

Advances in T Cells Based on Inflammation in Metabolic Diseases

 , , , ,
, , , , 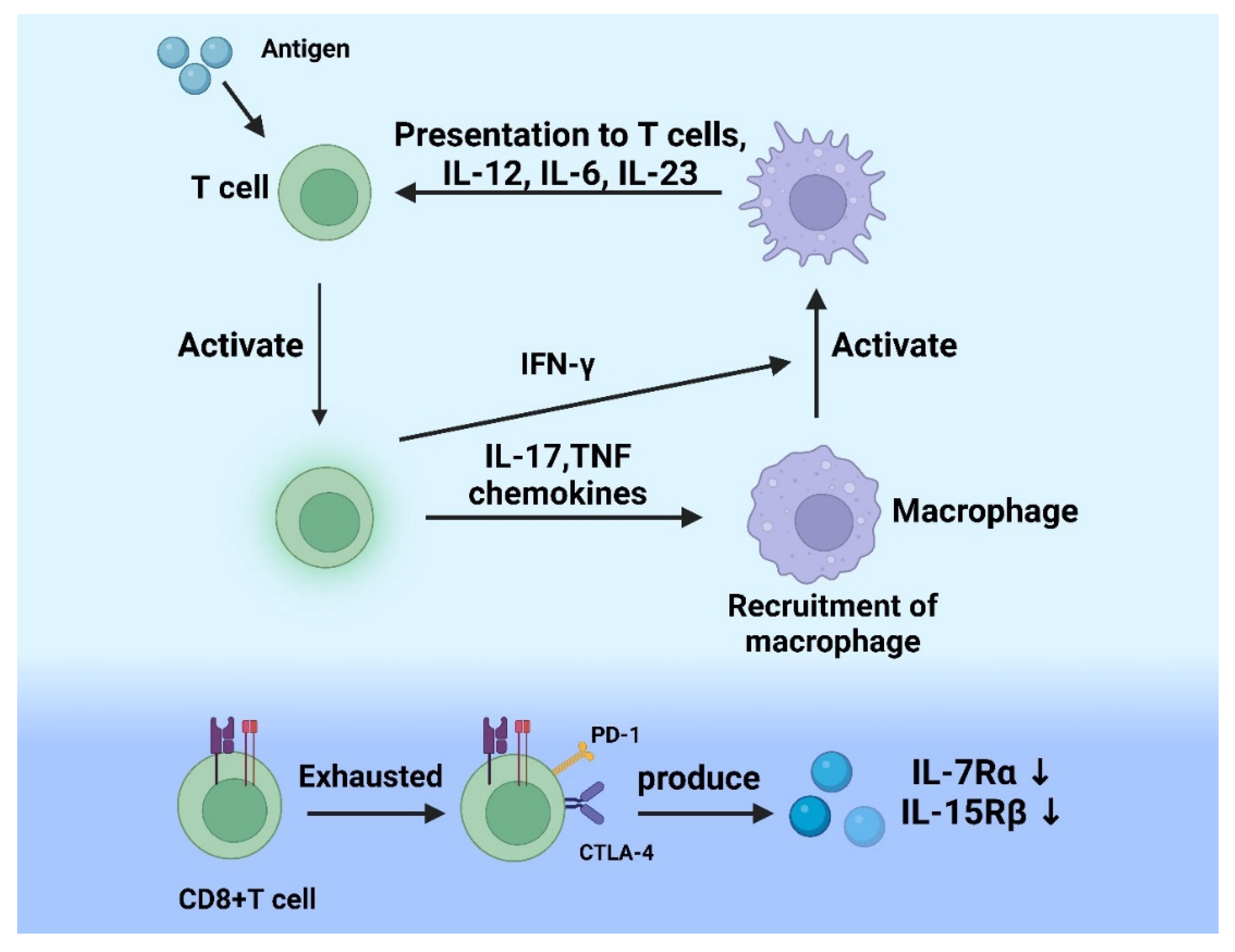{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Mechanism of Inflammatory Response and T Cell
2.1. T Cell
2.2. The Role of T Cells in Acute Inflammation
2.3. The Role of T Cells in Chronic Inflammation
3. The Role of Inflammatory Response in Metabolic Diseases
4. The Function of T Cells in Regulating Inflammatory Response in Metabolic Diseases
4.1. CD4+ T Cells
4.2. CD8+ T Cells
5. Signaling Pathway Regulates T Cells Involved in the Development of Metabolic Diseases
5.1. The mTOR Signaling Pathway
5.2. The Phosphatidylinositol 3-Kinase (PI3K)/Protein Kinase B (Akt) Pathway
5.3. The AMP-Activated Protein Kinase (AMPK) Pathway
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Akbar, N.; Azzimato, V.; Choudhury, R.P.; Aouadi, M. Extracellular vesicles in metabolic disease. Diabetologia 2019, 62, 2179–2187. [Google Scholar] [CrossRef] [PubMed]
- Srikanthan, K.; Feyh, A.; Visweshwar, H.; Shapiro, J.I.; Sodhi, K. Systematic Review of Metabolic Syndrome Biomarkers: A Panel for Early Detection, Management, and Risk Stratification in the West Virginian Population. Int. J. Med. Sci. 2016, 13, 25–38. [Google Scholar] [CrossRef] [PubMed]
- Saklayen, M.G. The Global Epidemic of the Metabolic Syndrome. Curr. Hypertens. Rep. 2018, 20, 12. [Google Scholar] [CrossRef]
- Berthoud, H.-R.; Neuhuber, W.L. Vagal mechanisms as neuromodulatory targets for the treatment of metabolic disease. Ann. N. Y. Acad. Sci. 2019, 1454, 42–55. [Google Scholar] [CrossRef]
- Baker, R.G.; Hayden, M.S.; Ghosh, S. NF-κB, inflammation, and metabolic disease. Cell Metab. 2011, 13, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Rani, V.; Deep, G.; Singh, R.K.; Palle, K.; Yadav, U.C.S. Oxidative stress and metabolic disorders: Pathogenesis and therapeutic strategies. Life Sci. 2016, 148, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Jia, J.; Rodrigues, B. Autophagy, Metabolic Disease, and Pathogenesis of Heart Dysfunction. Can. J. Cardiol. 2017, 33, 850–859. [Google Scholar] [CrossRef]
- Quarta, C.; Fioramonti, X.; Cota, D. POMC Neurons Dysfunction in Diet-induced Metabolic Disease: Hallmark or Mechanism of Disease? Neuroscience 2020, 447, 3–14. [Google Scholar] [CrossRef]
- Boulangé, C.L.; Neves, A.L.; Chilloux, J.; Nicholson, J.K.; Dumas, M.-E. Impact of the gut microbiota on inflammation, obesity, and metabolic disease. Genome Med. 2016, 8, 42. [Google Scholar] [CrossRef]
- Suzuki, K. Chronic Inflammation as an Immunological Abnormality and Effectiveness of Exercise. Biomolecules 2019, 9, 223. [Google Scholar] [CrossRef]
- Hotamisligil, G.S. Inflammation and metabolic disorders. Nature 2006, 444, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Gang, X.; Yang, S.; Cui, M.; Sun, L.; Li, Z.; Wang, G. The Alterations in and the Role of the Th17/Treg Balance in Metabolic Diseases. Front. Immunol. 2021, 12, 678355. [Google Scholar] [CrossRef] [PubMed]
- Touch, S.; Clément, K.; André, S. T Cell Populations and Functions Are Altered in Human Obesity and Type 2 Diabetes. Curr. Diabetes Rep. 2017, 17, 81. [Google Scholar] [CrossRef] [PubMed]
- Yi, H.-S.; Kim, S.Y.; Kim, J.T.; Lee, Y.-S.; Moon, J.S.; Kim, M.; Kang, Y.E.; Joung, K.H.; Lee, J.H.; Kim, H.J.; et al. T-cell senescence contributes to abnormal glucose homeostasis in humans and mice. Cell Death Dis. 2019, 10, 249. [Google Scholar] [CrossRef]
- Werlen, G.; Jain, R.; Jacinto, E. MTOR Signaling and Metabolism in Early T Cell Development. Genes 2021, 12, 728. [Google Scholar] [CrossRef]
- Kumar, B.V.; Connors, T.; Farber, D.L. Human T Cell Development, Localization, and Function throughout Life. Immunity 2018, 48, 202–213. [Google Scholar] [CrossRef]
- Mousset, C.M.; Hobo, W.; Woestenenk, R.; Preijers, F.; Dolstra, H.; Van Der Waart, A.B. Comprehensive Phenotyping of T Cells Using Flow Cytometry. Cytom. Part A 2019, 95, 647–654. [Google Scholar] [CrossRef]
- Hui, E. Understanding T cell signaling using membrane reconstitution. Immunol. Rev. 2019, 291, 44–56. [Google Scholar] [CrossRef]
- Wik, J.A.; Skålhegg, B.S. T Cell Metabolism in Infection. Front. Immunol. 2022, 13, 840610. [Google Scholar] [CrossRef]
- Santamaria, J.C.; Borelli, A.; Irla, M. Regulatory T Cell Heterogeneity in the Thymus: Impact on Their Functional Activities. Front. Immunol. 2021, 12, 643153. [Google Scholar] [CrossRef]
- Shiromizu, C.M.; Jancic, C.C. γδ T Lymphocytes: An Effector Cell in Autoimmunity and Infection. Front. Immunol. 2018, 9, 2389. [Google Scholar] [CrossRef]
- Sutton, C.E.; Mielke, L.A.; Mills, K.H. IL-17-producing γδ T cells and innate lymphoid cells. Eur. J. Immunol. 2012, 42, 2221–2231. [Google Scholar] [CrossRef]
- Chen, L.; Deng, H.; Cui, H.; Fang, J.; Zuo, Z.; Deng, J.; Li, Y.; Wang, X.; Zhao, L. Inflammatory responses and inflammation-associated diseases in organs. Oncotarget 2018, 9, 7204–7218. [Google Scholar] [CrossRef] [PubMed]
- Long, A.; Freeley, M. Protein kinase C: A regulator of cytoskeleton remodelling and T-cell migration. Biochem. Soc. Trans. 2014, 42, 1490–1497. [Google Scholar] [CrossRef] [PubMed]
- Sadasivam, M.; Noel, S.; Lee, S.A.; Gong, J.; Allaf, M.E.; Pierorazio, P.; Rabb, H.; Hamad, A.R.A. Activation and Proliferation of PD-1+ Kidney Double-Negative T Cells Is Dependent on Nonclassical MHC Proteins and IL-2. J. Am. Soc. Nephrol. 2019, 30, 277–292. [Google Scholar] [CrossRef] [PubMed]
- Moro-García, M.A.; Mayo, J.C.; Sainz, R.M.; Alonso-Arias, R. Influence of Inflammation in the Process of T Lymphocyte Differentiation: Proliferative, Metabolic, and Oxidative Changes. Front. Immunol. 2018, 9, 339. [Google Scholar] [CrossRef]
- Hashimoto, M.; Kamphorst, A.O.; Im, S.J.; Kissick, H.T.; Pillai, R.N.; Ramalingam, S.S.; Araki, K.; Ahmed, R. CD8 T Cell Exhaustion in Chronic Infection and Cancer: Opportunities for Interventions. Annu. Rev. Med. 2018, 69, 301–318. [Google Scholar] [CrossRef]
- Wherry, E.J.; Ahmed, R. Memory CD8 T-Cell Differentiation during Viral Infection. J. Virol. 2004, 78, 5535–5545. [Google Scholar] [CrossRef]
- Mueller, S.N.; Ahmed, R. High antigen levels are the cause of T cell exhaustion during chronic viral infection. Proc. Natl. Acad. Sci. USA 2009, 106, 8623–8628. [Google Scholar] [CrossRef]
- Wherry, E.J.; Kurachi, M. Molecular and cellular insights into T cell exhaustion. Nat. Rev. Immunol. 2015, 15, 486–499. [Google Scholar] [CrossRef]
- Barber, D.L.; Wherry, E.J.; Masopust, D.; Zhu, B.; Allison, J.P.; Sharpe, A.H.; Freeman, G.J.; Ahmed, R. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature 2006, 439, 682–687. [Google Scholar] [CrossRef] [PubMed]
- Sakuishi, K.; Apetoh, L.; Sullivan, J.M.; Blazar, B.R.; Kuchroo, V.K.; Anderson, A.C. Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. J. Exp. Med. 2010, 207, 2187–2194, Correction in J. Exp. Med. 2011, 208, 1331. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.-T.; Anderson, A.C.; Tan, W.G.; West, E.E.; Ha, S.-J.; Araki, K.; Freeman, G.J.; Kuchroo, V.K.; Ahmed, R. Cooperation of Tim-3 and PD-1 in CD8 T-cell exhaustion during chronic viral infection. Proc. Natl. Acad. Sci. USA 2010, 107, 14733–14738. [Google Scholar] [CrossRef]
- Shin, H.; Blackburn, S.D.; Blattman, J.N.; Wherry, E.J. Viral antigen and extensive division maintain virus-specific CD8 T cells during chronic infection. J. Exp. Med. 2007, 204, 941–949. [Google Scholar] [CrossRef] [PubMed]
- Ingram, J.T.; Yi, J.S.; Zajac, A.J. Exhausted CD8 T Cells Downregulate the IL-18 Receptor and Become Unresponsive to Inflammatory Cytokines and Bacterial Co-infections. PLOS Pathog. 2011, 7, e1002273. [Google Scholar] [CrossRef] [PubMed]
- Sharma, B.R.; Kanneganti, T.-D. NLRP3 inflammasome in cancer and metabolic diseases. Nat. Immunol. 2021, 22, 550–559. [Google Scholar] [CrossRef]
- Al Bander, Z.; Nitert, M.D.; Mousa, A.; Naderpoor, N. The Gut Microbiota and Inflammation: An Overview. Int. J. Environ. Res. Public Health 2020, 17, 7618. [Google Scholar] [CrossRef]
- Furman, D.; Campisi, J.; Verdin, E.; Carrera-Bastos, P.; Targ, S.; Franceschi, C.; Ferrucci, L.; Gilroy, D.W.; Fasano, A.; Miller, G.W.; et al. Chronic inflammation in the etiology of disease across the life span. Nat. Med. 2019, 25, 1822–1832. [Google Scholar] [CrossRef]
- Lee, Y.S.; Olefsky, J. Chronic tissue inflammation and metabolic disease. Genes Dev. 2021, 35, 307–328. [Google Scholar] [CrossRef]
- Saltiel, A.R.; Olefsky, J.M. Inflammatory mechanisms linking obesity and metabolic disease. J. Clin. Investig. 2017, 127, 1–4. [Google Scholar] [CrossRef]
- Arkan, M.C.; Hevener, A.L.; Greten, F.R.; Maeda, S.; Li, Z.W.; Long, J.M.; Wynshaw-Boris, A.; Poli, G.; Olefsky, J.; Karin, M. IKK-beta links inflammation to obesity-induced insulin resistance. Nat. Med. 2005, 11, 191–198. [Google Scholar] [CrossRef] [PubMed]
- Hirosumi, J.; Tuncman, G.; Chang, L.; Görgün, C.Z.; Uysal, K.T.; Maeda, K.; Karin, M.; Hotamisligil, G.S. A central role for JNK in obesity and insulin resistance. Nature 2002, 420, 333–336. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Kokoeva, M.V.; Inouye, K.; Tzameli, I.; Yin, H.; Flier, J.S. TLR4 links innate immunity and fatty acid–induced insulin resistance. J. Clin. Investig. 2006, 116, 3015–3025. [Google Scholar] [CrossRef] [PubMed]
- Vandanmagsar, B.; Youm, Y.-H.; Ravussin, A.; Galgani, J.E.; Stadler, K.; Mynatt, R.L.; Ravussin, E.; Stephens, J.M.; Dixit, V.D. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat. Med. 2011, 17, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Wellen, K.E.; Fucho, R.; Gregor, M.F.; Furuhashi, M.; Morgan, C.; Lindstad, T.; Vaillancourt, E.; Gorgun, C.Z.; Saatcioglu, F.; Hotamisligil, G.S. Coordinated Regulation of Nutrient and Inflammatory Responses by STAMP2 Is Essential for Metabolic Homeostasis. Cell 2007, 129, 537–548. [Google Scholar] [CrossRef]
- Stephens, J.; Pekala, P. Transcriptional repression of the GLUT4 and C/EBP genes in 3T3-L1 adipocytes by tumor necrosis factor-alpha. J. Biol. Chem. 1991, 266, 21839–21845. [Google Scholar] [CrossRef]
- Feinstein, R.; Kanety, H.; Papa, M.Z.; Lunenfeld, B.; Karasik, A. Tumor necrosis factor-alpha suppresses insulin-induced tyrosine phosphorylation of insulin receptor and its substrates. J. Biol. Chem. 1993, 268, 26055–26058. [Google Scholar] [CrossRef]
- Mahdi, T.; Hänzelmann, S.; Salehi, A.; Muhammed, S.J.; Reinbothe, T.M.; Tang, Y.; Axelsson, A.S.; Zhou, Y.; Jing, X.; Almgren, P.; et al. Secreted Frizzled-Related Protein 4 Reduces Insulin Secretion and Is Overexpressed in Type 2 Diabetes. Cell Metab. 2012, 16, 625–633. [Google Scholar] [CrossRef]
- Jourdan, T.; Godlewski, G.; Cinar, R.; Bertola, A.; Szanda, G.; Liu, J.; Tam, J.; Han, T.; Mukhopadhyay, B.; Skarulis, M.C.; et al. Activation of the Nlrp3 inflammasome in infiltrating macrophages by endocannabinoids mediates beta cell loss in type 2 diabetes. Nat. Med. 2013, 19, 1132–1140. [Google Scholar] [CrossRef]
- Hasnain, S.Z.; Borg, D.J.; Harcourt, B.E.; Tong, H.; Sheng, Y.H.; Ng, C.P.; Das, I.; Wang, R.; Chen, A.C.-H.; Loudovaris, T.; et al. Glycemic control in diabetes is restored by therapeutic manipulation of cytokines that regulate beta cell stress. Nat. Med. 2014, 20, 1417–1426. [Google Scholar] [CrossRef]
- Sun, K.; Kusminski, C.M.; Scherer, P.E. Adipose tissue remodeling and obesity. J. Clin. Investig. 2011, 121, 2094–2101. [Google Scholar] [CrossRef] [PubMed]
- O’Shea, J.J.; Paul, W.E. Mechanisms Underlying Lineage Commitment and Plasticity of Helper CD4 + T Cells. Science 2010, 327, 1098–1102. [Google Scholar] [CrossRef]
- Reiner, S.L. Development in Motion: Helper T Cells at Work. Cell 2007, 129, 33–36. [Google Scholar] [CrossRef]
- Hirahara, K.; Poholek, A.; Vahedi, G.; Laurence, A.; Kanno, Y.; Milner, J.D.; O’Shea, J.J. Mechanisms underlying helper T-cell plasticity: Implications for immune-mediated disease. J. Allergy Clin. Immunol. 2013, 131, 1276–1287. [Google Scholar] [CrossRef] [PubMed]
- Cosmi, L.; Maggi, L.; Santarlasci, V.; Liotta, F.; Annunziato, F. T helper cells plasticity in inflammation. Cytom. Part A 2013, 85, 36–42. [Google Scholar] [CrossRef] [PubMed]
- Oukka, M. Th17 cells in immunity and autoimmunity. Ann. Rheum. Dis. 2008, 67, iii26–iii29. [Google Scholar] [CrossRef] [PubMed]
- Schipper, H.S.; Prakken, B.; Kalkhoven, E.; Boes, M. Adipose tissue-resident immune cells: Key players in immunometabolism. Trends Endocrinol. Metab. 2012, 23, 407–415. [Google Scholar] [CrossRef]
- Ferrante, A.W., Jr. The immune cells in adipose tissue. Diabetes Obes. Metab. 2013, 15 (Suppl. 3), 34–38. [Google Scholar] [CrossRef]
- Rocha, V.Z.; Folco, E.J.; Sukhova, G.; Shimizu, K.; Gotsman, I.; Vernon, A.H.; Libby, P. Interferon-gamma, a Th1 cytokine, regulates fat inflammation: A role for adaptive immunity in obesity. Circ. Res. 2008, 103, 467–476. [Google Scholar] [CrossRef]
- Kintscher, U.; Hartge, M.; Hess, K.; Foryst-Ludwig, A.; Clemenz, M.; Wabitsch, M.; Fischer-Posovszky, P.; Barth, T.F.; Dragun, D.; Skurk, T.; et al. T-lymphocyte infiltration in visceral adipose tissue: A primary event in adipose tissue inflammation and the development of obesity-mediated insulin resistance. Arter. Thromb. Vasc. Biol. 2008, 28, 1304–1310. [Google Scholar] [CrossRef]
- Khan, I.M.; Perrard, X.-Y.D.; Perrard, J.L.; Mansoori, A.; Smith, C.W.; Wu, H.; Ballantyne, C.M. Attenuated adipose tissue and skeletal muscle inflammation in obese mice with combined CD4+ and CD8+ T cell deficiency. Atherosclerosis 2014, 233, 419–428. [Google Scholar] [CrossRef] [PubMed]
- Jaramillo, M.; Naccache, P.H.; Olivier, M. Monosodium urate crystals synergize with IFN-gamma to generate macrophage nitric oxide: Involvement of extracellular signal-regulated kinase 1/2 and NF-kappa B. J. Immunol. 2004, 172, 5734–5742. [Google Scholar] [CrossRef] [PubMed]
- Ricardo-Gonzalez, R.R.; Red Eagle, A.; Odegaard, J.I.; Jouihan, H.; Morel, C.R.; Heredia, J.E.; Mukundan, L.; Wu, D.; Locksley, R.M.; Chawla, A.; et al. IL-4/STAT6 immune axis regulates peripheral nutrient metabolism and insulin sensitivity. Proc. Natl. Acad. Sci. USA 2010, 107, 22617–22622. [Google Scholar] [CrossRef]
- Wensveen, F.M.; Valentić, S.; Šestan, M.; Turk Wensveen, T.; Polić, B. The “Big Bang” in obese fat: Events initiating obesity-induced adipose tissue inflammation. Eur. J. Immunol. 2015, 45, 2446–2456. [Google Scholar] [CrossRef] [PubMed]
- Endo, Y.; Yokote, K.; Nakayama, T. The obesity-related pathology and Th17 cells. Cell. Mol. Life Sci. 2017, 74, 1231–1245. [Google Scholar] [CrossRef]
- Ghannam, S.; Pène, J.; Torcy-Moquet, G.; Jorgensen, C.; Yssel, H. Mesenchymal Stem Cells Inhibit Human Th17 Cell Differentiation and Function and Induce a T Regulatory Cell Phenotype. J. Immunol. 2010, 185, 302–312. [Google Scholar] [CrossRef]
- Dalmas, E.; Venteclef, N.; Caer, C.; Poitou, C.; Cremer, I.; Aron-Wisnewsky, J.; Lacroix-Desmazes, S.; Bayry, J.; Kaveri, S.V.; Clément, K.; et al. T Cell–Derived IL-22 Amplifies IL-1β–Driven Inflammation in Human Adipose Tissue: Relevance to Obesity and Type 2 Diabetes. Diabetes 2014, 63, 1966–1977. [Google Scholar] [CrossRef]
- Sutti, S.; Albano, E. Adaptive immunity: An emerging player in the progression of NAFLD. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 81–92. [Google Scholar] [CrossRef]
- Rau, M.; Schilling, A.K.; Meertens, J.; Hering, I.; Weiss, J.; Jurowich, C.; Kudlich, T.; Hermanns, H.M.; Bantel, H.; Beyersdorf, N.; et al. Progression from Nonalcoholic Fatty Liver to Nonalcoholic Steatohepatitis Is Marked by a Higher Frequency of Th17 Cells in the Liver and an Increased Th17/Resting Regulatory T Cell Ratio in Peripheral Blood and in the Liver. J. Immunol. 2016, 196, 97–105. [Google Scholar] [CrossRef]
- Her, Z.; Tan, J.H.L.; Lim, Y.-S.; Tan, S.Y.; Chan, X.Y.; Tan, W.W.S.; Liu, M.; Yong, K.S.M.; Lai, F.; Ceccarello, E.; et al. CD4+ T Cells Mediate the Development of Liver Fibrosis in High Fat Diet-Induced NAFLD in Humanized Mice. Front. Immunol. 2020, 11, 580968. [Google Scholar] [CrossRef]
- Tang, Y.; Bian, Z.; Zhao, L.; Liu, Y.; Liang, S.; Wang, Q.; Han, X.; Peng, Y.; Chen, X.; Shen, L.; et al. Interleukin-17 exacerbates hepatic steatosis and inflammation in non-alcoholic fatty liver disease. Clin. Exp. Immunol. 2011, 166, 281–290. [Google Scholar] [CrossRef] [PubMed]
- Fossiez, F.; Djossou, O.; Chomarat, P.; Flores-Romo, L.; Ait-Yahia, S.; Maat, C.; Pin, J.J.; Garrone, P.; Garcia, E.; Saeland, S.; et al. T cell interleukin-17 induces stromal cells to produce proinflammatory and hematopoietic cytokines. J. Exp. Med. 1996, 183, 2593–2603. [Google Scholar] [CrossRef]
- Liu, Y.; Zhao, Q.; Yin, Y.; McNutt, M.A.; Zhang, T.; Cao, Y. Serum levels of IL-17 are elevated in patients with acute gouty arthritis. Biochem. Biophys. Res. Commun. 2018, 497, 897–902. [Google Scholar] [CrossRef]
- Yang, Q.-B.; He, Y.-L.; Zhang, Q.-B.; Mi, Q.-S.; Zhou, J.-G. Downregulation of Transcription Factor T-Bet as a Protective Strategy in Monosodium Urate-Induced Gouty Inflammation. Front. Immunol. 2019, 10, 1199. [Google Scholar] [CrossRef]
- Feuerer, M.; Herrero, L.; Cipolletta, D.; Naaz, A.; Wong, J.; Nayer, A.; Lee, J.; Goldfine, A.B.; Benoist, C.; Shoelson, S.; et al. Lean, but not obese, fat is enriched for a unique population of regulatory T cells that affect metabolic parameters. Nat. Med. 2009, 15, 930–939. [Google Scholar] [CrossRef] [PubMed]
- Deiuliis, J.; Shah, Z.; Shah, N.; Needleman, B.; Mikami, D.; Narula, V.; Perry, K.; Hazey, J.; Kampfrath, T.; Kollengode, M.; et al. Visceral Adipose Inflammation in Obesity Is Associated with Critical Alterations in Tregulatory Cell Numbers. PLoS ONE 2011, 6, e16376. [Google Scholar] [CrossRef] [PubMed]
- Kolodin, D.; van Panhuys, N.; Li, C.; Magnuson, A.M.; Cipolletta, D.; Miller, C.M.; Wagers, A.; Germain, R.N.; Benoist, C.; Mathis, D. Antigen- and Cytokine-Driven Accumulation of Regulatory T Cells in Visceral Adipose Tissue of Lean Mice. Cell Metab. 2015, 21, 543–557. [Google Scholar] [CrossRef]
- Ilan, Y.; Maron, R.; Tukpah, A.-M.; Maioli, T.U.; Murugaiyan, G.; Yang, K.; Wu, H.Y.; Weiner, H.L. Induction of regulatory T cells decreases adipose inflammation and alleviates insulin resistance in ob/ob mice. Proc. Natl. Acad. Sci. USA 2010, 107, 9765–9770. [Google Scholar] [CrossRef] [PubMed]
- Pettersson, U.S.; Waldén, T.B.; Carlsson, P.-O.; Jansson, L.; Phillipson, M. Female Mice are Protected against High-Fat Diet Induced Metabolic Syndrome and Increase the Regulatory T Cell Population in Adipose Tissue. PLoS ONE 2012, 7, e46057. [Google Scholar] [CrossRef]
- Ma, X.; Hua, J.; Mohamood, A.R.; Hamad, A.R.A.; Ravi, R.; Li, Z. A high-fat diet and regulatory T cells influence susceptibility to endotoxin-induced liver injury. Hepatology 2007, 46, 1519–1529. [Google Scholar] [CrossRef]
- Wang, H.; Zhang, H.; Wang, Y.; Brown, Z.J.; Xia, Y.; Huang, Z.; Shen, C.; Hu, Z.; Beane, J.; Ansa-Addo, E.A.; et al. Regulatory T-cell and neutrophil extracellular trap interaction contributes to carcinogenesis in non-alcoholic steatohepatitis. J. Hepatol. 2021, 75, 1271–1283. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, S.; Manabe, I.; Nagasaki, M.; Eto, K.; Yamashita, H.; Ohsugi, M.; Otsu, M.; Hara, K.; Ueki, K.; Sugiura, S.; et al. CD8+ effector T cells contribute to macrophage recruitment and adipose tissue inflammation in obesity. Nat. Med. 2009, 15, 914–920. [Google Scholar] [CrossRef] [PubMed]
- Jagannathan-Bogdan, M.; McDonnell, M.E.; Shin, H.; Rehman, Q.; Hasturk, H.; Apovian, C.M.; Nikolajczyk, B.S. Elevated Proinflammatory Cytokine Production by a Skewed T Cell Compartment Requires Monocytes and Promotes Inflammation in Type 2 Diabetes. J. Immunol. 2011, 186, 1162–1172. [Google Scholar] [CrossRef]
- Liu, X.; Li, Y.; Li, Z.; Wei, X.; Ma, Y.; Cheng, P.; Jiao, R.; Fang, J.; Xing, Y.; Tang, J.; et al. A novel IgG1 monoclonal antibody against xanthine oxidase alleviates inflammation induced by potassium oxonate in mice. Int. J. Biol. Macromol. 2018, 112, 537–547. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Sun, P.; Wu, Y.; Wang, L. Metabolic tissue-resident CD8+ T cells: A key player in obesity-related diseases. Obes. Rev. 2021, 22, e13133. [Google Scholar] [CrossRef] [PubMed]
- Rausch, M.E.; Weisberg, S.; Vardhana, P.; Tortoriello, D.V. Obesity in C57BL/6J mice is characterized by adipose tissue hypoxia and cytotoxic T-cell infiltration. Int. J. Obes. 2007, 32, 451–463. [Google Scholar] [CrossRef] [PubMed]
- Anderson, E.K.; Gutierrez, D.A.; Kennedy, A.; Hasty, A.H. Weight Cycling Increases T-Cell Accumulation in Adipose Tissue and Impairs Systemic Glucose Tolerance. Diabetes 2013, 62, 3180–3188. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharjee, J.; Kirby, M.; Softic, S.; Miles, L.; Salazar-Gonzalez, R.-M.; Shivakumar, P.; Kohli, R. Hepatic natural killer T-cell and CD8+ T-cell signatures in mice with nonalcoholic steatohepatitis. Hepatol. Commun. 2017, 1, 299–310. [Google Scholar] [CrossRef]
- Wolf, M.J.; Adili, A.; Piotrowitz, K.; Abdullah, Z.; Boege, Y.; Stemmer, K.; Ringelhan, M.; Simonavicius, N.; Egger, M.; Wohlleber, D.; et al. Metabolic Activation of Intrahepatic CD8+ T Cells and NKT Cells Causes Nonalcoholic Steatohepatitis and Liver Cancer via Cross-Talk with Hepatocytes. Cancer Cell 2014, 26, 549–564. [Google Scholar] [CrossRef]
- Van Herck, M.A.; Vonghia, L.; Kwanten, W.J.; Julé, Y.; Vanwolleghem, T.; Ebo, D.G.; Michielsen, P.P.; De Man, J.G.; Gama, L.; De Winter, B.Y.; et al. Diet Reversal and Immune Modulation Show Key Role for Liver and Adipose Tissue T Cells in Murine Nonalcoholic Steatohepatitis. Cell. Mol. Gastroenterol. Hepatol. 2020, 10, 467–490. [Google Scholar] [CrossRef]
- Ghazarian, M.; Revelo, X.S.; Nøhr, M.K.; Luck, H.; Zeng, K.; Lei, H.; Tsai, S.; Schroer, S.A.; Park, Y.J.; Chng, M.H.Y.; et al. Type I interferon responses drive intrahepatic T cells to promote metabolic syndrome. Sci. Immunol. 2017, 2, eaai7616. [Google Scholar] [CrossRef] [PubMed]
- Dudek, M.; Pfister, D.; Donakonda, S.; Filpe, P.; Schneider, A.; Laschinger, M.; Hartmann, D.; Hüser, N.; Meiser, P.; Bayerl, F.; et al. Auto-aggressive CXCR6+ CD8 T cells cause liver immune pathology in NASH. Nature 2021, 592, 444–449. [Google Scholar] [CrossRef]
- Pfister, D.; Núñez, N.G.; Pinyol, R.; Govaere, O.; Pinter, M.; Szydlowska, M.; Gupta, R.; Qiu, M.; Deczkowska, A.; Weiner, A.; et al. NASH limits anti-tumour surveillance in immunotherapy-treated HCC. Nature 2021, 592, 450–456. [Google Scholar] [CrossRef] [PubMed]
- Koda, Y.; Teratani, T.; Chu, P.-S.; Hagihara, Y.; Mikami, Y.; Harada, Y.; Tsujikawa, H.; Miyamoto, K.; Suzuki, T.; Taniki, N.; et al. CD8+ tissue-resident memory T cells promote liver fibrosis resolution by inducing apoptosis of hepatic stellate cells. Nat. Commun. 2021, 12, 4474. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.-H.; Kim, S.R.; Han, D.H.; Yu, H.T.; Han, Y.D.; Kim, J.H.; Kim, S.H.; Lee, C.J.; Min, B.-H.; Kim, D.-H.; et al. Senescent T Cells Predict the Development of Hyperglycemia in Humans. Diabetes 2018, 68, 156–162. [Google Scholar] [CrossRef] [PubMed]
- Zoncu, R.; Efeyan, A.; Sabatini, D.M. mTOR: From growth signal integration to cancer, diabetes and ageing. Nat. Rev. Mol. Cell Biol. 2011, 12, 21–35. [Google Scholar] [CrossRef]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 169, 361–371. [Google Scholar] [CrossRef]
- Yang, K.; Shrestha, S.; Zeng, H.; Karmaus, P.W.; Neale, G.; Vogel, P.; Guertin, D.A.; Lamb, R.F.; Chi, H. T cell exit from quiescence and differentiation into Th2 cells depend on Raptor-mTORC1-mediated metabolic reprogramming. Immunity 2013, 39, 1043–1056. [Google Scholar] [CrossRef]
- Ouyang, X.; Han, Y.; Qu, G.; Li, M.; Wu, N.; Liu, H.; Arojo, O.; Sun, H.; Liu, X.; Liu, U.; et al. Metabolic regulation of T cell development by Sin1–mTORC2 is mediated by pyruvate kinase M. J. Mol. Cell Biol. 2018, 11, 93–106. [Google Scholar] [CrossRef]
- Haxhinasto, S.; Mathis, D.; Benoist, C. The AKT-mTOR axis regulates de novo differentiation of CD4+Foxp3+ cells. J. Exp. Med. 2008, 205, 565–574. [Google Scholar] [CrossRef]
- Chen, H.; Zhang, L.; Zhang, H.; Xiao, Y.; Shao, L.; Li, H.; Yin, H.; Wang, R.; Liu, G.; Corley, D.; et al. Disruption of TSC1/2 signaling complex reveals a checkpoint governing thymic CD4+ CD25+ Foxp3+ regulatory T-cell development in mice. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2013, 27, 3979–3990. [Google Scholar] [CrossRef] [PubMed]
- Hoshii, T.; Kasada, A.; Hatakeyama, T.; Ohtani, M.; Tadokoro, Y.; Naka, K.; Ikenoue, T.; Ikawa, T.; Kawamoto, H.; Fehling, H.J.; et al. Loss of mTOR complex 1 induces developmental blockage in early T-lymphopoiesis and eradicates T-cell acute lymphoblastic leukemia cells. Proc. Natl. Acad. Sci. USA 2014, 111, 3805–3810. [Google Scholar] [CrossRef] [PubMed]
- Delgoffe, G.M.; Kole, T.P.; Zheng, Y.; Zarek, P.E.; Matthews, K.L.; Xiao, B.; Worley, P.F.; Kozma, S.C.; Powell, J.D. The mTOR Kinase Differentially Regulates Effector and Regulatory T Cell Lineage Commitment. Immunity 2009, 30, 832–844. [Google Scholar] [CrossRef] [PubMed]
- Delgoffe, G.M.; Pollizzi, K.N.; Waickman, A.T.; Heikamp, E.; Meyers, D.J.; Horton, M.R.; Xiao, B.; Worley, P.F.; Powell, J.D. The kinase mTOR regulates the differentiation of helper T cells through the selective activation of signaling by mTORC1 and mTORC. Nat. Immunol. 2011, 12, 295–303. [Google Scholar] [CrossRef]
- Shi, L.Z.; Wang, R.; Huang, G.; Vogel, P.; Neale, G.; Green, D.R.; Chi, H. HIF1α–dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. J. Exp. Med. 2011, 208, 1367–1376. [Google Scholar] [CrossRef]
- Yang, K.; Chi, H. mTOR and metabolic pathways in T cell quiescence and functional activation. Semin. Immunol. 2012, 24, 421–428. [Google Scholar] [CrossRef]
- Sauer, S.; Bruno, L.; Hertweck, A.; Finlay, D.; Leleu, M.; Spivakov, M.; Knight, Z.A.; Cobb, B.S.; Cantrell, D.; O’Connor, E.; et al. T cell receptor signaling controls Foxp3 expression via PI3K, Akt, and mTOR. Proc. Natl. Acad. Sci. USA 2008, 105, 7797–7802. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Bi, E.; Lu, Y.; Su, P.; Huang, C.; Liu, L.; Wang, Q.; Yang, M.; Kalady, M.F.; Qian, J.; et al. Cholesterol Induces CD8+ T Cell Exhaustion in the Tumor Microenvironment. Cell Metab. 2019, 30, 143–156.e5. [Google Scholar] [CrossRef]
- Kamyshnyi, O.; Matskevych, V.; Lenchuk, T.; Strilbytska, O.; Storey, K.; Lushchak, O. Metformin to decrease COVID-19 severity and mortality: Molecular mechanisms and therapeutic potential. Biomed. Pharmacother. 2021, 144, 112230. [Google Scholar] [CrossRef]
- Kalender, A.; Selvaraj, A.; Kim, S.Y.; Gulati, P.; Brûlé, S.; Viollet, B.; Kemp, B.E.; Bardeesy, N.; Dennis, P.; Schlager, J.J.; et al. Metformin, Independent of AMPK, Inhibits mTORC1 in a Rag GTPase-Dependent Manner. Cell Metab. 2010, 11, 390–401. [Google Scholar] [CrossRef]
- Vazirpanah, N.; Ottria, A.; van der Linden, M.; Wichers, C.G.K.; Schuiveling, M.; van Lochem, E.; Phipps-Green, A.; Merriman, T.; Zimmermann, M.; Jansen, M.; et al. mTOR inhibition by metformin impacts monosodium urate crystal-induced inflammation and cell death in gout: A prelude to a new add-on therapy? Ann. Rheum. Dis. 2019, 78, 663–671. [Google Scholar] [CrossRef] [PubMed]
- Barskova, V.G.; Eliseev, M.S.; Nasonov, E.L.; Volkov, A.; Tsapina, T.N.; Zilov, A.V.; Iakunina, I.A.; Il’Inykh, E.V.; Kudaeva, F.M. Use of metformin (siofor) in patients with gout and insulin resistance (pilot 6-month results). Ter. Arkhiv 2005, 77, 44–49. [Google Scholar]
- Barskova, V.G.; Eliseev, M.S.; Kudaeva, F.M.; Aleksandrova, E.; Volkov, A.; Nasonova, V.A.; Nasonov, E.L. Effect of metformin on the clinical course of gout and insulin resistance. Klin. Meditsina 2009, 87, 41–46. [Google Scholar]
- Makki, K.; Taront, S.; Molendi-Coste, O.; Bouchaert, E.; Neve, B.; Eury, E.; Lobbens, S.; Labalette, M.; Duez, H.; Staels, B.; et al. Beneficial Metabolic Effects of Rapamycin Are Associated with Enhanced Regulatory Cells in Diet-Induced Obese Mice. PLoS ONE 2014, 9, e92684. [Google Scholar] [CrossRef] [PubMed]
- Abeyrathna, P.; Su, Y. The critical role of Akt in cardiovascular function. Vasc. Pharmacol. 2015, 74, 38–48. [Google Scholar] [CrossRef]
- Molina, S.A.; Moriarty, H.K.; Infield, D.T.; Imhoff, B.R.; Vance, R.J.; Kim, A.H.; Hansen, J.M.; Hunt, W.R.; Koval, M.; McCarty, N.A. Insulin signaling via the PI3-kinase/Akt pathway regulates airway glucose uptake and barrier function in a CFTR-dependent manner. Am. J. Physiol. Cell. Mol. Physiol. 2017, 312, L688–L702. [Google Scholar] [CrossRef] [PubMed]
- Fruman, D.A.; Meyers, R.E.; Cantley, L.C. Phosphoinositide kinases. Annu. Rev. Biochem. 1998, 67, 481–507. [Google Scholar] [CrossRef]
- Guo, H.; German, P.; Bai, S.; Barnes, S.; Guo, W.; Qi, X.; Lou, H.; Liang, J.; Jonasch, E.; Mills, G.B.; et al. The PI3K/AKT Pathway and Renal Cell Carcinoma. J. Genet. Genom. 2015, 42, 343–353. [Google Scholar] [CrossRef]
- Krycer, J.R.; Sharpe, L.J.; Luu, W.; Brown, A.J. The Akt–SREBP nexus: Cell signaling meets lipid metabolism. Trends Endocrinol. Metab. 2010, 21, 268–276. [Google Scholar] [CrossRef]
- Soond, D.R.; Garçon, F.; Patton, D.T.; Rolf, J.; Turner, M.; Scudamore, C.; Garden, O.A.; Okkenhaug, K. Pten Loss in CD4 T Cells Enhances Their Helper Function but Does Not Lead to Autoimmunity or Lymphoma. J. Immunol. 2012, 188, 5935–5943. [Google Scholar] [CrossRef]
- Okkenhaug, K. Signaling by the Phosphoinositide 3-Kinase Family in Immune Cells. Annu. Rev. Immunol. 2013, 31, 675–704. [Google Scholar] [CrossRef] [PubMed]
- Rolf, J.; Bell, S.E.; Kovesdi, D.; Janas, M.L.; Soond, D.R.; Webb, L.M.C.; Santinelli, S.; Saunders, T.; Hebeis, B.; Killeen, N.; et al. Phosphoinositide 3-Kinase Activity in T Cells Regulates the Magnitude of the Germinal Center Reaction. J. Immunol. 2010, 185, 4042–4052. [Google Scholar] [CrossRef] [PubMed]
- So, L.; Fruman, D.A. PI3K signalling in B- and T-lymphocytes: New developments and therapeutic advances. Biochem. J. 2012, 442, 465–481. [Google Scholar] [CrossRef] [PubMed]
- Deane, J.A.; Kharas, M.G.; Oak, J.S.; Stiles, L.N.; Luo, J.; Moore, T.; Ji, H.; Rommel, C.; Cantley, L.; Lane, T.E.; et al. T-cell function is partially maintained in the absence of class IA phosphoinositide 3-kinase signaling. Blood 2006, 109, 2894–2902. [Google Scholar] [CrossRef]
- Okkenhaug, K.; Patton, D.T.; Bilancio, A.; Garçon, F.; Rowan, W.C.; Vanhaesebroeck, B. The p110delta isoform of phosphoinositide 3-kinase controls clonal expansion and differentiation of Th cells. J. Immunol. 2006, 177, 5122–5128. [Google Scholar] [CrossRef]
- Kerdiles, Y.M.; Stone, E.L.; Beisner, D.L.; McGargill, M.A.; Ch’En, I.L.; Stockmann, C.; Katayama, C.D.; Hedrick, S.M. Foxo Transcription Factors Control Regulatory T Cell Development and Function. Immunity 2010, 33, 890–904. [Google Scholar] [CrossRef]
- Ouyang, W.; Beckett, O.; Ma, Q.; Paik, J.-H.; DePinho, R.; Li, M. Foxo proteins cooperatively control the differentiation of Foxp3+ regulatory T cells. Nat. Immunol. 2010, 11, 618–627. [Google Scholar] [CrossRef]
- Harada, Y.; Harada, Y.; Elly, C.; Ying, G.; Paik, J.-H.; DePinho, R.A.; Liu, Y.-C. Transcription factors Foxo3a and Foxo1 couple the E3 ligase Cbl-b to the induction of Foxp3 expression in induced regulatory T cells. J. Exp. Med. 2010, 207, 1381–1391. [Google Scholar] [CrossRef]
- Zhang, T.T.; Okkenhaug, K.; Nashed, B.F.; Puri, K.D.; Knight, Z.A.; Shokat, K.M.; Vanhaesebroeck, B.; Marshall, A.J. Genetic or pharmaceutical blockade of p110delta phosphoinositide 3-kinase enhances IgE production. J. Allergy Clin. Immunol. 2008, 122, 811–819.e2. [Google Scholar] [CrossRef]
- Tan, C.L.; Kuchroo, J.R.; Sage, P.T.; Liang, D.; Francisco, L.M.; Buck, J.; Thaker, Y.R.; Zhang, Q.; McArdel, S.L.; Juneja, V.R.; et al. PD-1 restraint of regulatory T cell suppressive activity is critical for immune tolerance. J. Exp. Med. 2021, 218, e20182232. [Google Scholar] [CrossRef]
- Huang, X.; Liu, G.; Guo, J.; Su, Z. The PI3K/AKT pathway in obesity and type 2 diabetes. Int. J. Biol. Sci. 2018, 14, 1483–1496. [Google Scholar] [CrossRef] [PubMed]
- Carling, D. AMPK signalling in health and disease. Curr. Opin. Cell Biol. 2017, 45, 31–37. [Google Scholar] [CrossRef]
- Ma, E.H.; Poffenberger, M.C.; Wong, A.H.-T.; Jones, R.G. The role of AMPK in T cell metabolism and function. Curr. Opin. Immunol. 2017, 46, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Tamás, P.; Hawley, S.A.; Clarke, R.G.; Mustard, K.J.; Green, K.; Hardie, G.; Cantrell, D.A. Regulation of the energy sensor AMP-activated protein kinase by antigen receptor and Ca2+ in T lymphocytes. J. Exp. Med. 2006, 203, 1665–1670. [Google Scholar] [CrossRef] [PubMed]
- Blagih, J.; Coulombe, F.; Vincent, E.E.; Dupuy, F.; Galicia-Vázquez, G.; Yurchenko, E.; Raissi, T.C.; van der Windt, G.J.; Viollet, B.; Pearce, E.L.; et al. The Energy Sensor AMPK Regulates T Cell Metabolic Adaptation and Effector Responses In Vivo. Immunity 2015, 42, 41–54. [Google Scholar] [CrossRef] [PubMed]
- Pearce, E.L.; Walsh, M.C.; Cejas, P.J.; Harms, G.M.; Shen, H.; Wang, L.-S.; Jones, R.G.; Choi, Y. Enhancing CD8 T-cell memory by modulating fatty acid metabolism. Nature 2009, 460, 103–107. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.T.; Palanivel, V.R.; Kinjyo, I.; Schambach, F.; Intlekofer, A.M.; Banerjee, A.; Longworth, S.A.; Vinup, K.E.; Mrass, P.; Oliaro, J.; et al. Asymmetric T Lymphocyte Division in the Initiation of Adaptive Immune Responses. Science 2007, 315, 1687–1691. [Google Scholar] [CrossRef] [PubMed]
- Pollizzi, K.N.; Sun, I.-H.; Patel, C.H.; Lo, Y.-C.; Oh, M.-H.; Waickman, A.T.; Tam, A.J.; Blosser, R.L.; Wen, J.; Delgoffe, G.M.; et al. Asymmetric inheritance of mTORC1 kinase activity during division dictates CD8+ T cell differentiation. Nat. Immunol. 2016, 17, 704–711. [Google Scholar] [CrossRef]
- Nyambuya, T.M.; Dludla, P.V.; Mxinwa, V.; Mokgalaboni, K.; Ngcobo, S.R.; Tiano, L.; Nkambule, B.B. The impact of metformin and aspirin on T-cell mediated inflammation: A systematic review of in vitro and in vivo findings. Life Sci. 2020, 255, 117854. [Google Scholar] [CrossRef]
- Wang, B.; Chen, S.; Qian, H.; Zheng, Q.; Chen, R.; Liu, Y.; Shi, G. Role of T cells in the pathogenesis and treatment of gout. Int. Immunopharmacol. 2020, 88, 106877. [Google Scholar] [CrossRef]
- O’Rourke, R.W. Inflammation, obesity, and the promise of immunotherapy for metabolic disease. Surg. Obes. Relat. Dis. 2012, 9, 609–616. [Google Scholar] [CrossRef] [PubMed]
- Malozowski, S.; Sahlroot, J.T. Interleukin-1–Receptor Antagonist in Type 2 Diabetes Mellitus. New Engl. J. Med. 2007, 357, 302–303. [Google Scholar] [CrossRef] [PubMed]
- Weisberg, S.P.; Hunter, D.; Huber, R.; Lemieux, J.; Slaymaker, S.; Vaddi, K.; Charo, I.; Leibel, R.L.; Ferrante, A.W., Jr. CCR2 modulates inflammatory and metabolic effects of high-fat feeding. J. Clin. Investig. 2006, 116, 115–124. [Google Scholar] [CrossRef] [PubMed]
- O’Rourke, R.W.; White, A.E.; Metcalf, M.D.; Winters, B.R.; Diggs, B.S.; Zhu, X.; Marks, D.L. Systemic inflammation and insulin sensitivity in obese IFN-γ knockout mice. Metabolism 2012, 61, 1152–1161. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, W.; Li, C.; Zhang, D.; Li, Z.; Xia, P.; Liu, X.; Cai, X.; Yang, P.; Ling, J.; Zhang, J.; et al. Advances in T Cells Based on Inflammation in Metabolic Diseases. Cells 2022, 11, 3554. https://doi.org/10.3390/cells11223554
Yu W, Li C, Zhang D, Li Z, Xia P, Liu X, Cai X, Yang P, Ling J, Zhang J, et al. Advances in T Cells Based on Inflammation in Metabolic Diseases. Cells. 2022; 11(22):3554. https://doi.org/10.3390/cells11223554
Chicago/Turabian StyleYu, Wenlu, Chunxiu Li, Deju Zhang, Zhangwang Li, Panpan Xia, Xiao Liu, Xia Cai, Pingping Yang, Jitao Ling, Jing Zhang, and et al. 2022. "Advances in T Cells Based on Inflammation in Metabolic Diseases" Cells 11, no. 22: 3554. https://doi.org/10.3390/cells11223554
APA StyleYu, W., Li, C., Zhang, D., Li, Z., Xia, P., Liu, X., Cai, X., Yang, P., Ling, J., Zhang, J., Zhang, M., & Yu, P. (2022). Advances in T Cells Based on Inflammation in Metabolic Diseases. Cells, 11(22), 3554. https://doi.org/10.3390/cells11223554