, which represents the most abundant SERCA isoform in MDA-MB-231 cells [59]. Therefore, the increase in SERCA2B expression might be responsible for the increase in the ER Ca2+ load unmasked by CPA-evoked intracellular Ca2+ mobilization. Similarly, the increase in SERCA2B expression underlies the larger Ca2+ releasing ability of colorectal cancer cells lacking the oncogenic K-Ras isoform, K-RasG13D [54]. The increase in ER Ca2+ content in PEV-treated MDA-MB-231 cells is associated to an increase in InsP3R1 and InsP3R2 transcript and protein expression. The Ca2+ add-back protocol confirmed that both physiological (with ATP) and pharmacological (with InsP3-BM) stimulation of InsP3Rs resulted in a significant elevation in InsP3-dependent ER Ca2+ mobilization in MDA-MB-231 cells exposed to PEVs. An increase in InsP3R1 expression may contribute to multiple oncological processes, including apoptosis resistance in prostate cancer [74], autophagy induction in clear cell renal cell carcinoma [75], and proliferation, invasion, and migration in osteosarcoma [76]. Similarly, InsP3R2 regulates migration in non-small cell lung cancer [77], maintains the self-renewal ability of liver cancer stem cells [78], and prevents apoptosis in B-cell lymphoma and chronic lymphocytic leukemia [79]. Interestingly, both InsP3R1 and InsP3R2 proteins drive migration in MDA-MB-231 cells [26], whereas InsP3R1 has also been shown to promote proliferation [80]. Therefore, the combined overexpression of SERCA2B and InsP3R1/InsP3R2 proteins nicely correlates with the increased ER Ca2+ content and higher level of InsP3-induced ER Ca2+ mobilization induced by long-term exposure to PEVs and could potentiate migration in MDA-MB-231 cells [16]. PEVs can transfer specific cargo molecules, such as mRNA, DNA, cytokines, and membrane receptors or enzymes, to recipient cells and thereby increase the capability of invasive behaviour in cancer cells [3,6,13]. Nonetheless, we could not find detectable levels of SERCA2B and InsP3R1/InsP3R2 transcripts in the mRNA content of PEVs. Therefore, we can conclude that long-term exposure to PEVs stimulates the partial remodelling of the Ca2+ handling machinery in MDA-MB-231 cells by boosting the expression of genes encoding for SERCA2B, InsP3R1, and InsP3R2.
4.1.2. PEV-Induced SOCE Potentiation Does Not Involve STIM1 and ORA1 Up-Regulation
The larger ER Ca2+ release could also lead to an increase in SOCE activation even though the expression of its underlying molecular components, i.e., STIM1 and Orai1, is not altered by PEVs [60,61,62], as shown in the present investigation. Although some studies have provided evidence against a straightforward relationship between the extent of ER Ca2+ depletion and SOCE amplitude [81,82], other investigations have clearly shown a roughly linear relationship between the magnitude of InsP3-induced ER Ca2+ release and SOCE activation [60,61,62,83,84]. In agreement with these observations, SOCE amplitude in MDA-MB-231 cells treated with PEVs was always significantly larger than in untreated cells whatever the stimulus inducing ER Ca2+ release, i.e., CPA, ATP, or InsP3-BM. Interestingly, SOCE has also been shown to drive metastasis and invasion in MDA-MB-231 cells [24,30,31]. Therefore, the overall remodelling of intracellular Ca2+ dynamics, i.e., the enhancement of InsP3-induced ER Ca2+ release and SOCE activation, is predicted to stimulate migration in PEV-treated MDA-MB-231.
4.2. PEV-Dependent Increase in InsP3-Induced Ca2+ Release Potentiates Migration in MDA-MB-231 Cells: The Role of p38 MAPK and MLC2 Signalling Pathways
An increase in [Ca2+]i has long been known to mediate the pro-migratory effect of serum on cancer cells [26,30,31,52,53]. In line with this evidence, early work showed that PLCγ1 is recruited by serum to stimulate motility and adhesion [85] and that genetic silencing of Orai1 prevents serum-induced migration in MDA-MB-231 cells [31]. Herein, we provided the first characterization of the molecular mechanisms whereby serum triggers an increase in [Ca2+]i in this highly migrating breast cancer cell line. Pharmacological manipulation confirmed that serum-evoked intracellular Ca2+ release in MDA-MB-231 cells was inhibited by interfering with PLC activity with U73122 by inhibiting InsP3Rs with 2-APB and by depleting the ER Ca2+ pool with CPA. Furthermore, serum-evoked extracellular Ca2+ entry was dampened by BTP-2 and Pyr6, two highly specific inhibitors of Orai1. These findings concur with the involvement of InsP3Rs and SOCE in the Ca2+ signal evoked by serum in MDA-MB-231 cells, as also documented in other types of cancer cells [40,53,86], including low-migrating MCF-7 breast cancer cells [87]. As expected by the preliminary characterization with CPA, ATP, and InsP3-BM, both phases of serum-evoked intracellular Ca2+ signals (i.e., ER Ca2+ mobilization and SOCE) were significantly increased upon exposure to PEVs. Previous work has shown that both InsP3Rs and SOCE drive migration in highly migrating MDA-MB-231 breast cancer cells [24,26,32]. Therefore, the potentiation of serum-evoked intracellular Ca2+ signals could contribute to the enhanced migration rate that we recently reported in MDA-MB-231 cells exposed to PEVs [16]. In agreement with this hypothesis, blocking InsP3Rs with 2-APB significantly reduced serum-induced migration, a finding that is entirely consistent with the reported involvement InsP3R1 and InsP3R2 in MDA-MB-231 cell migration [26]. Surprisingly, inhibiting SOCE with BTP-2 did not affect motility in PEV-treated MDA-MB-231 cells. Nevertheless, both 2-APB and BTP-2 prevented serum-induced MLC2 and p38 MAPK phosphorylation, which drives cancer cell migration [17,18,19,20]. These data confirm our recent evidence that the long-term exposure to PEVs potentiates p38 MAPK and MLC2 phosphorylation in MDA-MB-231 cells [16] and further shows that the recruitment of these signalling pathways requires an increase in [Ca2+]i. Under the same conditions, we failed to detect any effect with regard to PEVs on the activation of the Ca2+-sensitive focal adhesion kinase Pyk2, whose role in driving the breast cancer metastatic outgrowth was previously documented [16]. The seeming discrepancy between the divergent effects of 2-APB and BTP-2 on p38 MAPK and MLC2 phosphorylation and cell motility could reflect some degree of redundancy between multiple Ca2+ sources which can engage the same Ca2+-dependent effectors [88,89,90]. However, the phosphorylation cascades triggered by SOCE do not seem to play a major role in MDA-MB-231 cell migration, which is instead finely tuned by the p38 MAPK and MLC2 pathways engaged by InsP3Rs. Alternatively, InsP3-induced ER Ca2+ release could recruit additional Ca2+-dependent effectors of cell motility that are not coupled to SOCE and remain to be identified. Future work is mandatory to assess this issue, but InsP3-induced ER Ca2+ release is clearly required to potentiate p38 MAPK and MLC2 phosphorylation and to stimulate migration in PEV-treated MDA-MB-231 cells.
5. Conclusions
Herein, we provide the first evidence that long-term exposure to PEVs induces a remarkable alteration in the Ca2+ transport system in the highly aggressive TNBC cell line, MDA-MB-231, thereby leading to an increase in InsP3-induced Ca2+ release and SOCE amplitude. In particular, the larger Ca2+ mobilization from the ER is required to potentiate serum-induced migration through the recruitment of p38 MAPK and MLC2. These findings lay the foundation for targeting the Ca2+ handling machinery not only to prevent breast cancer cell stimulation by pro-oncogenic chemical mediators and physical signals [23,24,29,37,55,56] but also by PEVs.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/cells11193120/s1, Figure S1: Long-term exposure of MDA-MB-231 cells with PEVs did not affect basal [Ca2+]i and constitutive Ca2+ entry; Figure S2: InsP3-dependent ER Ca2+ mobilization supports ATP-induced intracellular Ca2+ release in MDA-MB-231 cells; Figure S3: Further evidence that long-term exposure of MDA-MB-231 cells with PEVs increases FBS-induced extracellular Ca2+ entry. Figure S4: Long-term exposure of MDA-MB-231 cells with PEVs did not increase Pyk2 phosphorylation; Table S1: Primers used for real time qPCRs.
Author Contributions
Conceptualization, G.F.G. and F.M.; investigation, M.V., S.N., F.S., V.B., S.M.G.T., P.F., L.G. and T.S.; data curation, M.V., S.N. and F.S.; writing—original draft preparation, F.M. and G.F.G.; writing—review and editing, F.M., G.F.G., I.C., R.B.-R. and M.T.; supervision, G.F.G. and F.M.; project administration, G.F.G. and F.M.; funding acquisition, M.T., G.F.G. and F.M. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by: Italian Ministry of Education, University and Research (MIUR): Dipartimenti di Eccellenza Program (2018–2022)-Dept. of Biology and Biotechnology “L. Spallanzani”, University of Pavia (I.C., M.T., G.F.G. and F.M.); Fondo Ricerca Giovani from the University of Pavia (I.C., M.T., G.F.G. and F.M.), and the EU Horizon 2020 FETOPEN-2018-2020 Program under Grant Agreement N. 828984, LION-HEARTED (F.M.).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data supporting reported results can be obtained upon reasonable request to the authors.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Won, K.A.; Spruck, C. Triplenegative breast cancer therapy: Current and future perspectives (Review). Int. J. Oncol. 2020, 57, 1245–1261. [Google Scholar] [CrossRef] [PubMed]
- Dong, M.; Liu, Q.; Xu, Y.; Zhang, Q. Extracellular Vesicles: The Landscape in the Progression, Diagnosis, and Treatment of Triple-Negative Breast Cancer. Front. Cell Dev. Biol. 2022, 10, 842898. [Google Scholar] [CrossRef] [PubMed]
- Zara, M.; Guidetti, G.F.; Camera, M.; Canobbio, I.; Amadio, P.; Torti, M.; Tremoli, E.; Barbieri, S.S. Biology and Role of Extracellular Vesicles (EVs) in the Pathogenesis of Thrombosis. Int. J. Mol. Sci. 2019, 20, 2840. [Google Scholar] [CrossRef] [PubMed]
- Zara, M.; Amadio, P.; Campodonico, J.; Sandrini, L.; Barbieri, S.S. Exosomes in Cardiovascular Diseases. Diagnostics 2020, 10, 943. [Google Scholar] [CrossRef]
- Zmigrodzka, M.; Witkowska-Pilaszewicz, O.; Winnicka, A. Platelets Extracellular Vesicles as Regulators of Cancer Progression—An Updated Perspective. Int. J. Mol. Sci. 2020, 21, 5195. [Google Scholar] [CrossRef]
- Taus, F.; Meneguzzi, A.; Castelli, M.; Minuz, P. Platelet-Derived Extracellular Vesicles as Target of Antiplatelet Agents. What Is the Evidence? Front. Pharmacol. 2019, 10, 1256. [Google Scholar] [CrossRef]
- Laresche, C.; Pelletier, F.; Garnache-Ottou, F.; Lihoreau, T.; Biichle, S.; Mourey, G.; Saas, P.; Humbert, P.; Seilles, E.; Aubin, F. Increased levels of circulating microparticles are associated with increased procoagulant activity in patients with cutaneous malignant melanoma. J. Investig. Dermatol. 2014, 134, 176–182. [Google Scholar] [CrossRef] [PubMed]
- Hron, G.; Kollars, M.; Weber, H.; Sagaster, V.; Quehenberger, P.; Eichinger, S.; Kyrle, P.A.; Weltermann, A. Tissue factor-positive microparticles: Cellular origin and association with coagulation activation in patients with colorectal cancer. Thromb. Haemost. 2007, 97, 119–123. [Google Scholar] [CrossRef]
- Tseng, C.C.; Wang, C.C.; Chang, H.C.; Tsai, T.H.; Chang, L.T.; Huang, K.T.; Leu, S.; Yen, C.H.; Liu, S.F.; Chen, C.H.; et al. Levels of circulating microparticles in lung cancer patients and possible prognostic value. Dis. Markers 2013, 35, 301–310. [Google Scholar] [CrossRef] [PubMed]
- Chaari, M.; Ayadi, I.; Rousseau, A.; Lefkou, E.; Van Dreden, P.; Sidibe, F.; Ketatni, H.; Galea, V.; Khaterchi, A.; Bouzguenda, R.; et al. Impact of breast cancer stage, time from diagnosis and chemotherapy on plasma and cellular biomarkers of hypercoagulability. BMC Cancer 2014, 14, 991. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.; Wu, Y.W.; Blyth, C.; Lichtfuss, G.; Goubran, H.; Burnouf, T. Prospective Therapeutic Applications of Platelet Extracellular Vesicles. Trends Biotechnol. 2021, 39, 598–612. [Google Scholar] [CrossRef] [PubMed]
- Dovizio, M.; Bruno, A.; Contursi, A.; Grande, R.; Patrignani, P. Platelets and extracellular vesicles in cancer: Diagnostic and therapeutic implications. Cancer Metastasis Rev. 2018, 37, 455–467. [Google Scholar] [CrossRef] [PubMed]
- Michael, J.V.; Wurtzel, J.G.T.; Mao, G.F.; Rao, A.K.; Kolpakov, M.A.; Sabri, A.; Hoffman, N.E.; Rajan, S.; Tomar, D.; Madesh, M.; et al. Platelet microparticles infiltrating solid tumors transfer miRNAs that suppress tumor growth. Blood 2017, 130, 567–580. [Google Scholar] [CrossRef]
- Gasperi, V.; Vangapandu, C.; Savini, I.; Ventimiglia, G.; Adorno, G.; Catani, M.V. Polyunsaturated fatty acids modulate the delivery of platelet microvesicle-derived microRNAs into human breast cancer cell lines. J. Nutr. Biochem. 2019, 74, 108242. [Google Scholar] [CrossRef]
- Vismara, M.; Zara, M.; Negri, S.; Canino, J.; Canobbio, I.; Barbieri, S.S.; Moccia, F.; Torti, M.; Guidetti, G.F. Platelet-derived extracellular vesicles regulate cell cycle progression and cell migration in breast cancer cells. Biochim. Biophys. Acta Mol. Cell Res. 2021, 1868, 118886. [Google Scholar] [CrossRef]
- Faris, P.; Pellavio, G.; Ferulli, F.; Di Nezza, F.; Shekha, M.; Lim, D.; Maestri, M.; Guerra, G.; Ambrosone, L.; Pedrazzoli, P.; et al. Nicotinic Acid Adenine Dinucleotide Phosphate (NAADP) Induces Intracellular Ca2+ Release through the Two-Pore Channel TPC1 in Metastatic Colorectal Cancer Cells. Cancers 2019, 11, 542. [Google Scholar] [CrossRef]
- Stadler, S.; Nguyen, C.H.; Schachner, H.; Milovanovic, D.; Holzner, S.; Brenner, S.; Eichsteininger, J.; Stadler, M.; Senfter, D.; Krenn, L.; et al. Colon cancer cell-derived 12(S)-HETE induces the retraction of cancer-associated fibroblast via MLC2, RHO/ROCK and Ca2+ signalling. Cell. Mol. Life Sci. 2017, 74, 1907–1921. [Google Scholar] [CrossRef] [PubMed]
- Chao, F.; Song, Z.; Wang, S.; Ma, Z.; Zhuo, Z.; Meng, T.; Xu, G.; Chen, G. Novel circular RNA circSOBP governs amoeboid migration through the regulation of the miR-141-3p/MYPT1/p-MLC2 axis in prostate cancer. Clin. Transl. Med. 2021, 11, e360. [Google Scholar] [CrossRef]
- Kong, X.; Li, M.; Shao, K.; Yang, Y.; Wang, Q.; Cai, M. Progesterone induces cell apoptosis via the CACNA2D3/Ca2+/p38 MAPK pathway in endometrial cancer. Oncol. Rep. 2020, 43, 121–132. [Google Scholar] [CrossRef]
- Prevarskaya, N.; Skryma, R.; Shuba, Y. Ion Channels in Cancer: Are Cancer Hallmarks Oncochannelopathies? Physiol. Rev. 2018, 98, 559–621. [Google Scholar] [CrossRef]
- Monteith, G.R.; Prevarskaya, N.; Roberts-Thomson, S.J. The calcium-cancer signalling nexus. Nat. Rev. Cancer 2017, 17, 367–380. [Google Scholar] [CrossRef] [PubMed]
- Azimi, I.; Bong, A.H.; Poo, G.X.H.; Armitage, K.; Lok, D.; Roberts-Thomson, S.J.; Monteith, G.R. Pharmacological inhibition of store-operated calcium entry in MDA-MB-468 basal A breast cancer cells: Consequences on calcium signalling, cell migration and proliferation. Cell. Mol. Life Sci. 2018, 75, 4525–4537. [Google Scholar] [CrossRef] [PubMed]
- Jardin, I.; Lopez, J.J.; Salido, G.M.; Rosado, J.A. Store-Operated Ca2+ Entry in Breast Cancer Cells: Remodeling and Functional Role. Int. J. Mol. Sci. 2018, 19, 4053. [Google Scholar] [CrossRef] [PubMed]
- Faris, P.; Shekha, M.; Montagna, D.; Guerra, G.; Moccia, F. Endolysosomal Ca2+ Signalling and Cancer Hallmarks: Two-Pore Channels on the Move, TRPML1 Lags Behind! Cancers 2018, 11, 27. [Google Scholar] [CrossRef]
- Mound, A.; Vautrin-Glabik, A.; Foulon, A.; Botia, B.; Hague, F.; Parys, J.B.; Ouadid-Ahidouch, H.; Rodat-Despoix, L. Downregulation of type 3 inositol (1,4,5)-trisphosphate receptor decreases breast cancer cell migration through an oscillatory Ca2+ signal. Oncotarget 2017, 8, 72324–72341. [Google Scholar] [CrossRef]
- Motiani, R.K.; Abdullaev, I.F.; Trebak, M. A novel native store-operated calcium channel encoded by Orai3: Selective requirement of Orai3 versus Orai1 in estrogen receptor-positive versus estrogen receptor-negative breast cancer cells. J. Biol. Chem. 2010, 285, 19173–19183. [Google Scholar] [CrossRef]
- McAndrew, D.; Grice, D.M.; Peters, A.A.; Davis, F.M.; Stewart, T.; Rice, M.; Smart, C.E.; Brown, M.A.; Kenny, P.A.; Roberts-Thomson, S.J.; et al. ORAI1-mediated calcium influx in lactation and in breast cancer. Mol. Cancer Ther. 2011, 10, 448–460. [Google Scholar] [CrossRef]
- Moccia, F.; Zuccolo, E.; Poletto, V.; Turin, I.; Guerra, G.; Pedrazzoli, P.; Rosti, V.; Porta, C.; Montagna, D. Targeting Stim and Orai Proteins as an Alternative Approach in Anticancer Therapy. Curr. Med. Chem. 2016, 23, 3450–3480. [Google Scholar] [CrossRef]
- Yang, S.; Zhang, J.J.; Huang, X.Y. Orai1 and STIM1 are critical for breast tumor cell migration and metastasis. Cancer Cell 2009, 15, 124–134. [Google Scholar] [CrossRef]
- Hammadi, M.; Chopin, V.; Matifat, F.; Dhennin-Duthille, I.; Chasseraud, M.; Sevestre, H.; Ouadid-Ahidouch, H. Human ether a-gogo K+ channel 1 (hEag1) regulates MDA-MB-231 breast cancer cell migration through Orai1-dependent calcium entry. J. Cell. Physiol. 2012, 227, 3837–3846. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Collado, J.; Lopez, J.J.; Jardin, I.; Camello, P.J.; Falcon, D.; Regodon, S.; Salido, G.M.; Smani, T.; Rosado, J.A. Adenylyl Cyclase Type 8 Overexpression Impairs Phosphorylation-Dependent Orai1 Inactivation and Promotes Migration in MDA-MB-231 Breast Cancer Cells. Cancers 2019, 11, 1624. [Google Scholar] [CrossRef]
- Parys, J.B.; Bultynck, G.; Vervliet, T. IP3 Receptor Biology and Endoplasmic Reticulum Calcium Dynamics in Cancer. Prog. Mol. Subcell. Biol. 2021, 59, 215–237. [Google Scholar] [CrossRef]
- Morciano, G.; Marchi, S.; Morganti, C.; Sbano, L.; Bittremieux, M.; Kerkhofs, M.; Corricelli, M.; Danese, A.; Karkucinska-Wieckowska, A.; Wieckowski, M.R.; et al. Role of Mitochondria-Associated ER Membranes in Calcium Regulation in Cancer-Specific Settings. Neoplasia 2018, 20, 510–523. [Google Scholar] [CrossRef] [PubMed]
- Faris, P.; Rumolo, A.; Tapella, L.; Tanzi, M.; Metallo, A.; Conca, F.; Negri, S.; Lefkimmiatis, K.; Pedrazzoli, P.; Lim, D.; et al. Store-Operated Ca2+ Entry Is Up-Regulated in Tumour-Infiltrating Lymphocytes from Metastatic Colorectal Cancer Patients. Cancers 2022, 14, 3312. [Google Scholar] [CrossRef]
- Moccia, F. Endothelial Ca2+ Signaling and the Resistance to Anticancer Treatments: Partners in Crime. Int. J. Mol. Sci. 2018, 19, 217. [Google Scholar] [CrossRef] [PubMed]
- Lodola, F.; Laforenza, U.; Cattaneo, F.; Ruffinatti, F.A.; Poletto, V.; Massa, M.; Tancredi, R.; Zuccolo, E.; Khdar, A.D.; Riccardi, A.; et al. VEGF-induced intracellular Ca2+ oscillations are down-regulated and do not stimulate angiogenesis in breast cancer-derived endothelial colony forming cells. Oncotarget 2017, 8, 95223–95246. [Google Scholar] [CrossRef] [PubMed]
- Milano, S.; Gerbino, A.; Schena, G.; Carmosino, M.; Svelto, M.; Procino, G. Human beta3-Adrenoreceptor is Resistant to Agonist-Induced Desensitization in Renal Epithelial Cells. Cell. Physiol. Biochem. 2018, 48, 847–862. [Google Scholar] [CrossRef]
- Procino, G.; Gerbino, A.; Milano, S.; Nicoletti, M.C.; Mastrofrancesco, L.; Carmosino, M.; Svelto, M. Rosiglitazone promotes AQP2 plasma membrane expression in renal cells via a Ca-dependent/cAMP-independent mechanism. Cell. Physiol. Biochem. 2015, 35, 1070–1085. [Google Scholar] [CrossRef] [PubMed]
- Zuccolo, E.; Laforenza, U.; Ferulli, F.; Pellavio, G.; Scarpellino, G.; Tanzi, M.; Turin, I.; Faris, P.; Lucariello, A.; Maestri, M.; et al. Stim and Orai mediate constitutive Ca2+ entry and control endoplasmic reticulum Ca2+ refilling in primary cultures of colorectal carcinoma cells. Oncotarget 2018, 9, 31098–31119. [Google Scholar] [CrossRef]
- Feng, M.; Grice, D.M.; Faddy, H.M.; Nguyen, N.; Leitch, S.; Wang, Y.; Muend, S.; Kenny, P.A.; Sukumar, S.; Roberts-Thomson, S.J.; et al. Store-independent activation of Orai1 by SPCA2 in mammary tumors. Cell 2010, 143, 84–98. [Google Scholar] [CrossRef] [PubMed]
- Zuccolo, E.; Bottino, C.; Diofano, F.; Poletto, V.; Codazzi, A.C.; Mannarino, S.; Campanelli, R.; Fois, G.; Marseglia, G.L.; Guerra, G.; et al. Constitutive Store-Operated Ca2+ Entry Leads to Enhanced Nitric Oxide Production and Proliferation in Infantile Hemangioma-Derived Endothelial Colony-Forming Cells. Stem Cells Dev. 2016, 25, 301–319. [Google Scholar] [CrossRef] [PubMed]
- Gerbino, A.; Bottillo, I.; Milano, S.; Lipari, M.; Zio, R.; Morlino, S.; Mola, M.G.; Procino, G.; Re, F.; Zachara, E.; et al. Functional Characterization of a Novel Truncating Mutation in Lamin A/C Gene in a Family with a Severe Cardiomyopathy with Conduction Defects. Cell. Physiol. Biochem. 2017, 44, 1559–1577. [Google Scholar] [CrossRef]
- Pienimaeki-Roemer, A.; Konovalova, T.; Musri, M.M.; Sigruener, A.; Boettcher, A.; Meister, G.; Schmitz, G. Transcriptomic profiling of platelet senescence and platelet extracellular vesicles. Transfusion 2017, 57, 144–156. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Coulouris, G.; Zaretskaya, I.; Cutcutache, I.; Rozen, S.; Madden, T.L. Primer-BLAST: A tool to design target-specific primers for polymerase chain reaction. BMC Bioinform. 2012, 13, 134. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Pfaffl, M.W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001, 29, e45. [Google Scholar] [CrossRef] [PubMed]
- Guidetti, G.F.; Zara, M.; Canobbio, I.; Visconte, C.; Di Nunzio, G.; Torti, M. Novel pharmacological inhibitors demonstrate the role of the tyrosine kinase Pyk2 in adhesion and aggregation of human platelets. Thromb. Haemost. 2016, 116, 904–917. [Google Scholar] [CrossRef] [PubMed]
- Zara, M.; Guidetti, G.F.; Boselli, D.; Villa, C.; Canobbio, I.; Seppi, C.; Visconte, C.; Canino, J.; Torti, M. Release of Prometastatic Platelet-Derived Microparticles Induced by Breast Cancer Cells: A Novel Positive Feedback Mechanism for Metastasis. TH Open Companion J. Thromb. Haemost. 2017, 1, e155–e163. [Google Scholar] [CrossRef] [PubMed]
- Davis, F.M.; Peters, A.A.; Grice, D.M.; Cabot, P.J.; Parat, M.O.; Roberts-Thomson, S.J.; Monteith, G.R. Non-stimulated, agonist-stimulated and store-operated Ca2+ influx in MDA-MB-468 breast cancer cells and the effect of EGF-induced EMT on calcium entry. PLoS ONE 2012, 7, e36923. [Google Scholar] [CrossRef] [PubMed]
- Moccia, F.; Negri, S.; Faris, P.; Perna, A.; De Luca, A.; Soda, T.; Romani, R.B.; Guerra, G. Targeting Endolysosomal Two-Pore Channels to Treat Cardiovascular Disorders in the Novel COronaVIrus Disease 2019. Front. Physiol. 2021, 12, 629119. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Zhang, H.; Jin, F.; Fang, M.; Huang, M.; Yang, C.S.; Chen, T.; Fu, L.; Pan, Z. Elevated Orai1 expression mediates tumor-promoting intracellular Ca2+ oscillations in human esophageal squamous cell carcinoma. Oncotarget 2014, 5, 3455–3471. [Google Scholar] [CrossRef] [PubMed]
- Dragoni, S.; Turin, I.; Laforenza, U.; Potenza, D.M.; Bottino, C.; Glasnov, T.N.; Prestia, M.; Ferulli, F.; Saitta, A.; Mosca, A.; et al. Store-operated Ca2+ entry does not control proliferation in primary cultures of human metastatic renal cellular carcinoma. BioMed Res. Int. 2014, 2014, 739494. [Google Scholar] [CrossRef] [PubMed]
- Pierro, C.; Cook, S.J.; Foets, T.C.; Bootman, M.D.; Roderick, H.L. Oncogenic K-Ras suppresses IP(3)-dependent Ca2+ release through remodelling of the isoform composition of IP(3)Rs and ER luminal Ca2+ levels in colorectal cancer cell lines. J. Cell Sci. 2014, 127, 1607–1619. [Google Scholar] [CrossRef]
- So, C.L.; Meinert, C.; Xia, Q.; Robitaille, M.; Roberts-Thomson, S.J.; Monteith, G.R. Increased matrix stiffness suppresses ATP-induced sustained Ca2+ influx in MDA-MB-231 breast cancer cells. Cell Calcium 2022, 104, 102569. [Google Scholar] [CrossRef]
- Bong, A.H.L.; Bassett, J.J.; Roberts-Thomson, S.J.; Monteith, G.R. Assessment of doxorubicin-induced remodeling of Ca2+ signaling and associated Ca2+ regulating proteins in MDA-MB-231 breast cancer cells. Biochem. Biophys. Res. Commun. 2020, 522, 532–538. [Google Scholar] [CrossRef]
- Astesana, V.; Faris, P.; Ferrari, B.; Siciliani, S.; Lim, D.; Biggiogera, M.; De Pascali, S.A.; Fanizzi, F.P.; Roda, E.; Moccia, F.; et al. [Pt(O,O’-acac)(γ-acac)(DMS)]: Alternative Strategies to Overcome Cisplatin-Induced Side Effects and Resistance in T98G Glioma Cells. Cell. Mol. Neurobiol. 2021, 41, 563–587. [Google Scholar] [CrossRef]
- Fierro, L.; Lund, P.E.; Parekh, A.B. Comparison of the activation of the Ca2+ release-activated Ca2+ current ICRAC to InsP3 in Jurkat T-lymphocytes, pulmonary artery endothelia and RBL-1 cells. Pflüg. Arch. 2000, 440, 580–587. [Google Scholar] [CrossRef]
- Contreras-Leal, E.; Hernandez-Oliveras, A.; Flores-Peredo, L.; Zarain-Herzberg, A.; Santiago-Garcia, J. Histone deacetylase inhibitors promote the expression of ATP2A3 gene in breast cancer cell lines. Mol. Carcinog. 2016, 55, 1477–1485. [Google Scholar] [CrossRef]
- Parekh, A.B.; Putney, J.W., Jr. Store-operated calcium channels. Physiol. Rev. 2005, 85, 757–810. [Google Scholar] [CrossRef]
- Michalak, M.; Groenendyk, J.; Szabo, E.; Gold, L.I.; Opas, M. Calreticulin, a multi-process calcium-buffering chaperone of the endoplasmic reticulum. Biochem. J. 2009, 417, 651–666. [Google Scholar] [CrossRef] [PubMed]
- Pick, T.; Beck, A.; Gamayun, I.; Schwarz, Y.; Schirra, C.; Jung, M.; Krause, E.; Niemeyer, B.A.; Zimmermann, R.; Lang, S.; et al. Remodelling of Ca2+ homeostasis is linked to enlarged endoplasmic reticulum in secretory cells. Cell Calcium 2021, 99, 102473. [Google Scholar] [CrossRef] [PubMed]
- Park, S.J.; Lee, K.P.; Kang, S.; Chung, H.Y.; Bae, Y.S.; Okajima, F.; Im, D.S. Lysophosphatidylethanolamine utilizes LPA(1) and CD97 in MDA-MB-231 breast cancer cells. Cell. Signal. 2013, 25, 2147–2154. [Google Scholar] [CrossRef] [PubMed]
- Lazar, S.; Goldfinger, L.E. Platelets and extracellular vesicles and their cross talk with cancer. Blood 2021, 137, 3192–3200. [Google Scholar] [CrossRef] [PubMed]
- Ramirez-Ricardo, J.; Leal-Orta, E.; Martinez-Baeza, E.; Ortiz-Mendoza, C.; Breton-Mora, F.; Herrera-Torres, A.; Elizalde-Acosta, I.; Cortes-Reynosa, P.; Thompson-Bonilla, R.; Perez Salazar, E. Circulating extracellular vesicles from patients with breast cancer enhance migration and invasion via a Srcdependent pathway in MDAMB231 breast cancer cells. Mol. Med. Rep. 2020, 22, 1932–1948. [Google Scholar] [CrossRef]
- Zara, M.; Canobbio, I.; Visconte, C.; Canino, J.; Torti, M.; Guidetti, G.F. Molecular mechanisms of platelet activation and aggregation induced by breast cancer cells. Cell. Signal. 2018, 48, 45–53. [Google Scholar] [CrossRef]
- Lu, F.; Sun, J.; Zheng, Q.; Li, J.; Hu, Y.; Yu, P.; He, H.; Zhao, Y.; Wang, X.; Yang, S.; et al. Imaging elemental events of store-operated Ca2+ entry in invading cancer cells with plasmalemmal targeted sensors. J. Cell Sci. 2019, 132, jcs224923. [Google Scholar] [CrossRef]
- Vautrin-Glabik, A.; Botia, B.; Kischel, P.; Ouadid-Ahidouch, H.; Rodat-Despoix, L. IP3R3 silencing induced actin cytoskeletal reorganization through ARHGAP18/RhoA/mDia1/FAK pathway in breast cancer cell lines. Biochim. Biophys. Acta. Mol. Cell Res. 2018, 1865, 945–958. [Google Scholar] [CrossRef]
- Moccia, F.; Dragoni, S.; Poletto, V.; Rosti, V.; Tanzi, F.; Ganini, C.; Porta, C. Orai1 and Transient Receptor Potential Channels as novel molecular targets to impair tumor neovascularisation in renal cell carcinoma and other malignancies. Anticancer Agents Med. Chem. 2014, 14, 296–312. [Google Scholar] [CrossRef]
- Negri, S.; Faris, P.; Berra-Romani, R.; Guerra, G.; Moccia, F. Endothelial Transient Receptor Potential Channels and Vascular Remodeling: Extracellular Ca2+ Entry for Angiogenesis, Arteriogenesis and Vasculogenesis. Front. Physiol. 2019, 10, 1618. [Google Scholar] [CrossRef]
- Li, S.S.; Gao, S.; Chen, Y.; Bao, H.; Li, Z.T.; Yao, Q.P.; Liu, J.T.; Wang, Y.; Qi, Y.X. Platelet-derived microvesicles induce calcium oscillations and promote VSMC migration via TRPV4. Theranostics 2021, 11, 2410–2423. [Google Scholar] [CrossRef] [PubMed]
- Vanden Abeele, F.; Skryma, R.; Shuba, Y.; Van Coppenolle, F.; Slomianny, C.; Roudbaraki, M.; Mauroy, B.; Wuytack, F.; Prevarskaya, N. Bcl-2-dependent modulation of Ca2+ homeostasis and store-operated channels in prostate cancer cells. Cancer Cell 2002, 1, 169–179. [Google Scholar] [CrossRef]
- Humez, S.; Legrand, G.; Vanden-Abeele, F.; Monet, M.; Marchetti, P.; Lepage, G.; Crepin, A.; Dewailly, E.; Wuytack, F.; Prevarskaya, N. Role of endoplasmic reticulum calcium content in prostate cancer cell growth regulation by IGF and TNFalpha. J. Cell. Physiol. 2004, 201, 201–213. [Google Scholar] [CrossRef]
- Boutin, B.; Tajeddine, N.; Monaco, G.; Molgo, J.; Vertommen, D.; Rider, M.; Parys, J.B.; Bultynck, G.; Gailly, P. Endoplasmic reticulum Ca2+ content decrease by PKA-dependent hyperphosphorylation of type 1 IP3 receptor contributes to prostate cancer cell resistance to androgen deprivation. Cell Calcium 2015, 57, 312–320. [Google Scholar] [CrossRef] [PubMed]
- Messai, Y.; Noman, M.Z.; Hasmim, M.; Janji, B.; Tittarelli, A.; Boutet, M.; Baud, V.; Viry, E.; Billot, K.; Nanbakhsh, A.; et al. ITPR1 protects renal cancer cells against natural killer cells by inducing autophagy. Cancer Res. 2014, 74, 6820–6832. [Google Scholar] [CrossRef]
- Yu, M.; Lu, W.; Cao, Z.; Xuan, T. LncRNA LINC00662 Exerts an Oncogenic Effect on Osteosarcoma by the miR-16-5p/ITPR1 Axis. J. Oncol. 2021, 2021, 8493431. [Google Scholar] [CrossRef]
- Huang, X.; Jin, M.; Chen, Y.X.; Wang, J.; Zhai, K.; Chang, Y.; Yuan, Q.; Yao, K.T.; Ji, G. ERP44 inhibits human lung cancer cell migration mainly via IP3R2. Aging 2016, 8, 1276–1286. [Google Scholar] [CrossRef][Green Version]
- Sun, C.; Shui, B.; Zhao, W.; Liu, H.; Li, W.; Lee, J.C.; Doran, R.; Lee, F.K.; Sun, T.; Shen, Q.S.; et al. Central role of IP3R2-mediated Ca2+ oscillation in self-renewal of liver cancer stem cells elucidated by high-signal ER sensor. Cell Death Dis. 2019, 10, 396. [Google Scholar] [CrossRef] [PubMed]
- Bittremieux, M.; La Rovere, R.M.; Akl, H.; Martines, C.; Welkenhuyzen, K.; Dubron, K.; Baes, M.; Janssens, A.; Vandenberghe, P.; Laurenti, L.; et al. Constitutive IP3 signaling underlies the sensitivity of B-cell cancers to the Bcl-2/IP3 receptor disruptor BIRD-2. Cell Death Differ. 2019, 26, 531–547. [Google Scholar] [CrossRef] [PubMed]
- Chovancova, B.; Liskova, V.; Miklikova, S.; Hudecova, S.; Babula, P.; Penesova, A.; Sevcikova, A.; Durinikova, E.; Novakova, M.; Matuskova, M.; et al. Calcium signaling affects migration and proliferation differently in individual cancer cells due to nifedipine treatment. Biochem. Pharmacol. 2020, 171, 113695. [Google Scholar] [CrossRef]
- Parekh, A.B.; Fleig, A.; Penner, R. The store-operated calcium current I(CRAC): Nonlinear activation by InsP3 and dissociation from calcium release. Cell 1997, 89, 973–980. [Google Scholar] [CrossRef]
- Ribeiro, C.M.; Putney, J.W. Differential effects of protein kinase C activation on calcium storage and capacitative calcium entry in NIH 3T3 cells. J. Biol. Chem. 1996, 271, 21522–21528. [Google Scholar] [CrossRef]
- Sedova, M.; Klishin, A.; Huser, J.; Blatter, L.A. Capacitative Ca2+ entry is graded with degree of intracellular Ca2+ store depletion in bovine vascular endothelial cells. J. Physiol. 2000, 523 Pt 3, 549–559. [Google Scholar] [CrossRef]
- Luik, R.M.; Wang, B.; Prakriya, M.; Wu, M.M.; Lewis, R.S. Oligomerization of STIM1 couples ER calcium depletion to CRAC channel activation. Nature 2008, 454, 538–542. [Google Scholar] [CrossRef]
- Piccolo, E.; Innominato, P.F.; Mariggio, M.A.; Maffucci, T.; Iacobelli, S.; Falasca, M. The mechanism involved in the regulation of phospholipase Cgamma1 activity in cell migration. Oncogene 2002, 21, 6520–6529. [Google Scholar] [CrossRef][Green Version]
- Sun, J.; Lu, F.; He, H.; Shen, J.; Messina, J.; Mathew, R.; Wang, D.; Sarnaik, A.A.; Chang, W.C.; Kim, M.; et al. STIM1- and Orai1-mediated Ca2+ oscillation orchestrates invadopodium formation and melanoma invasion. J. Cell Biol. 2014, 207, 535–548. [Google Scholar] [CrossRef]
- Szatkowski, C.; Parys, J.B.; Ouadid-Ahidouch, H.; Matifat, F. Inositol 1,4,5-trisphosphate-induced Ca2+ signalling is involved in estradiol-induced breast cancer epithelial cell growth. Mol. Cancer 2010, 9, 156. [Google Scholar] [CrossRef]
- Zuccolo, E.; Di Buduo, C.; Lodola, F.; Orecchioni, S.; Scarpellino, G.; Kheder, D.A.; Poletto, V.; Guerra, G.; Bertolini, F.; Balduini, A.; et al. Stromal Cell-Derived Factor-1alpha Promotes Endothelial Colony-Forming Cell Migration Through the Ca2+-Dependent Activation of the Extracellular Signal-Regulated Kinase 1/2 and Phosphoinositide 3-Kinase/AKT Pathways. Stem Cells Dev. 2018, 27, 23–34. [Google Scholar] [CrossRef]
- Di Buduo, C.A.; Abbonante, V.; Marty, C.; Moccia, F.; Rumi, E.; Pietra, D.; Soprano, P.M.; Lim, D.; Cattaneo, D.; Iurlo, A.; et al. Defective interaction of mutant calreticulin and SOCE in megakaryocytes from patients with myeloproliferative neoplasms. Blood 2020, 135, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Di Buduo, C.A.; Moccia, F.; Battiston, M.; De Marco, L.; Mazzucato, M.; Moratti, R.; Tanzi, F.; Balduini, A. The importance of calcium in the regulation of megakaryocyte function. Haematologica 2014, 99, 769–778. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).