
Amyloidogenesis and Neurotrophic Dysfunction in Alzheimer’s Disease: Do They have a Common Regulating Pathway?


Abstract
:1. Introduction
2. Amyloidogenesis and AD
3. NTFs and AD
4. Mutual Influence of Amyloidogenic Process and Neurotrophic Pathways
5. Is There Common Pathway to Control Both the Amyloidogenic Process and the Neurotrophic Pathways?
6. Targeting the Common Pathway for Preventing Both Amyloidogenesis and Neurotrophic Dysfunction
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gulisano, W.; Maugeri, D.; Baltrons, M.A.; Fa, M.; Amato, A.; Palmeri, A.; D’Adamio, L.; Grassi, C.; Devanand, D.P.; Honig, L.S.; et al. Role of Amyloid-beta and Tau Proteins in Alzheimer’s Disease: Confuting the Amyloid Cascade. J. Alzheimers Dis. JAD 2018, 64, S611–S631. [Google Scholar] [CrossRef] [PubMed]
- Appel, S.H. A unifying hypothesis for the cause of amyotrophic lateral sclerosis, parkinsonism, and Alzheimer disease. Ann. Neurol. 1981, 10, 499–505. [Google Scholar] [CrossRef] [PubMed]
- Allen, S.J.; Watson, J.J.; Dawbarn, D. The neurotrophins and their role in Alzheimer’s disease. Curr. Neuropharmacol. 2011, 9, 559–573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iulita, M.F.; Cuello, A.C. Nerve growth factor metabolic dysfunction in Alzheimer’s disease and Down syndrome. Trends Pharmacol. Sci. 2014, 35, 338–348. [Google Scholar] [CrossRef]
- Cattaneo, A.; Calissano, P. Nerve growth factor and Alzheimer’s disease: New facts for an old hypothesis. Mol. Neurobiol. 2012, 46, 588–604. [Google Scholar] [CrossRef] [PubMed]
- Capsoni, S.; Ugolini, G.; Comparini, A.; Ruberti, F.; Berardi, N.; Cattaneo, A. Alzheimer-like neurodegeneration in aged antinerve growth factor transgenic mice. Proc. Natl. Acad. Sci. USA 2000, 97, 6826–6831. [Google Scholar] [CrossRef] [Green Version]
- Pentz, R.; Iulita, M.F.; Ducatenzeiler, A.; Bennett, D.A.; Cuello, A.C. The human brain NGF metabolic pathway is impaired in the pre-clinical and clinical continuum of Alzheimers disease. Mol. Psychiatry 2020, 26, 6023–6037. [Google Scholar] [CrossRef]
- Hefti, F.; Weiner, W.J. Nerve growth factor and Alzheimer’s disease. Ann. Neurol. 1986, 20, 275–281. [Google Scholar] [CrossRef] [PubMed]
- Bartus, R.T.; Dean, R.L., III; Beer, B.; Lippa, A.S. The cholinergic hypothesis of geriatric memory dysfunction. Science 1982, 217, 408–414. [Google Scholar] [CrossRef] [PubMed]
- Aghourian, M.; Legault-Denis, C.; Soucy, J.P.; Rosa-Neto, P.; Gauthier, S.; Kostikov, A.; Gravel, P.; Bedard, M.A. Quantification of brain cholinergic denervation in Alzheimer’s disease using PET imaging with [F-18]-FEOBV. Mol. Psychiatry 2017, 22, 1531–1538. [Google Scholar] [CrossRef]
- Kimura, N.; Yanagisawa, K. Traffic jam hypothesis: Relationship between endocytic dysfunction and Alzheimer’s disease. Neurochem. Int. 2018, 119, 35–41. [Google Scholar] [CrossRef]
- Kent, S.A.; Spires-Jones, T.L.; Durrant, C.S. The physiological roles of tau and Abeta: Implications for Alzheimer’s disease pathology and therapeutics. Acta Neuropathol. 2020, 140, 417–447. [Google Scholar] [CrossRef] [PubMed]
- Guo, T.; Noble, W.; Hanger, D.P. Roles of tau protein in health and disease. Acta Neuropathol. 2017, 133, 665–704. [Google Scholar] [CrossRef] [Green Version]
- Long, J.M.; Holtzman, D.M. Alzheimer Disease: An Update on Pathobiology and Treatment Strategies. Cell 2019, 179, 312–339. [Google Scholar] [CrossRef]
- Budni, J.; Bellettini-Santos, T.; Mina, F.; Garcez, M.L.; Zugno, A.I. The involvement of BDNF, NGF and GDNF in aging and Alzheimer’s disease. Aging Dis. 2015, 6, 331–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mufson, E.J.; Counts, S.E.; Ginsberg, S.D.; Mahady, L.; Perez, S.E.; Massa, S.M.; Longo, F.M.; Ikonomovic, M.D. Nerve Growth Factor Pathobiology during the Progression of Alzheimer’s Disease. Front. Neurosci. 2019, 13, 533. [Google Scholar] [CrossRef]
- Matrone, C.; Ciotti, M.T.; Mercanti, D.; Marolda, R.; Calissano, P. NGF and BDNF signaling control amyloidogenic route and Abeta production in hippocampal neurons. Proc. Natl. Acad. Sci. USA 2008, 105, 13139–13144. [Google Scholar] [CrossRef] [Green Version]
- Zou, L.; Wang, Z.; Shen, L.; Bao, G.B.; Wang, T.; Kang, J.H.; Pei, G. Receptor tyrosine kinases positively regulate BACE activity and Amyloid-beta production through enhancing BACE internalization. Cell Res. 2007, 17, 389–401. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.W.; Chen, Y.; Liu, Y.; Zhao, Y.; Liao, F.F.; Xu, H. APP regulates NGF receptor trafficking and NGF-mediated neuronal differentiation and survival. PLoS ONE 2013, 8, e80571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matrone, C.; Barbagallo, A.P.; La Rosa, L.R.; Florenzano, F.; Ciotti, M.T.; Mercanti, D.; Chao, M.V.; Calissano, P.; D’Adamio, L. APP is phosphorylated by TrkA and regulates NGF/TrkA signaling. J. Neurosci. 2011, 31, 11756–11761. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, H.G.; Stricker, R.; Lendeckel, U.; Bertram, I.; Dobrowolny, H.; Steiner, J.; Bogerts, B.; Reiser, G. Reduced neuronal co-localisation of nardilysin and the putative alpha-secretases ADAM10 and ADAM17 in Alzheimer’s disease and Down syndrome brains. Age 2009, 31, 11–25. [Google Scholar] [CrossRef] [Green Version]
- Vassar, R.; Kuhn, P.H.; Haass, C.; Kennedy, M.E.; Rajendran, L.; Wong, P.C.; Lichtenthaler, S.F. Function, therapeutic potential and cell biology of BACE proteases: Current status and future prospects. J. Neurochem. 2014, 130, 4–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pasternak, S.H.; Callahan, J.W.; Mahuran, D.J. The role of the endosomal/lysosomal system in amyloid-beta production and the pathophysiology of Alzheimer’s disease: Reexamining the spatial paradox from a lysosomal perspective. J. Alzheimers Dis. 2004, 6, 53–65. [Google Scholar] [CrossRef]
- Sideris, D.I.; Danial, J.S.H.; Emin, D.; Ruggeri, F.S.; Xia, Z.; Zhang, Y.P.; Lobanova, E.; Dakin, H.; De, S.; Miller, A.; et al. Soluble amyloid beta-containing aggregates are present throughout the brain at early stages of Alzheimer’s disease. Brain Commun. 2021, 3, fcab147. [Google Scholar] [CrossRef]
- Donohue, M.C.; Sperling, R.A.; Petersen, R.; Sun, C.K.; Weiner, M.W.; Aisen, P.S.; Alzheimer’s Disease Neuroimaging Initiative. Association between Elevated Brain Amyloid and Subsequent Cognitive Decline among Cognitively Normal Persons. JAMA 2017, 317, 2305–2316. [Google Scholar] [CrossRef]
- Baker, J.E.; Lim, Y.Y.; Pietrzak, R.H.; Hassenstab, J.; Snyder, P.J.; Masters, C.L.; Maruff, P. Cognitive impairment and decline in cognitively normal older adults with high amyloid-beta: A meta-analysis. Alzheimers Dement. Diagn. Assess. Dis. Monit. 2017, 6, 108–121. [Google Scholar] [CrossRef] [Green Version]
- Sofroniew, M.V.; Howe, C.L.; Mobley, W.C. Nerve growth factor signaling, neuroprotection, and neural repair. Annu. Rev. Neurosci. 2001, 24, 1217–1281. [Google Scholar] [CrossRef]
- Lubke, J.H.; Idoon, F.; Mohasel-Roodi, M.; Alipour, F.; Hami, J.; Ehteshampour, A.; Mostafaee, H.; Sadeghi, A. Neurotrophic factors in Alzheimer’s disease: Pathogenesis and therapy. Acta Neurobiol. Exp. 2021, 81, 314–327. [Google Scholar]
- Gao, L.; Zhang, Y.; Sterling, K.; Song, W. Brain-derived neurotrophic factor in Alzheimer’s disease and its pharmaceutical potential. Transl. Neurodegener. 2022, 11, 34. [Google Scholar] [CrossRef]
- Nasrolahi, A.; Javaherforooshzadeh, F.; Jafarzadeh-Gharehziaaddin, M.; Mahmoudi, J.; Asl, K.D.; Shabani, Z. Therapeutic potential of neurotrophic factors in Alzheimer’s Disease. Mol. Biol. Rep. 2022, 49, 2345–2357. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Wu, H.T.; Qin, X.Y.; Cao, C.; Liu, Y.; Cao, Z.Z.; Cheng, Y. Postmortem Brain, Cerebrospinal Fluid, and Blood Neurotrophic Factor Levels in Alzheimer’s Disease: A Systematic Review and Meta-Analysis. J. Mol. Neurosci. 2018, 65, 289–300. [Google Scholar] [CrossRef] [PubMed]
- Williams, D.M.; Karlsson, I.K.; Pedersen, N.L.; Hagg, S. Circulating insulin-like growth factors and Alzheimer disease: A mendelian randomization study. Neurology 2018, 90, e291–e297. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Y.; Cawley, N.X.; Yanik, T.; Murthy, S.R.; Liu, C.; Kasikci, F.; Abebe, D.; Loh, Y.P. A human carboxypeptidase E/NF-alpha1 gene mutation in an Alzheimer’s disease patient leads to dementia and depression in mice. Transl. Psychiatry 2016, 6, e973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuello, A.C.; Pentz, R.; Hall, H. The Brain NGF Metabolic Pathway in Health and in Alzheimer’s Pathology. Front. Neurosci. 2019, 13, 62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, X.Y.; Cao, C.; Cawley, N.X.; Liu, T.T.; Yuan, J.; Loh, Y.P.; Cheng, Y. Decreased peripheral brain-derived neurotrophic factor levels in Alzheimer’s disease: A meta-analysis study (N = 7277). Mol. Psychiatry 2017, 22, 312–320. [Google Scholar] [CrossRef] [PubMed]
- Lim, Y.Y.; Villemagne, V.L.; Laws, S.M.; Ames, D.; Pietrzak, R.H.; Ellis, K.A.; Harrington, K.D.; Bourgeat, P.; Salvado, O.; Darby, D.; et al. BDNF Val66Met, Abeta amyloid, and cognitive decline in preclinical Alzheimer’s disease. Neurobiol. Aging 2013, 34, 2457–2464. [Google Scholar] [CrossRef] [PubMed]
- Narisawa-Saito, M.; Wakabayashi, K.; Tsuji, S.; Takahashi, H.; Nawa, H. Regional specificity of alterations in NGF, BDNF and NT-3 levels in Alzheimer’s disease. Neuroreport 1996, 7, 2925–2928. [Google Scholar] [CrossRef]
- Hock, C.; Heese, K.; Hulette, C.; Rosenberg, C.; Otten, U. Region-specific neurotrophin imbalances in Alzheimer disease: Decreased levels of brain-derived neurotrophic factor and increased levels of nerve growth factor in hippocampus and cortical areas. Arch. Neurol. 2000, 57, 846–851. [Google Scholar] [CrossRef] [Green Version]
- Airavaara, M.; Pletnikova, O.; Doyle, M.E.; Zhang, Y.E.; Troncoso, J.C.; Liu, Q.R. Identification of Novel GDNF Isoforms and cis-Antisense GDNFOS Gene and Their Regulation in Human Middle Temporal Gyrus of Alzheimer Disease. J. Biol. Chem. 2011, 286, 45093–45102. [Google Scholar] [CrossRef] [Green Version]
- Sharif, M.; Noroozian, M.; Hashemian, F. Do serum GDNF levels correlate with severity of Alzheimer’s disease? Neurol. Sci. 2021, 42, 2865–2872. [Google Scholar] [CrossRef]
- Forlenza, O.V.; Miranda, A.S.; Barbosa, I.G.; Talib, L.L.; Diniz, B.S.; Gattaz, W.F.; Teixeira, A.L. Decreased Neurotrophic Support is Associated with Cognitive Decline in Non-Demented Subjects. J. Alzheimers Dis. 2015, 46, 423–429. [Google Scholar] [CrossRef] [PubMed]
- Straten, G.; Eschweiler, G.W.; Maetzler, W.; Laske, C.; Leyhe, T. Glial cell-line derived neurotrophic factor (GDNF) concentrations in cerebrospinal fluid and serum of patients with early Alzheimer’s disease and normal controls. J. Alzheimers Dis. 2009, 18, 331–337. [Google Scholar] [CrossRef]
- Marksteiner, J.; Kemmler, G.; Weiss, E.M.; Knaus, G.; Ullrich, C.; Mechtcheriakov, S.; Oberbauer, H.; Auffinger, S.; Hinterholzl, J.; Hinterhuber, H.; et al. Five out of 16 plasma signaling proteins are enhanced in plasma of patients with mild cognitive impairment and Alzheimer’s disease. Neurobiol. Aging 2011, 32, 539–540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, C.; Brown, R.W.; Malone, H.M.; Burgess, K.C.; Gill, W.D.; Keasey, M.P.; Hagg, T. Ciliary neurotrophic factor is a key sex-specific regulator of depressive-like behavior in mice. Psychoneuroendocrinology 2019, 100, 96–105. [Google Scholar] [CrossRef] [PubMed]
- Soilu-Hanninen, M.; Broberg, E.; Roytta, M.; Mattila, P.; Rinne, J.; Hukkanen, V. Expression of LIF and LIF receptor beta in Alzheimer’s and Parkinson’s diseases. Acta Neurol. Scand. 2010, 121, 44–50. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Li, X.; Gao, K.; Lu, D.; Zhang, X.; Ma, C.; Ye, F.; Zhang, L. Cardiotrophin-1 (CTF1) ameliorates glucose-uptake defects and improves memory and learning deficits in a transgenic mouse model of Alzheimer’s disease. Pharm. Biochem. Behav. 2013, 107, 48–57. [Google Scholar] [CrossRef]
- Joshi, H.; Shah, J.; Abu-Hijleh, F.A.; Patel, V.; Rathbone, M.; Gabriele, S.; Gabriele, J.; Baranowski, D.; Molloy, D.; Frey, B.N.; et al. Decreased Expression of Cerebral Dopamine Neurotrophic Factor in Platelets of Probable Alzheimer Patients. Alzheimer Dis. Assoc. Disord. 2021, 36, 269–271. [Google Scholar] [CrossRef]
- Liu, X.C.; Qi, X.H.; Fang, H.; Zhou, K.Q.; Wang, Q.S.; Chen, G.H. Increased MANF Expression in the Inferior Temporal Gyrus in Patients with Alzheimer Disease. Front. Aging Neurosci. 2021, 13, 639318. [Google Scholar] [CrossRef]
- Kimura, K.; Wakamatsu, A.; Suzuki, Y.; Ota, T.; Nishikawa, T.; Yamashita, R.; Yamamoto, J.; Sekine, M.; Tsuritani, K.; Wakaguri, H.; et al. Diversification of transcriptional modulation: Large-scale identification and characterization of putative alternative promoters of human genes. Genome Res. 2006, 16, 55–65. [Google Scholar] [CrossRef] [Green Version]
- Mahoney, E.R.; Dumitrescu, L.; Moore, A.M.; Cambronero, F.E.; De Jager, P.L.; Koran, M.E.I.; Petyuk, V.A.; Robinson, R.A.S.; Goyal, S.; Schneider, J.A.; et al. Brain expression of the vascular endothelial growth factor gene family in cognitive aging and alzheimer’s disease. Mol. Psychiatry 2021, 26, 888–896. [Google Scholar] [CrossRef] [Green Version]
- Thomas, T.; Miners, S.; Love, S. Post-mortem assessment of hypoperfusion of cerebral cortex in Alzheimer’s disease and vascular dementia. Brain 2015, 138, 1059–1069. [Google Scholar] [CrossRef] [PubMed]
- Provias, J.; Jeynes, B. Reduction in vascular endothelial growth factor expression in the superior temporal, hippocampal, and brainstem regions in Alzheimer’s disease. Curr. Neurovascular Res. 2014, 11, 202–209. [Google Scholar] [CrossRef] [PubMed]
- Inai, T.; Mancuso, M.; Hashizume, H.; Baffert, F.; Haskell, A.; Baluk, P.; Hu-Lowe, D.D.; Shalinsky, D.R.; Thurston, G.; Yancopoulos, G.D.; et al. Inhibition of vascular endothelial growth factor (VEGF) signaling in cancer causes loss of endothelial fenestrations, regression of tumor vessels, and appearance of basement membrane ghosts. Am. J. Pathol. 2004, 165, 35–52. [Google Scholar] [CrossRef] [Green Version]
- Chakraborty, A.; Chatterjee, M.; Twaalfhoven, H.; Del Campo Milan, M.; Teunissen, C.E.; Scheltens, P.; Fontijn, R.D.; van Der Flier, W.M.; de Vries, H.E. Vascular Endothelial Growth Factor remains unchanged in cerebrospinal fluid of patients with Alzheimer’s disease and vascular dementia. Alzheimers Res. Ther. 2018, 10, 58. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.H.; Alexopoulos, P.; Perneczky, R. Heart-type fatty acid binding protein and vascular endothelial growth factor: Cerebrospinal fluid biomarker candidates for Alzheimer’s disease. Eur. Arch. Psychiatry Clin. Neurosci. 2013, 263, 553–560. [Google Scholar] [CrossRef]
- Zhang, J.B.; Li, M.F.; Zhang, H.X.; Li, Z.G.; Sun, H.R.; Zhang, J.S.; Wang, P.F. Association of serum vascular endothelial growth factor levels and cerebral microbleeds in patients with Alzheimer’s disease. Eur. J. Neurol. 2016, 23, 1337–1342. [Google Scholar] [CrossRef]
- Mateo, I.; Llorca, J.; Infante, J.; Rodriguez-Rodriguez, E.; Fernandez-Viadero, C.; Pena, N.; Berciano, J.; Combarros, O. Low serum VEGF levels are associated with Alzheimer’s disease. Acta Neurol. Scand. 2007, 116, 56–58. [Google Scholar] [CrossRef]
- Liu, S.Y.; Zeng, F.F.; Chen, Z.W.; Wang, C.Y.; Zhao, B.; Li, K.S. Vascular endothelial growth factor gene promoter polymorphisms and Alzheimer’s disease risk: A meta-analysis. CNS Neurosci. Ther. 2013, 19, 469–476. [Google Scholar] [CrossRef]
- Bjorkqvist, M.; Ohlsson, M.; Minthon, L.; Hansson, O. Evaluation of a previously suggested plasma biomarker panel to identify Alzheimer’s disease. PLoS ONE 2012, 7, e29868. [Google Scholar] [CrossRef]
- Sagare, A.P.; Bell, R.D.; Zhao, Z.; Ma, Q.; Winkler, E.A.; Ramanathan, A.; Zlokovic, B.V. Pericyte loss influences Alzheimer-like neurodegeneration in mice. Nat. Commun. 2013, 4, 2932. [Google Scholar] [CrossRef] [Green Version]
- Sagare, A.P.; Sweeney, M.D.; Makshanoff, J.; Zlokovic, B.V. Shedding of soluble platelet-derived growth factor receptor-beta from human brain pericytes. Neurosci. Lett. 2015, 607, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Stopa, E.G.; Gonzalez, A.M.; Chorsky, R.; Corona, R.J.; Alvarez, J.; Bird, E.D.; Baird, A. Basic fibroblast growth factor in Alzheimer’s disease. Biochem. Biophys. Res. Commun. 1990, 171, 690–696. [Google Scholar] [CrossRef]
- Mocali, A.; Cedrola, S.; Della Malva, N.; Bontempelli, M.; Mitidieri, V.A.; Bavazzano, A.; Comolli, R.; Paoletti, F.; La Porta, C.A. Increased plasma levels of soluble CD40, together with the decrease of TGF beta 1, as possible differential markers of Alzheimer disease. Exp. Gerontol. 2004, 39, 1555–1561. [Google Scholar] [CrossRef]
- Tesseur, I.; Zou, K.; Esposito, L.; Bard, F.; Berber, E.; Can, J.V.; Lin, A.H.; Crews, L.; Tremblay, P.; Mathews, P.; et al. Deficiency in neuronal TGF-beta signaling promotes neurodegeneration and Alzheimer’s pathology. J. Clin. Investig. 2006, 116, 3060–3069. [Google Scholar] [CrossRef] [Green Version]
- Decourt, B.; Lahiri, D.K.; Sabbagh, M.N. Targeting Tumor Necrosis Factor Alpha for Alzheimer’s Disease. Curr. Alzheimer Res. 2017, 14, 412–425. [Google Scholar] [CrossRef] [Green Version]
- Ostrowski, P.P.; Barszczyk, A.; Forstenpointner, J.; Zheng, W.; Feng, Z.P. Meta-Analysis of Serum Insulin-Like Growth Factor 1 in Alzheimer’s Disease. PLoS ONE 2016, 11, e0155733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fenton, H.; Finch, P.W.; Rubin, J.S.; Rosenberg, J.M.; Taylor, W.G.; Kuo-Leblanc, V.; Rodriguez-Wolf, M.; Baird, A.; Schipper, H.M.; Stopa, E.G. Hepatocyte growth factor (HGF/SF) in Alzheimer’s disease. Brain Res. 1998, 779, 262–270. [Google Scholar] [CrossRef]
- Tuszynski, M.H.; Thal, L.; Pay, M.; Salmon, D.P.; U, H.S.; Bakay, R.; Patel, P.; Blesch, A.; Vahlsing, H.L.; Ho, G.; et al. A phase 1 clinical trial of nerve growth factor gene therapy for Alzheimer disease. Nat. Med. 2005, 11, 551–555. [Google Scholar] [CrossRef]
- Tuszynski, M.H.; Yang, J.H.; Barba, D.; U, H.S.; Bakay, R.A.; Pay, M.M.; Masliah, E.; Conner, J.M.; Kobalka, P.; Roy, S.; et al. Nerve Growth Factor Gene Therapy: Activation of Neuronal Responses in Alzheimer Disease. JAMA Neurol. 2015, 72, 1139–1147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fahnestock, M.; Shekari, A. ProNGF and Neurodegeneration in Alzheimer’s Disease. Front. Neurosci. 2019, 13, 129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmitz, T.W.; Spreng, R.N. Alzheimer’s Disease Neuroimaging Initiative. Basal forebrain degeneration precedes and predicts the cortical spread of Alzheimer’s pathology. Nat. Commun. 2016, 7, 13249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilcock, G.K.; Esiri, M.M.; Bowen, D.M.; Smith, C.C. Alzheimer’s disease. Correlation of cortical choline acetyltransferase activity with the severity of dementia and histological abnormalities. J. Neurol. Sci. 1982, 57, 407–417. [Google Scholar] [CrossRef]
- Sobreviela, T.; Clary, D.O.; Reichardt, L.F.; Brandabur, M.M.; Kordower, J.H.; Mufson, E.J. TrkA-immunoreactive profiles in the central nervous system: Colocalization with neurons containing p75 nerve growth factor receptor, choline acetyltransferase, and serotonin. J. Comp. Neurol. 1994, 350, 587–611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ginsberg, S.D.; Che, S.; Wuu, J.; Counts, S.E.; Mufson, E.J. Down regulation of trk but not p75NTR gene expression in single cholinergic basal forebrain neurons mark the progression of Alzheimer’s disease. J. Neurochem. 2006, 97, 475–487. [Google Scholar] [CrossRef]
- Counts, S.E.; Nadeem, M.; Wuu, J.; Ginsberg, S.D.; Saragovi, H.U.; Mufson, E.J. Reduction of cortical TrkA but not p75(NTR) protein in early-stage Alzheimer’s disease. Ann. Neurol. 2004, 56, 520–531. [Google Scholar] [CrossRef]
- Crutcher, K.A.; Scott, S.A.; Liang, S.; Everson, W.V.; Weingartner, J. Detection of NGF-like activity in human brain tissue: Increased levels in Alzheimer’s disease. J. Neurosci. 1993, 13, 2540–2550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scott, S.A.; Mufson, E.J.; Weingartner, J.A.; Skau, K.A.; Crutcher, K.A. Nerve growth factor in Alzheimer’s disease: Increased levels throughout the brain coupled with declines in nucleus basalis. J. Neurosci. 1995, 15, 6213–6221. [Google Scholar] [CrossRef] [Green Version]
- Mufson, E.J.; Conner, J.M.; Kordower, J.H. Nerve growth factor in Alzheimer’s disease: Defective retrograde transport to nucleus basalis. Neuroreport 1995, 6, 1063–1066. [Google Scholar] [CrossRef]
- Shekari, A.; Fahnestock, M. Retrograde axonal transport of BDNF and proNGF diminishes with age in basal forebrain cholinergic neurons. Neurobiol. Aging 2019, 84, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.; Kermani, P.; Teng, K.K.; Hempstead, B.L. Regulation of cell survival by secreted proneurotrophins. Science 2001, 294, 1945–1948. [Google Scholar] [CrossRef] [Green Version]
- Fahnestock, M.; Yu, G.; Coughlin, M.D. ProNGF: A neurotrophic or an apoptotic molecule? Prog. Brain Res. 2004, 146, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Fahnestock, M.; Michalski, B.; Xu, B.; Coughlin, M.D. The precursor pro-nerve growth factor is the predominant form of nerve growth factor in brain and is increased in Alzheimer’s disease. Mol. Cell. Neurosci. 2001, 18, 210–220. [Google Scholar] [CrossRef] [Green Version]
- Masoudi, R.; Ioannou, M.S.; Coughlin, M.D.; Pagadala, P.; Neet, K.E.; Clewes, O.; Allen, S.J.; Dawbarn, D.; Fahnestock, M. Biological activity of nerve growth factor precursor is dependent upon relative levels of its receptors. J. Biol. Chem. 2009, 284, 18424–18433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, L.F.; Doherty, D.H.; Lile, J.D.; Bektesh, S.; Collins, F. GDNF: A glial cell line-derived neurotrophic factor for midbrain dopaminergic neurons. Science 1993, 260, 1130–1132. [Google Scholar] [CrossRef]
- Allen, S.J.; Watson, J.J.; Shoemark, D.K.; Barua, N.U.; Patel, N.K. GDNF, NGF and BDNF as therapeutic options for neurodegeneration. Pharmacol. Ther. 2013, 138, 155–175. [Google Scholar] [CrossRef] [PubMed]
- Pertusa, M.; Garcia-Matas, S.; Mammeri, H.; Adell, A.; Rodrigo, T.; Mallet, J.; Cristofol, R.; Sarkis, C.; Sanfeliu, C. Expression of GDNF transgene in astrocytes improves cognitive deficits in aged rats. Neurobiol. Aging 2008, 29, 1366–1379. [Google Scholar] [CrossRef] [Green Version]
- Yan, Z.; Shi, X.; Wang, H.; Si, C.; Liu, Q.; Du, Y. Neurotrophin-3 Promotes the Neuronal Differentiation of BMSCs and Improves Cognitive Function in a Rat Model of Alzheimer’s Disease. Front. Cell. Neurosci. 2021, 15, 629356. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, K.; Grundke-Iqbal, I.; Zaidi, T.; Merz, P.A.; Wen, G.Y.; Shaikh, S.S.; Wisniewski, H.M.; Alafuzoff, I.; Winblad, B. Defective brain microtubule assembly in Alzheimer’s disease. Lancet 1986, 2, 421–426. [Google Scholar] [CrossRef]
- Adalbert, R.; Milde, S.; Durrant, C.; Ando, K.; Stygelbout, V.; Yilmaz, Z.; Gould, S.; Brion, J.P.; Coleman, M.P. Interaction between a MAPT variant causing frontotemporal dementia and mutant APP affects axonal transport. Neurobiol. Aging 2018, 68, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Yaar, M.; Zhai, S.; Pilch, P.F.; Doyle, S.M.; Eisenhauer, P.B.; Fine, R.E.; Gilchrest, B.A. Binding of beta-amyloid to the p75 neurotrophin receptor induces apoptosis. A possible mechanism for Alzheimer’s disease. J. Clin. Investig. 1997, 100, 2333–2340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geetha, T.; Zheng, C.; McGregor, W.C.; White, B.D.; Diaz-Meco, M.T.; Moscat, J.; Babu, J.R. TRAF6 and p62 inhibit amyloid beta-induced neuronal death through p75 neurotrophin receptor. Neurochem. Int. 2012, 61, 1289–1293. [Google Scholar] [CrossRef] [PubMed]
- Zheng, C.; Geetha, T.; Gearing, M.; Babu, J.R. Amyloid beta-abrogated TrkA ubiquitination in PC12 cells analogous to Alzheimer’s disease. J. Neurochem. 2015, 133, 919–925. [Google Scholar] [CrossRef]
- Sen, A.; Nelson, T.J.; Alkon, D.L. ApoE4 and Abeta Oligomers Reduce BDNF Expression via HDAC Nuclear Translocation. J. Neurosci. 2015, 35, 7538–7551. [Google Scholar] [CrossRef] [Green Version]
- Rosa, E.; Fahnestock, M. CREB expression mediates amyloid beta-induced basal BDNF downregulation. Neurobiol. Aging 2015, 36, 2406–2413. [Google Scholar] [CrossRef] [PubMed]
- Kemppainen, S.; Rantamaki, T.; Jeronimo-Santos, A.; Lavasseur, G.; Autio, H.; Karpova, N.; Karkkainen, E.; Staven, S.; Vicente Miranda, H.; Outeiro, T.F.; et al. Impaired TrkB receptor signaling contributes to memory impairment in APP/PS1 mice. Neurobiol. Aging 2012, 33, 1122.e23–1122.e39. [Google Scholar] [CrossRef]
- Jeronimo-Santos, A.; Vaz, S.H.; Parreira, S.; Rapaz-Lerias, S.; Caetano, A.P.; Buee-Scherrer, V.; Castren, E.; Valente, C.A.; Blum, D.; Sebastiao, A.M.; et al. Dysregulation of TrkB Receptors and BDNF Function by Amyloid-beta Peptide is Mediated by Calpain. Cereb. Cortex 2015, 25, 3107–3121. [Google Scholar] [CrossRef] [Green Version]
- Matrone, C.; Marolda, R.; Ciafre, S.; Ciotti, M.T.; Mercanti, D.; Calissano, P. Tyrosine kinase nerve growth factor receptor switches from prosurvival to proapoptotic activity via Abeta-mediated phosphorylation. Proc. Natl. Acad. Sci. USA 2009, 106, 11358–11363. [Google Scholar] [CrossRef] [Green Version]
- Cattaneo, A.; Capsoni, S.; Paoletti, F. Towards non invasive nerve growth factor therapies for Alzheimer’s disease. J. Alzheimers Dis. JAD 2008, 15, 255–283. [Google Scholar] [CrossRef] [Green Version]
- Capsoni, S.; Marinelli, S.; Ceci, M.; Vignone, D.; Amato, G.; Malerba, F.; Paoletti, F.; Meli, G.; Viegi, A.; Pavone, F.; et al. Intranasal “painless” human Nerve Growth Factor [corrected] slows amyloid neurodegeneration and prevents memory deficits in App X PS1 mice. PLoS ONE 2012, 7, e37555. [Google Scholar] [CrossRef] [Green Version]
- Tian, L.; Guo, R.; Yue, X.; Lv, Q.; Ye, X.; Wang, Z.; Chen, Z.; Wu, B.; Xu, G.; Liu, X. Intranasal administration of nerve growth factor ameliorate beta-amyloid deposition after traumatic brain injury in rats. Brain Res. 2012, 1440, 47–55. [Google Scholar] [CrossRef]
- Xu, C.J.; Wang, J.L.; Jin, W.L. The Emerging Therapeutic Role of NGF in Alzheimer’s Disease. Neurochem. Res. 2016, 41, 1211–1218. [Google Scholar] [CrossRef] [PubMed]
- Fragkouli, A.; Tzinia, A.K.; Charalampopoulos, I.; Gravanis, A.; Tsilibary, E.C. Matrix metalloproteinase-9 participates in NGF-induced alpha-secretase cleavage of amyloid-beta protein precursor in PC12 cells. J. Alzheimers Dis. JAD 2011, 24, 705–719. [Google Scholar] [CrossRef]
- Yang, C.; Liu, Y.L.; Ni, X.Q.; Li, N.; Zhang, B.H.; Fang, X.B. Enhancement of the nonamyloidogenic pathway by exogenous NGF in an Alzheimer transgenic mouse model. Neuropeptides 2014, 48, 233–238. [Google Scholar] [CrossRef] [PubMed]
- Xie, H.; Xiao, Z.; Huang, J. C6 Glioma-Secreted NGF and FGF2 Regulate Neuronal APP Processing Through Up-Regulation of ADAM10 and Down-Regulation of BACE1, Respectively. J. Mol. Neurosci. 2016, 59, 334–342. [Google Scholar] [CrossRef]
- Rizzi, C.; Tiberi, A.; Giustizieri, M.; Marrone, M.C.; Gobbo, F.; Carucci, N.M.; Meli, G.; Arisi, I.; D’Onofrio, M.; Marinelli, S.; et al. NGF steers microglia toward a neuroprotective phenotype. Glia 2018, 66, 1395–1416. [Google Scholar] [CrossRef] [Green Version]
- Chang, K.A.; Kim, H.S.; Ha, T.Y.; Ha, J.W.; Shin, K.Y.; Jeong, Y.H.; Lee, J.P.; Park, C.H.; Kim, S.; Baik, T.K.; et al. Phosphorylation of amyloid precursor protein (APP) at Thr668 regulates the nuclear translocation of the APP intracellular domain and induces neurodegeneration. Mol. Cell. Biol. 2006, 26, 4327–4338. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, T.; Nakaya, T. Regulation of amyloid beta-protein precursor by phosphorylation and protein interactions. J. Biol. Chem. 2008, 283, 29633–29637. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.S.; Kao, S.C.; Lemere, C.A.; Xia, W.M.; Tseng, H.C.; Zhou, Y.; Neve, R.; Ahlijanian, M.K.; Tsai, L.H. APP processing is regulated by cytoplasmic phosphorylation. J. Cell Biol. 2003, 163, 83–95. [Google Scholar] [CrossRef]
- Tamayev, R.; Zhou, D.W.; D’Adamio, L. The interactome of the amyloid beta precursor protein family members is shaped by phosphorylation of their intracellular domains. Mol. Neurodegener. 2009, 4, 28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canu, N.; Amadoro, G.; Triaca, V.; Latina, V.; Sposato, V.; Corsetti, V.; Severini, C.; Ciotti, M.T.; Calissano, P. The Intersection of NGF/TrkA Signaling and Amyloid Precursor Protein Processing in Alzheimer’s Disease Neuropathology. Int. J. Mol. Sci. 2017, 18, 1319. [Google Scholar] [CrossRef] [Green Version]
- Triaca, V.; Sposato, V.; Bolasco, G.; Ciotti, M.T.; Pelicci, P.; Bruni, A.C.; Cupidi, C.; Maletta, R.; Feligioni, M.; Nistico, R.; et al. NGF controls APP cleavage by downregulating APP phosphorylation at Thr668: Relevance for Alzheimer’s disease. Aging Cell 2016, 15, 661–672. [Google Scholar] [CrossRef]
- Canu, N.; Pagano, I.; La Rosa, L.R.; Pellegrino, M.; Ciotti, M.T.; Mercanti, D.; Moretti, F.; Sposato, V.; Triaca, V.; Petrella, C.; et al. Association of TrkA and APP Is Promoted by NGF and Reduced by Cell Death-Promoting Agents. Front. Mol. Neurosci. 2017, 10, 15. [Google Scholar] [CrossRef]
- Costantini, C.; Scrable, H.; Puglielli, L. An aging pathway controls the TrkA to p75NTR receptor switch and amyloid beta-peptide generation. EMBO J. 2006, 25, 1997–2006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossner, S.; Ueberham, U.; Schliebs, R.; Perez-Polo, J.R.; Bigl, V. The regulation of amyloid precursor protein metabolism by cholinergic mechanisms and neurotrophin receptor signaling. Prog. Neurobiol. 1998, 56, 541–569. [Google Scholar] [CrossRef]
- Costantini, C.; Weindruch, R.; Della Valle, G.; Puglielli, L. A TrkA-to-p75NTR molecular switch activates amyloid beta-peptide generation during aging. Biochem. J. 2005, 391, 59–67. [Google Scholar] [CrossRef] [Green Version]
- Saadipour, K.; Tiberi, A.; Lombardo, S.; Grajales, E.; Montroull, L.; Manucat-Tan, N.B.; LaFrancois, J.; Cammer, M.; Mathews, P.M.; Scharfman, H.E.; et al. Regulation of BACE1 expression after injury is linked to the p75 neurotrophin receptor. Mol. Cell. Neurosci. 2019, 99, 103395. [Google Scholar] [CrossRef] [PubMed]
- Fukumoto, H.; Tokuda, T.; Kasai, T.; Ishigami, N.; Hidaka, H.; Kondo, M.; Allsop, D.; Nakagawa, M. High-molecular-weight beta-amyloid oligomers are elevated in cerebrospinal fluid of Alzheimer patients. FASEB J. 2010, 24, 2716–2726. [Google Scholar] [CrossRef]
- Iulita, M.F.; Millon, M.B.B.; Pentz, R.; Aguilar, L.F.; Do Carmo, S.; Allard, S.; Michalski, B.; Wilson, E.N.; Ducatenzeiler, A.; Bruno, M.A.; et al. Differential deregulation of NGF and BDNF neurotrophins in a transgenic rat model of Alzheimer’s disease. Neurobiol. Dis. 2017, 108, 307–323. [Google Scholar] [CrossRef]
- Small, S.A.; Petsko, G.A. Endosomal recycling reconciles the Alzheimer’s disease paradox. Sci. Transl. Med. 2020, 12, eabb1717. [Google Scholar] [CrossRef] [PubMed]
- Cataldo, A.M.; Peterhoff, C.M.; Troncoso, J.C.; Gomez-Isla, T.; Hyman, B.T.; Nixon, R.A. Endocytic pathway abnormalities precede amyloid beta deposition in sporadic Alzheimer’s disease and Down syndrome: Differential effects of APOE genotype and presenilin mutations. Am. J. Pathol. 2000, 157, 277–286. [Google Scholar] [CrossRef]
- Offe, K.; Dodson, S.E.; Shoemaker, J.T.; Fritz, J.J.; Gearing, M.; Levey, A.I.; Lah, J.J. The lipoprotein receptor LR11 regulates amyloid beta production and amyloid precursor protein traffic in endosomal compartments. J. Neurosci. 2006, 26, 1596–1603. [Google Scholar] [CrossRef] [PubMed]
- Small, S.A.; Simoes-Spassov, S.; Mayeux, R.; Petsko, G.A. Endosomal Traffic Jams Represent a Pathogenic Hub and Therapeutic Target in Alzheimer’s Disease. Trends Neurosci. 2017, 40, 592–602. [Google Scholar] [CrossRef]
- Chai, A.B.; Lam, H.H.J.; Kockx, M.; Gelissen, I.C. Apolipoprotein E isoform-dependent effects on the processing of Alzheimer’s amyloid-beta. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2021, 1866, 158980. [Google Scholar] [CrossRef]
- Corder, E.H.; Saunders, A.M.; Strittmatter, W.J.; Schmechel, D.E.; Gaskell, P.C.; Small, G.W.; Roses, A.D.; Haines, J.L.; Pericak-Vance, M.A. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 1993, 261, 921–923. [Google Scholar] [CrossRef]
- Yamazaki, Y.; Zhao, N.; Caulfield, T.R.; Liu, C.C.; Bu, G. Apolipoprotein E and Alzheimer disease: Pathobiology and targeting strategies. Nat. Rev. Neurol. 2019, 15, 501–518. [Google Scholar] [CrossRef] [PubMed]
- Scherzer, C.R.; Offe, K.; Gearing, M.; Rees, H.D.; Fang, G.; Heilman, C.J.; Schaller, C.; Bujo, H.; Levey, A.I.; Lah, J.J. Loss of apolipoprotein E receptor LR11 in Alzheimer disease. Arch. Neurol. 2004, 61, 1200–1205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andersen, O.M.; Rudolph, I.M.; Willnow, T.E. Risk factor SORL1: From genetic association to functional validation in Alzheimer’s disease. Acta Neuropathol. 2016, 132, 653–665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andersen, O.M.; Reiche, J.; Schmidt, V.; Gotthardt, M.; Spoelgen, R.; Behlke, J.; von Arnim, C.A.; Breiderhoff, T.; Jansen, P.; Wu, X.; et al. Neuronal sorting protein-related receptor sorLA/LR11 regulates processing of the amyloid precursor protein. Proc. Natl. Acad. Sci. USA 2005, 102, 13461–13466. [Google Scholar] [CrossRef] [Green Version]
- Fjorback, A.W.; Andersen, O.M. SorLA is a molecular link for retromer-dependent sorting of the Amyloid precursor protein. Commun. Integr. Biol. 2012, 5, 616–619. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, M.S.; Gustafsen, C.; Madsen, P.; Nyengaard, J.R.; Hermey, G.; Bakke, O.; Mari, M.; Schu, P.; Pohlmann, R.; Dennes, A.; et al. Sorting by the cytoplasmic domain of the amyloid precursor protein binding receptor SorLA. Mol. Cell. Biol. 2007, 27, 6842–6851. [Google Scholar] [CrossRef] [Green Version]
- Huang, T.Y.; Zhao, Y.; Li, X.; Wang, X.; Tseng, I.C.; Thompson, R.; Tu, S.; Willnow, T.E.; Zhang, Y.W.; Xu, H. SNX27 and SORLA Interact to Reduce Amyloidogenic Subcellular Distribution and Processing of Amyloid Precursor Protein. J. Neurosci. 2016, 36, 7996–8011. [Google Scholar] [CrossRef]
- Schmidt, V.; Baum, K.; Lao, A.; Rateitschak, K.; Schmitz, Y.; Teichmann, A.; Wiesner, B.; Petersen, C.M.; Nykjaer, A.; Wolf, J.; et al. Quantitative modelling of amyloidogenic processing and its influence by SORLA in Alzheimer’s disease. EMBO J. 2012, 31, 187–200. [Google Scholar] [CrossRef]
- Spoelgen, R.; von Arnim, C.A.; Thomas, A.V.; Peltan, I.D.; Koker, M.; Deng, A.; Irizarry, M.C.; Andersen, O.M.; Willnow, T.E.; Hyman, B.T. Interaction of the cytosolic domains of sorLA/LR11 with the amyloid precursor protein (APP) and beta-secretase beta-site APP-cleaving enzyme. J. Neurosci. 2006, 26, 418–428. [Google Scholar] [CrossRef] [Green Version]
- Dumanis, S.B.; Burgert, T.; Caglayan, S.; Fuchtbauer, A.; Fuchtbauer, E.M.; Schmidt, V.; Willnow, T.E. Distinct Functions for Anterograde and Retrograde Sorting of SORLA in Amyloidogenic Processes in the Brain. J. Neurosci. 2015, 35, 12703–12713. [Google Scholar] [CrossRef] [Green Version]
- Glerup, S.; Lume, M.; Olsen, D.; Nyengaard, J.R.; Vaegter, C.B.; Gustafsen, C.; Christensen, E.I.; Kjolby, M.; Hay-Schmidt, A.; Bender, D.; et al. SorLA controls neurotrophic activity by sorting of GDNF and its receptors GFRalpha1 and RET. Cell Rep. 2013, 3, 186–199. [Google Scholar] [CrossRef] [Green Version]
- Rohe, M.; Hartl, D.; Fjorback, A.N.; Klose, J.; Willnow, T.E. SORLA-mediated trafficking of TrkB enhances the response of neurons to BDNF. PLoS ONE 2013, 8, e72164. [Google Scholar] [CrossRef]
- Stupack, J.; Xiong, X.P.; Jiang, L.L.; Zhang, T.M.; Zhou, L.S.; Campos, A.; Ranscht, B.; Mobley, W.; Pasquale, E.B.; Xu, H.X.; et al. Soluble SORLA Enhances Neurite Outgrowth and Regeneration through Activation of the EGF Receptor/ERK Signaling Axis. J. Neurosci. 2020, 40, 5908–5921. [Google Scholar] [CrossRef]
- Saadipour, K.; Yang, M.; Lim, Y.; Georgiou, K.; Sun, Y.; Keating, D.; Liu, J.; Wang, Y.R.; Gai, W.P.; Zhong, J.H.; et al. Amyloid beta(1)(-)(4)(2) (Abeta(4)(2)) up-regulates the expression of sortilin via the p75(NTR)/RhoA signaling pathway. J. Neurochem. 2013, 127, 152–162. [Google Scholar] [CrossRef]
- Andersson, C.H.; Hansson, O.; Minthon, L.; Andreasen, N.; Blennow, K.; Zetterberg, H.; Skoog, I.; Wallin, A.; Nilsson, S.; Kettunen, P. A Genetic Variant of the Sortilin 1 Gene is Associated with Reduced Risk of Alzheimer’s Disease. J. Alzheimers Dis. JAD 2016, 53, 1353–1363. [Google Scholar] [CrossRef] [Green Version]
- Finan, G.M.; Okada, H.; Kim, T.W. BACE1 retrograde trafficking is uniquely regulated by the cytoplasmic domain of sortilin. J. Biol. Chem. 2011, 286, 12602–12616. [Google Scholar] [CrossRef] [Green Version]
- Tan, J.; Evin, G. Beta-site APP-cleaving enzyme 1 trafficking and Alzheimer’s disease pathogenesis. J. Neurochem. 2012, 120, 869–880. [Google Scholar] [CrossRef]
- Gustafsen, C.; Glerup, S.; Pallesen, L.T.; Olsen, D.; Andersen, O.M.; Nykjaer, A.; Madsen, P.; Petersen, C.M. Sortilin and SorLA display distinct roles in processing and trafficking of amyloid precursor protein. J. Neurosci. 2013, 33, 64–71. [Google Scholar] [CrossRef] [Green Version]
- Carlo, A.S. Sortilin, a novel APOE receptor implicated in Alzheimer disease. Prion 2013, 7, 378–382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carlo, A.S.; Gustafsen, C.; Mastrobuoni, G.; Nielsen, M.S.; Burgert, T.; Hartl, D.; Rohe, M.; Nykjaer, A.; Herz, J.; Heeren, J.; et al. The pro-neurotrophin receptor sortilin is a major neuronal apolipoprotein E receptor for catabolism of amyloid-beta peptide in the brain. J. Neurosci. 2013, 33, 358–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takamura, A.; Sato, Y.; Watabe, D.; Okamoto, Y.; Nakata, T.; Kawarabayashi, T.; Oddo, S.; Laferla, F.M.; Shoji, M.; Matsubara, E. Sortilin is required for toxic action of Abeta oligomers (AbetaOs): Extracellular AbetaOs trigger apoptosis, and intraneuronal AbetaOs impair degradation pathways. Life Sci. 2012, 91, 1177–1186. [Google Scholar] [CrossRef] [PubMed]
- Vaegter, C.B.; Jansen, P.; Fjorback, A.W.; Glerup, S.; Skeldal, S.; Kjolby, M.; Richner, M.; Erdmann, B.; Nyengaard, J.R.; Tessarollo, L.; et al. Sortilin associates with Trk receptors to enhance anterograde transport and neurotrophin signaling. Nat. Neurosci. 2011, 14, 54–61. [Google Scholar] [CrossRef] [Green Version]
- Capsoni, S.; Amato, G.; Vignone, D.; Criscuolo, C.; Nykjaer, A.; Cattaneo, A. Dissecting the role of sortilin receptor signaling in neurodegeneration induced by NGF deprivation. Biochem. Biophys. Res. Commun. 2013, 431, 579–585. [Google Scholar] [CrossRef] [Green Version]
- Skeldal, S.; Matusica, D.; Nykjaer, A.; Coulson, E.J. Proteolytic processing of the p75 neurotrophin receptor: A prerequisite for signalling? Bioessays 2011, 33, 614–625. [Google Scholar] [CrossRef]
- Nykjaer, A.; Lee, R.; Teng, K.K.; Jansen, P.; Madsen, P.; Nielsen, M.S.; Jacobsen, C.; Kliemannel, M.; Schwarz, E.; Willnow, T.E.; et al. Sortilin is essential for proNGF-induced neuronal cell death. Nature 2004, 427, 843–848. [Google Scholar] [CrossRef]
- Chen, Z.Y.; Ieraci, A.; Teng, H.; Dall, H.; Meng, C.X.; Herrera, D.G.; Nykjaer, A.; Hempstead, B.L.; Lee, F.S. Sortilin controls intracellular sorting of brain-derived neurotrophic factor to the regulated secretory pathway. J. Neurosci. 2005, 25, 6156–6166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tesco, G.; Koh, Y.H.; Kang, E.L.; Cameron, A.N.; Das, S.; Sena-Esteves, M.; Hiltunen, M.; Yang, S.H.; Zhong, Z.Y.; Shen, Y.; et al. Depletion of GGA3 stabilizes BACE and enhances beta-secretase activity. Neuron 2007, 54, 721–737. [Google Scholar] [CrossRef] [PubMed]
- Lomoio, S.; Willen, R.; Kim, W.; Ho, K.Z.; Robinson, E.K.; Prokopenko, D.; Kennedy, M.E.; Tanzi, R.E.; Tesco, G. Gga3 deletion and a GGA3 rare variant associated with late onset Alzheimer’s disease trigger BACE1 accumulation in axonal swellings. Sci. Transl. Med. 2020, 12, eaba1871. [Google Scholar] [CrossRef]
- Kim, W.; Ma, L.; Lomoio, S.; Willen, R.; Lombardo, S.; Dong, J.; Haydon, P.G.; Tesco, G. BACE1 elevation engendered by GGA3 deletion increases beta-amyloid pathology in association with APP elevation and decreased CHL1 processing in 5XFAD mice. Mol. Neurodegener. 2018, 13, 6. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Lavigne, P.; Lavoie, C. GGA3 mediates TrkA endocytic recycling to promote sustained Akt phosphorylation and cell survival. Mol. Biol. Cell 2015, 26, 4412–4426. [Google Scholar] [CrossRef] [PubMed]
- Hickman, F.E.; Stanley, E.M.; Carter, B.D. Neurotrophin Responsiveness of Sympathetic Neurons Is Regulated by Rapid Mobilization of the p75 Receptor to the Cell Surface through TrkA Activation of Arf6. J. Neurosci. 2018, 38, 5606–5619. [Google Scholar] [CrossRef]
- Tang, W.; Tam, J.H.; Seah, C.; Chiu, J.; Tyrer, A.; Cregan, S.P.; Meakin, S.O.; Pasternak, S.H. Arf6 controls beta-amyloid production by regulating macropinocytosis of the Amyloid Precursor Protein to lysosomes. Mol. Brain 2015, 8, 41. [Google Scholar] [CrossRef] [Green Version]
- Perdigao, C.; Barata, M.A.; Burrinha, T.; Almeida, C.G. Alzheimer’s disease BIN1 coding variants increase intracellular Abeta levels by interfering with BACE1 recycling. J. Biol. Chem. 2021, 297, 101056. [Google Scholar] [CrossRef]
- Tan, M.S.; Yu, J.T.; Jiang, T.; Zhu, X.C.; Guan, H.S.; Tan, L. Genetic variation in BIN1 gene and Alzheimer’s disease risk in Han Chinese individuals. Neurobiol. Aging 2014, 35, 1781.e1–1781.e8. [Google Scholar] [CrossRef]
- Vardarajan, B.N.; Ghani, M.; Kahn, A.; Sheikh, S.; Sato, C.; Barral, S.; Lee, J.H.; Cheng, R.; Reitz, C.; Lantigua, R.; et al. Rare coding mutations identified by sequencing of Alzheimer disease genome-wide association studies loci. Ann. Neurol. 2015, 78, 487–498. [Google Scholar] [CrossRef]
- Ubelmann, F.; Burrinha, T.; Salavessa, L.; Gomes, R.; Ferreira, C.; Moreno, N.; Almeida, C.G. Bin1 and CD2AP polarise the endocytic generation of beta-amyloid. EMBO Rep. 2017, 18, 102–122. [Google Scholar] [CrossRef] [Green Version]
- De Rossi, P.; Nomura, T.; Andrew, R.J.; Masse, N.Y.; Sampathkumar, V.; Musial, T.F.; Sudwarts, A.; Recupero, A.J.; Le Metayer, T.; Hansen, M.T.; et al. Neuronal BIN1 Regulates Presynaptic Neurotransmitter Release and Memory Consolidation. Cell Rep. 2020, 30, 3520–3535.e3527. [Google Scholar] [CrossRef]
- Chen, H.; Wu, G.; Jiang, Y.; Feng, R.; Liao, M.; Zhang, L.; Ma, G.; Chen, Z.; Zhao, B.; Li, K.; et al. Analyzing 54,936 Samples Supports the Association between CD2AP rs9349407 Polymorphism and Alzheimer’s Disease Susceptibility. Mol. Neurobiol. 2015, 52, 1–7. [Google Scholar] [CrossRef]
- Harrison, B.J.; Venkat, G.; Lamb, J.L.; Hutson, T.H.; Drury, C.; Rau, K.K.; Bunge, M.B.; Mendell, L.M.; Gage, F.H.; Johnson, R.D.; et al. The Adaptor Protein CD2AP is a Coordinator of Neurotrophin Signaling-Mediated Axon Arbor Plasticity. J. Neurosci. 2016, 36, 4259–4275. [Google Scholar] [CrossRef] [Green Version]
- Small, S.A.; Kent, K.; Pierce, A.; Leung, C.; Kang, M.S.; Okada, H.; Honig, L.; Vonsattel, J.P.; Kim, T.W. Model-guided microarray implicates the retromer complex in Alzheimer’s disease. Ann. Neurol. 2005, 58, 909–919. [Google Scholar] [CrossRef] [PubMed]
- Wen, L.; Tang, F.L.; Hong, Y.; Luo, S.W.; Wang, C.L.; He, W.; Shen, C.; Jung, J.U.; Xiong, F.; Lee, D.H.; et al. VPS35 haploinsufficiency increases Alzheimer’s disease neuropathology. J. Cell Biol. 2011, 195, 765–779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, E.; Lee, Y.; Lee, H.J.; Kim, J.S.; Song, B.S.; Huh, J.W.; Lee, S.R.; Kim, S.U.; Kim, S.H.; Hong, Y.; et al. Implication of mouse Vps26b-Vps29-Vps35 retromer complex in sortilin trafficking. Biochem. Biophys. Res. Commun. 2010, 403, 167–171. [Google Scholar] [CrossRef]
- Zhang, X.; Huang, T.Y.; Yancey, J.; Luo, H.; Zhang, Y.W. Role of Rab GTPases in Alzheimer’s Disease. ACS Chem. Neurosci. 2019, 10, 828–838. [Google Scholar] [CrossRef]
- Faustino, C.; Rijo, P.; Reis, C.P. Nanotechnological strategies for nerve growth factor delivery: Therapeutic implications in Alzheimer’s disease. Pharmacol. Res. 2017, 120, 68–87. [Google Scholar] [CrossRef] [PubMed]
- Amadoro, G.; Latina, V.; Balzamino, B.O.; Squitti, R.; Varano, M.; Calissano, P.; Micera, A. Nerve Growth Factor-Based Therapy in Alzheimer’s Disease and Age-Related Macular Degeneration. Front. Neurosci. 2021, 15, 735928. [Google Scholar] [CrossRef]
- Fjord-Larsen, L.; Kusk, P.; Tornoe, J.; Juliusson, B.; Torp, M.; Bjarkam, C.R.; Nielsen, M.S.; Handberg, A.; Sorensen, J.C.; Wahlberg, L.U. Long-term delivery of nerve growth factor by encapsulated cell biodelivery in the Gottingen minipig basal forebrain. Mol. Ther. 2010, 18, 2164–2172. [Google Scholar] [CrossRef] [PubMed]
- Eriksdotter-Jonhagen, M.; Linderoth, B.; Lind, G.; Aladellie, L.; Almkvist, O.; Andreasen, N.; Blennow, K.; Bogdanovic, N.; Jelic, V.; Kadir, A.; et al. Encapsulated cell biodelivery of nerve growth factor to the Basal forebrain in patients with Alzheimer’s disease. Dement. Geriatr. Cogn. Disord. 2012, 33, 18–28. [Google Scholar] [CrossRef]
- Qureshi, Y.H.; Berman, D.E.; Marsh, S.E.; Klein, R.L.; Patel, V.M.; Simoes, S.; Kannan, S.; Petsko, G.A.; Stevens, B.; Small, S.A. The neuronal retromer can regulate both neuronal and microglial phenotypes of Alzheimer’s disease. Cell Rep. 2022, 38, 110262. [Google Scholar] [CrossRef]
- Li, J.G.; Chiu, J.; Pratico, D. Full recovery of the Alzheimer’s disease phenotype by gain of function of vacuolar protein sorting 35. Mol. Psychiatry 2020, 25, 2630–2640. [Google Scholar] [CrossRef]
- Young, J.E.; Fong, L.K.; Frankowski, H.; Petsko, G.A.; Small, S.A.; Goldstein, L.S.B. Stabilizing the Retromer Complex in a Human Stem Cell Model of Alzheimer’s Disease Reduces TAU Phosphorylation Independently of Amyloid Precursor Protein. Stem. Cell Rep. 2018, 10, 1046–1058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Temkin, P.; Morishita, W.; Goswami, D.; Arendt, K.; Chen, L.; Malenka, R. The Retromer Supports AMPA Receptor Trafficking during LTP. Neuron 2017, 94, 74–82.e75. [Google Scholar] [CrossRef] [Green Version]
- Mecozzi, V.J.; Berman, D.E.; Simoes, S.; Vetanovetz, C.; Awal, M.R.; Patel, V.M.; Schneider, R.T.; Petsko, G.A.; Ringe, D.; Small, S.A. Pharmacological chaperones stabilize retromer to limit APP processing. Nat. Chem. Biol. 2014, 10, 443–449. [Google Scholar] [CrossRef] [Green Version]
- Berman, D.E.; Ringe, D.; Petsko, G.A.; Small, S.A. The Use of Pharmacological Retromer Chaperones in Alzheimer’s Disease and other Endosomal-related Disorders. Neurotherapeutics 2015, 12, 12–18. [Google Scholar] [CrossRef] [Green Version]
- Agola, J.O.; Jim, P.A.; Ward, H.H.; BasuRay, S.; Wandinger-Ness, A. Rab GTPases as regulators of endocytosis, targets of disease and therapeutic opportunities. Clin. Genet. 2011, 80, 305–318. [Google Scholar] [CrossRef]
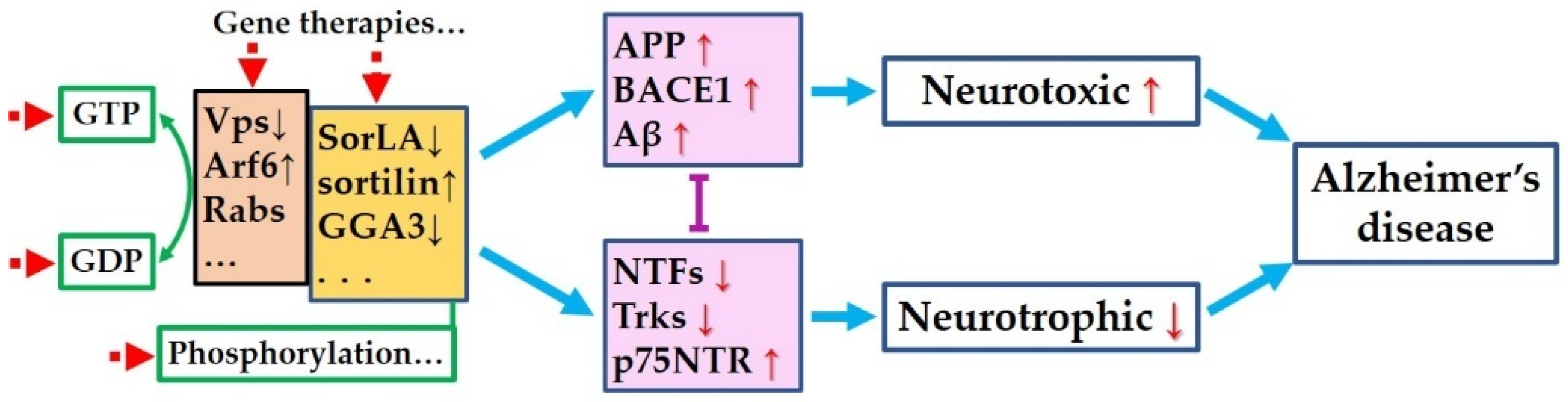

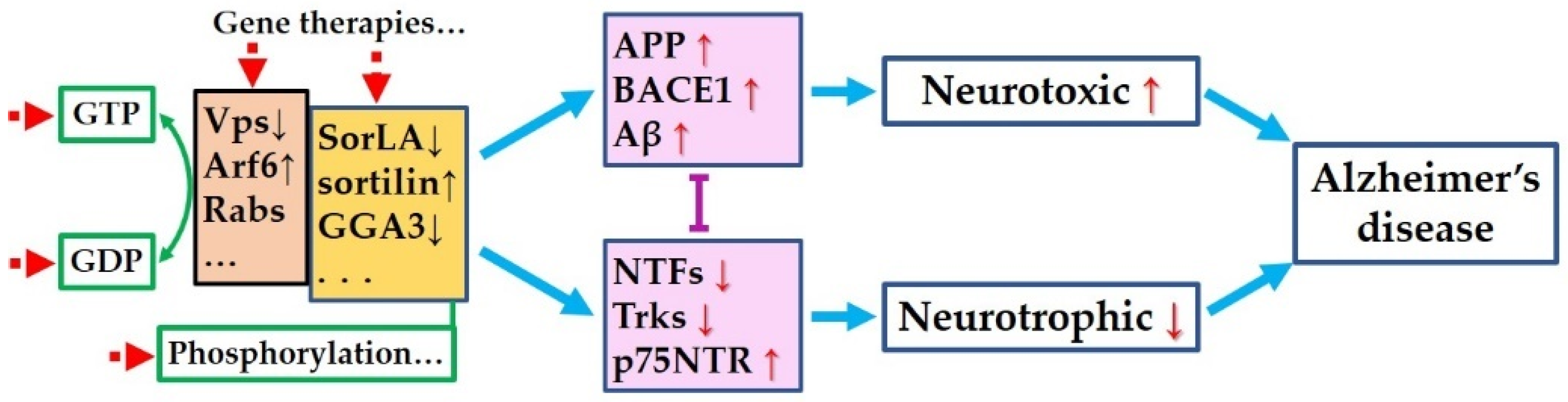{kind=link}
| Abbreviation | Full Name | In AD |
|---|---|---|
| NGF | Nerve growth factor | Mature NGF↓ and proNGF↑ in brain [7,34]; (pro)NGF in hippocampus and neocortex↑ [31] |
| BDNF | Brain-derived neurotrophic factor | mRNA and protein levels in specific brain areas↓ [29]; decreased BDNF in hippocampus and neocortex↓ [31]; peripheral levels↓ [35]; Val66Met polymorphism [36] |
| NT-3 | Neurotrophin-3 | Motor cortex↓ [37]; no change in all brain regions [38] |
| NT-4/5 | Neurotrophin-4/5 | Hippocampus and cerebellum↓ [38] |
| GDNF | Glial cell-line-derived neurotrophic factor | Middle temporal gyrus↓ [39]; serum↓ [40,41]; cerebrospinal fluid↑ and serum↓ [42]; serum and brain of early stage AD↑ [43] |
| CNTF | Ciliary neurotrophic factor | Increases following brain injury in mice [44] |
| LIF | Leukemia inhibitory factor | Hippocampus and temporal cortex↑ [45] |
| CTF-1 | Cardiotrophin-1 | Brain of APPswe/PS1dE9 mice (AD model)↓ [46] |
| CDNF | Cerebral dopamine neurotrophic factor | Platelets of probable AD patients↓ [47] |
| MANF | Mesencephalic astrocyte-derived neurotrophic factor | Inferior temporal gyrus of the cortex↑ [48] |
| CPE/NF-α1 | Carboxypeptidase E/neurotrophic factor-α1 | Mutation [33,49] |
| VEGF | Vascular endothelial growth factor | Prefrontal cortex RNA↑ [50]; frontal cortex and parahippocampal↑ [51]; cerebral capillaries in postmortem brain↓ [52,53]; controversial in cerebrospinal fluid [54,55] and serum [56,57]; 2578C/A and 1154G/A polymorphisms [58] |
| PDGFs | Platelet-derived growth factors | Controversial in plasma and cerebrospinal fluid [59,60,61] |
| bFGF | Basic fibroblast growth factors | Brain (Brodmann areas 10/11 and 20/21)↑ [62] |
| TGFβ1 | Transforming growth factors β1 | Plasmatic levels↓ [63]; receptor (TGFβRII) in brain↓ [64] |
| TNF-α | Tumor necrosis factor α | Plasma levels↑; postmortem brain of early-stage AD↑; G308A mutant [65] |
| CNTF | Ciliary neurotrophic factor | Increased following brain injury in mice [44] |
| IGF | Insulin-like growth factors | Controversial [66] |
| HGF | Hepatocyte growth factor | Prefrontal cortex↑ [67] |
| Name | Deficits in AD | Effects |
|---|---|---|
| ApoE | ApoE2 and ApoE4 gene dose-dependent AD risk [123,124,125] | ApoE4 increases Aβ aggregation, synthesis, deposition, reuptake, clearance and degradation [123], while it suppresses BDNF mRNA expression [93]. |
| SorLA | brain↓ [126]; 13 SNPs associated with sporadic AD [127] | APP trafficking in endosomal compartments and Aβ production [121,128,129,130,131,132,133]; Aβ42 degradation [134]; sorting of GDNF and GFRα1/RET [135], trafficking of TrkB [136], activation of the EGFR/ERK/Fos pathway [137]. |
| Sortilin | brain↑ [138]; rs17646665 and other SNPs [139] | BACE1 and APP trafficking [140,141,142]; ApoE/Aβ lysosomal degradation [143,144]; receptor for oligomerized Aβ [145]; anterograde transport of Trk receptors [146]; co-receptor with p75NTR for proNGF [147,148,149], BDNF secretion [150]. |
| GGA3 | temporal cortex↓ [151]; gene depletion or rare variant [152] | BACE1 degradation, recycling and axon transport [151,152,153]; TrkA recycling [154]; rapid recruitment of p75NTR to the plasma membrane upon NGF activation of TrkA [155]. |
| Arf6 | hippocampus↑ [156] | Regulating macropinocytosis of APP in lysosomes [156]; TrkA post-endocytic recycling [154]; rapid recruitment of p75NTR to the plasma membrane upon NGF activation of TrkA [155]. |
| BIN1 | Controversial [157]; SNP rs754834233 [158] and rs138047593 [159] | Endocytic BACE1 recycling [160]; presynaptic neurotransmitter release [161]. |
| CD2AP | rs9349407 [162] | Endocytic BACE1 degradation [160]; TrkA location to endosomes and TrkA-induced AKT pathway [163]. |
| VPS | VPS35 and VPS26 in entorhinal cortex↓ [164] | Promotes BACE1 endosome-to-Golgi retrieval to inhibit BACE1 activation and Aβ production [165]; VPS26b/VPS29/VPS35 retromer complex controls p75NTR/sortilin interaction [166]. |
| Rabs | Rab5, Rab6, Rab7 and Rab10, with abnormal expression or activation | controls amyloidogenesis and the neurotrophic pathway at multiple trafficking stages (recently reviewed by Zhang X. et al.) [167]. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jiao, F.; Jiang, D.; Li, Y.; Mei, J.; Wang, Q.; Li, X. Amyloidogenesis and Neurotrophic Dysfunction in Alzheimer’s Disease: Do They have a Common Regulating Pathway? Cells 2022, 11, 3201. https://doi.org/10.3390/cells11203201
Jiao F, Jiang D, Li Y, Mei J, Wang Q, Li X. Amyloidogenesis and Neurotrophic Dysfunction in Alzheimer’s Disease: Do They have a Common Regulating Pathway? Cells. 2022; 11(20):3201. https://doi.org/10.3390/cells11203201
Chicago/Turabian StyleJiao, Fengjuan, Dongjun Jiang, Yingshuai Li, Juan Mei, Qinqin Wang, and Xuezhi Li. 2022. "Amyloidogenesis and Neurotrophic Dysfunction in Alzheimer’s Disease: Do They have a Common Regulating Pathway?" Cells 11, no. 20: 3201. https://doi.org/10.3390/cells11203201