

MST4: A Potential Oncogene and Therapeutic Target in Breast Cancer

 , , , , ,
, , , , ,  , , and
, , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines and Transfection
2.2. Generation of Stable Cell Lines
2.3. MST4 Wild Type and MST4 Kinase Defective Mutant Stable Cell Line Construction
2.4. Biological Phenotype Assays
2.5. EMT Gene PCR Array
2.6. Western Blot Analysis
2.7. Tissue Microarray Analysis
2.8. TCGA Data Analysis
2.9. Statistical Analysis
3. Results
3.1. MST4 Increases Breast Cancer Cell Growth
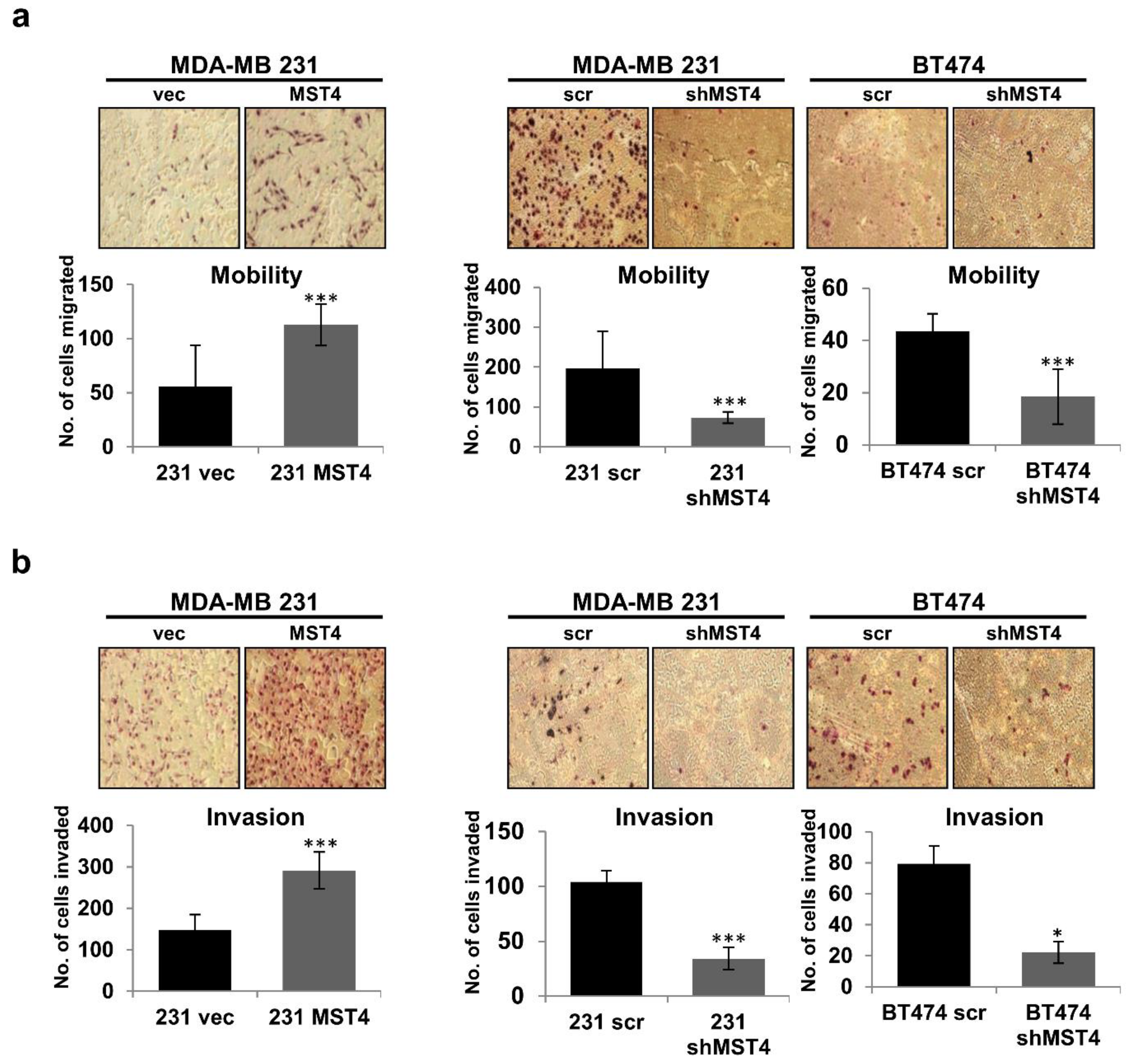
3.2. MST4 Promotes the Invasive Properties of Breast Cancer Cells
3.3. Activation of MST4 Is Required for Breast Cancer Cell Survival and Enhances Chemoresistance
3.4. MST4 Regulates Epithelial-Mesenchymal Transition in Breast Cancer
3.5. MST4 Regulates AKT Pathway
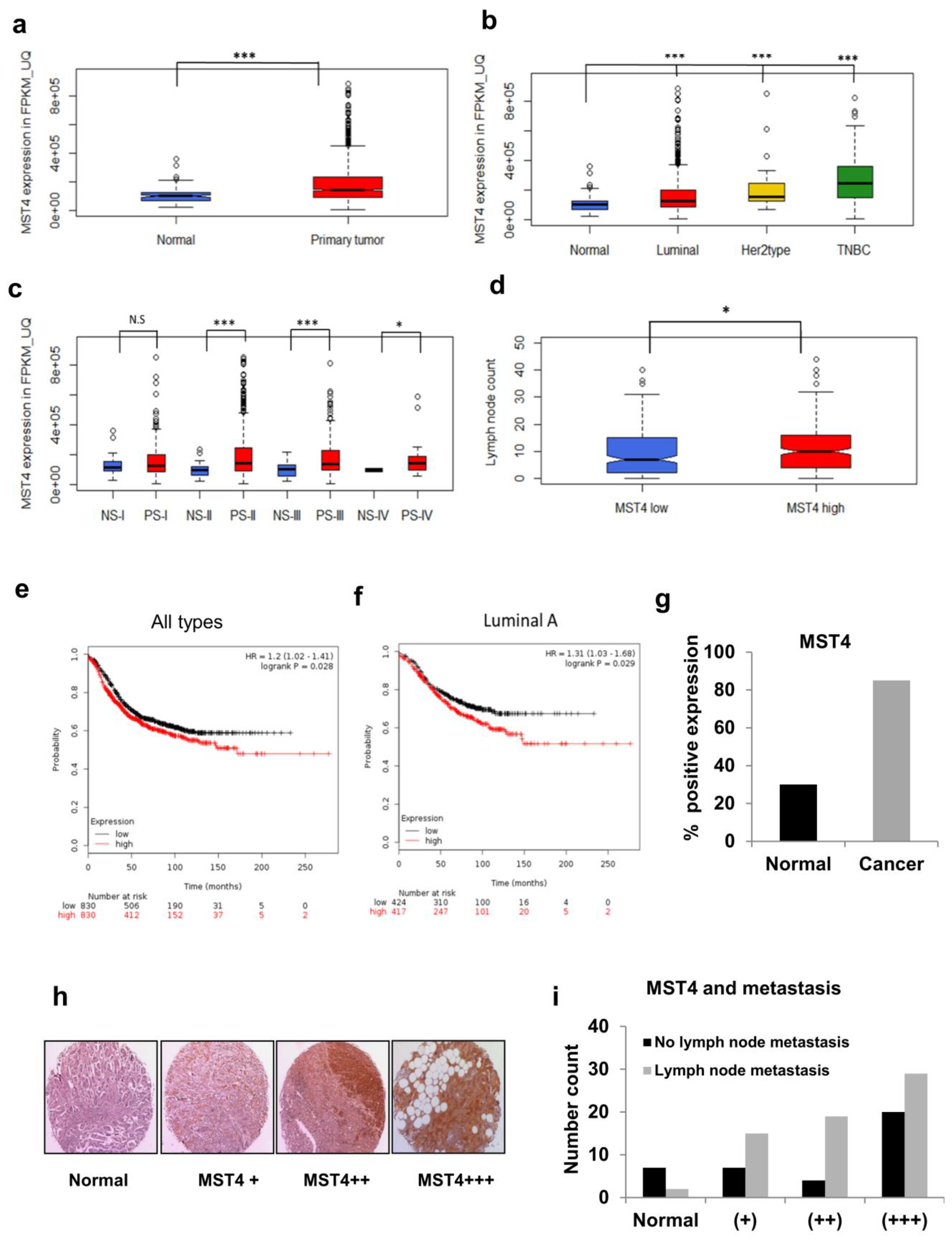
3.6. TCGA and Patient Tissue Microarray Analyses of MST4 Expression
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Prat, A.; Perou, C.M. Deconstructing the molecular portraits of breast cancer. Mol. Oncol. 2011, 5, 5–23. [Google Scholar] [CrossRef] [PubMed]
- Taube, J.H.; Herschkowitz, J.I.; Komurov, K.; Zhou, A.Y.; Gupta, S.; Yang, J.; Hartwell, K.; Onder, T.T.; Gupta, P.B.; Evans, K.W.; et al. Core epithelial-to-mesenchymal transition interactome gene-expression signature is associated with claudin-low and metaplastic breast cancer subtypes. Proc. Natl. Acad. Sci. USA 2010, 107, 15449–15454. [Google Scholar] [CrossRef] [PubMed]
- Micalizzi, D.S.; Ford, H.L. Epithelial-mesenchymal transition in development and cancer. Future Oncol. 2009, 5, 1129–1143. [Google Scholar] [CrossRef] [PubMed]
- Scimeca, M.; Antonacci, C.; Colombo, D.; Bonfiglio, R.; Buonomo, O.C.; Bonanno, E. Emerging prognostic markers related to mesenchymal characteristics of poorly differentiated breast cancers. Tumour Biol. 2016, 37, 5427–5435. [Google Scholar] [CrossRef] [PubMed]
- Hazan, R.B.; Qiao, R.; Keren, R.; Badano, I.; Suyama, K. Cadherin switch in tumor progression. Ann. N. Y. Acad. Sci. 2004, 1014, 155–163. [Google Scholar] [CrossRef]
- Maeda, M.; Johnson, K.R.; Wheelock, M.J. Cadherin switching: Essential for behavioral but not morphological changes during an epithelium-to-mesenchyme transition. J. Cell Sci. 2005, 118, 873–887. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, X.; Shang, M.; Zhang, Y.; Xia, B.; Niu, M.; Liu, Y.; Pang, D. Dysregulated expression of Slug, vimentin, and E-cadherin correlates with poor clinical outcome in patients with basal-like breast cancer. J. Surg. Oncol. 2013, 107, 188–194. [Google Scholar] [CrossRef]
- Joannes, A.; Grelet, S.; Duca, L.; Gilles, C.; Kileztky, C.; Dalstein, V.; Birembaut, P.; Polette, M.; Nawrocki-Raby, B. Fhit regulates EMT targets through an EGFR/Src/ERK/Slug signaling axis in human bronchial cells. Mol. Cancer Res. 2014, 12, 775–783. [Google Scholar] [CrossRef]
- Shah, P.P.; Fong, M.Y.; Kakar, S.S. PTTG induces EMT through integrin alphaVbeta3-focal adhesion kinase signaling in lung cancer cells. Oncogene 2012, 31, 3124–3135. [Google Scholar] [CrossRef]
- Guo, Y.H.; Wang, L.Q.; Li, B.; Xu, H.; Yang, J.H.; Zheng, L.S.; Yu, P.; Zhou, A.D.; Zhang, Y.; Xie, S.J.; et al. Wnt/beta-catenin pathway transactivates microRNA-150 that promotes EMT of colorectal cancer cells by suppressing CREB signaling. Oncotarget 2016, 7, 42513–42526. [Google Scholar] [CrossRef]
- Liu, X.; Li, Z.; Song, Y.; Wang, R.; Han, L.; Wang, Q.; Jiang, K.; Kang, C.; Zhang, Q. AURKA induces EMT by regulating histone modification through Wnt/beta-catenin and PI3K/Akt signaling pathway in gastric cancer. Oncotarget 2016, 7, 33152–33164. [Google Scholar] [CrossRef] [PubMed]
- Espinoza, I.; Miele, L. Deadly crosstalk: Notch signaling at the intersection of EMT and cancer stem cells. Cancers Lett. 2013, 341, 41–45. [Google Scholar] [CrossRef] [PubMed]
- Nath, A.K.; Brown, R.M.; Michaud, M.; Sierra-Honigmann, M.R.; Snyder, M.; Madri, J.A. Leptin affects endocardial cushion formation by modulating EMT and migration via Akt signaling cascades. J. Cell Biol. 2008, 181, 367–380. [Google Scholar] [CrossRef] [PubMed]
- Wolf, D.; Witte, V.; Laffert, B.; Blume, K.; Stromer, E.; Trapp, S.; d’Aloja, P.; Schurmann, A.; Baur, A.S. HIV-1 Nef associated PAK and PI3-kinases stimulate Akt-independent Bad-phosphorylation to induce anti-apoptotic signals. Nat. Med. 2001, 7, 1217–1224. [Google Scholar] [CrossRef] [PubMed]
- Mikami, I.; Zhang, F.; Hirata, T.; Okamoto, J.; Koizumi, K.; Shimizu, K.; Jablons, D.; He, B. Inhibition of activated phosphatidylinositol 3-kinase/AKT pathway in malignant pleural mesothelioma leads to G1 cell cycle arrest. Oncol. Rep. 2010, 24, 1677–1681. [Google Scholar]
- Taniguchi, C.M.; Kondo, T.; Sajan, M.; Luo, J.; Bronson, R.; Asano, T.; Farese, R.; Cantley, L.C.; Kahn, C.R. Divergent regulation of hepatic glucose and lipid metabolism by phosphoinositide 3-kinase via Akt and PKClambda/zeta. Cell Metab. 2006, 3, 343–353. [Google Scholar] [CrossRef]
- Joassard, O.R.; Amirouche, A.; Gallot, Y.S.; Desgeorges, M.M.; Castells, J.; Durieux, A.C.; Berthon, P.; Freyssenet, D.G. Regulation of Akt-mTOR, ubiquitin-proteasome and autophagy-lysosome pathways in response to formoterol administration in rat skeletal muscle. Int. J. Biochem. Cell Biol. 2013, 45, 2444–2455. [Google Scholar] [CrossRef]
- Xue, X.; Wang, X.; Liu, Y.; Teng, G.; Wang, Y.; Zang, X.; Wang, K.; Zhang, J.; Xu, Y.; Wang, J.; et al. SchA-p85-FAK complex dictates isoform-specific activation of Akt2 and subsequent PCBP1-mediated post-transcriptional regulation of TGFbeta-mediated epithelial to mesenchymal transition in human lung cancer cell line A549. Tumour Biol. 2014, 35, 7853–7859. [Google Scholar] [CrossRef]
- Liu, A.X.; Testa, J.R.; Hamilton, T.C.; Jove, R.; Nicosia, S.V.; Cheng, J.Q. AKT2, a member of the protein kinase B family, is activated by growth factors, v-Ha-ras, and v-src through phosphatidylinositol 3-kinase in human ovarian epithelial cancer cells. Cancer Res. 1998, 58, 2973–2977. [Google Scholar]
- Zhu, J.; Wang, M.; He, J.; Zhu, M.; Wang, J.C.; Jin, L.; Wang, X.F.; Yang, Y.J.; Xiang, J.Q.; Wei, Q. Polymorphisms in the AKT1 and AKT2 genes and oesophageal squamous cell carcinoma risk in an Eastern Chinese population. J. Cell. Mol. Med. 2016, 20, 666–677. [Google Scholar] [CrossRef]
- Schmitt, D.C.; Madeira da Silva, L.; Zhang, W.; Liu, Z.; Arora, R.; Lim, S.; Schuler, A.M.; McClellan, S.; Andrews, J.F.; Kahn, A.G.; et al. ErbB2-intronic microRNA-4728: A novel tumor suppressor and antagonist of oncogenic MAPK signaling. Cell Death Dis. 2015, 6, e1742. [Google Scholar] [CrossRef] [PubMed]
- Qian, Z.; Lin, C.; Espinosa, R.; LeBeau, M.; Rosner, M.R. Cloning and characterization of MST4, a novel Ste20-like kinase. J. Biol. Chem. 2001, 276, 22439–22445. [Google Scholar] [CrossRef] [PubMed]
- Ceccarelli, D.F.; Laister, R.C.; Mulligan, V.K.; Kean, M.J.; Goudreault, M.; Scott, I.C.; Derry, W.B.; Chakrabartty, A.; Gingras, A.C.; Sicheri, F. CCM3/PDCD10 heterodimerizes with germinal center kinase III (GCKIII) proteins using a mechanism analogous to CCM3 homodimerization. J. Biol. Chem. 2011, 286, 25056–25064. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.L.; Chen, H.C.; Fang, H.I.; Robinson, D.; Kung, H.J.; Shih, H.M. MST4, a new Ste20-related kinase that mediates cell growth and transformation via modulating ERK pathway. Oncogene 2001, 20, 6559–6569. [Google Scholar] [CrossRef][Green Version]
- Jiang, H.; Wang, W.; Zhang, Y.; Yao, W.W.; Jiang, J.; Qin, B.; Yao, W.Y.; Liu, F.; Wu, H.; Ward, T.L.; et al. Cell Polarity Kinase MST4 Cooperates with cAMP-dependent Kinase to Orchestrate Histamine-stimulated Acid Secretion in Gastric Parietal Cells. J. Biol. Chem. 2015, 290, 28272–28285. [Google Scholar] [CrossRef]
- Lin, Z.H.; Wang, L.; Zhang, J.B.; Liu, Y.; Li, X.Q.; Guo, L.; Zhang, B.; Zhu, W.W.; Ye, Q.H. MST4 promotes hepatocellular carcinoma epithelial-mesenchymal transition and metastasis via activation of the p-ERK pathway. Int. J Oncol. 2014, 45, 629–640. [Google Scholar] [CrossRef]
- ten Klooster, J.P.; Jansen, M.; Yuan, J.; Oorschot, V.; Begthel, H.; Di Giacomo, V.; Colland, F.; de Koning, J.; Maurice, M.M.; Hornbeck, P.; et al. Mst4 and Ezrin induce brush borders downstream of the Lkb1/Strad/Mo25 polarization complex. Dev. Cell 2009, 16, 551–562. [Google Scholar] [CrossRef]
- Ma, X.; Zhao, H.; Shan, J.; Long, F.; Chen, Y.; Chen, Y.; Zhang, Y.; Han, X.; Ma, D. PDCD10 interacts with Ste20-related kinase MST4 to promote cell growth and transformation via modulation of the ERK pathway. Mol. Biol. Cell 2007, 18, 1965–1978. [Google Scholar] [CrossRef]
- Sung, V.; Luo, W.; Qian, D.; Lee, I.; Jallal, B.; Gishizky, M. The Ste20 kinase MST4 plays a role in prostate cancer progression. Cancer Res. 2003, 63, 3356–3363. [Google Scholar]
- Xiong, W.; Matheson, C.J.; Xu, M.; Backos, D.S.; Mills, T.S.; Salian-Mehta, S.; Kiseljak-Vassiliades, K.; Reigan, P.; Wierman, M.E. Structure-Based Screen Identification of a Mammalian Ste20-like Kinase 4 (MST4) Inhibitor with Therapeutic Potential for Pituitary Tumors. Mol. Cancer Ther. 2016, 15, 412–420. [Google Scholar] [CrossRef]
- Perez, E.A. Paclitaxel in Breast Cancer. Oncologist 1998, 3, 373–389. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.; Massague, J. Epithelial-mesenchymal transitions: Twist in development and metastasis. Cells 2004, 118, 277–279. [Google Scholar] [CrossRef] [PubMed]
- Tania, M.; Khan, M.A.; Fu, J. Epithelial to mesenchymal transition inducing transcription factors and metastatic cancer. Tumour Biol. 2014, 35, 7335–7342. [Google Scholar] [CrossRef] [PubMed]
- Xiong, W.; Knox, A.J.; Xu, M.; Kiseljak-Vassiliades, K.; Colgan, S.P.; Brodsky, K.S.; Kleinschmidt-Demasters, B.K.; Lillehei, K.O.; Wierman, M.E. Mammalian Ste20-like kinase 4 promotes pituitary cell proliferation and survival under hypoxia. Mol. Endocrinol. 2015, 29, 460–472. [Google Scholar] [CrossRef] [PubMed]
- Toson, B.; Fortes, I.S.; Roesler, R.; Andrade, S.F. Targeting Akt/PKB in pediatric tumors: A review from preclinical to clinical trials. Pharmacol. Res. 2022, 183, 106403. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Lin, M.; Dong, C.; Tang, Y.; An, L.; Ju, J.; Wen, F.; Chen, F.; Wang, M.; Wang, W.; et al. An MST4-pβ-Catenin(Thr40) Signaling Axis Controls Intestinal Stem Cell and Tumorigenesis. Adv. Sci. 2021, 8, e2004850. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Deng, L.; He, X.; Jiang, G.; Hu, F.; Ye, S.; You, Y.; Duanmu, J.; Dai, H.; Huang, G.; et al. MST4 Predicts Poor Prognosis and Promotes Metastasis by Facilitating Epithelial-Mesenchymal Transition In Gastric Cancer. Cancer Manag. Res. 2019, 11, 9353–9369. [Google Scholar] [CrossRef]
- Huang, T.; Kim, C.K.; Alvarez, A.A.; Pangeni, R.P.; Wan, X.; Song, X.; Shi, T.; Yang, Y.; Sastry, N.; Horbinski, C.M.; et al. MST4 Phosphorylation of ATG4B Regulates Autophagic Activity, Tumorigenicity, and Radioresistance in Glioblastoma. Cancer Cell 2017, 32, 840–855. [Google Scholar] [CrossRef]
- Dian, M.J.; Li, J.; Zhang, X.L.; Li, Z.J.; Zhou, Y.; Zhou, W.; Zhong, Q.L.; Pang, W.Q.; Lin, X.L.; Liu, T.; et al. MST4 negatively regulates the EMT, invasion and metastasis of HCC cells by inactivating PI3K/AKT/Snail1 axis. J. Cancer 2021, 12, 4463–4477. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arora, R.; Kim, J.-H.; Getu, A.A.; Angajala, A.; Chen, Y.-L.; Wang, B.; Kahn, A.G.; Chen, H.; Reshi, L.; Lu, J.; et al. MST4: A Potential Oncogene and Therapeutic Target in Breast Cancer. Cells 2022, 11, 4057. https://doi.org/10.3390/cells11244057
Arora R, Kim J-H, Getu AA, Angajala A, Chen Y-L, Wang B, Kahn AG, Chen H, Reshi L, Lu J, et al. MST4: A Potential Oncogene and Therapeutic Target in Breast Cancer. Cells. 2022; 11(24):4057. https://doi.org/10.3390/cells11244057
Chicago/Turabian StyleArora, Ritu, Jin-Hwan Kim, Ayechew A. Getu, Anusha Angajala, Yih-Lin Chen, Bin Wang, Andrea G. Kahn, Hong Chen, Latif Reshi, Jianrong Lu, and et al. 2022. "MST4: A Potential Oncogene and Therapeutic Target in Breast Cancer" Cells 11, no. 24: 4057. https://doi.org/10.3390/cells11244057
APA StyleArora, R., Kim, J.-H., Getu, A. A., Angajala, A., Chen, Y.-L., Wang, B., Kahn, A. G., Chen, H., Reshi, L., Lu, J., Zhang, W., Zhou, M., & Tan, M. (2022). MST4: A Potential Oncogene and Therapeutic Target in Breast Cancer. Cells, 11(24), 4057. https://doi.org/10.3390/cells11244057