

Ferroptosis-Related Gene GCLC Is a Novel Prognostic Molecular and Correlates with Immune Infiltrates in Lung Adenocarcinoma



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Data Collection
2.2. Identification of Differentially Expressed Genes in the TCGA Datasets
2.3. Stepwise Construction of Prognosis Signature
2.4. External Validation of the Prognostic Signature
2.5. Independence of the Prognostic Signature from Traditional Clinical Characteristics
2.6. Construction and Evaluation of a Predictive Nomogram Integrating FRG Signature and Clinical Data
2.7. Functional and Pathway Enrichment Analysis
2.8. Tumor Microenvironment and Immune Infiltration Analysis
2.9. Comprehensive Bioinformatics Evaluation of Key Gene GCLC
2.10. The Human Protein Atlas (HPA)
2.11. Cell Culture and Transfection
2.12. Cell Proliferation
2.13. Colony Formation Assay
2.14. Cell Death Assays
2.15. Iron Assay
2.16. Lipid Peroxidation Assay
2.17. Glutathione Quantification
2.18. MDA Assay
2.19. Western Blotting
2.20. Statistical Analysis
3. Results
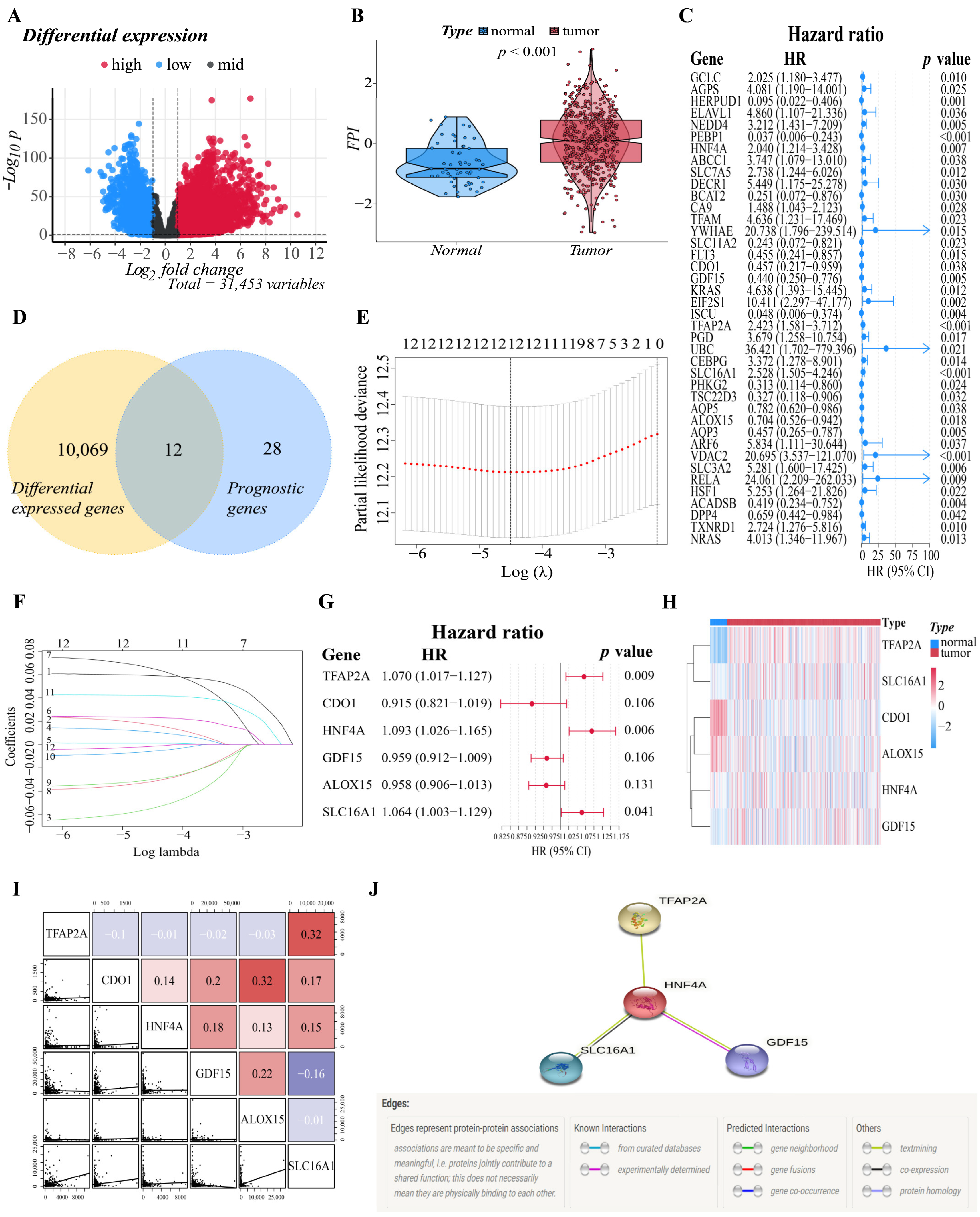
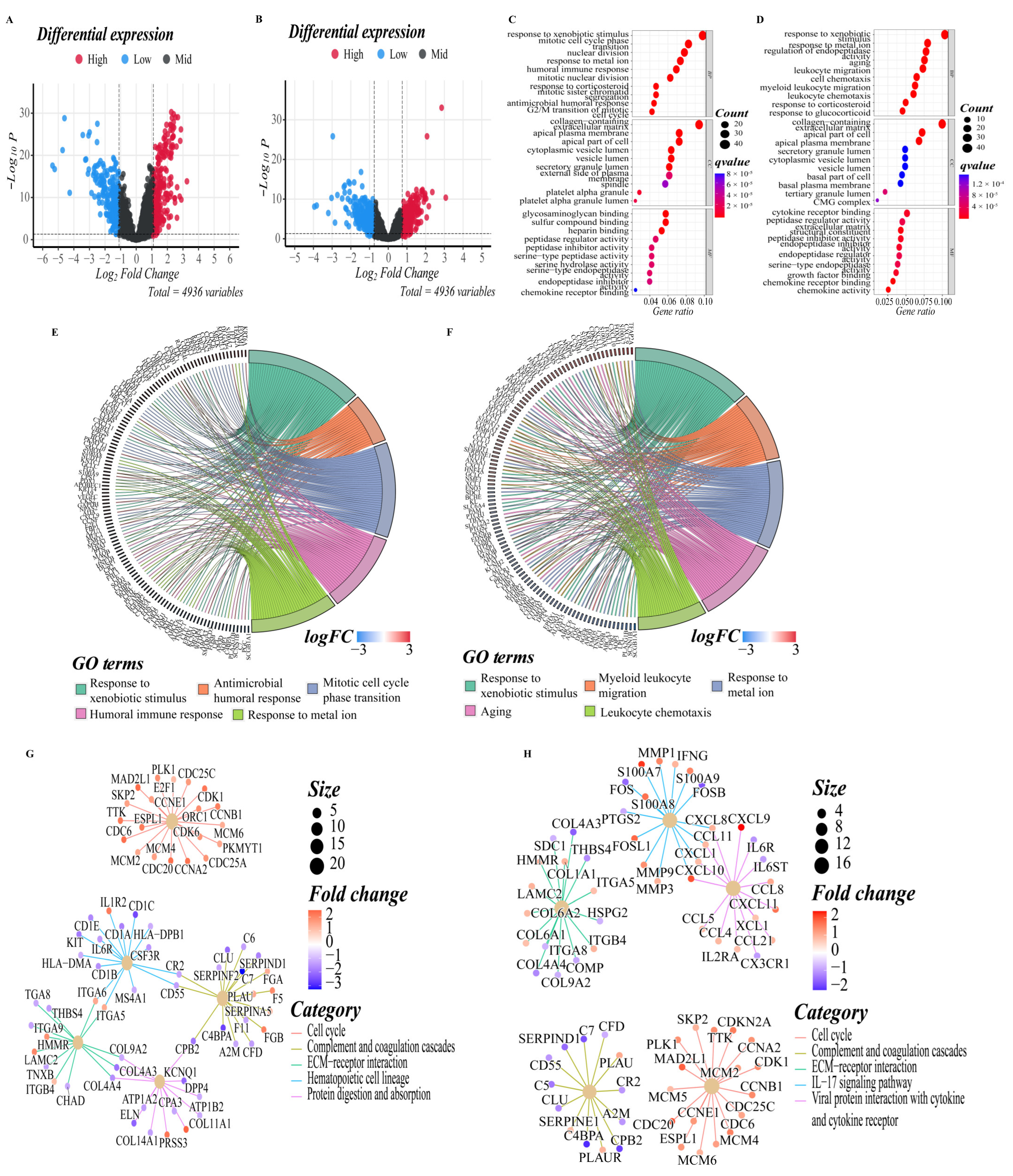
3.1. Identification of Prognostic Ferroptosis-Related DEGs in the Training Cohort
3.2. Construction of the Six-Gene Signatures Related to Ferroptosis
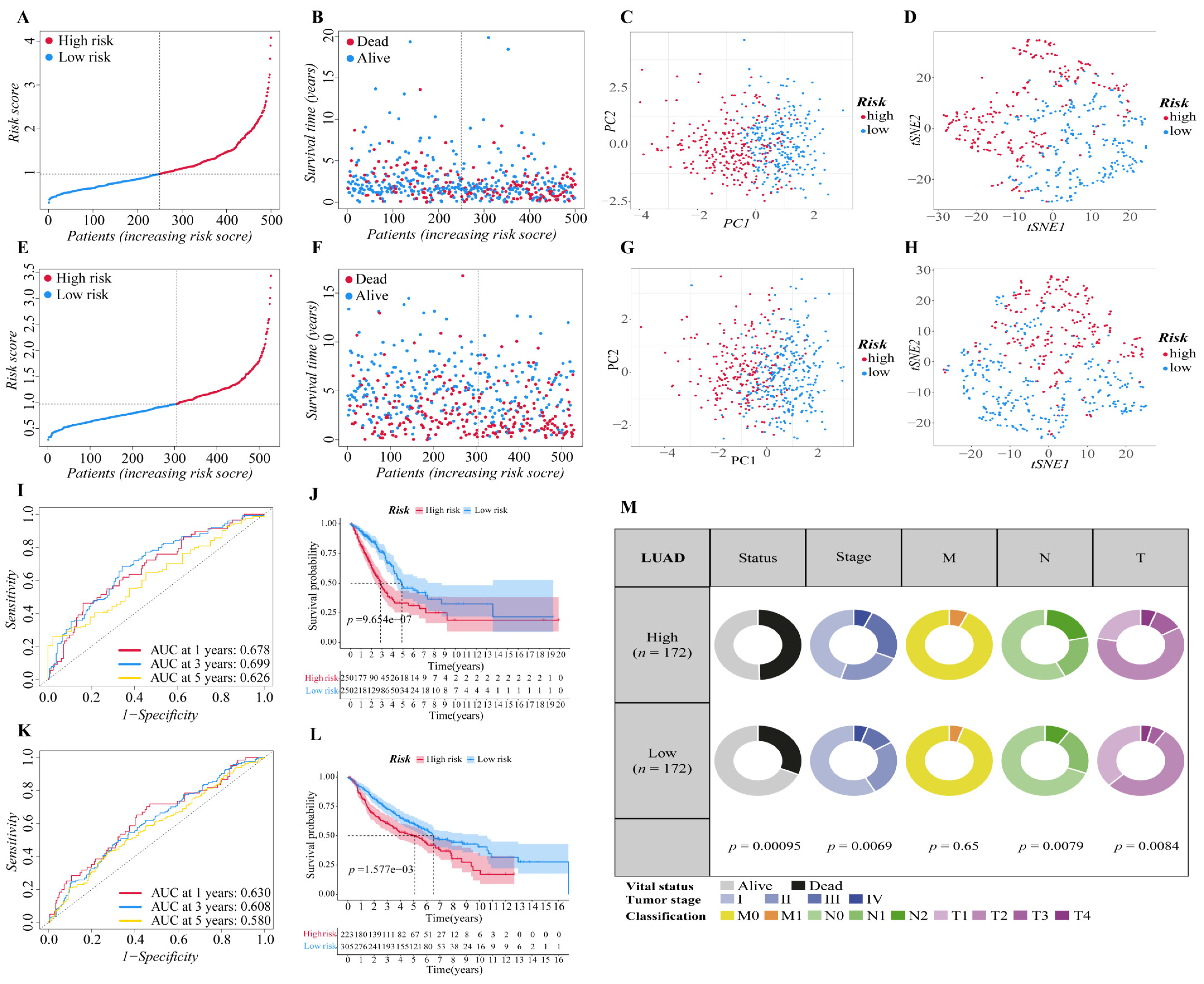
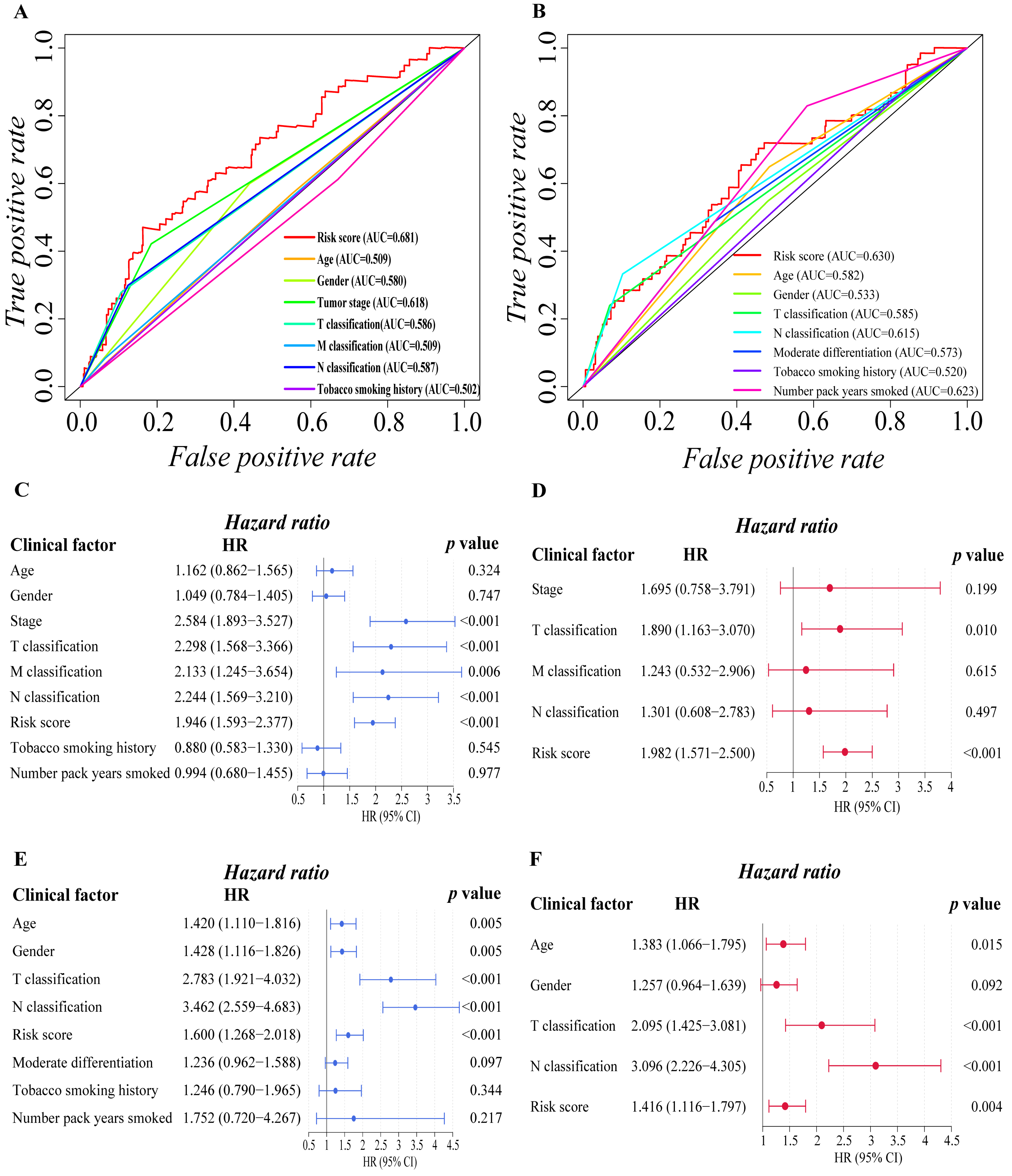
3.3. Independent Prognostic Predictive Efficacy of the Six-Gene Signature
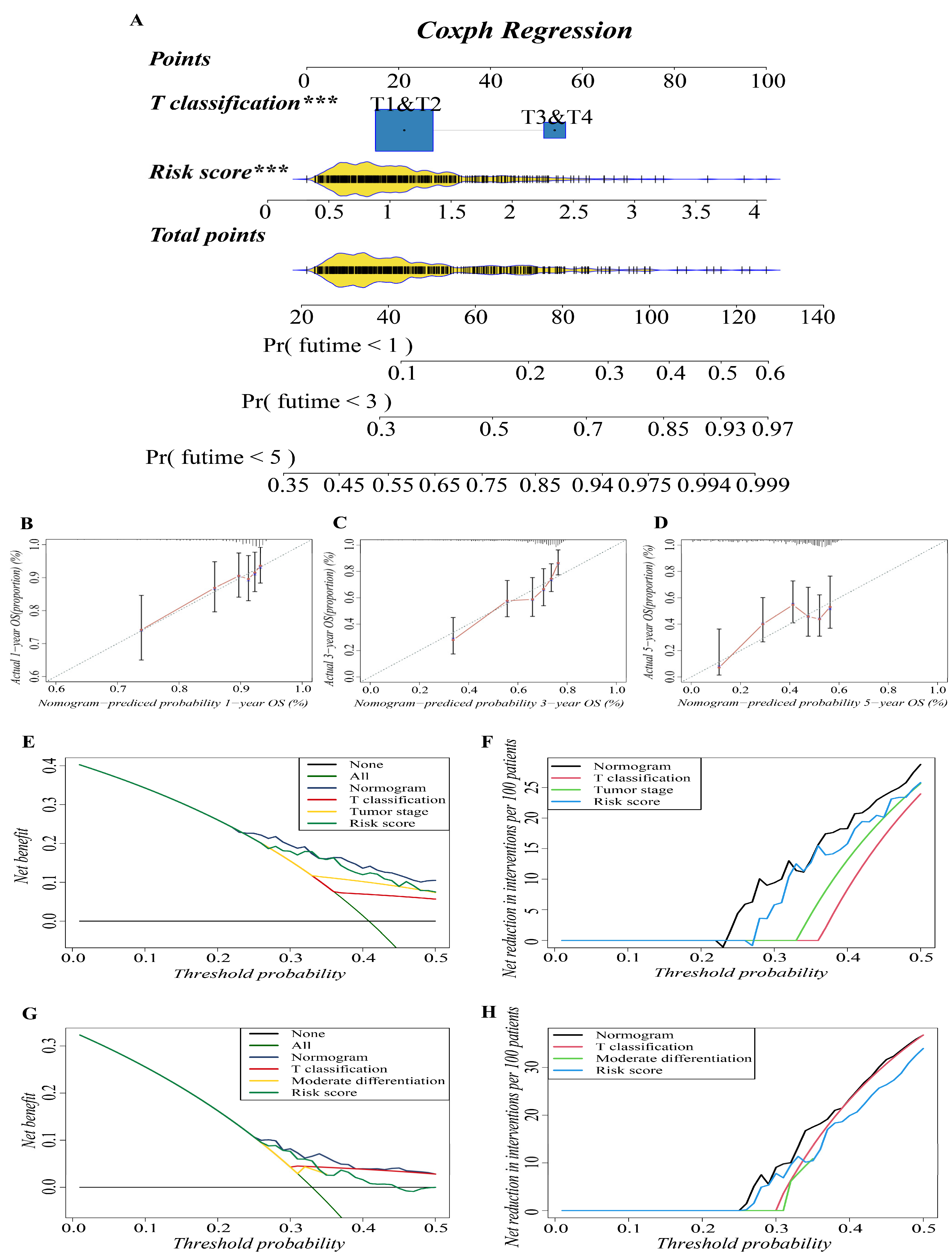
3.4. Construction and Validation of the Predictive Nomogram
3.5. Functional Enrichment Analyses of Different Risk Groups
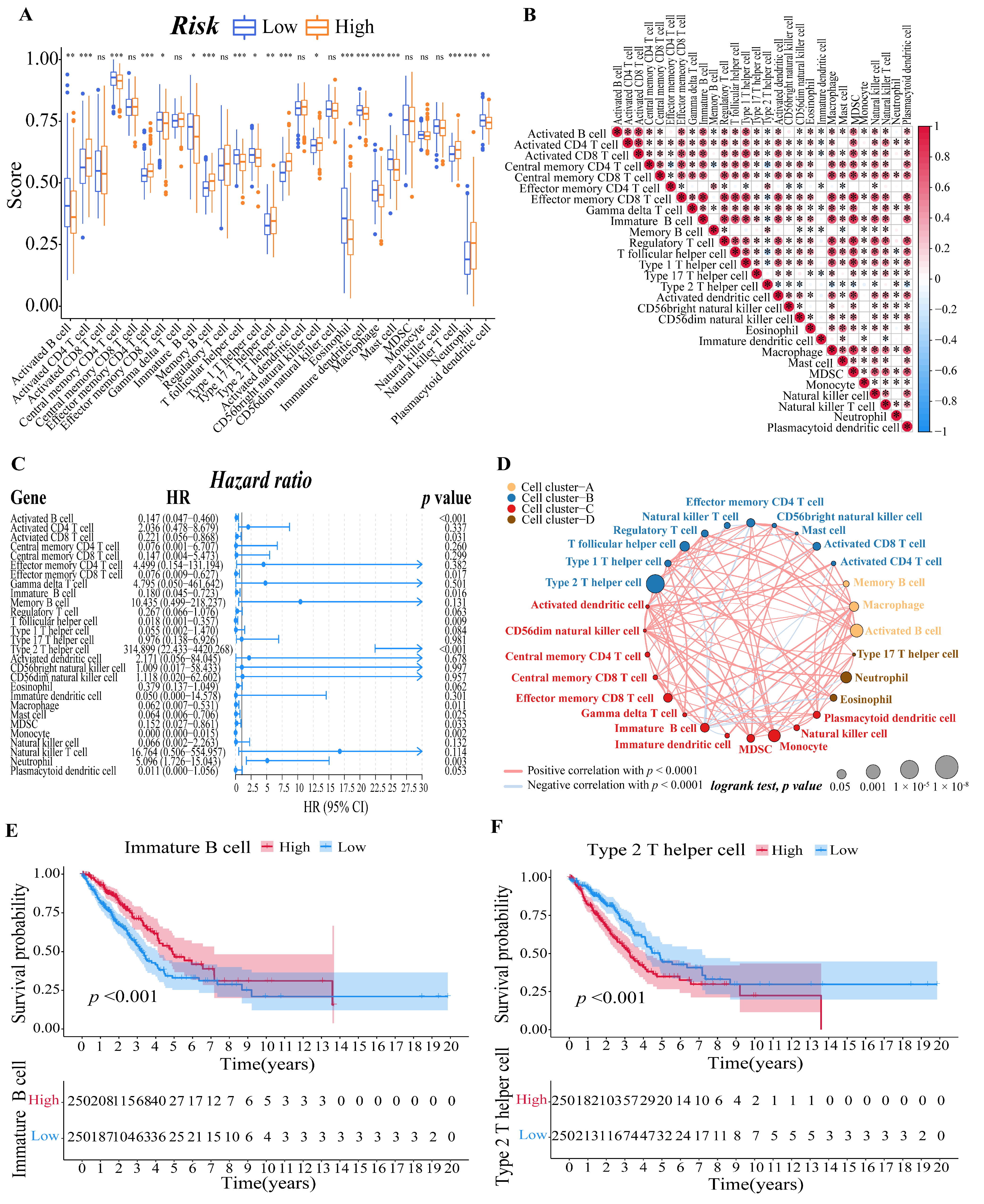
3.6. Evaluation of the LUAD Patients’ Immune Microenvironment
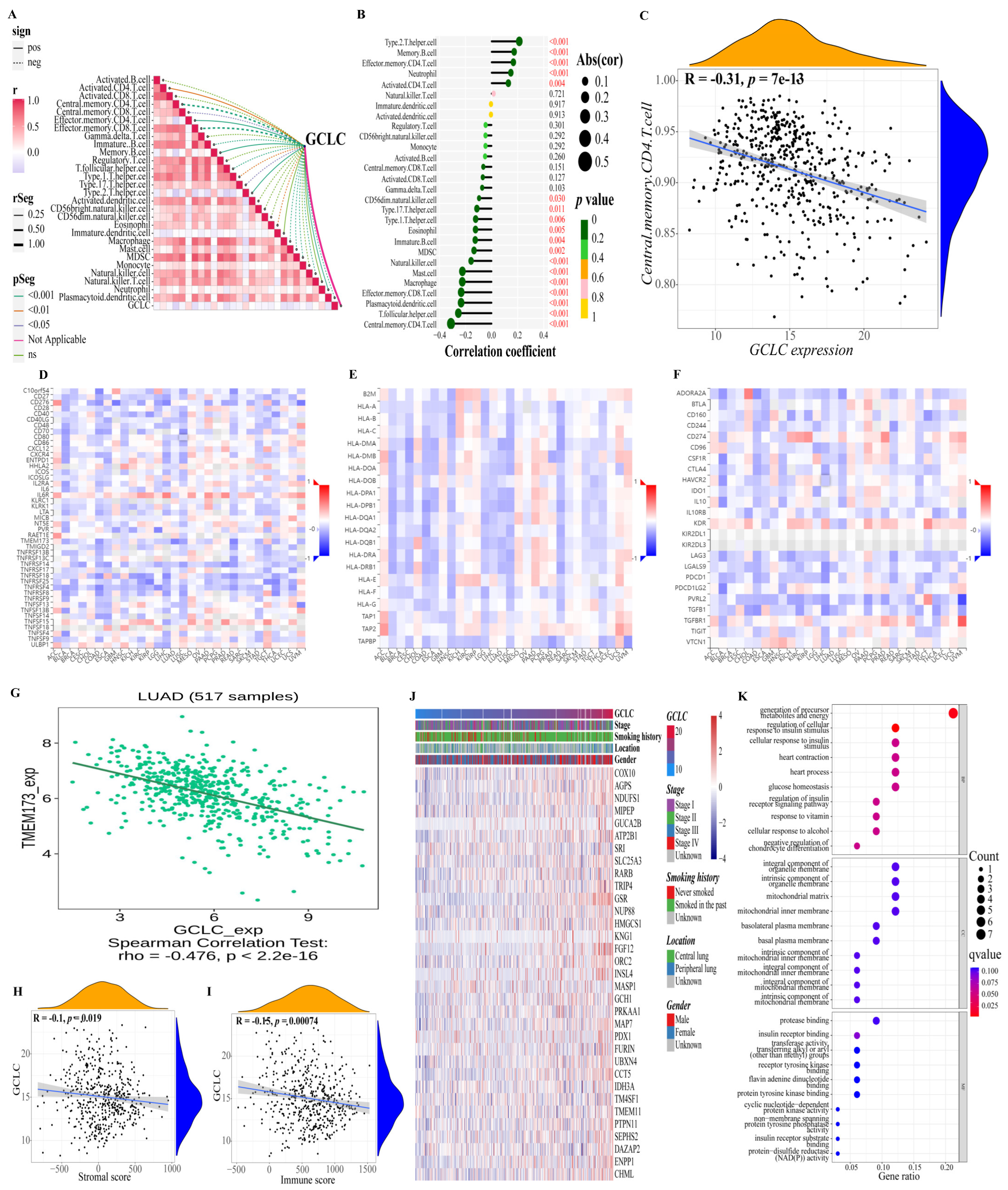
3.7. GCLC Could Be a Significant Prognostic Predictor
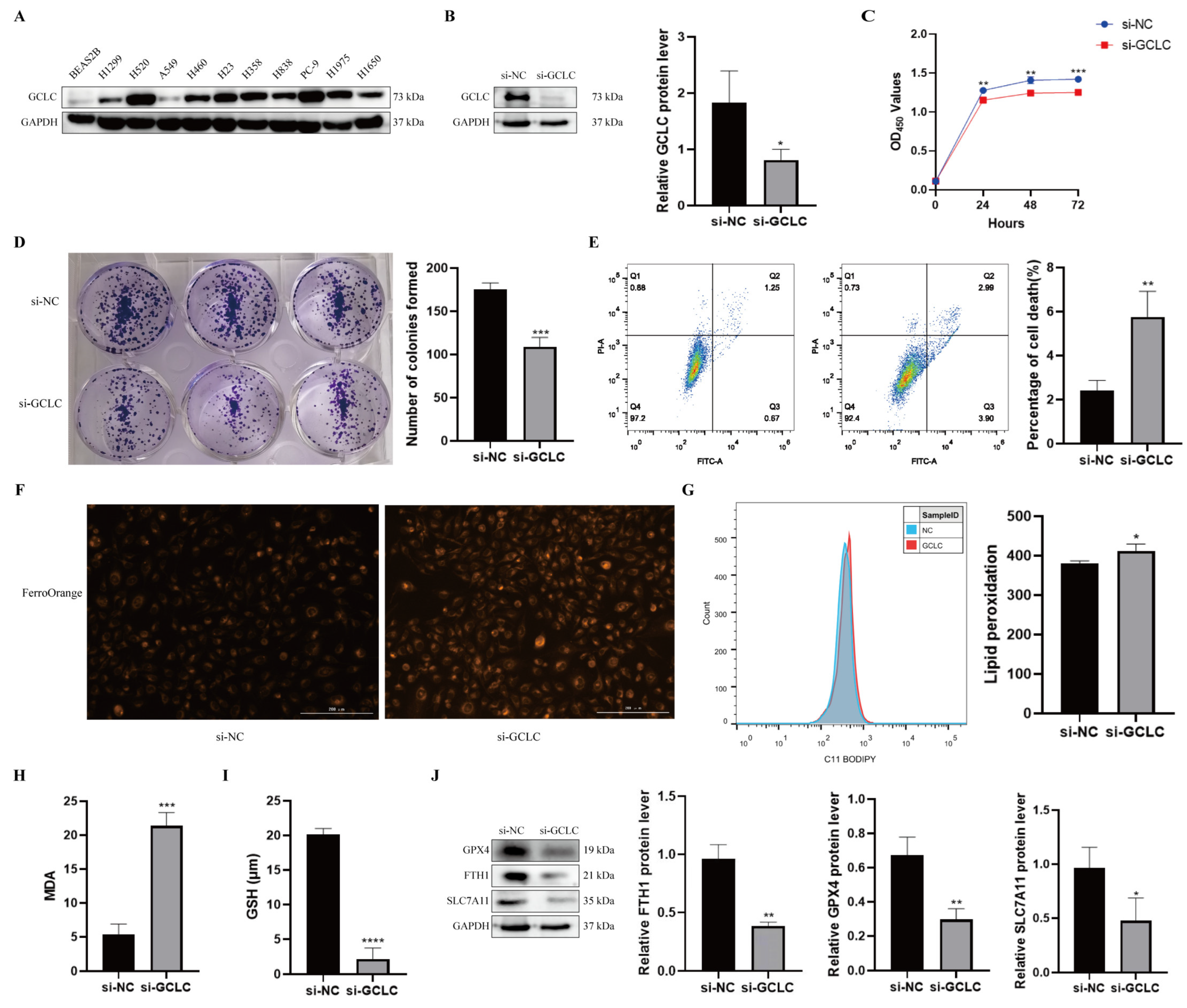
3.8. GCLC Silencing Promotes Ferroptosis of H358 Cells
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shukla, S.; Evans, J.R.; Malik, R.; Feng, F.Y.; Dhanasekaran, S.M.; Cao, X.; Chen, G.; Beer, D.G.; Jiang, H.; Chinnaiyan, A.M. Development of a RNA-Seq Based Prognostic Signature in Lung Adenocarcinoma. J. Natl. Cancer Inst. 2017, 109, djw200. [Google Scholar] [CrossRef] [PubMed]
- Zappa, C.; Mousa, S.A. Non-small cell lung cancer: Current treatment and future advances. Transl. Lung Cancer Res. 2016, 5, 288–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herbst, R.S.; Morgensztern, D.; Boshoff, C. The biology and management of non-small cell lung cancer. Nature 2018, 553, 446–454. [Google Scholar] [CrossRef]
- Miller, K.D.; Siegel, R.L.; Lin, C.C.; Mariotto, A.B.; Kramer, J.L.; Rowland, J.H.; Stein, K.D.; Alteri, R.; Jemal, A. Cancer treatment and survivorship statistics, 2016. CA Cancer J. Clin. 2016, 66, 271–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Cao, F.; Yin, H.L.; Huang, Z.J.; Lin, Z.T.; Mao, N.; Sun, B.; Wang, G. Ferroptosis: Past, present and future. Cell Death Dis. 2020, 11, 88. [Google Scholar] [CrossRef] [Green Version]
- Liang, C.; Zhang, X.; Yang, M.; Dong, X. Recent Progress in Ferroptosis Inducers for Cancer Therapy. Adv. Mater 2019, 31, e1904197. [Google Scholar] [CrossRef]
- Xu, T.; Ding, W.; Ji, X.; Ao, X.; Liu, Y.; Yu, W.; Wang, J. Molecular mechanisms of ferroptosis and its role in cancer therapy. J. Cell Mol. Med. 2019, 23, 4900–4912. [Google Scholar] [CrossRef] [Green Version]
- Stockwell, B.R.; Friedmann Angeli, J.P.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascon, S.; Hatzios, S.K.; Kagan, V.E.; et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef] [Green Version]
- Xia, X.; Fan, X.; Zhao, M.; Zhu, P. The Relationship between Ferroptosis and Tumors: A Novel Landscape for Therapeutic Approach. Curr. Gene Ther. 2019, 19, 117–124. [Google Scholar] [CrossRef]
- Hassannia, B.; Vandenabeele, P.; Vanden Berghe, T. Targeting Ferroptosis to Iron Out Cancer. Cancer Cell 2019, 35, 830–849. [Google Scholar] [CrossRef] [PubMed]
- Bersuker, K.; Hendricks, J.M.; Li, Z.; Magtanong, L.; Ford, B.; Tang, P.H.; Roberts, M.A.; Tong, B.; Maimone, T.J.; Zoncu, R.; et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature 2019, 575, 688–692. [Google Scholar] [CrossRef] [PubMed]
- Doll, S.; Freitas, F.P.; Shah, R.; Aldrovandi, M.; da Silva, M.C.; Ingold, I.; Goya Grocin, A.; Xavier da Silva, T.N.; Panzilius, E.; Scheel, C.H.; et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature 2019, 575, 693–698. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.J.; Hu, H.M.; Li, G.Z.; Zhang, Y.; Wu, F.; Liu, X.; Wang, K.Y.; Zhang, C.B.; Jiang, T. Ferroptosis-Related Gene Signature Predicts Glioma Cell Death and Glioma Patient Progression. Front Cell Dev. Biol. 2020, 8, 538. [Google Scholar] [CrossRef]
- Ji, X.; Qian, J.; Rahman, S.M.J.; Siska, P.J.; Zou, Y.; Harris, B.K.; Hoeksema, M.D.; Trenary, I.A.; Heidi, C.; Eisenberg, R.; et al. xCT (SLC7A11)-mediated metabolic reprogramming promotes non-small cell lung cancer progression. Oncogene 2018, 37, 5007–5019. [Google Scholar] [CrossRef]
- Lai, Y.; Zhang, Z.; Li, J.; Li, W.; Huang, Z.; Zhang, C.; Li, X.; Zhao, J. STYK1/NOK correlates with ferroptosis in non-small cell lung carcinoma. Biochem. Biophys. Res. Commun. 2019, 519, 659–666. [Google Scholar] [CrossRef]
- Ma, L.; Chen, T.; Zhang, X.; Miao, Y.; Tian, X.; Yu, K.; Xu, X.; Niu, Y.; Guo, S.; Zhang, C.; et al. The m(6)A reader YTHDC2 inhibits lung adenocarcinoma tumorigenesis by suppressing SLC7A11-dependent antioxidant function. Redox Biol. 2021, 38, 101801. [Google Scholar] [CrossRef]
- Hu, K.; Li, K.; Lv, J.; Feng, J.; Chen, J.; Wu, H.; Cheng, F.; Jiang, W.; Wang, J.; Pei, H.; et al. Suppression of the SLC7A11/glutathione axis causes synthetic lethality in KRAS-mutant lung adenocarcinoma. J. Clin. Invest. 2020, 130, 1752–1766. [Google Scholar] [CrossRef] [Green Version]
- Lei, G.; Zhuang, L.; Gan, B. Targeting ferroptosis as a vulnerability in cancer. Nat. Rev. Cancer 2022, 22, 381–396. [Google Scholar] [CrossRef]
- Huang, F.; Zheng, Y.; Li, X.; Luo, H.; Luo, L. Ferroptosis-related gene AKR1C1 predicts the prognosis of non-small cell lung cancer. Cancer Cell Int. 2021, 21, 567. [Google Scholar] [CrossRef]
- Sun, R.; Liu, M.; Xu, K.; Pu, Y.; Huang, J.; Liu, J.; Zhang, J.; Yin, L.; Pu, Y. Ferroptosis is involved in the benzene-induced hematotoxicity in mice via iron metabolism, oxidative stress and NRF2 signaling pathway. Chem. Biol. Interact. 2022, 362, 110004. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Li, Y.; Li, J.; Chen, W. Ethyl carbamate triggers ferroptosis in liver through inhibiting GSH synthesis and suppressing Nrf2 activation. Redox Biol. 2022, 53, 102349. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.P.; Mockabee-Macias, A.; Jiang, C.; Falzone, A.; Prieto-Farigua, N.; Stone, E.; Harris, I.S.; DeNicola, G.M. Non-canonical Glutamate-Cysteine Ligase Activity Protects agaInst Ferroptosis. Cell Metab. 2021, 33, 174–189.e177. [Google Scholar] [CrossRef]
- Stockwell, B.R.; Jiang, X. A Physiological Function for Ferroptosis in Tumor Suppression by the Immune System. Cell Metab. 2019, 30, 14–15. [Google Scholar] [CrossRef] [PubMed]
- Mou, Y.; Wang, J.; Wu, J.; He, D.; Zhang, C.; Duan, C.; Li, B. Ferroptosis, a new form of cell death: Opportunities and challenges in cancer. J. Hematol. Oncol. 2019, 12, 34. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Sun, Z.; Bi, Q.; Wang, W. A Ferroptosis-Related Genes Model Allows for Prognosis and Treatment Stratification of Clear Cell Renal Cell Carcinoma: A Bioinformatics Analysis and Experimental Verification. Front Oncol. 2022, 12, 815223. [Google Scholar] [CrossRef]
- Lung, A.; Shedden, K.; Taylor, J.M.; Enkemann, S.A.; Tsao, M.S.; Yeatman, T.J.; Gerald, W.L.; Eschrich, S.; Jurisica, I.; Giordano, T.J.; et al. Gene expression-based survival prediction in lung adenocarcinoma: A multi-site, blinded validation study. Nat. Med. 2008, 14, 822–827. [Google Scholar] [CrossRef]
- Beer, D.G.; Kardia, S.L.; Huang, C.C.; Giordano, T.J.; Levin, A.M.; Misek, D.E.; Lin, L.; Chen, G.; Gharib, T.G.; Thomas, D.G.; et al. Gene-expression profiles predict survival of patients with lung adenocarcinoma. Nat. Med. 2002, 8, 816–824. [Google Scholar] [CrossRef]
- Zhou, N.; Bao, J. FerrDb: A manually curated resource for regulators and markers of ferroptosis and ferroptosis-disease associations. Database 2020, 2020, baaa021. [Google Scholar] [CrossRef]
- Liu, Z.; Zhao, Q.; Zuo, Z.X.; Yuan, S.Q.; Yu, K.; Zhang, Q.; Zhang, X.; Sheng, H.; Ju, H.Q.; Cheng, H.; et al. Systematic Analysis of the Aberrances and Functional Implications of Ferroptosis in Cancer. iScience 2020, 23, 101302. [Google Scholar] [CrossRef]
- Tibshirani, R. The lasso method for variable selection in the Cox model. Stat. Med. 1997, 16, 385–395. [Google Scholar] [CrossRef]
- Li, T.; Fu, J.; Zeng, Z.; Cohen, D.; Li, J.; Chen, Q.; Li, B.; Liu, X.S. TIMER2.0 for analysis of tumor-infiltrating immune cells. Nucleic Acids Res. 2020, 48, W509–W514. [Google Scholar] [CrossRef] [PubMed]
- Yoshihara, K.; Shahmoradgoli, M.; Martinez, E.; Vegesna, R.; Kim, H.; Torres-Garcia, W.; Trevino, V.; Shen, H.; Laird, P.W.; Levine, D.A.; et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat. Commun. 2013, 4, 2612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ru, B.; Wong, C.N.; Tong, Y.; Zhong, J.Y.; Zhong, S.S.W.; Wu, W.C.; Chu, K.C.; Wong, C.Y.; Lau, C.Y.; Chen, I.; et al. TISIDB: An integrated repository portal for tumor-immune system interactions. Bioinformatics 2019, 35, 4200–4202. [Google Scholar] [CrossRef] [PubMed]
- Detterbeck, F.C.; Boffa, D.J.; Kim, A.W.; Tanoue, L.T. The Eighth Edition Lung Cancer Stage Classification. Chest 2017, 151, 193–203. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Liu, J.; Hou, W.; Kang, R.; Tang, D. STING1 Promotes Ferroptosis Through MFN1/2-Dependent Mitochondrial Fusion. Front Cell Dev. Biol. 2021, 9, 698679. [Google Scholar] [CrossRef]
- Zhao, J.; Guo, C.; Ma, Z.; Liu, H.; Yang, C.; Li, S. Identification of a novel gene expression signature associated with overall survival in patients with lung adenocarcinoma: A comprehensive analysis based on TCGA and GEO databases. Lung Cancer 2020, 149, 90–96. [Google Scholar] [CrossRef]
- Sun, S.; Guo, W.; Wang, Z.; Wang, X.; Zhang, G.; Zhang, H.; Li, R.; Gao, Y.; Qiu, B.; Tan, F.; et al. Development and validation of an immune-related prognostic signature in lung adenocarcinoma. Cancer Med. 2020, 9, 5960–5975. [Google Scholar] [CrossRef]
- Badgley, M.A.; Kremer, D.M.; Maurer, H.C.; DelGiorno, K.E.; Lee, H.J.; Purohit, V.; Sagalovskiy, I.R.; Ma, A.; Kapilian, J.; Firl, C.E.M.; et al. Cysteine depletion induces pancreatic tumor ferroptosis in mice. Science 2020, 368, 85–89. [Google Scholar] [CrossRef]
- Leu, J.I.; Murphy, M.E.; George, D.L. Mechanistic basis for impaired ferroptosis in cells expressing the African-centric S47 variant of p53. Proc. Natl. Acad. Sci. USA 2019, 116, 8390–8396. [Google Scholar] [CrossRef]
- Alvarez, S.W.; Sviderskiy, V.O.; Terzi, E.M.; Papagiannakopoulos, T.; Moreira, A.L.; Adams, S.; Sabatini, D.M.; Birsoy, K.; Possemato, R. NFS1 undergoes positive selection in lung tumours and protects cells from ferroptosis. Nature 2017, 551, 639–643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cramer, S.L.; Saha, A.; Liu, J.; Tadi, S.; Tiziani, S.; Yan, W.; Triplett, K.; Lamb, C.; Alters, S.E.; Rowlinson, S.; et al. Systemic depletion of L-cyst(e)ine with cyst(e)inase increases reactive oxygen species and suppresses tumor growth. Nat. Med. 2017, 23, 120–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Tan, H.; Daniels, J.D.; Zandkarimi, F.; Liu, H.; Brown, L.M.; Uchida, K.; O’Connor, O.A.; Stockwell, B.R. Imidazole Ketone Erastin Induces Ferroptosis and Slows Tumor Growth in a Mouse Lymphoma Model. Cell Chem. Biol. 2019, 26, 623–633.e629. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.P.; Torrente, L.; Falzone, A.; Elkins, C.M.; Liu, M.; Asara, J.M.; Dibble, C.C.; DeNicola, G.M. Cysteine dioxygenase 1 is a metabolic liability for non-small cell lung cancer. Elife 2019, 8, e45572. [Google Scholar] [CrossRef]
- Fiorillo, M.; Sotgia, F.; Sisci, D.; Cappello, A.R.; Lisanti, M.P. Mitochondrial “power” drives tamoxifen resistance: NQO1 and GCLC are new therapeutic targets in breast cancer. Oncotarget 2017, 8, 20309–20327. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.; Zhou, C.; Ma, Q.; Chen, W.; Atyah, M.; Yin, Y.; Fu, P.; Liu, S.; Hu, B.; Ren, N.; et al. High GCLC level in tumor tissues is associated with poor prognosis of hepatocellular carcinoma after curative resection. J. Cancer 2019, 10, 3333–3343. [Google Scholar] [CrossRef] [Green Version]
- Kim, A.D.; Zhang, R.; Han, X.; Kang, K.A.; Piao, M.J.; Maeng, Y.H.; Chang, W.Y.; Hyun, J.W. Involvement of glutathione and glutathione metabolizing enzymes in human colorectal cancer cell lines and tissues. Mol. Med. Rep. 2015, 12, 4314–4319. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.H.; Jang, J.H.; Kwon, O.S.; Cha, H.J.; Youn, H.J.; Chun, K.S.; Surh, Y.J. Nuclear Factor Erythroid-Derived 2-Like 2-Induced Reductive Stress Favors Self-Renewal of Breast Cancer Stem-Like Cells via the FoxO3a-Bmi-1 Axis. Antioxid Redox Signal 2020, 32, 1313–1329. [Google Scholar] [CrossRef]
- Leon, B.; Ballesteros-Tato, A. Modulating Th2 Cell Immunity for the Treatment of Asthma. Front Immunol. 2021, 12, 637948. [Google Scholar] [CrossRef]
- Anichini, A.; Perotti, V.E.; Sgambelluri, F.; Mortarini, R. Immune Escape Mechanisms in Non Small Cell Lung Cancer. Cancers 2020, 12. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Luo, L.; Zhang, Z.; Weng, Y.; Zeng, J. Ferroptosis-Related Gene GCLC Is a Novel Prognostic Molecular and Correlates with Immune Infiltrates in Lung Adenocarcinoma. Cells 2022, 11, 3371. https://doi.org/10.3390/cells11213371
Luo L, Zhang Z, Weng Y, Zeng J. Ferroptosis-Related Gene GCLC Is a Novel Prognostic Molecular and Correlates with Immune Infiltrates in Lung Adenocarcinoma. Cells. 2022; 11(21):3371. https://doi.org/10.3390/cells11213371
Chicago/Turabian StyleLuo, Lianxiang, Zhentao Zhang, Yanmin Weng, and Jiayan Zeng. 2022. "Ferroptosis-Related Gene GCLC Is a Novel Prognostic Molecular and Correlates with Immune Infiltrates in Lung Adenocarcinoma" Cells 11, no. 21: 3371. https://doi.org/10.3390/cells11213371
APA StyleLuo, L., Zhang, Z., Weng, Y., & Zeng, J. (2022). Ferroptosis-Related Gene GCLC Is a Novel Prognostic Molecular and Correlates with Immune Infiltrates in Lung Adenocarcinoma. Cells, 11(21), 3371. https://doi.org/10.3390/cells11213371