


Aberrant DNA and RNA Methylation Occur in Spinal Cord and Skeletal Muscle of Human SOD1 Mouse Models of ALS and in Human ALS: Targeting DNA Methylation Is Therapeutic


Abstract
:1. Introduction
2. Materials and Methods
2.1. Transgenic (tg) Mice
2.2. Human CNS Tissues
2.3. DNMT Enzyme Activity
2.4. 5mC and 6mA Assays
2.5. Genome-Wide CpG DNA Methylation Profiling in Skeletal Muscle of ALS Mice by Methylated DNA-Immunoprecipitation (MeDIP)-Chip Analysis
2.6. Immunohistochemistry (IHC) and Immunofluorescence (IF)
2.7. Cell Counting
2.8. Electron Microscopy (EM)
2.9. Immunoblotting and IP
2.10. RG108 Therapeutic Testing in Mouse ALS
2.11. Data Analysis
3. Results
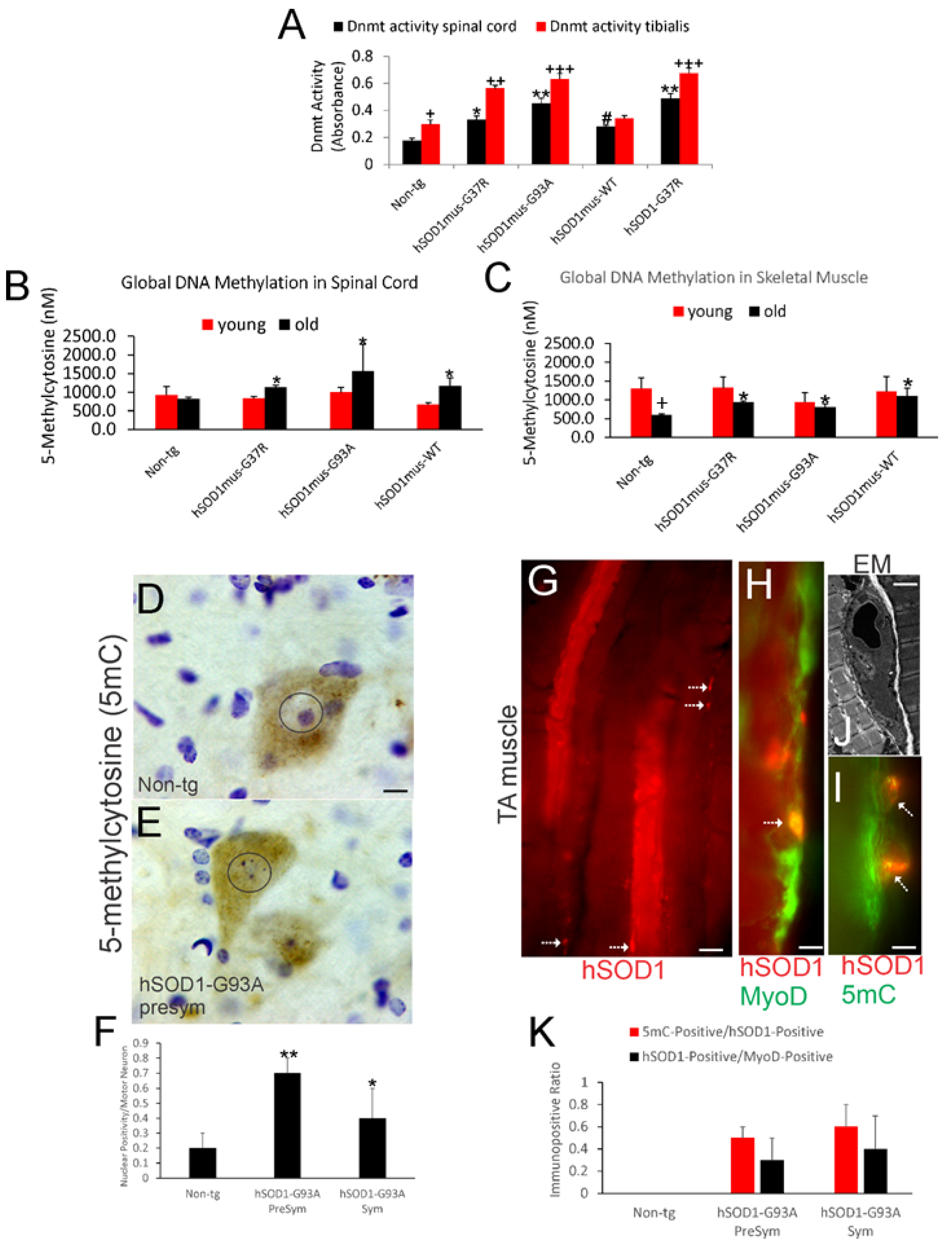
3.1. DNMT Enzyme Activity Is Increased in Spinal Cord and Skeletal Muscle of hSOD1 tg Mice
3.2. Global DNA Methylation Is Increased in Spinal Cord and Skeletal Muscle of hSOD1 tg Mice
3.3. 5mC Is Localized to Spinal Cord MNs and Skeletal Muscle Satellite Cells
3.4. Gene Promoter CpG Hypermethylation Occurs in Skeletal Muscle of hSOD1 tg Mice
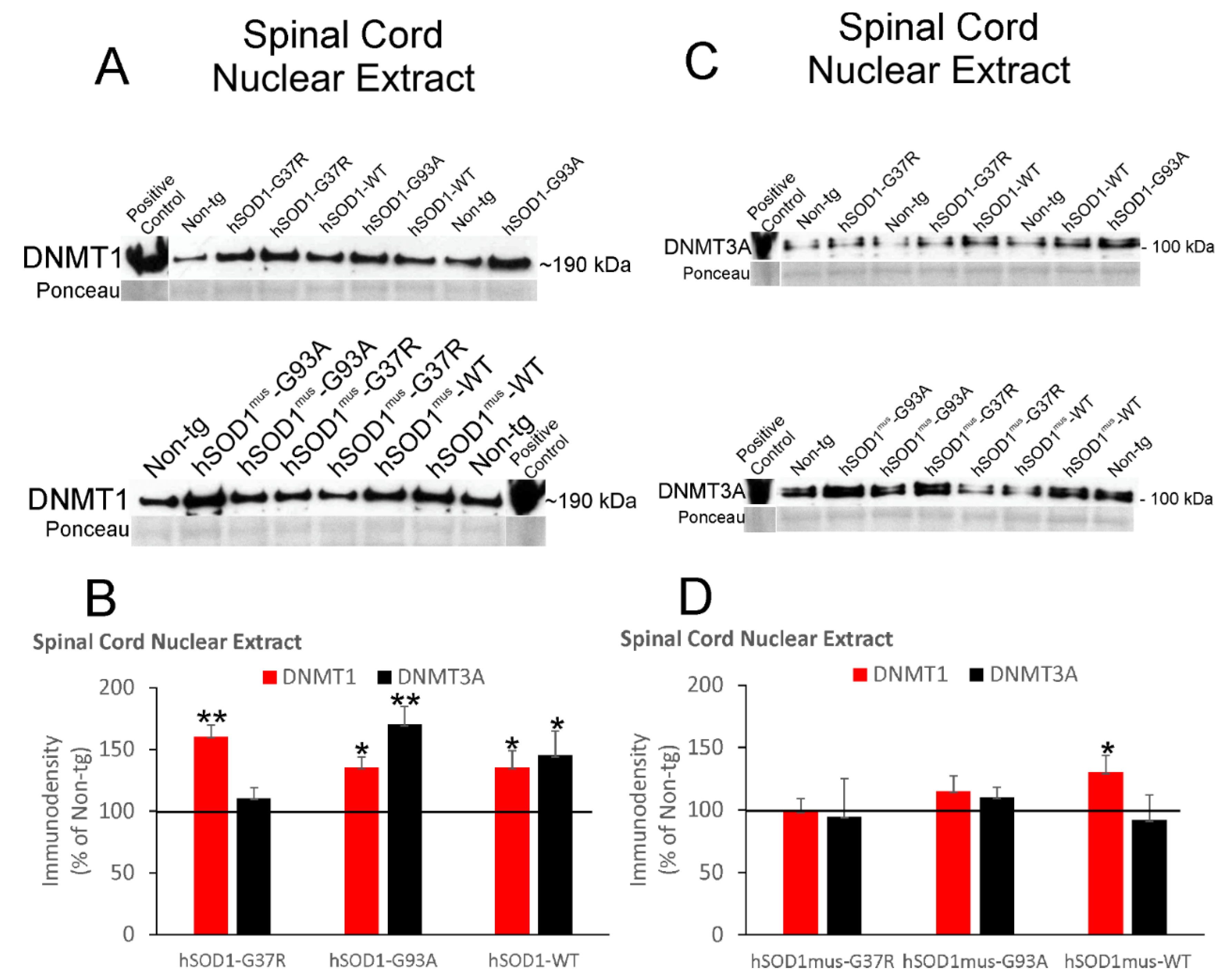
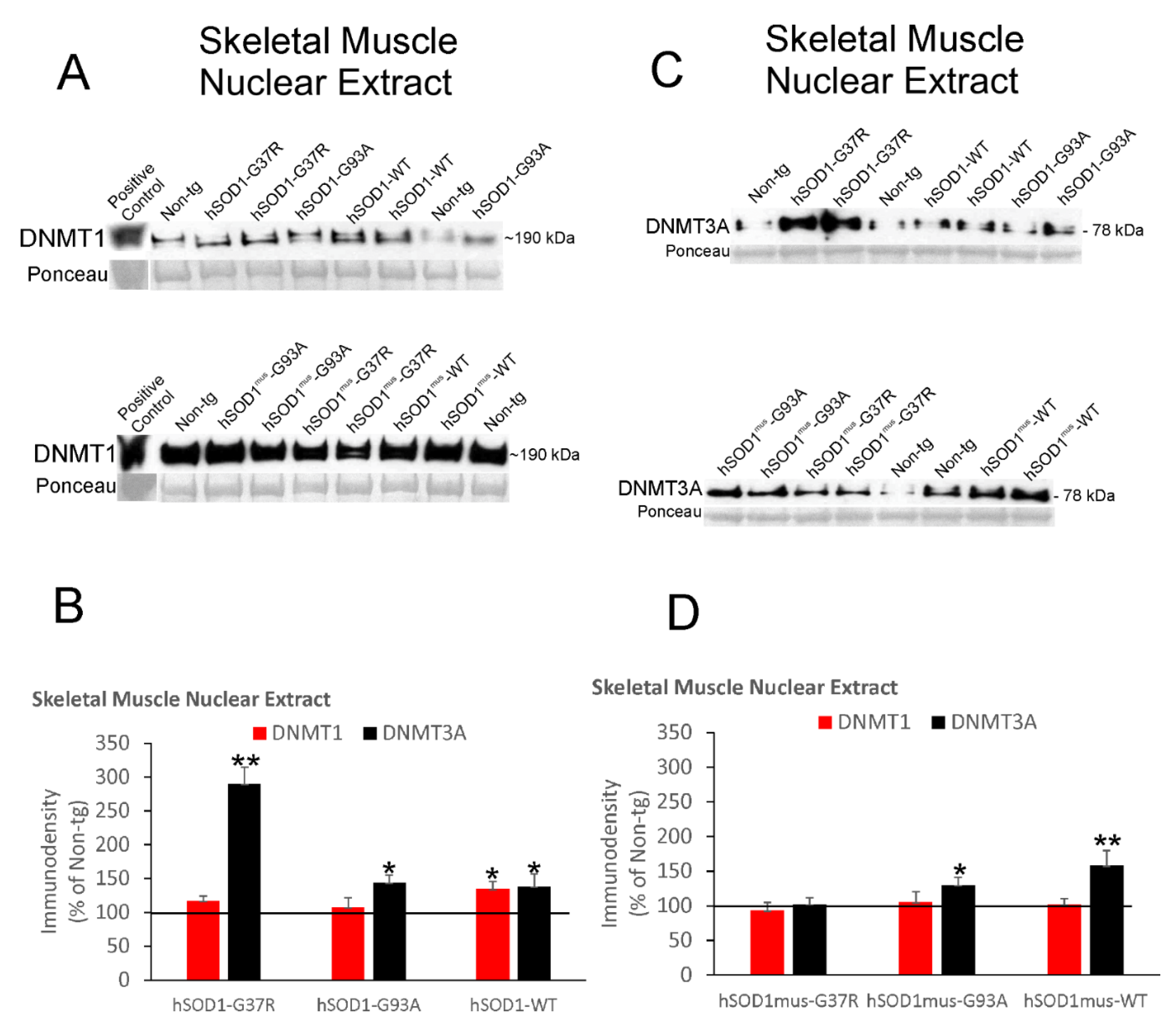
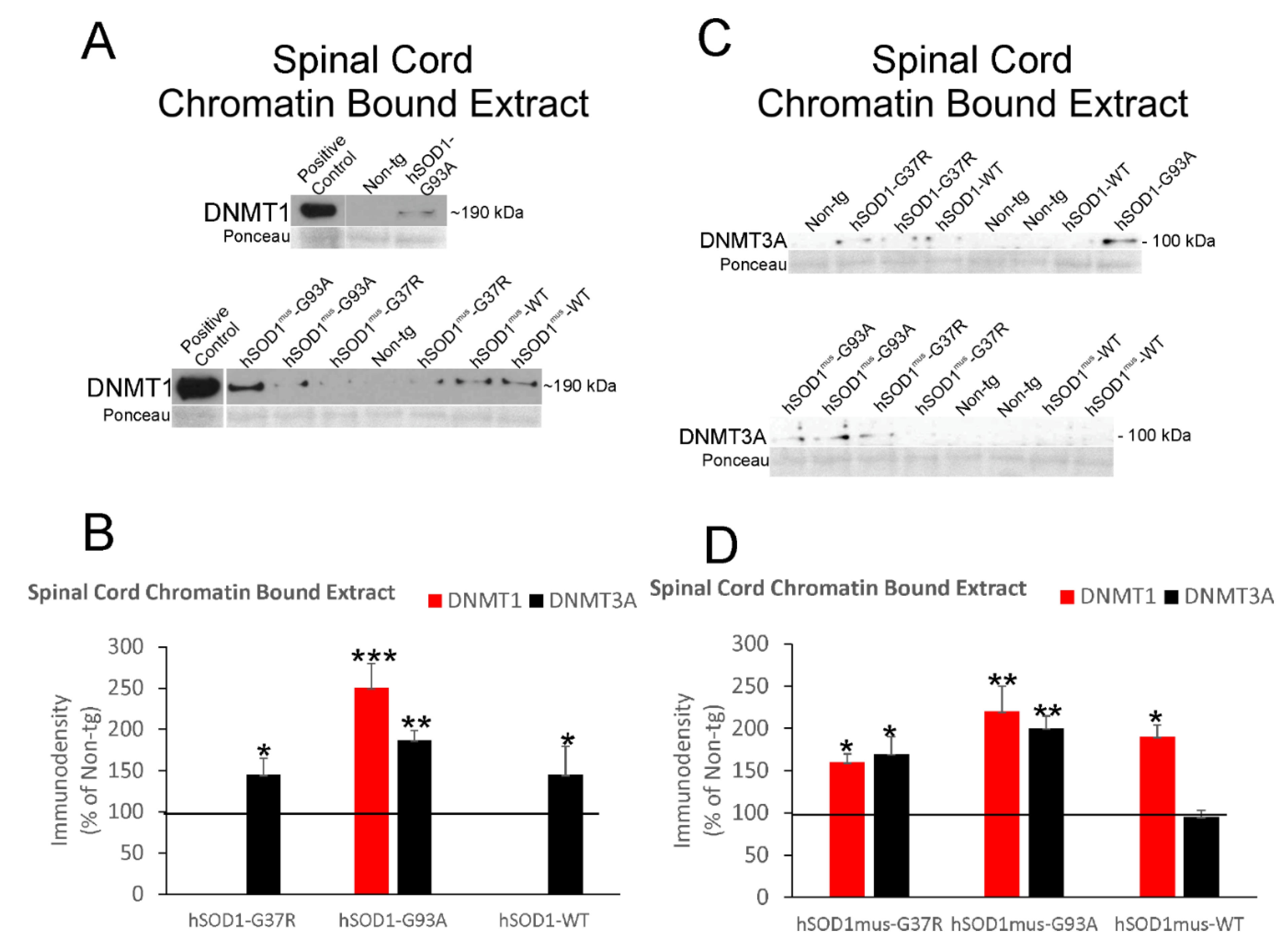
3.5. DNMT Levels Increase in Spinal Cord and Skeletal Muscle Nuclear Fractions of hSOD1 tg Mice
3.6. DNMT Levels Increase in Chromatin Bound Fractions of hSOD1 tg Mouse Spinal Cord and Skeletal Muscle
3.7. hSOD1 Interacts with DNMT3A in Skeletal Muscle of tg Mice
3.8. DNA Methylation Is Increased in Human ALS CNS
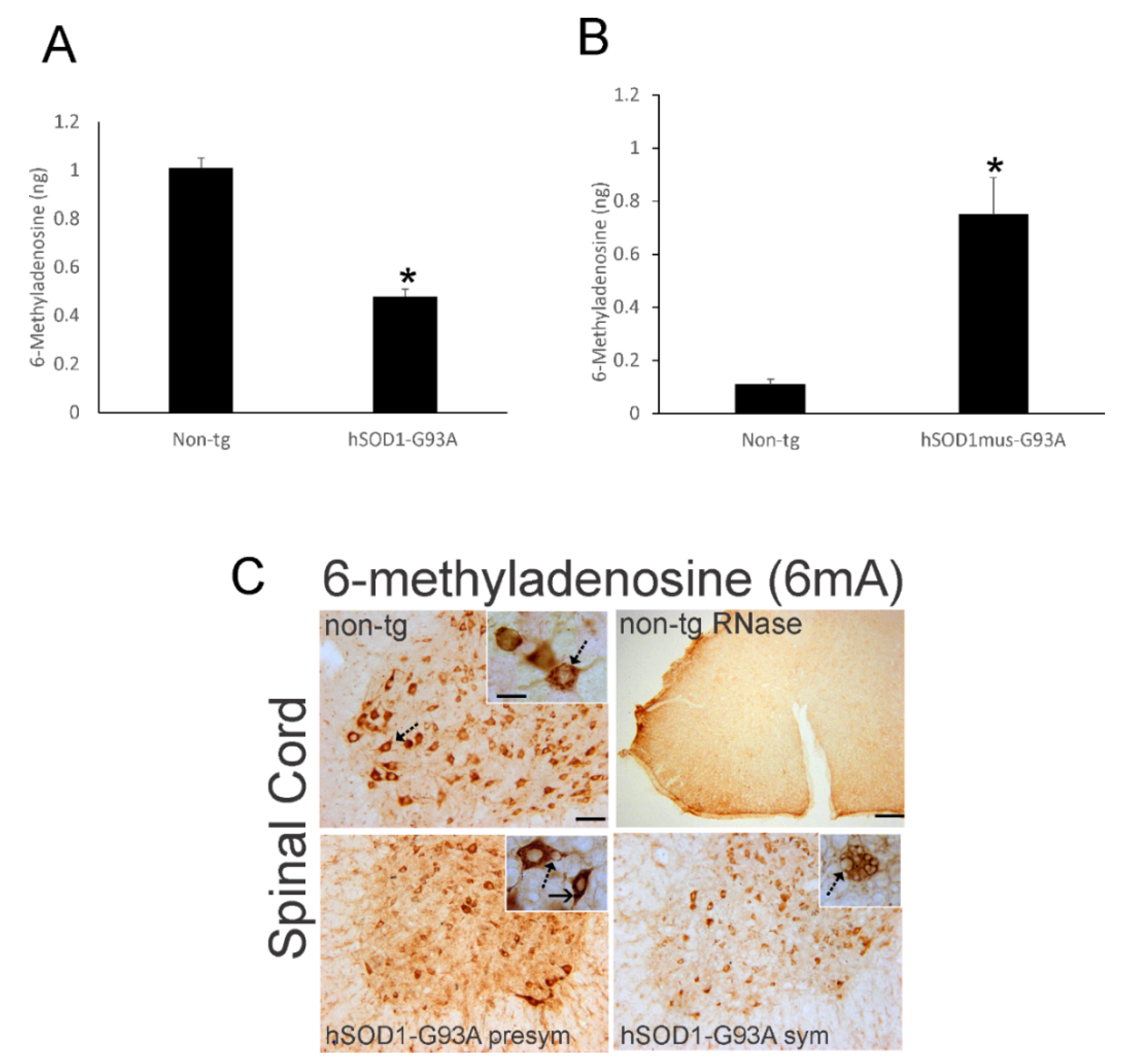
3.9. RNA Methylation Is Aberrant in ALS Mouse Spinal Cord and Skeletal Muscle
3.10. RNA Methylation Is Depleted Severely in Human ALS CNS
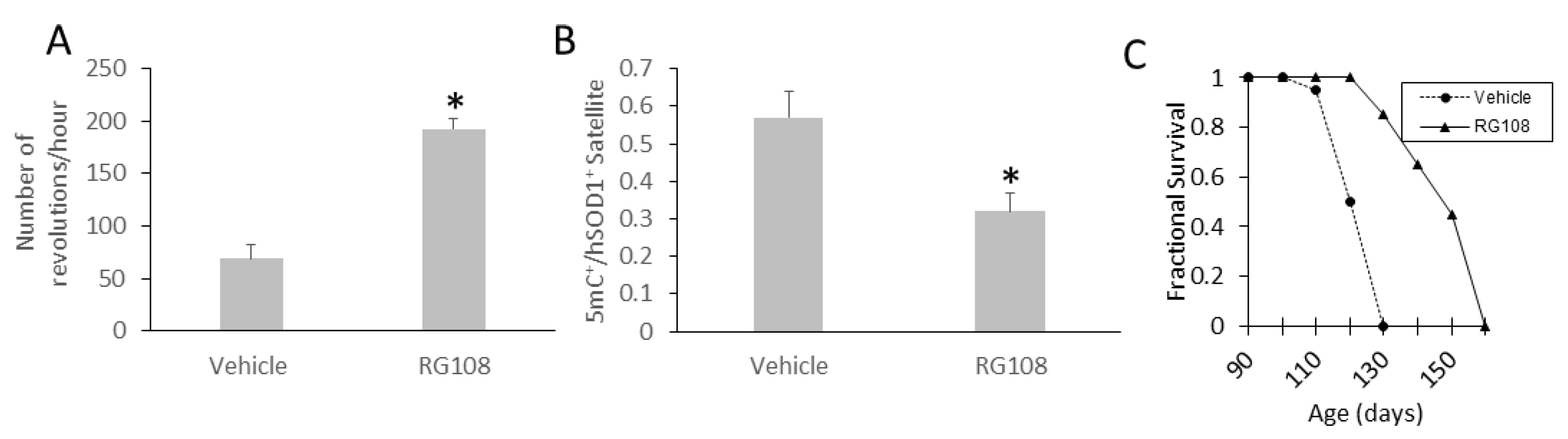
3.11. RG108 Delays Disease Onset and Extends the Lifespan of hSOD1-G93A ALS Mice
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rowland, L.P.; Shneider, N.A. Amyotrophic lateral sclerosis. N. Engl. J. Med. 2001, 344, 1688–1700. [Google Scholar] [CrossRef] [PubMed]
- Petrov, D.; Mansfield, C.; Moussy, A.; Hermine, O. ALS clinical trials review: 20 years of failure. Are we any closer to registering a new treatment? Front. Aging Neurosci. 2017, 9, 68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heath, P.R.; Shaw, P.J. Update on the glutamatergic neurotransmitter system and the role of excitotoxicity in amyotrophic lateral sclerosis. Muscle Nerve 2002, 26, 438–458. [Google Scholar] [CrossRef] [PubMed]
- Alonso, A.; Logroscino, G.; Jick, S.S.; Hernán, M.A. Incidence and lifetime risk of motor neuron disease in the United Kingdom: A population-based study. Eur. J. Neurol. 2009, 16, 745–751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCombe, P.A.; Henderson, R.D. Effects of gender in amyotrophic lateral sclerosis. Gender Med. 2010, 7, 57–570. [Google Scholar] [CrossRef]
- Finsterer, J.; Burgunder, J.M. Recent progress in the genetics of motor neuron disease. Eur. J. Med. Genet. 2014, 57, 103–112. [Google Scholar] [CrossRef]
- Martin, L.J. Mitochondrial and cell death mechanisms in neurodegenerative disease. Pharmaceuticals 2010, 3, 839–915. [Google Scholar] [CrossRef] [Green Version]
- Tarr, I.S.; McCann, E.P.; Benyamin, B.; Peters, T.J.; Twine, N.A.; Zhang, K.Y.; Zhao, Q.; Zhang, Z.H.; Rowe, D.B.; Nicholson, G.A.; et al. Monozygotic twins and triplets discordant for amyotrophic lateral sclerosis display differential methylation and gene expression. Sci. Rep. 2019, 9, 8254. [Google Scholar] [CrossRef] [Green Version]
- Wolffe, A.P.; Matzke, M.A. Epigenetics: Regulation through repression. Science 1999, 286, 481–486. [Google Scholar] [CrossRef]
- Jaenisch, R.; Bird, A. Epigenetic regulation of gene expression: How the genome integrates intrinsic and environmental signals. Nat. Genet. 2003, 33, 245–254. [Google Scholar] [CrossRef]
- Brenner, C.; Fuks, F. DNA methyltransferases: Facts, clues, mysteries. Curr. Top. Microbiol. Immunol. 2006, 301, 45–66. [Google Scholar] [PubMed]
- Bird, A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002, 16, 6–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Curradi, M.; Izzo, A.; Badaracco, G.; Landsberger, N. Molecular mechanisms of gene silencing mediated by DNA methylation. Mol. Cell. Biol. 2002, 22, 3157–3173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, X.; Yan, L.; Davidson, N. DNA methylation in breast cancer. Endocr. Relat. Cancer 2001, 8, 115–127. [Google Scholar] [CrossRef]
- Luczak, M.W.; Jagodzinski, P.P. The role of DNA methylation in cancer development. Folia Histochem. Cytobiol. 2006, 44, 43–154. [Google Scholar]
- Levenson, J.M.; Roth, T.L.; Lubin, F.D.; Miller, C.A.; Huang, I.C.; Desai, P.; Malone, L.M.; Sweatt, J.D. Evidence that DNA (cytosine-5) methyltransferase regulates synaptic plasticity in the hippocampus. J. Biol. Chem. 2006, 281, 5763–15773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, L.J.; Wong, M. Aberrant regulation of DNA methylation in amyotrophic lateral sclerosis: A new target of disease mechanisms. Neurotherapeutics 2013, 10, 722–733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, B.W.; Jeong, Y.E.; Wong, M.; Martin, L.J. DNA damage accumulates and responses are engaged in human ALS brain and spinal motor neurons and DNA repair is activatable in iPSC-derived motor neurons with SOD1 mutations. Acta Neuropathol. Commun. 2020, 8, 1–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, X. Structure and function of DNA methyltransferases. Annu. Rev. Biophys. Biomol. Struct. 1995, 24, 293–318. [Google Scholar] [CrossRef]
- Pradhan, S.; Bacolla, A.; Wells, R.D.; Roberts, R.J. Recombinant human DNA (cytosine-5) methyltransferase. I. expression, purification, and comparison of de novo and maintenance methylation. J. Biol. Chem. 1999, 274, 33002–33010. [Google Scholar] [CrossRef] [Green Version]
- Bestor, T.H. The DNA methyltransferases of mammals. Hum. Mol. Genet. 2000, 9, 2395–2402. [Google Scholar] [CrossRef] [PubMed]
- Szyf, M. DNA methylation and demethylation as targets for anticancer therapy. Biochemistry 2005, 70, 533–549. [Google Scholar] [CrossRef] [PubMed]
- Robertson, K.D. DNA methylation, methyltransferases, and cancer. Oncogene 2002, 20, 3139–3155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mortusewicz, O.; Schermelleh, L.; Walter, J.; Cardoso, M.C.; Leonhardt, H. Recruitment of DNA methyltransferase I to DNA repair sites. Proc. Natl. Acad. Sci. USA 2005, 102, 8905–8909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klein, C.J.; Botuyan, M.-V.; Wu, Y.; Ward, C.J.; Nicholson, G.A.; Hammans, S.; Hojo, K.; Yamanishi, H.; Karpf, A.R.; Wallace, D.C.; et al. Mutations in DNMT1 cause hereditary sensory neuropathy with dementia and hearing loss. Nat. Genet. 2011, 43, 595–600. [Google Scholar] [CrossRef] [PubMed]
- Winkelman, J.; Lin, L.; Schormair, B.; Kornum, B.R.; Faraco, J.; Plazzi, G.; Melberg, A.; Cornelio, F.; Urban, A.E.; Pizza, F.; et al. Mutations in DNMT1 cause autosomal dominant cerebellar ataxia, deafness and narcolepsy. Hum. Mol. Genet. 2012, 21, 2205–2210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okano, M.; Bell, D.W.; Haber, D.A.; Li, E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 1999, 99, 247–257. [Google Scholar] [CrossRef] [Green Version]
- Xie, S.; Wang, Z.; Okano, M.; Nogami, M.; Li, Y.; He, W.W.; Okumura, K.; Li, E. Cloning, expression and chromosome locations of the human DNMT3 gene family. Gene 1999, 236, 87–95. [Google Scholar] [CrossRef]
- Chestnut, B.A.; Chang, Q.; Price, A.; Lesuisse, C.; Wong, M.; Martin, L.J. Epigenetic regulation of motor neuron cell death through DNA methylation. J. Neurosci. 2011, 31, 16619–16636. [Google Scholar] [CrossRef] [Green Version]
- Wong, M.; Gertz, B.; Chestnut, B.A.; Martin, L.J. Mitochondrial DNMT3A and DNA methylation in skeletal muscle and CNS of transgenic mouse models of ALS. Front. Cell. Neurosci. 2013, 7, 279. [Google Scholar] [CrossRef] [Green Version]
- Tatton-Brown, K.; Seal, S.; Ruark, E.; Harmer, J.; Ramsay, E.; Del Vecchio Duarte, S.; Zachariou, A.; Hanks, S.; O’Brien, E.; Aksglaede, L.; et al. Mutations in the DNA methyltransferase gene DNMT3A cause an overgrowth syndrome with intellectual disability. Nat. Genet. 2014, 46, 385–388. [Google Scholar] [CrossRef] [PubMed]
- Shock, L.S.; Thakkar, P.V.; Peterson, E.J.; Moran, R.G.; Taylor, S.M. DNA methyltransferase 1, cytosine methylation, and cytosine hydroxymethylation in mammalian mitochondria. Proc. Natl. Acad. Sci. USA 2011, 108, 3630–3635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, C. RNA epigenetics? Nat. Chem. Biol. 2010, 6, 863–865. [Google Scholar] [CrossRef] [PubMed]
- Darnell, R.B.; Ke, S.; Darnell, J.E., Jr. Pre-mRNA processing includes N6 methylation of adenosine residues that are retained in mRNA exons and the fallacy of “RNA epigenetics”. RNA 2018, 24, 262–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaefer, M.; Lyko., F. Solving the Dnmt2 enigma. Chromosoma 2010, 119, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Zhong, H.; Tang, H.F.; Kai, Y. N6-methyladenine RNA modification (m6A): An emerging regulator of metabolic diseases. Curr. Drug Targets 2020, 21, 1056–1067. [Google Scholar] [CrossRef]
- Huang, H.; Weng, H.; Chen, J. The biogenesis and precise control of RNA m6A methylation. Trends Genet. 2020, 36, 44–52. [Google Scholar] [CrossRef]
- Zhang, N.; Ding, C.; Zuo, Y.; Peng, Y.; Zuo, L. N6-methyladenosine and neurological diseases. Mol. Neurobiol. 2022, 59, 1925–1937. [Google Scholar] [CrossRef]
- Schaefer, M.; Pollex, T.; Hanna, K.; Tuorto, F.; Meusburger, M.; Helm, M.; Lykom, F. RNA methylation by Dnmt2 protects transfer RNAs against stress-induced cleavage. Genes Dev. 2010, 24, 1590–1595. [Google Scholar] [CrossRef] [Green Version]
- Tuorto, F.; Liebers, R.; Musch, T.; Schaefer, M.; Hofmann, S.; Kellner, S.; Frye, M.; Helm, M.; Stoecklin, G.; Lyko, F. RNA cytosine methylation by Dnmt2 and NSun2 promotes tRNA stability and protein synthesis. Nat. Struct. Mol. Biol. 2012, 19, 900–905. [Google Scholar] [CrossRef]
- Ke, S.; Pandya-Jones, A.; Saito, Y.; Fak, J.J.; Vågbø, C.B.; Geula, S.; Hanna, J.H.; Black, D.L.; Darnell, J.E., Jr.; Darnell, R.B. m6A mRNA modifications are deposited in nascent pre-mRNA and are not required for splicing but do specify cytoplasmic turnover. Genes Dev. 2017, 31, 990–1006. [Google Scholar] [CrossRef] [PubMed]
- Hop, P.J.; Zwamborn, R.A.J.; Hannon, E.; Shireby, G.L.; Nabais, M.F.; Walker, E.M.; van Rheenen, W.; van Vugt, J.J.F.A.; Dekker, A.M.; Westeneng, H.J.; et al. Genome-wide study of DNA methylation shows alterations in metabolic, inflammatory, and cholesterol pathways in ALS. Sci. Transl. Med. 2022, 14, eabj0264. [Google Scholar] [CrossRef] [PubMed]
- Dobrowolny, G.; Aucello, M.; Rizzuto, E.; Beccaficao, S.; Mammucari, C.; Bonconpagni, S.; Belia, S.; Wannenes, F.; Nicoletti, C.; Del Prete, Z.; et al. Skeletal muscle is a primary target of SOD1G93A-mediated toxicity. Cell Metab. 2008, 8, 425–436. [Google Scholar] [CrossRef] [PubMed]
- Wong, M.; Martin, L.J. Skeletal muscle-restricted expression of human SOD1 causes motor neuron degeneration in transgenic mice. Hum. Mol Genet. 2010, 19, 2284–2302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, L.J.; Wong, M. Skeletal muscle-restricted expression of human SOD1 in transgenic mice causes a fatal ALS-like syndrome. Front. Neurol. 2020, 11, 592851. [Google Scholar] [CrossRef]
- Pikatza-Menoio, O.; Elicegui, A.; Bengoetxea, X.; Naldaiz-Gastesi, N.; López de Munain, A.; Gerenu, G.; Gil-Bea, F.J.; Alonso-Martín, S. The skeletal muscle emerges as a new disease target in amyotrophic lateral sclerosis. J. Pers. Med. 2021, 11, 671. [Google Scholar] [CrossRef]
- Steyn, F.J.; Ioannides, Z.A.; van Eijk, R.P.A.; Heggie, S.; Thorpe, K.A.; Ceslis, A.; Heshmat, S.; Henders, A.K.; Wray, N.R.; van den Berg, L.H.; et al. Hypermetabolism in ALS is associated with greater functional decline and shorter survival. J. Neurol. Neurosurg. Psychiatry 2018, 89, 1016–1023. [Google Scholar] [CrossRef] [Green Version]
- Dupuis, L.; Oudart, H.; René, F.; Gonzalez de Aguilar, J.L.; Loeffler, J.P. Evidence for defective energy homeostasis in amyotrophic lateral sclerosis: Benefit of a high-energy diet in a transgenic mouse model. Proc. Natl. Acad. Sci. USA 2004, 101, 11159–11164. [Google Scholar] [CrossRef] [Green Version]
- Dupuis, L.; Petersen, A.; Wedt, P. Thermoregulation in amyotrophic lateral sclerosis. In Thermoregulation: From Basic Neuroscience to Clinical Neurology; Part II; Romanovsky, A.A., Ed.; Elsevier: Amsterdam, The Netherlands, 2018; Volume 157, pp. 749–760. [Google Scholar]
- Mann, D.M.A.; Yates, P.O. Motor neurone disease: The nature of the pathogenic mechanism. J. Neurol. Neurosurg. Psychiatry 1974, 37, 1036–1046. [Google Scholar] [CrossRef] [Green Version]
- Hartmann, H.A.; McMahon, S.; Sun, D.Y.; Abbs, J.H.; Uemura, E. Neuronal RNA in nucleus ambiguus and nucleus hypoglossus of patients with amyotrophic lateral sclerosis. J. Neuropath. Exp. Neurol. 1989, 48, 669–673. [Google Scholar] [CrossRef]
- Davidson, T.J.; Hartmann, H.A.; Johnson, P.C. RNA content and volume of motor neurons in amyotrophic lateral sclerosis. I. The cervical swelling. J. Neuropathol. Exp. Neurol. 1981, 40, 32–36. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, H.A.; Sun, D.Y. Regional mRNA changes in brain stem motor neurons from patients with amyotrophic lateral sclerosis. Mol. Chem. Neuropath. 1992, 17, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Gurney, M.E.; Pu, H.; Chiu, A.Y.; Dal Canto, M.C.; Polchow, C.Y.; Alexander, D.D.; Caliendo, J.; Hentati, A.; Kwon, Y.W.; Deng, H.X.; et al. Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science 1994, 264, 1772–1775. [Google Scholar] [CrossRef] [PubMed]
- Martin, L.J.; Liu, Z.; Chen, K.; Price, A.C.; Pan, Y.; Swaby, J.A.; Golden, W.C. Motor neuron degeneration in ALS mutant superoxide dismutase-1 transgenic mice: Mechanisms of mitochondriopathy and cell death. J. Comp. Neurol. 2007, 500, 20–46. [Google Scholar] [CrossRef] [PubMed]
- Martin, L.J.; Gertz, B.; Pan, Y.; Price, A.C.; Molkentin, J.D.; Chang, Q. The mitochondrial permeability transition pore in motor neurons: Involvement in the pathobiology of ALS mice. Exp. Neurol. 2009, 218, 333–346. [Google Scholar] [CrossRef] [Green Version]
- Martin, L.J.; Fancelli, D.; Wong, M.; Niedzwiecki, M.; Ballarini, M.; Plyte, S.; Chang, Q. GNX-4728, a novel small molecule drug inhibitor of mitochondrial permeability transition, is therapeutic in a mouse model of amyotrophic lateral sclerosis. Front. Cell. Neurosci. 2014, 8, 433. [Google Scholar] [CrossRef] [Green Version]
- Gertz, B.; Wong, M.; Martin, L.J. Nuclear localization of human SOD1 and mutant SOD1-specific disruption of survival motor neuron protein complex in transgenic amyotrophic lateral sclerosis mice. J. Neuropathol. Exp. Neurol. 2012, 71, 162–177. [Google Scholar] [CrossRef] [Green Version]
- Chen, K.; Northington, F.J.; Martin, L.J. Inducible nitric oxide synthase is present in motor neuron mitochondria and Schwann cells and contributes to disease mechanisms in ALS mice. Brain Struct. Funct. 2010, 214, 219–234. [Google Scholar] [CrossRef] [Green Version]
- Clerc, P.; Lipnick, S.; Willett, C. A look into the future if ALS research. Drug Discov. Today 2016, 21, 939949. [Google Scholar] [CrossRef]
- Jaarsma, D.; Haasdijk, E.D.; Grashorn, J.A.; Hawkins, R.; van Duijn, W.; Verspaget, H.W.; London, J.; Holstege, J.C. Human Cu/Zn superoxide dismutase (SOD1) overexpression in mice causes mitochondrial vacuolization, axonal degeneration, and premature motoneuron death and accelerates motoneuron disease in mice expressing a familial amyotrophic lateral sclerosis mutant SOD1. Neurobiol. Dis. 2000, 7, 623–643. [Google Scholar] [CrossRef]
- Martin, L.J. Neuronal death in amyotrophic lateral sclerosis is apoptosis: Possible contribution of a programmed cell death mechanism. J. Neuropathol. Exp. Neurol. 1999, 58, 459–471. [Google Scholar] [CrossRef] [PubMed]
- Martin, L.J. p53 is abnormally elevated and active in the CNS of patients with amyotrophic lateral sclerosis. Neurobiol. Dis. 2000, 7, 613–622. [Google Scholar] [CrossRef] [PubMed]
- Shaikh, A.Y.; Martin, L.J. DNA base-excision repair enzyme apurinic/apyrimidinic endonuclease/redox factor-1 is increased and competent in the brain and spinal cord of individuals with amyotrophic lateral sclerosis. Neuromolecular. Med. 2002, 2, 47–60. [Google Scholar] [CrossRef] [PubMed]
- Fayzullina, S.; Martin, L.J. DNA damage response and DNA repair in skeletal myocytes from a mouse model of spinal muscular atrophy. J. Neuropathol. Exp. Neurol. 2016, 75, 889–902. [Google Scholar] [CrossRef] [Green Version]
- Martin, L.J.; Blackstone, C.D.; Huganir, R.L.; Price, D.L. Cellular localization of a metabotropic glutamate receptor in rat brain. Neuron 1992, 9, 259–270. [Google Scholar] [CrossRef]
- Furuta, A.; Martin, L.J. Laminar segregation of the cortical plate during corticogenesis is accompanied by changes in glutamate receptor expression. J. Neurobiol. 1999, 39, 67–80. [Google Scholar] [CrossRef]
- Furuta, A.; Price, D.L.; Pardo, C.A.; Troncoso, J.C.; Xu, Z.S.; Taniguchi, N.; Martin, L.J. Localization of superoxide dismutases in Alzheimer’s disease and Down’s syndrome neocortex and hippocampus. Am. J. Pathol. 1995, 146, 357–367. [Google Scholar]
- Martin, L.J.; Pardo, C.A.; Cork, L.C.; Price, D.L. Synaptic pathology and glial responses to neuronal injury precede the formation of senile plaques and amyloid deposits in the aging cerebral cortex. Am. J. Pathol. 1994, 145, 1358–1381. [Google Scholar]
- Martin, L.J.; Price, A.C.; McClendon, K.B.; Al-Abdulla, N.A.; Subramaniam, J.R.; Wong, P.C.; Liu, Z. Early events of target deprivation/axotomy-induced neuronal apoptosis in vivo: Oxidative stress, DNA damage, p53 phosphorylation and subcellular redistribution of death proteins. J. Neurochem. 2003, 85, 234–247. [Google Scholar] [CrossRef]
- Siedlecki, P.; Garcia Boy, R.; Much, T.; Brueckner, B.; Suhai, S.; Lyko, F.; Zielenkiewicz, P. Discovery of two novel, small-molecule inhibitors of DNA methylation. J. Med. Chem. 2006, 49, 678–683. [Google Scholar] [CrossRef]
- Suzuki, T.; Tanaka, R.; Hamada, S.; Nakagawa, H.; Miyata, N. Design, synthesis, inhibitory activity, and binding mode study of novel DNA methyltransferase 1 inhibitors. Bioorg. Med. Chem. Lett. 2010, 20, 1124–1127. [Google Scholar] [CrossRef] [PubMed]
- Weber, M.; Hellmann, I.; Stadler, M.; Ramos, L.; Paabo, S.; Rebhan, M.; Schubeler, D. Distribution, silencing potential and evolutionary impact of promoter DNA methylation in the human genome. Nat. Genet. 2007, 39, 457–466. [Google Scholar] [CrossRef] [PubMed]
- Chang, Q.; Martin, L.J. Glycinergic innervation of motoneurons is deficient in amyotrophic lateral sclerosis mice: A quantitative confocal analysis. Am. J. Pathol. 2009, 174, 574–585. [Google Scholar] [CrossRef] [PubMed]
- McTaggart, J.S.; Lee, S.; Iberl, M.; Church, C.; Cox, R.D.; Ashcroft, F.M. FTO is expressed in neurones throughout the brain and its expression is unaltered by fasting. PLoS ONE 2011, 6, e27968. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Hu, J.; Sun, X.; Yang, K.; Yang, L.; Kong, L.; Zhang, B.; Li, F.; Li, C.; Shi, B.; et al. Loss of m6A demethylase ALKBH5 promotes post-ischemic angiogenesis via post-transcriptional stabilization of WNT5A. Clin. Transl. Med. 2021, 11, e402. [Google Scholar] [CrossRef]
- Du, Y.; Hou, G.; Zhang, H.; Dou, J.; He, J.; Guo, Y.; Li, L.; Chen, R.; Wang, Y.; Deng, R.; et al. SUMOylation of the m6A-RNA methyltransferase METTL3 modulates its function. Nucleic Acids Res. 2018, 46, 5195–5208. [Google Scholar] [CrossRef]
- Liu, Y.; Zhou, T.; Wang, Q.; Fu, R.; Zhang, Z.; Chen, N.; Li, Z.; Gao, G.; Peng, S.; Yang, D. m6A demethylase ALKBH5 drives denervation-induced muscle atrophy by targeting HDAC4 to activate FoxO3 signalling. J. Cachexia Sarcopenia Muscle 2022, 13, 1210–1223. [Google Scholar] [CrossRef]
- Horvath, S. DNA methylation age of human tissues and cell types. Genome Biol. 2013, 14, 3156. [Google Scholar] [CrossRef] [Green Version]
- Horvath, S.; Raj, K. DNA methylation-based biomarkers and the epigenetic clock theory of ageing. Nat. Rev. Genet. 2018, 19, 371–384. [Google Scholar] [CrossRef]
- Levine, M.E.; Lu, A.T.; Quach, A.; Chen, B.H.; Assimes, T.L.; Bandinelli, S.; Hou, L.; Baccarelli, A.A.; Stewart, J.D.; Li, Y.; et al. An epigenetic biomarker of aging for lifespan and healthspan. Aging 2018, 1810, 573–591. [Google Scholar] [CrossRef] [Green Version]
- Govender, P.; Ghai, M.; Okpeku, M. Sex-specific DNA methylation: Impact on human health and development. Mol. Genet. Genom. 2022, 297, 1451–1466. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Tartaglia, M.C.; Moreno, D.; Sato, C.; McKeever, P.; Weichert, A.; Keith, J.; Robertson, J.; Zinman, L.; Rogaeva, E. DNA methylation age-acceleration is associated with disease duration and age at onset in C9orf72 patients. Acta Neuropathol. 2017, 134, 271–279. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; McKeever, P.M.; Xi, Z.; Moreno, D.; Sato, C.; Bergsma, T.; McGoldrick, P.; Keith, J.; Robertson, J.; Zinman, L.; et al. DNA methylation age acceleration is associated with ALS age of onset and survival. Acta Neuropathol. 2020, 139, 943–946. [Google Scholar] [CrossRef] [PubMed]
- Migliore, L.; Coppedè, F. Genetics, environmental factors and the emerging role of epigenetics in neurodegenerative diseases. Mutat. Res. 2009, 667, 82–97. [Google Scholar] [CrossRef] [PubMed]
- Freydenzon, A.; Nabais, M.F.; Lin, T.; Williams, K.L.; Wallace, L.; Henders, A.K.; Blair, I.P.; Wray, N.R.; Pamphlett, R.; McRae, A.E. Association between DNA methylation variability and self-reported exposure to heavy metals. Sci. Rep. 2022, 12, 10582. [Google Scholar] [CrossRef] [PubMed]
- Chiò, A.; Benzi, G.; Dossena, M.; Mutani, R.; Mora, G. Severely increased risk of amyotrophic lateral sclerosis among Italian professional football players. Brain 2005, 128, 472–476. [Google Scholar] [CrossRef] [Green Version]
- Pupillo, E.; Bianchi, E.; Vanacore, N.; Montalto, C.; Ricca, G.; Della Cuna, R.S.; Fumagalli, F.; Castellani, M.; Poli, F.; Romeo, F.; et al. Increased risk and early onset of ALS in professional players from Italian soccer teams. Amyotrop. Lateral Scler. Frontotemporal Degener. 2020, 21, 403–409. [Google Scholar] [CrossRef] [PubMed]
- Dobrowolny, G.; Barbiera, A.; Sica, G.; Scicchitano, B.M. Age-related alterations at neuromuscular junction: Role of oxidative stress and epigenetic modifications. Cells 2021, 10, 1307. [Google Scholar] [CrossRef]
- Oates, N.; Pamphlett, R. An epigenetic analysis of SOD1 and VEGF in ALS. Amyotroph Lateral Scler. 2007, 8, 3–86. [Google Scholar] [CrossRef]
- Morahan, J.M.; Yu, B.; Trent, R.J.; Pamphlett, R. Are metallothionein genes silenced in ALS? Toxicol. Lett. 2007, 168, 83–87. [Google Scholar] [CrossRef]
- Yang, Y.; Gozen, O.; Vidensky, S.; Robinson, M.B.; Rothstein, J.D. Epigenetic regulation of neuron-dependent induction of astroglial synaptic protein GLT1. Glia 2010, 58, 277–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morahan, J.M.; Yu, B.; Trent, R.J.; Pamphlett, R. A genome-wide analysis of brain DNA methylation identifies new candidate genes for sporadic amyotrophic lateral sclerosis. Amyotroph Lateral Scler. 2009, 10, 418–429. [Google Scholar] [CrossRef] [PubMed]
- Figueroa-Romero, C.; Hur, J.; Bender, D.E.; Delaney, C.E.; Cataldo, M.D.; Smith, A.L.; Yung, R.; Ruden, D.M.; Callaghan, B.C.; Feldman, E.L. Identification of epigenetically altered genes in sporadic amyotrophic lateral sclerosis. PLoS ONE 2012, 7, e52672. [Google Scholar] [CrossRef] [PubMed]
- Majchrzak-Celińska, A.; Warych, A.; Szoszkiewicz, M. Novel approaches to epigenetic therapies: From drug combinations to epigenetic editing. Genes 2021, 12, 208. [Google Scholar] [CrossRef]
- Price, D.L.; Porter, K.R. The response of ventral horn neurons to axonal transection. J. Cell Biol. 1972, 53, 24–37. [Google Scholar] [CrossRef]
- Sen, S.; Lagas, S.; Roy, A.; Kumar, H. Cytoskeleton saga: Its regulation in normal physiology and modulation in neurodegenerative disorders. Eur. J. Pharm. 2022, 925, 175001. [Google Scholar] [CrossRef]
- Shefner, J.M.; Cudkowicz, M.E.; Hardiman, O.; Cockroft, B.M.; Lee, J.H.; Malik, F.I.; Meng, L.; Rudnicki, S.A.; Wolff, A.A.; Andrews, J.A.; et al. A phase III trial of tirasemtiv as a potential treatment for amyotrophic lateral sclerosis. Amyotroph Lateral Scler. Front. Degener. 2019, 20, 584–594. [Google Scholar] [CrossRef] [Green Version]
- Jin, J.P.; Brotto, M.A.; Hossain, M.M.; Huang, Q.Q.; Brotto, L.S.; Nosek, T.M.; Morton, D.H.; Crawford, T.O. Truncation by Glu180 nonsense mutation results in complete loss of slow skeletal muscle troponin T in a lethal nemaline myopathy. J. Biol. Chem. 2003, 278, 26159–26165. [Google Scholar] [CrossRef] [Green Version]
- Maksin-Matveev, A.; Kanfi, Y.; Hochhauser, E.; Isak, A.; Cohen, H.Y.; Shainberg, A. Sirtuin 6 protects the heart from hypoxic damage. Exp. Cell Res. 2015, 330, 81–90. [Google Scholar] [CrossRef]
- Cui, X.; Yao, L.; Yang, X.; Gao, Y.; Fang, F.; Zhang, J.; Wang, Q.; Chang, Y. SIRT6 regulates metabolic homeostasis in skeletal muscle through activation of AMPK. Am. J. Physiol. Endocrinol. Metab. 2017, 313, E493–E505. [Google Scholar] [CrossRef]
- Yao, L.; Cui, X.; Chen, Q.; Yang, X.; Fang, F.; Zhang, J.; Liu, G.; Jin, W.; Chang, Y. Cold-inducible SIRT6 regulates thermogenesis of brown and beige fat. Cell Rep. 2017, 20, 641–654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, L.J.; Niedzwiecki, M.V.; Wong, M. Chronic intermittent mild whole-body hypothermia is therapeutic in a mouse model of ALS. Cells 2021, 10, 320. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.; Chen, X.; Cai, X.; Lin, H.; Xu, J.; Cheng, X. RNA-binding protein MEX3D promotes cervical carcinoma tumorigenesis by destabilizing TSC22D1 mRNA. Cell Death Discov. 2022, 8, 250. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Zhang, P.; Liu, Z.; Xing, Y.; Xiao, Y. NXPH4 promotes gemcitabine resistance in bladder cancer by enhancing reactive oxygen species and glycolysis activation through modulating NDUFA4L2. Cancers 2022, 14, 3782. [Google Scholar] [CrossRef]
- Brunet, M.; Vargas, C.; Larrieu, D.; Torrisani, J.; Dufresne, M. E3 Ubiquitin Ligase TRIP12: Regulation, Structure, and Physiopathological Functions. Int. J. Mol. Sci. 2020, 21, 8515. [Google Scholar] [CrossRef]
- An, C.I.; Ganio, E.; Hagiwara, N. Trip12, a HECT domain E3 ubiquitin ligase, targets Sox6 for proteasomal degradation and affects fiber type-specific gene expression in muscle cells. Skeletal Muscle 2013, 3, 11. [Google Scholar] [CrossRef] [Green Version]
- Sangari, S.; Peyre, I.; Lackmy-Vallée, A.; Bayen, E.; Pradat, P.-F.; Marchand-Pauvert, V. Transient increase in recurrent inhibition in amyotrophic lateral sclerosis as a putative protection from neurodegeneration. Acta Physiol. 2022, 234, e13758. [Google Scholar] [CrossRef]
- Oyanagi, K.; Ikuta, F.; Horikawa, Y. Evidence for sequential degeneration of the neurons in the intermediate zone of the spinal cord in amyotrophic lateral sclerosis: A topographic and quantitative investigation. Acta Neuropathol. 1989, 77, 343–349. [Google Scholar] [CrossRef]
- Brueckner, B.; Garcia Boy, R.; Siedlecki, P.; Much, T.; Kliem, H.C.; Zielenkiewicz, P.; Suhai, S.; Wiessler, M.; Lyko, F. Epigenetic reactivation of tumor suppressor genes by a novel small-molecule inhibitor of human DNA methyltransferases. Cancer Res. 2005, 65, 6305–6311. [Google Scholar] [CrossRef] [Green Version]
- Oh, Y.S.; Kim, S.H.; Cho, G.W. Functional restoration of amyotrophic lateral sclerosis patient-derived mesenchymal stromal cells through inhibition of DNA methyltransferase. Cell. Mol. Neurobiol. 2016, 36, 613–620. [Google Scholar] [CrossRef]
- Kondo, N.; Tohnai, G.; Sahashi, K.; Iida, M.; Kataoka, M.; Nakatsuji, H.; Tsutsumi, Y.; Hashizume, A.; Adachi, H.; Koike, H.; et al. DNA methylation inhibitor attenuates polyglutamine-induced neurodegeneration by regulating Hes5. EMBO Mol. Med. 2019, 11, e8547. [Google Scholar] [CrossRef] [PubMed]
- Martin, L.J.; Kaiser, A.; Price, A.C. Motor neuron degeneration after sciatic nerve avulsion in adult rat evolves with oxidative stress and is apoptosis. J. Neurobiol. 1999, 40, 185–201. [Google Scholar] [CrossRef]
- Martin, L.J.; Liu, Z. Injury-induced spinal motor neuron apoptosis is preceded by DNA single-strand breaks and is p53- and bax-dependent. J. Neurobiol. 2002, 50, 181–197. [Google Scholar] [CrossRef] [PubMed]
- Martin, L.J.; Chen, K.; Liu, Z. Adult motor neuron apoptosis is mediated by nitric oxide and Fas death receptor linked by DNA damage and p53 activation. J. Neurosci. 2005, 25, 6449–6459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cook, A.M.; Mieure, K.D.; Owen, R.D.; Pesaturo, A.B.; Hatton, J. Intracerebroventricular administration of drugs. Pharmacotherapy 2009, 29, 832–845. [Google Scholar] [CrossRef] [PubMed]
- Sarda, N.; Reynaud, D.; Gharib, A. S-adenosylmethionine, S-adenosylhomocysteine and adenosine system. Age-dependent availability in rat brain. Dev. Pharmacol. Ther. 1989, 13, 104–112. [Google Scholar] [CrossRef] [PubMed]
- Stramentinoli, G.; Gualano, M.; Catto, E.; Algeri, S. Tissue levels of S-adenosylmethionine in aging rats. J. Gerontol. 1977, 32, 392–394. [Google Scholar] [CrossRef] [PubMed]
- Gharib, A.; Sarda, N.; Chabannes, B.; Cronenberger, L.; Pacheco, H. The regional concentrations of S-adenosyl-L-methionine, S-adenosyl-L-homocysteine, and adenosine in rat brain. J. Neurochem. 1982, 38, 810–815. [Google Scholar] [CrossRef]
- Davidson, T.J.; Hartmann, H.A. Base composition of RNA obtained from motor neurons in amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. 1981, 40, 193–198. [Google Scholar] [CrossRef] [Green Version]
- Tank, E.M.; Figueroa-Romero, C.; Hinder, L.M.; Bedi, K.; Archbold, H.C.; Li, X.; Weskamp, K.; Safren, N.; Paez-Colasante, X.; Pacut, C.; et al. Abnormal RNA stability in amyotrophic lateral sclerosis. Nat. Commun. 2018, 9, 2845. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target Protein | Commercial Source | Assay |
|---|---|---|
| Human SOD1 (human-specific, not reactive with mouse SOD1) | MBL International, clone1G2 | Immunoprecipitation (IP), IF |
| MyoD | BD Pharmingen, clone MoAb5.8A | IF |
| 5mC | Calbiochem, clone 16233D3 | IF, IHC |
| 6mA | Proteintech, clone 1D5E10 | IHC |
| DNMT1 | Enzo, clone 60B12220.1 | Western blotting (WB) |
| DNMT3A | Enzo, clone 64B1446 | WB |
| DNMT2/TRDMT1 | LSBio, rabbit polyclonal | WB |
| FTO | LSBio, rabbit polyclonal | WB |
| METTL3 | Bethyl Labs, rabbit polyclonal | WB |
| ALKBH5 | Proteintech, rabbit polyclonal | WB |
| Parvalbumin | Sigma, clone PARV-19 | IHC |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martin, L.J.; Adams, D.A.; Niedzwiecki, M.V.; Wong, M. Aberrant DNA and RNA Methylation Occur in Spinal Cord and Skeletal Muscle of Human SOD1 Mouse Models of ALS and in Human ALS: Targeting DNA Methylation Is Therapeutic. Cells 2022, 11, 3448. https://doi.org/10.3390/cells11213448
Martin LJ, Adams DA, Niedzwiecki MV, Wong M. Aberrant DNA and RNA Methylation Occur in Spinal Cord and Skeletal Muscle of Human SOD1 Mouse Models of ALS and in Human ALS: Targeting DNA Methylation Is Therapeutic. Cells. 2022; 11(21):3448. https://doi.org/10.3390/cells11213448
Chicago/Turabian StyleMartin, Lee J., Danya A. Adams, Mark V. Niedzwiecki, and Margaret Wong. 2022. "Aberrant DNA and RNA Methylation Occur in Spinal Cord and Skeletal Muscle of Human SOD1 Mouse Models of ALS and in Human ALS: Targeting DNA Methylation Is Therapeutic" Cells 11, no. 21: 3448. https://doi.org/10.3390/cells11213448
APA StyleMartin, L. J., Adams, D. A., Niedzwiecki, M. V., & Wong, M. (2022). Aberrant DNA and RNA Methylation Occur in Spinal Cord and Skeletal Muscle of Human SOD1 Mouse Models of ALS and in Human ALS: Targeting DNA Methylation Is Therapeutic. Cells, 11(21), 3448. https://doi.org/10.3390/cells11213448