
Repeated Administration of Cisplatin Transforms Kidney Fibroblasts through G2/M Arrest and Cellular Senescence

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
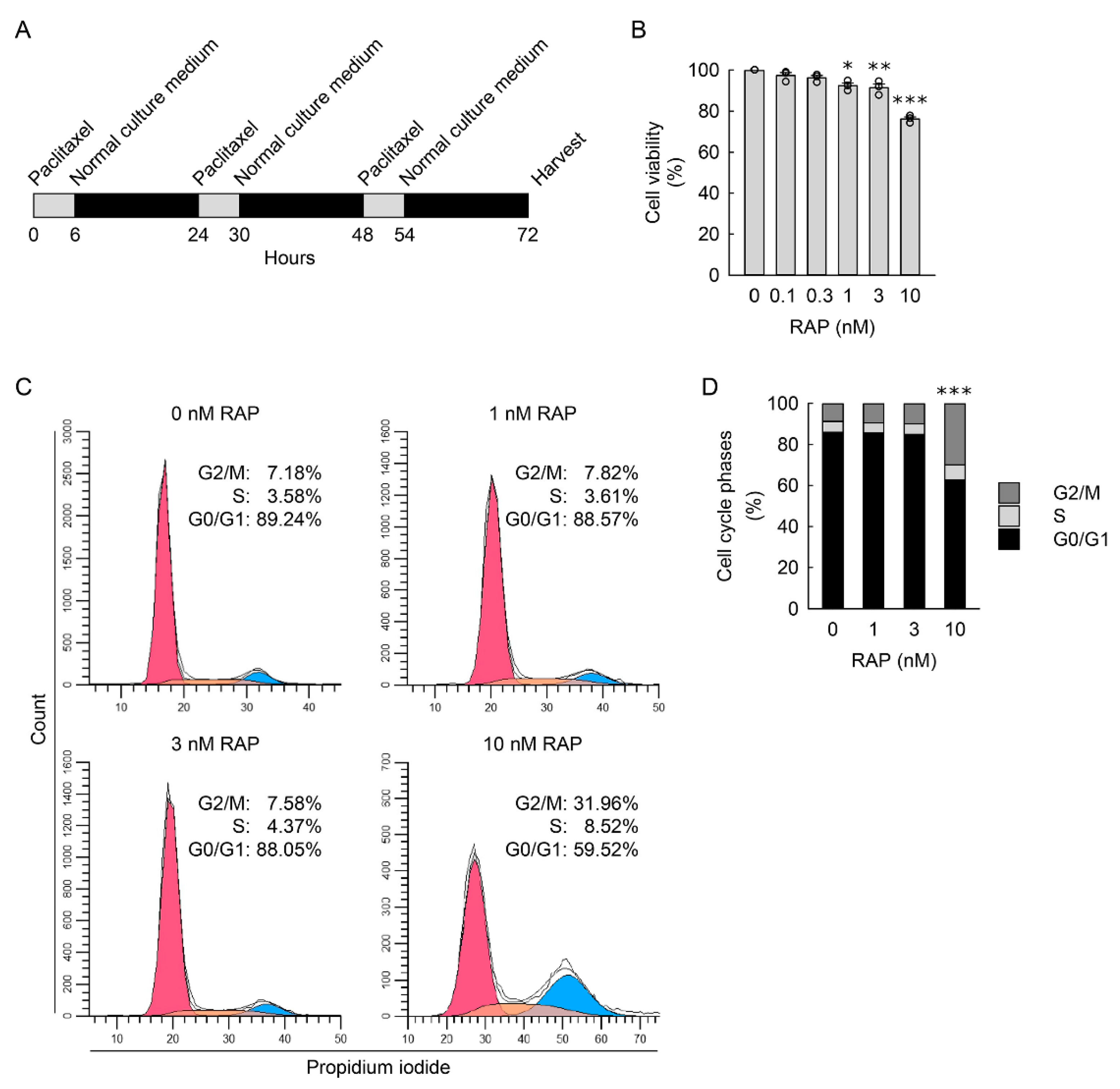
2.1. Cell Culture and Treatment
2.2. Cell Viability
2.3. Western Blot Analysis
2.4. Immunocytochemistry
2.5. Cell Size
2.6. Senescence-Associated β-Galactosidase (SA-β-Gal) Activity
2.7. Cell Cycle Assessment
2.8. Statistical Analysis
3. Results
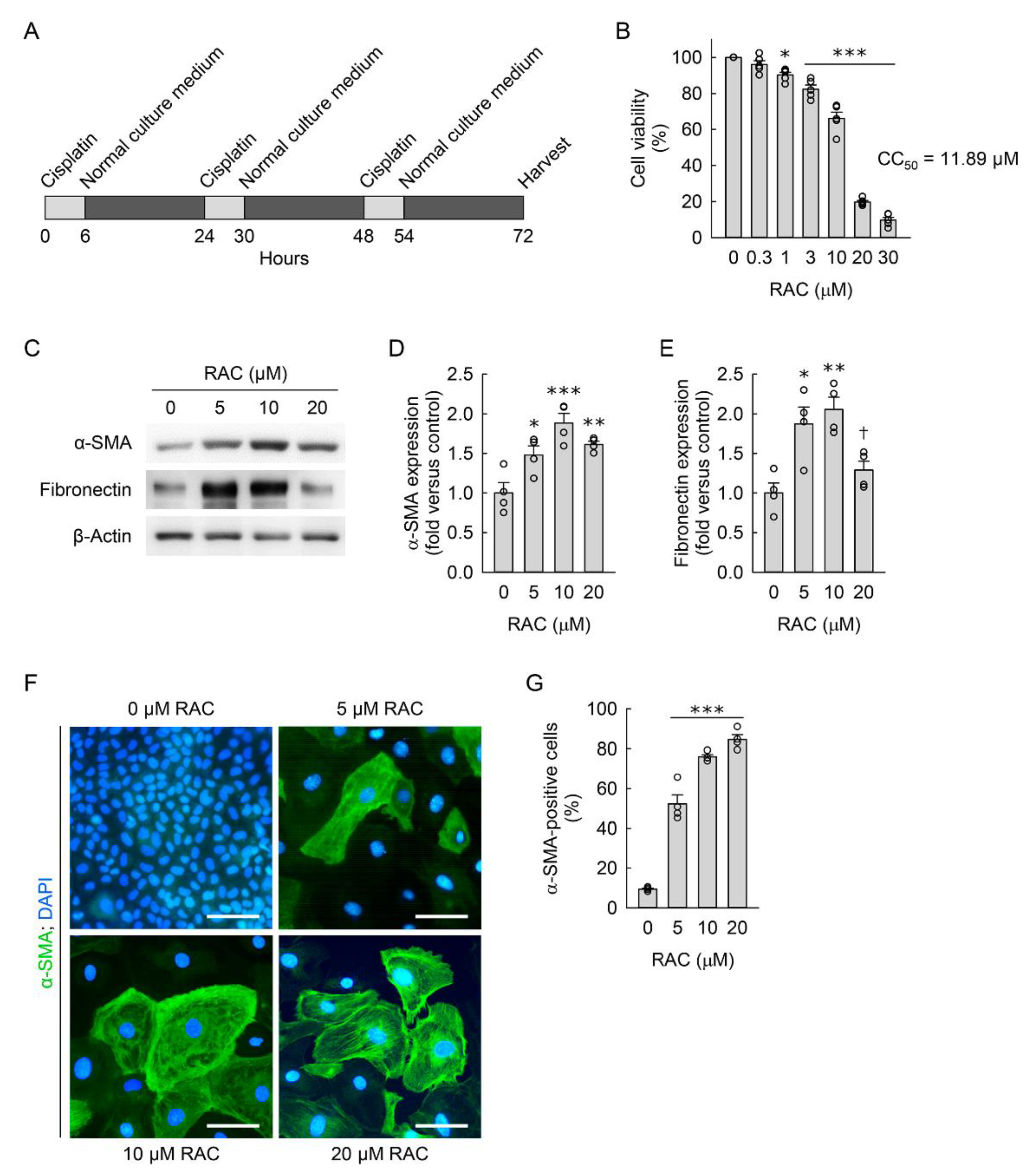
3.1. RAC Transforms Kidney Fibroblasts into Myofibroblasts
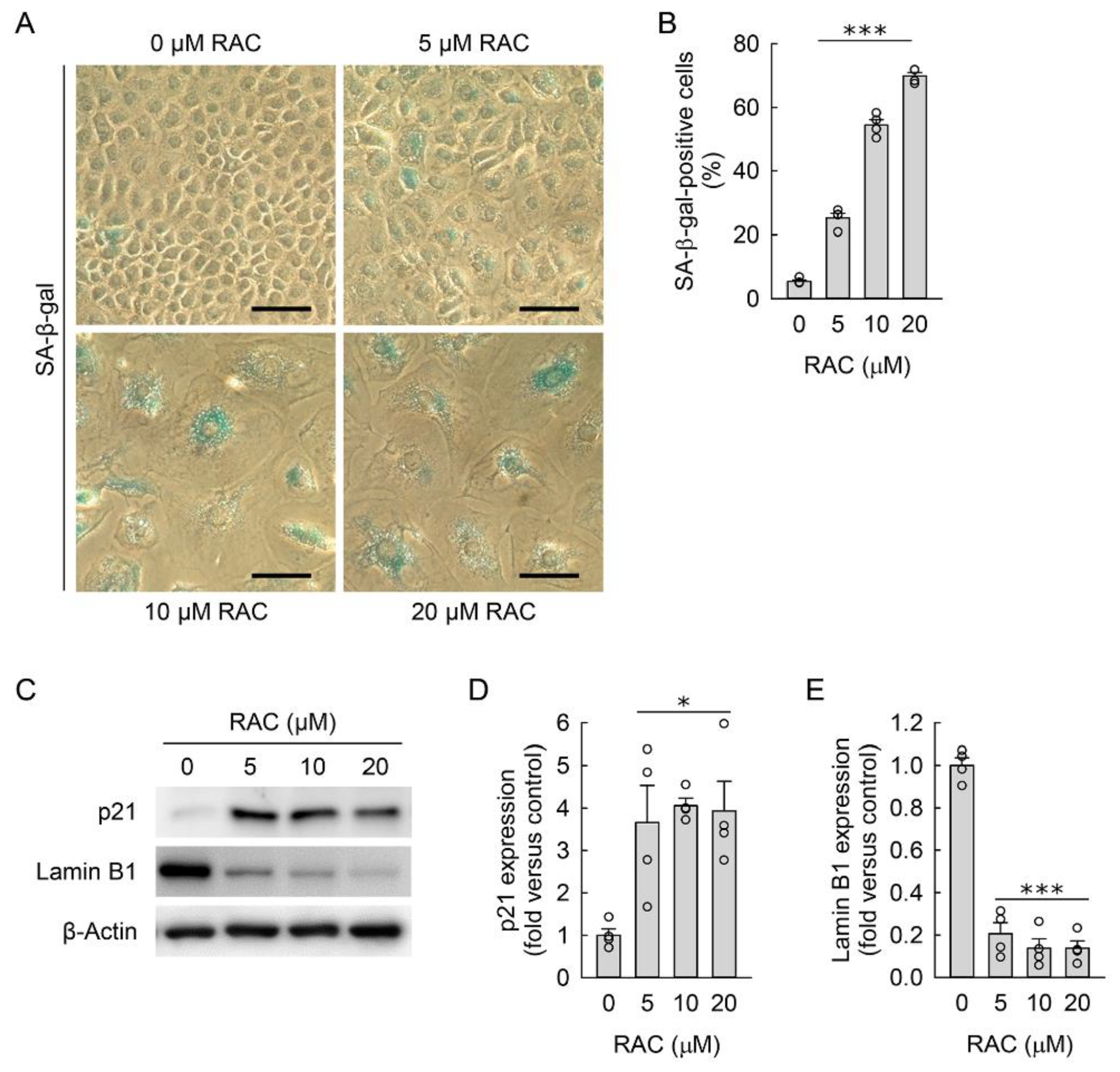
3.2. RAC Induces Cellular Hypertrophy and Senescence in Kidney Fibroblast Cells
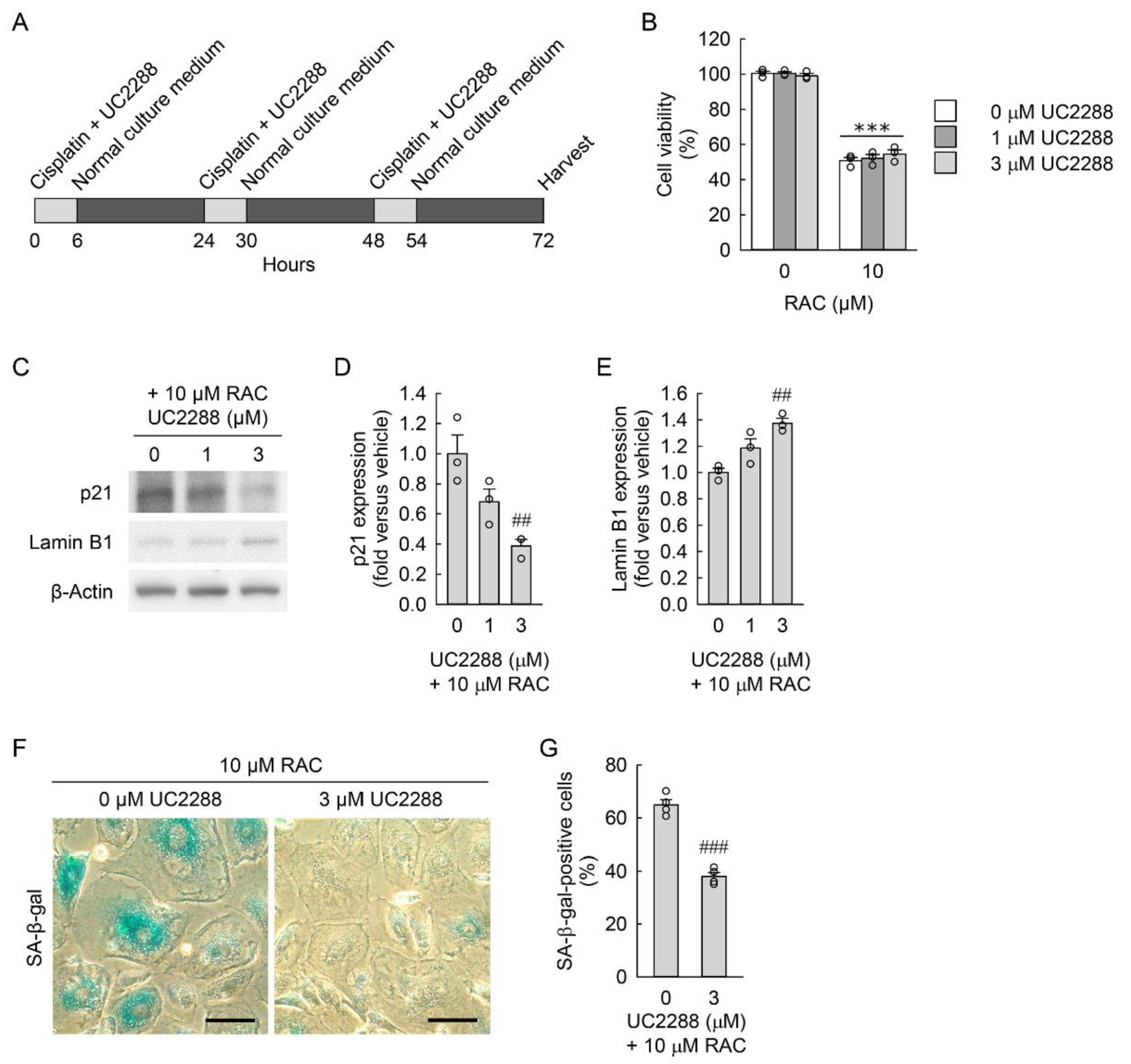
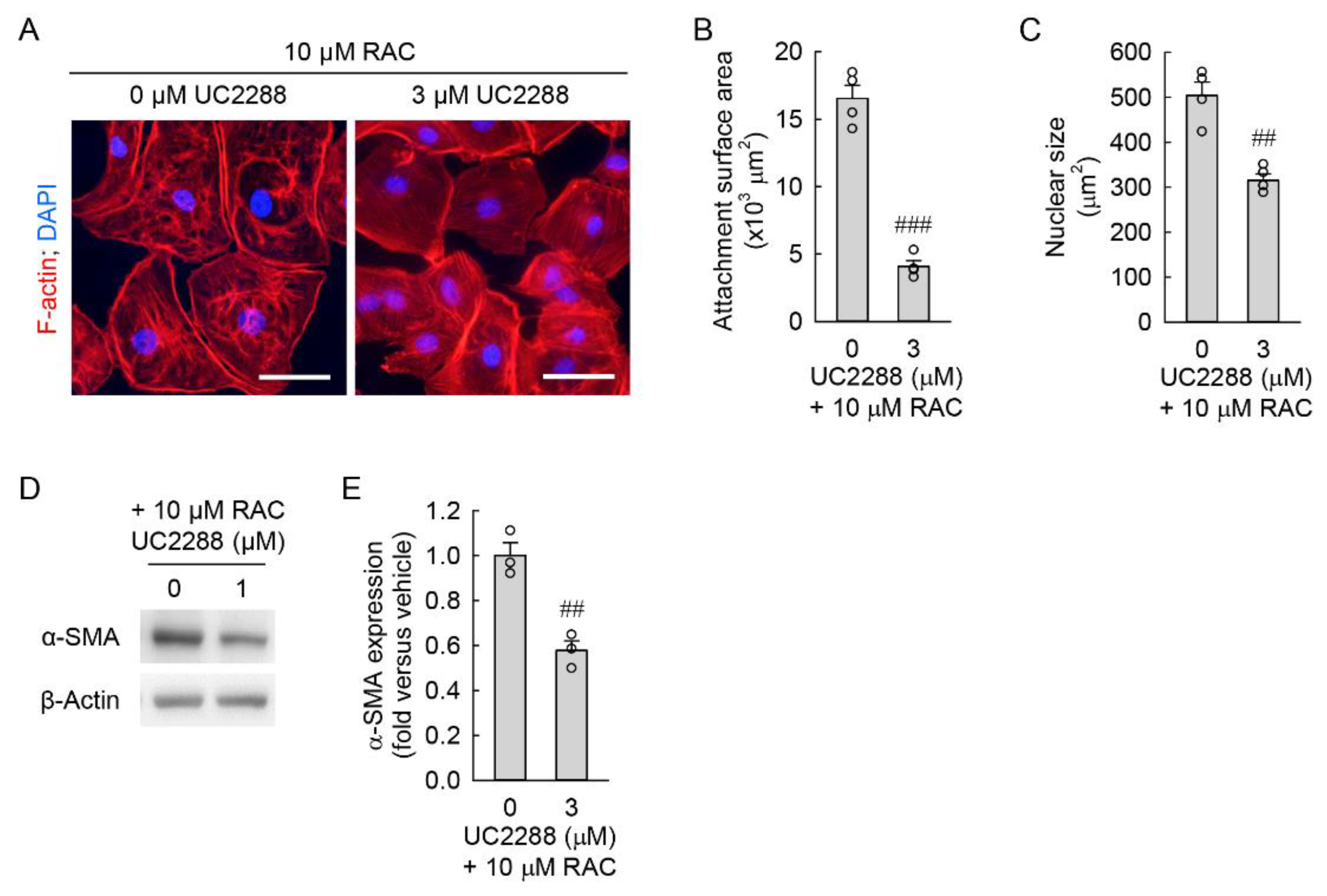
3.3. Pharmacological Inhibition of p21 Attenuates RAC-Induced Myofibroblast Transformation in Kidney Fibroblast Cells through Reduction in Cellular Senescence
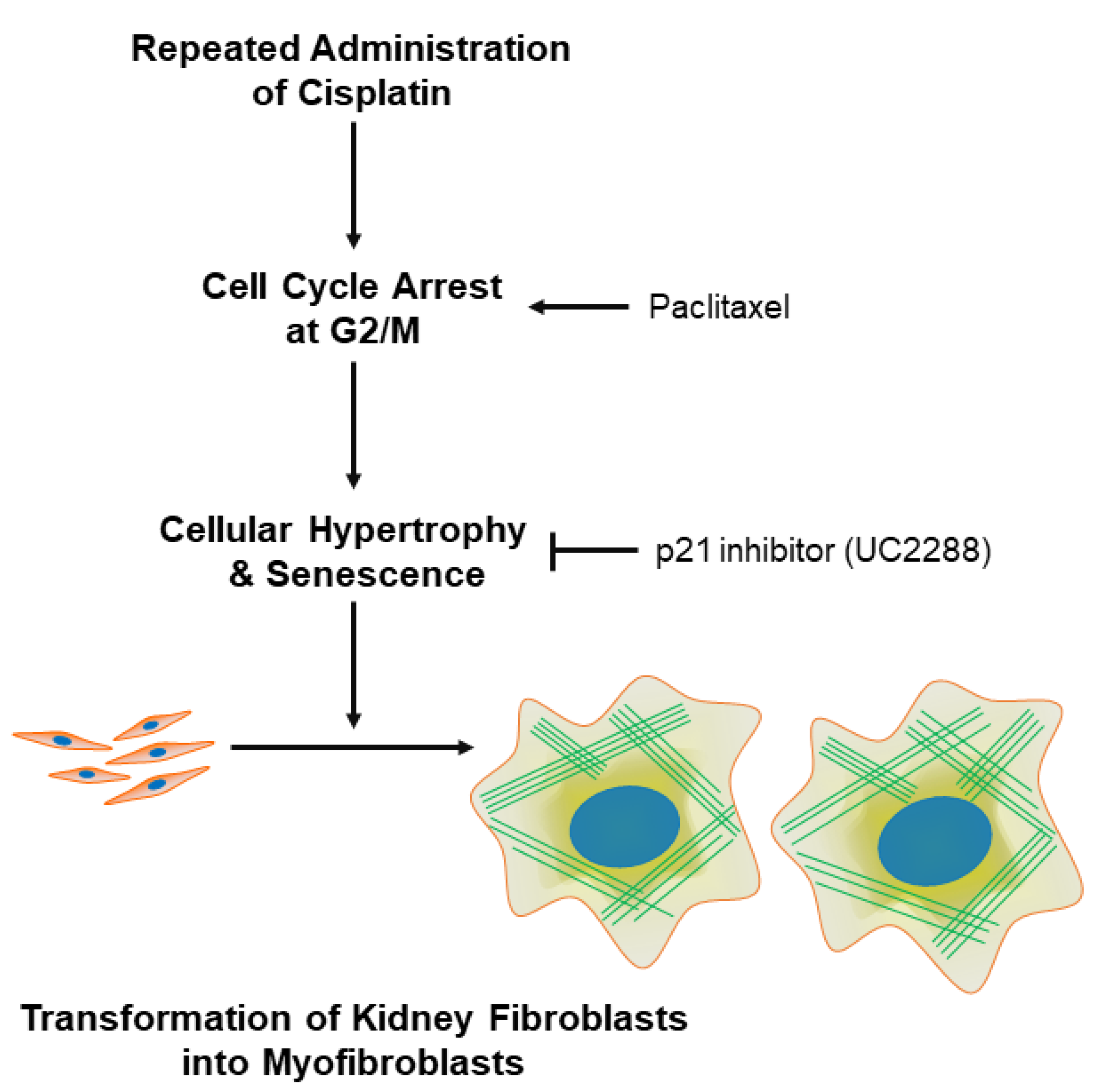
3.4. G2/M Cell Cycle Arrest Contributes to Cellular Hypertrophy, Senescence, and Myofibroblast Transformation in Kidney Fibroblasts
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Pabla, N.; Dong, Z. Cisplatin nephrotoxicity: Mechanisms and renoprotective strategies. Kidney Int. 2008, 73, 994–1007. [Google Scholar] [CrossRef] [Green Version]
- Arany, I.; Safirstein, R.L. Cisplatin nephrotoxicity. Semin. Nephrol. 2003, 23, 460–464. [Google Scholar] [CrossRef]
- Ciccarelli, R.B.; Solomon, M.J.; Varshavsky, A.; Lippard, S.J. In vivo effects of cis- and trans-diamminedichloroplatinum(II) on SV40 chromosomes: Differential repair, DNA-protein cross-linking, and inhibition of replication. Biochemistry 1985, 24, 7533–7540. [Google Scholar] [CrossRef]
- Lebwohl, D.; Canetta, R. Clinical development of platinum complexes in cancer therapy: An historical perspective and an update. Eur. J. Cancer 1998, 34, 1522–1534. [Google Scholar] [CrossRef]
- Miller, R.P.; Tadagavadi, R.K.; Ramesh, G.; Reeves, W.B. Mechanisms of Cisplatin nephrotoxicity. Toxins 2010, 2, 2490–2518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bennis, Y.; Savry, A.; Rocca, M.; Gauthier-Villano, L.; Pisano, P.; Pourroy, B. Cisplatin dose adjustment in patients with renal impairment, which recommendations should we follow? Int. J. Clin. Pharm 2014, 36, 420–429. [Google Scholar] [CrossRef] [PubMed]
- Ferenbach, D.A.; Bonventre, J.V. Mechanisms of maladaptive repair after AKI leading to accelerated kidney ageing and CKD. Nat. Rev. Nephrol. 2015, 11, 264–276. [Google Scholar] [CrossRef] [Green Version]
- Sharp, C.N.; Doll, M.A.; Dupre, T.V.; Shah, P.P.; Subathra, M.; Siow, D.; Arteel, G.E.; Megyesi, J.; Beverly, L.J.; Siskind, L.J. Repeated administration of low-dose cisplatin in mice induces fibrosis. Am. J. Physiol. Ren. Physiol. 2016, 310, F560–F568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, Y.; Cai, J.; Li, F.; Liu, Z.; Shu, S.; Wang, Y.; Liu, Y.; Tang, C.; Dong, Z. Chronic effects of repeated low-dose cisplatin treatment in mouse kidneys and renal tubular cells. Am. J. Physiol. Ren. Physiol. 2019, 317, F1582–F1592. [Google Scholar] [CrossRef]
- Black, L.M.; Lever, J.M.; Traylor, A.M.; Chen, B.; Yang, Z.; Esman, S.K.; Jiang, Y.; Cutter, G.R.; Boddu, R.; George, J.F.; et al. Divergent effects of AKI to CKD models on inflammation and fibrosis. Am. J. Physiol. Ren. Physiol. 2018, 315, F1107–F1118. [Google Scholar] [CrossRef]
- Latcha, S.; Jaimes, E.A.; Patil, S.; Glezerman, I.G.; Mehta, S.; Flombaum, C.D. Long-Term Renal Outcomes after Cisplatin Treatment. Clin. J. Am. Soc. Nephrol 2016, 11, 1173–1179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaissling, B.; Le Hir, M. The renal cortical interstitium: Morphological and functional aspects. Histochem. Cell Biol. 2008, 130, 247–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Hir, M.; Kaissling, B. Distribution and regulation of renal ecto-5’-nucleotidase: Implications for physiological functions of adenosine. Am. J. Physiol. 1993, 264, F377–F387. [Google Scholar] [CrossRef]
- Bachmann, S.; Le Hir, M.; Eckardt, K.U. Co-localization of erythropoietin mRNA and ecto-5’-nucleotidase immunoreactivity in peritubular cells of rat renal cortex indicates that fibroblasts produce erythropoietin. J. Histochem. Cytochem. 1993, 41, 335–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eddy, A.A. Overview of the cellular and molecular basis of kidney fibrosis. Kidney Int. Suppl. 2014, 4, 2–8. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.B.Y.; Qu, X.; Caruana, G.; Li, J. The origin of renal fibroblasts/myofibroblasts and the signals that trigger fibrosis. Differentiation 2016, 92, 102–107. [Google Scholar] [CrossRef]
- Wada, T.; Sakai, N.; Matsushima, K.; Kaneko, S. Fibrocytes: A new insight into kidney fibrosis. Kidney Int. 2007, 72, 269–273. [Google Scholar] [CrossRef] [Green Version]
- Fan, J.M.; Ng, Y.Y.; Hill, P.A.; Nikolic-Paterson, D.J.; Mu, W.; Atkins, R.C.; Lan, H.Y. Transforming growth factor-beta regulates tubular epithelial-myofibroblast transdifferentiation in vitro. Kidney Int. 1999, 56, 1455–1467. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Qu, X.; Bertram, J.F. Endothelial-myofibroblast transition contributes to the early development of diabetic renal interstitial fibrosis in streptozotocin-induced diabetic mice. Am. J. Pathol. 2009, 175, 1380–1388. [Google Scholar] [CrossRef] [Green Version]
- Vierhout, M.; Ayoub, A.; Naiel, S.; Yazdanshenas, P.; Revill, S.D.; Reihani, A.; Dvorkin-Gheva, A.; Shi, W.; Ask, K. Monocyte and macrophage derived myofibroblasts: Is it fate? A review of the current evidence. Wound Repair Regen 2021, 29, 548–562. [Google Scholar] [CrossRef]
- Eddy, A.A. The origin of scar-forming kidney myofibroblasts. Nat. Med. 2013, 19, 964–966. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Xiang, Y.; Wu, W.; Cai, J.; Tang, C.; Dong, Z. Persistent Activation of Autophagy After Cisplatin Nephrotoxicity Promotes Renal Fibrosis and Chronic Kidney Disease. Front. Pharmacol. 2022, 13, 918732. [Google Scholar] [CrossRef]
- Fu, S.; Hu, X.; Ma, Z.; Wei, Q.; Xiang, X.; Li, S.; Wen, L.; Liang, Y.; Dong, Z. p53 in Proximal Tubules Mediates Chronic Kidney Problems after Cisplatin Treatment. Cells 2022, 11, 712. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Padanilam, B.J. Loss of poly(ADP-ribose) polymerase 1 attenuates renal fibrosis and inflammation during unilateral ureteral obstruction. Am. J. Physiology. Ren. Physiol. 2011, 301, F450–F459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wettersten, H.I.; Hee Hwang, S.; Li, C.; Shiu, E.Y.; Wecksler, A.T.; Hammock, B.D.; Weiss, R.H. A novel p21 attenuator which is structurally related to sorafenib. Cancer Biol. Ther. 2013, 14, 278–285. [Google Scholar] [CrossRef] [Green Version]
- Schiff, P.B.; Fant, J.; Horwitz, S.B. Promotion of microtubule assembly in vitro by taxol. Nature 1979, 277, 665–667. [Google Scholar] [CrossRef]
- Moon, D.; Yoon, S.-P.; Jang, H.-S.; Noh, M.R.; Ha, L.; Padanilam, B.J.; Kim, J. Extraneural CGRP Induces Oxidative Stress in Kidney Proximal Tubule Epithelial Cells. Anat. Biol. Anthropol. 2019, 32, 121. [Google Scholar] [CrossRef]
- Moon, D.; Kim, J. Cyclosporin A aggravates hydrogen peroxide-induced cell death in kidney proximal tubule epithelial cells. Anat. Cell Biol. 2019, 52, 312–323. [Google Scholar] [CrossRef]
- Kim, J. Spermidine rescues proximal tubular cells from oxidative stress and necrosis after ischemic acute kidney injury. Arch. Pharmacal Res. 2017, 40, 1197–1208. [Google Scholar] [CrossRef]
- Kim, M.J.; Moon, D.; Jung, S.; Lee, J.; Kim, J. Cisplatin nephrotoxicity is induced via poly(ADP-ribose) polymerase activation in adult zebrafish and mice. Am. J. Physiology. Regul. Integr. Comp. Physiol. 2020, 318, R843–R854. [Google Scholar] [CrossRef]
- Yoon, S.P.; Kim, J. Exogenous spermidine ameliorates tubular necrosis during cisplatin nephrotoxicity. Anat. Cell Biol. 2018, 51, 189–199. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.P.; Kim, J. Exogenous CGRP upregulates profibrogenic growth factors through PKC/JNK signaling pathway in kidney proximal tubular cells. Cell Biol. Toxicol. 2018, 34, 251–262. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Yoon, S.P.; Kim, J. Cisplatin induces primary necrosis through poly(ADP-ribose) polymerase 1 activation in kidney proximal tubular cells. Anat. Cell Biol. 2015, 48, 66–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Devalaraja-Narashimha, K.; Padanilam, B.J. TIGAR regulates glycolysis in ischemic kidney proximal tubules. Am. J. Physiol. Ren. Physiol. 2015, 308, F298–F308. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Padanilam, B.J. Renal nerves drive interstitial fibrogenesis in obstructive nephropathy. J. Am. Soc. Nephrol. 2013, 24, 229–242. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Park, J.W.; Park, K.M. Increased superoxide formation induced by irradiation preconditioning triggers kidney resistance to ischemia-reperfusion injury in mice. Am. J. Physiology. Ren. Physiol. 2009, 296, F1202–F1211. [Google Scholar] [CrossRef] [Green Version]
- Jang, H.S.; Kim, J.; Park, Y.K.; Park, K.M. Infiltrated macrophages contribute to recovery after ischemic injury but not to ischemic preconditioning in kidneys. Transplantation 2008, 85, 447–455. [Google Scholar] [CrossRef]
- Latham, S.L.; Ehmke, N.; Reinke, P.Y.A.; Taft, M.H.; Eicke, D.; Reindl, T.; Stenzel, W.; Lyons, M.J.; Friez, M.J.; Lee, J.A.; et al. Variants in exons 5 and 6 of ACTB cause syndromic thrombocytopenia. Nat. Commun. 2018, 9, 4250. [Google Scholar] [CrossRef] [Green Version]
- Debacq-Chainiaux, F.; Erusalimsky, J.D.; Campisi, J.; Toussaint, O. Protocols to detect senescence-associated beta-galactosidase (SA-betagal) activity, a biomarker of senescent cells in culture and in vivo. Nat. Protoc. 2009, 4, 1798–1806. [Google Scholar] [CrossRef]
- Kim, J.; Padanilam, B.J. Renal denervation prevents long-term sequelae of ischemic renal injury. Kidney Int. 2015, 87, 350–358. [Google Scholar] [CrossRef]
- Moon, D.; Padanilam, B.J.; Jang, H.S.; Kim, J. 2-Mercaptoethanol protects against DNA double-strand breaks after kidney ischemia and reperfusion injury through GPX4 upregulation. Pharmacol. Rep. PR 2022, 74, 1041–1053. [Google Scholar] [CrossRef]
- Uhal, B.D.; Ramos, C.; Joshi, I.; Bifero, A.; Pardo, A.; Selman, M. Cell size, cell cycle, and alpha-smooth muscle actin expression by primary human lung fibroblasts. Am. J. Physiol. 1998, 275, L998–L1005. [Google Scholar] [CrossRef]
- Sasaki, N.; Itakura, Y.; Toyoda, M. Sialylation regulates myofibroblast differentiation of human skin fibroblasts. Stem. Cell Res. Ther. 2017, 8, 81. [Google Scholar] [CrossRef] [Green Version]
- Roche, P.L.; Nagalingam, R.S.; Bagchi, R.A.; Aroutiounova, N.; Belisle, B.M.; Wigle, J.T.; Czubryt, M.P. Role of scleraxis in mechanical stretch-mediated regulation of cardiac myofibroblast phenotype. Am. J. Physiol. Cell Physiol. 2016, 311, C297–C307. [Google Scholar] [CrossRef] [Green Version]
- Angello, J.C.; Pendergrass, W.R.; Norwood, T.H.; Prothero, J. Cell enlargement: One possible mechanism underlying cellular senescence. J. Cell. Physiol. 1989, 140, 288–294. [Google Scholar] [CrossRef]
- Wang, Y.; Blandino, G.; Givol, D. Induced p21waf expression in H1299 cell line promotes cell senescence and protects against cytotoxic effect of radiation and doxorubicin. Oncogene 1999, 18, 2643–2649. [Google Scholar] [CrossRef] [Green Version]
- Shtutman, M.; Chang, B.D.; Schools, G.P.; Broude, E.V. Cellular Model of p21-Induced Senescence. Methods Mol. Biol. 2017, 1534, 31–39. [Google Scholar] [CrossRef]
- Poulsen, R.C.; Watts, A.C.; Murphy, R.J.; Snelling, S.J.; Carr, A.J.; Hulley, P.A. Glucocorticoids induce senescence in primary human tenocytes by inhibition of sirtuin 1 and activation of the p53/p21 pathway: In vivo and in vitro evidence. Ann. Rheum. Dis. 2014, 73, 1405–1413. [Google Scholar] [CrossRef] [Green Version]
- da Silva, F.M.; Guimaraes, E.L.; Grivicich, I.; Trindade, V.M.; Guaragna, R.M.; Borojevic, R.; Guma, F.C. Hepatic stellate cell activation in vitro: Cell cycle arrest at G2/M and modification of cell motility. J. Cell. Biochem. 2003, 90, 387–396. [Google Scholar] [CrossRef]
- White, T.L.; Deshpande, N.; Kumar, V.; Gauthier, A.G.; Jurkunas, U.V. Cell cycle re-entry and arrest in G2/M phase induces senescence and fibrosis in Fuchs Endothelial Corneal Dystrophy. Free. Radic. Biol. Med. 2021, 164, 34–43. [Google Scholar] [CrossRef]
- Koyano, T.; Namba, M.; Kobayashi, T.; Nakakuni, K.; Nakano, D.; Fukushima, M.; Nishiyama, A.; Matsuyama, M. The p21 dependent G2 arrest of the cell cycle in epithelial tubular cells links to the early stage of renal fibrosis. Sci. Rep. 2019, 9, 12059. [Google Scholar] [CrossRef] [Green Version]
- Chawla, L.S.; Eggers, P.W.; Star, R.A.; Kimmel, P.L. Acute kidney injury and chronic kidney disease as interconnected syndromes. N. Engl. J. Med. 2014, 371, 58–66. [Google Scholar] [CrossRef] [Green Version]
- Sato, Y.; Takahashi, M.; Yanagita, M. Pathophysiology of AKI to CKD progression. Semin. Nephrol. 2020, 40, 206–215. [Google Scholar] [CrossRef]
- Paz, Z.; Shoenfeld, Y. Antifibrosis: To reverse the irreversible. Clin. Rev. Allergy Immunol. 2010, 38, 276–286. [Google Scholar] [CrossRef] [PubMed]
- Hinz, B.; Phan, S.H.; Thannickal, V.J.; Galli, A.; Bochaton-Piallat, M.L.; Gabbiani, G. The myofibroblast: One function, multiple origins. Am. J. Pathol. 2007, 170, 1807–1816. [Google Scholar] [CrossRef]
- Grgic, I.; Campanholle, G.; Bijol, V.; Wang, C.; Sabbisetti, V.S.; Ichimura, T.; Humphreys, B.D.; Bonventre, J.V. Targeted proximal tubule injury triggers interstitial fibrosis and glomerulosclerosis. Kidney Int. 2012, 82, 172–183. [Google Scholar] [CrossRef] [Green Version]
- Sears, S.; Siskind, L. Potential Therapeutic Targets for Cisplatin-Induced Kidney Injury: Lessons from Other Models of AKI and Fibrosis. J. Am. Soc. Nephrol. 2021, 32, 1559–1567. [Google Scholar] [CrossRef]
- McKay, B.C.; Becerril, C.; Ljungman, M. P53 plays a protective role against UV- and cisplatin-induced apoptosis in transcription-coupled repair proficient fibroblasts. Oncogene 2001, 20, 6805–6808. [Google Scholar] [CrossRef] [Green Version]
- Dijt, F.J.; Fichtinger-Schepman, A.M.; Berends, F.; Reedijk, J. Formation and repair of cisplatin-induced adducts to DNA in cultured normal and repair-deficient human fibroblasts. Cancer Res. 1988, 48, 6058–6062. [Google Scholar]
- Lee, B.Y.; Han, J.A.; Im, J.S.; Morrone, A.; Johung, K.; Goodwin, E.C.; Kleijer, W.J.; DiMaio, D.; Hwang, E.S. Senescence-associated beta-galactosidase is lysosomal beta-galactosidase. Aging Cell 2006, 5, 187–195. [Google Scholar] [CrossRef]
- Freund, A.; Laberge, R.M.; Demaria, M.; Campisi, J. Lamin B1 loss is a senescence-associated biomarker. Mol. Biol. Cell 2012, 23, 2066–2075. [Google Scholar] [CrossRef] [PubMed]
- Stein, G.H.; Drullinger, L.F.; Soulard, A.; Dulic, V. Differential roles for cyclin-dependent kinase inhibitors p21 and p16 in the mechanisms of senescence and differentiation in human fibroblasts. Mol. Cell. Biol. 1999, 19, 2109–2117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yosef, R.; Pilpel, N.; Papismadov, N.; Gal, H.; Ovadya, Y.; Vadai, E.; Miller, S.; Porat, Z.; Ben-Dor, S.; Krizhanovsky, V. p21 maintains senescent cell viability under persistent DNA damage response by restraining JNK and caspase signaling. EMBO J. 2017, 36, 2280–2295. [Google Scholar] [CrossRef]
- Al-Douahji, M.; Brugarolas, J.; Brown, P.A.; Stehman-Breen, C.O.; Alpers, C.E.; Shankland, S.J. The cyclin kinase inhibitor p21WAF1/CIP1 is required for glomerular hypertrophy in experimental diabetic nephropathy. Kidney Int. 1999, 56, 1691–1699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, S.; Swaffer, M.; Skotheim, J.M. Eukaryotic Cell Size Control and Its Relation to Biosynthesis and Senescence. Annu. Rev. Cell Dev. Biol. 2022, 38, 291–319. [Google Scholar] [CrossRef]
- Biran, A.; Zada, L.; Abou Karam, P.; Vadai, E.; Roitman, L.; Ovadya, Y.; Porat, Z.; Krizhanovsky, V. Quantitative identification of senescent cells in aging and disease. Aging Cell 2017, 16, 661–671. [Google Scholar] [CrossRef]
- Lanz, M.C.; Zatulovskiy, E.; Swaffer, M.P.; Zhang, L.; Ilerten, I.; Zhang, S.; You, D.S.; Marinov, G.; McAlpine, P.; Elias, J.E.; et al. Increasing cell size remodels the proteome and promotes senescence. Mol. Cell 2022, 82, 3255–3269. [Google Scholar] [CrossRef]
- Karimian, A.; Ahmadi, Y.; Yousefi, B. Multiple functions of p21 in cell cycle, apoptosis and transcriptional regulation after DNA damage. DNA Repair 2016, 42, 63–71. [Google Scholar] [CrossRef]
- Demidenko, Z.N.; Blagosklonny, M.V. Growth stimulation leads to cellular senescence when the cell cycle is blocked. Cell Cycle 2008, 7, 3355–3361. [Google Scholar] [CrossRef]
- Neurohr, G.E.; Terry, R.L.; Lengefeld, J.; Bonney, M.; Brittingham, G.P.; Moretto, F.; Miettinen, T.P.; Vaites, L.P.; Soares, L.M.; Paulo, J.A.; et al. Excessive Cell Growth Causes Cytoplasm Dilution And Contributes to Senescence. Cell 2019, 176, 1083–1097.e1018. [Google Scholar] [CrossRef] [Green Version]
- Goldstein, S. Replicative senescence: The human fibroblast comes of age. Science 1990, 249, 1129–1133. [Google Scholar] [CrossRef] [PubMed]
- Camps, J.; Erdos, M.R.; Ried, T. The role of lamin B1 for the maintenance of nuclear structure and function. Nucleus 2015, 6, 8–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veitia, R.A. DNA Content, Cell Size, and Cell Senescence. Trends Biochem. Sci. 2019, 44, 645–647. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zohar, R.; McCulloch, C.A. Multiple roles of alpha-smooth muscle actin in mechanotransduction. Exp. Cell Res. 2006, 312, 205–214. [Google Scholar] [CrossRef]
- Komuro, I.; Yazaki, Y. Control of cardiac gene expression by mechanical stress. Annu. Rev. Physiol 1993, 55, 55–75. [Google Scholar] [CrossRef]
- Hinz, B.; Mastrangelo, D.; Iselin, C.E.; Chaponnier, C.; Gabbiani, G. Mechanical tension controls granulation tissue contractile activity and myofibroblast differentiation. Am. J. Pathol. 2001, 159, 1009–1020. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.S.; Lim, J.Y.; Kim, J. Mechanical stretch induces angiotensinogen expression through PARP1 activation in kidney proximal tubular cells. Vitr. Cell. Dev. Biol. Anim. 2015, 51, 72–78. [Google Scholar] [CrossRef]
- Chaqour, B.; Goppelt-Struebe, M. Mechanical regulation of the Cyr61/CCN1 and CTGF/CCN2 proteins. FEBS J. 2006, 273, 3639–3649. [Google Scholar] [CrossRef]
- Follonier Castella, L.; Gabbiani, G.; McCulloch, C.A.; Hinz, B. Regulation of myofibroblast activities: Calcium pulls some strings behind the scene. Exp. Cell Res. 2010, 316, 2390–2401. [Google Scholar] [CrossRef]
- Hara, E.; Smith, R.; Parry, D.; Tahara, H.; Stone, S.; Peters, G. Regulation of p16CDKN2 expression and its implications for cell immortalization and senescence. Mol. Cell. Biol. 1996, 16, 859–867. [Google Scholar] [CrossRef] [Green Version]
- Dulic, V.; Drullinger, L.F.; Lees, E.; Reed, S.I.; Stein, G.H. Altered regulation of G1 cyclins in senescent human diploid fibroblasts: Accumulation of inactive cyclin E-Cdk2 and cyclin D1-Cdk2 complexes. Proc. Natl. Acad. Sci. USA 1993, 90, 11034–11038. [Google Scholar] [CrossRef] [PubMed]
- Krenning, L.; Feringa, F.M.; Shaltiel, I.A.; van den Berg, J.; Medema, R.H. Transient activation of p53 in G2 phase is sufficient to induce senescence. Mol. Cell 2014, 55, 59–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lossaint, G.; Horvat, A.; Gire, V.; Bacevic, K.; Mrouj, K.; Charrier-Savournin, F.; Georget, V.; Fisher, D.; Dulic, V. Reciprocal regulation of p21 and Chk1 controls the cyclin D1-RB pathway to mediate senescence onset after G2 arrest. J. Cell Sci. 2022, 135, jcs259114. [Google Scholar] [CrossRef]
- El-Daly, S.M.; Gouhar, S.A.; Gamal-Eldeen, A.M.; Abdel Hamid, F.F.; Ashour, M.N.; Hassan, N.S. Synergistic Effect of alpha-Solanine and Cisplatin Induces Apoptosis and Enhances Cell Cycle Arrest in Human Hepatocellular Carcinoma Cells. Anticancer. Agents Med. Chem. 2019, 19, 2197–2210. [Google Scholar] [CrossRef]
- Sarin, N.; Engel, F.; Kalayda, G.V.; Mannewitz, M.; Cinatl, J., Jr.; Rothweiler, F.; Michaelis, M.; Saafan, H.; Ritter, C.A.; Jaehde, U.; et al. Cisplatin resistance in non-small cell lung cancer cells is associated with an abrogation of cisplatin-induced G2/M cell cycle arrest. PLoS ONE 2017, 12, e0181081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, W.R.; Stark, G.R. Regulation of the G2/M transition by p53. Oncogene 2001, 20, 1803–1815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bunz, F.; Dutriaux, A.; Lengauer, C.; Waldman, T.; Zhou, S.; Brown, J.P.; Sedivy, J.M.; Kinzler, K.W.; Vogelstein, B. Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science 1998, 282, 1497–1501. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Besschetnova, T.Y.; Brooks, C.R.; Shah, J.V.; Bonventre, J.V. Epithelial cell cycle arrest in G2/M mediates kidney fibrosis after injury. Nat. Med. 2010, 16, 535–543. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, J.-B.; Lee, D.-S.; Padanilam, B.J.; Kim, J. Repeated Administration of Cisplatin Transforms Kidney Fibroblasts through G2/M Arrest and Cellular Senescence. Cells 2022, 11, 3472. https://doi.org/10.3390/cells11213472
Yu J-B, Lee D-S, Padanilam BJ, Kim J. Repeated Administration of Cisplatin Transforms Kidney Fibroblasts through G2/M Arrest and Cellular Senescence. Cells. 2022; 11(21):3472. https://doi.org/10.3390/cells11213472
Chicago/Turabian StyleYu, Jia-Bin, Dong-Sun Lee, Babu J. Padanilam, and Jinu Kim. 2022. "Repeated Administration of Cisplatin Transforms Kidney Fibroblasts through G2/M Arrest and Cellular Senescence" Cells 11, no. 21: 3472. https://doi.org/10.3390/cells11213472