
Genotype-Phenotype Correlations in Human Diseases Caused by Mutations of LINC Complex-Associated Genes: A Systematic Review and Meta-Summary


Abstract
1. Introduction
2. Methods
2.1. Overview of Methods
2.2. Eligibility Criteria
Inclusion Criteria
2.3. Exclusion Criteria
2.4. Information Sources
2.5. Search Strategy
2.6. Selection Process
2.7. Data Collection Process
2.8. Risk of Bias and Certainty Assessment
2.9. Limitations of the Study
3. Results
3.1. Study Selection
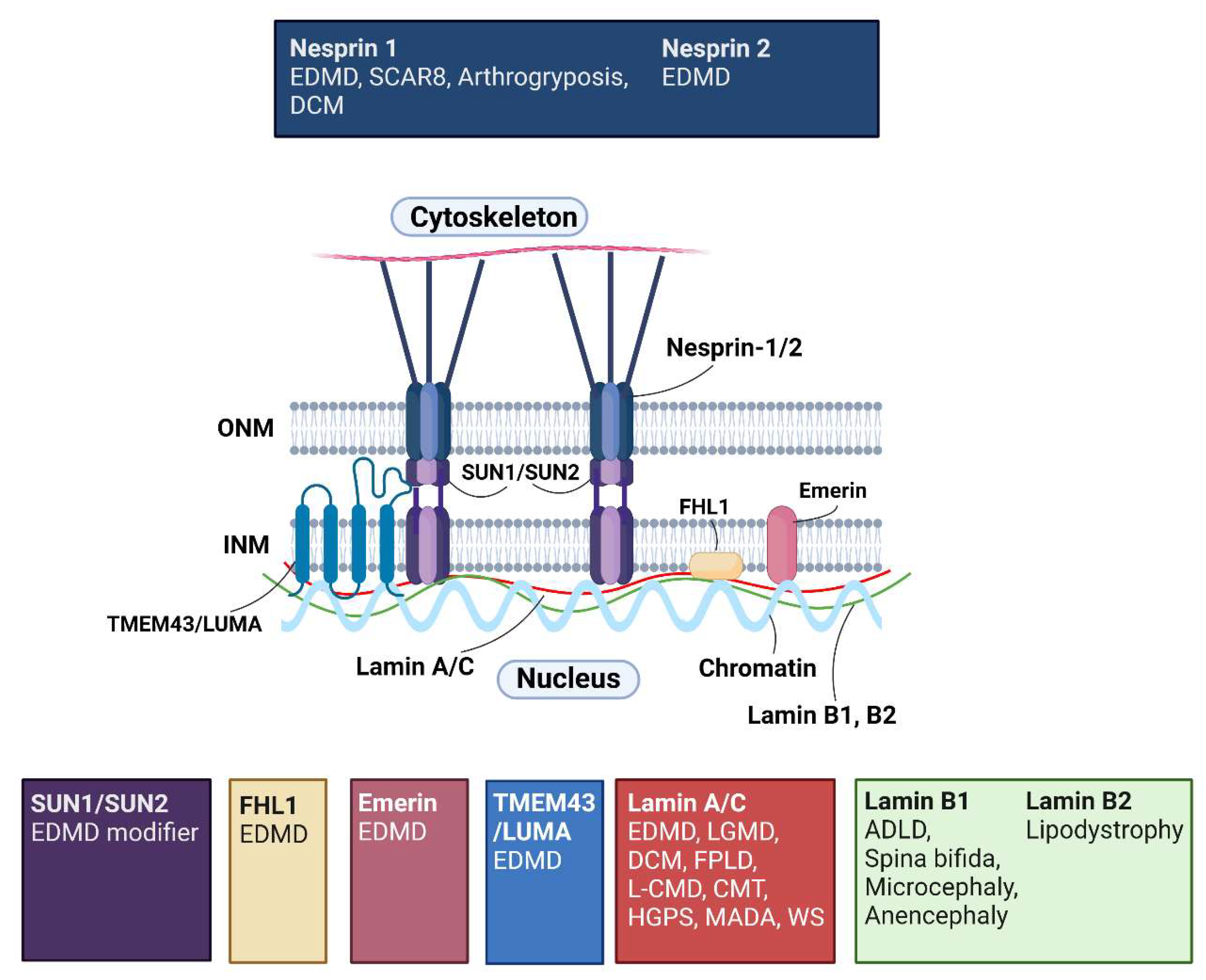
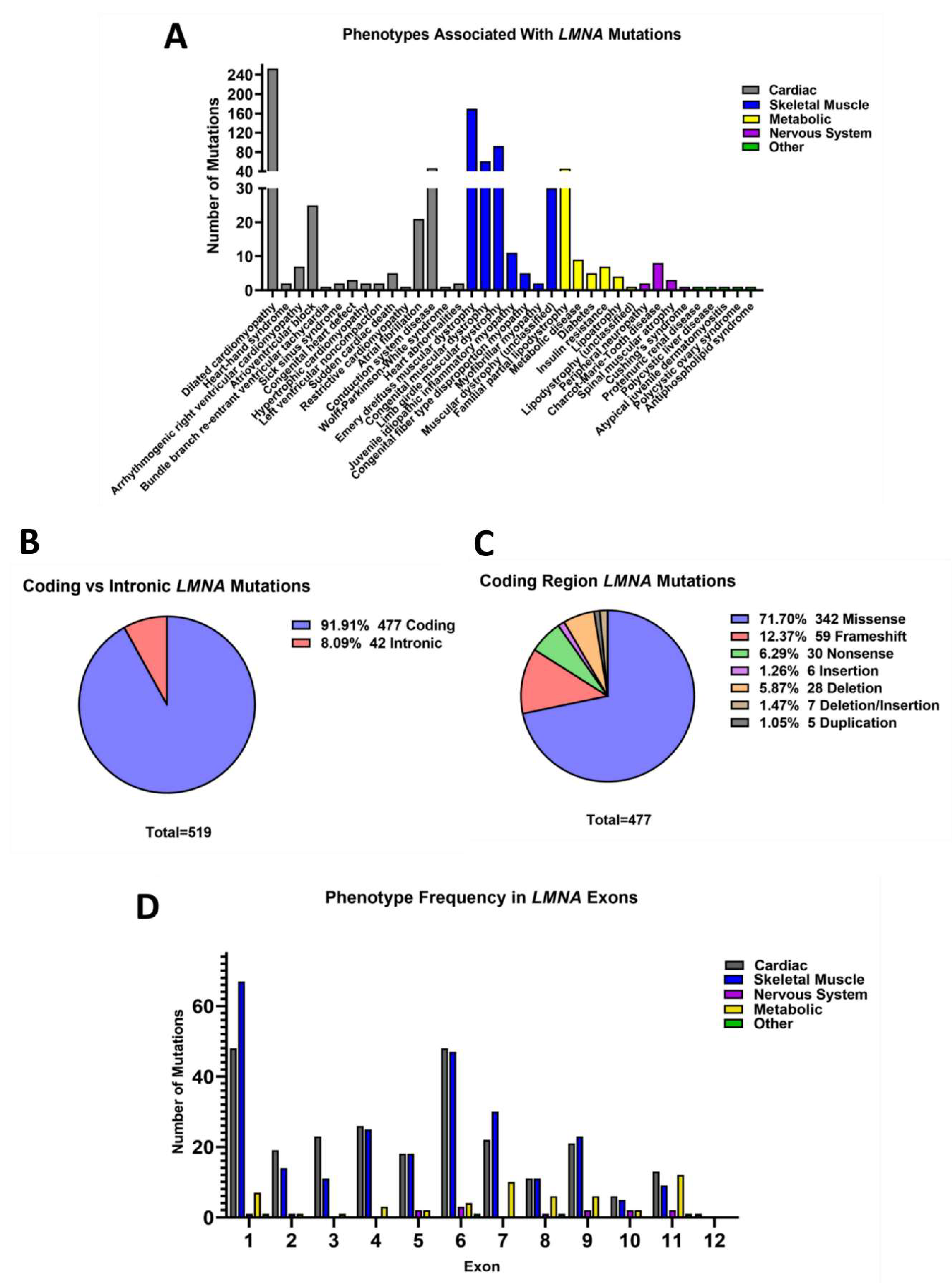
3.2. Nuclear Lamins
3.3. Fewer Mutations Are Reported in B-Type Lamins Compared to Lamin A/C
3.4. Inner Nuclear Membrane
3.4.1. EMD Is Implicated in Diseases Other Than Emery-Dreifuss Muscular Dystrophy
3.4.2. Mutations in Inner Nuclear Membrane Protein, TMEM43, and Integral LINC Complex Components SUN1/SUN2, Are Also Linked to Emery-Dreifuss Muscular Dystrophy
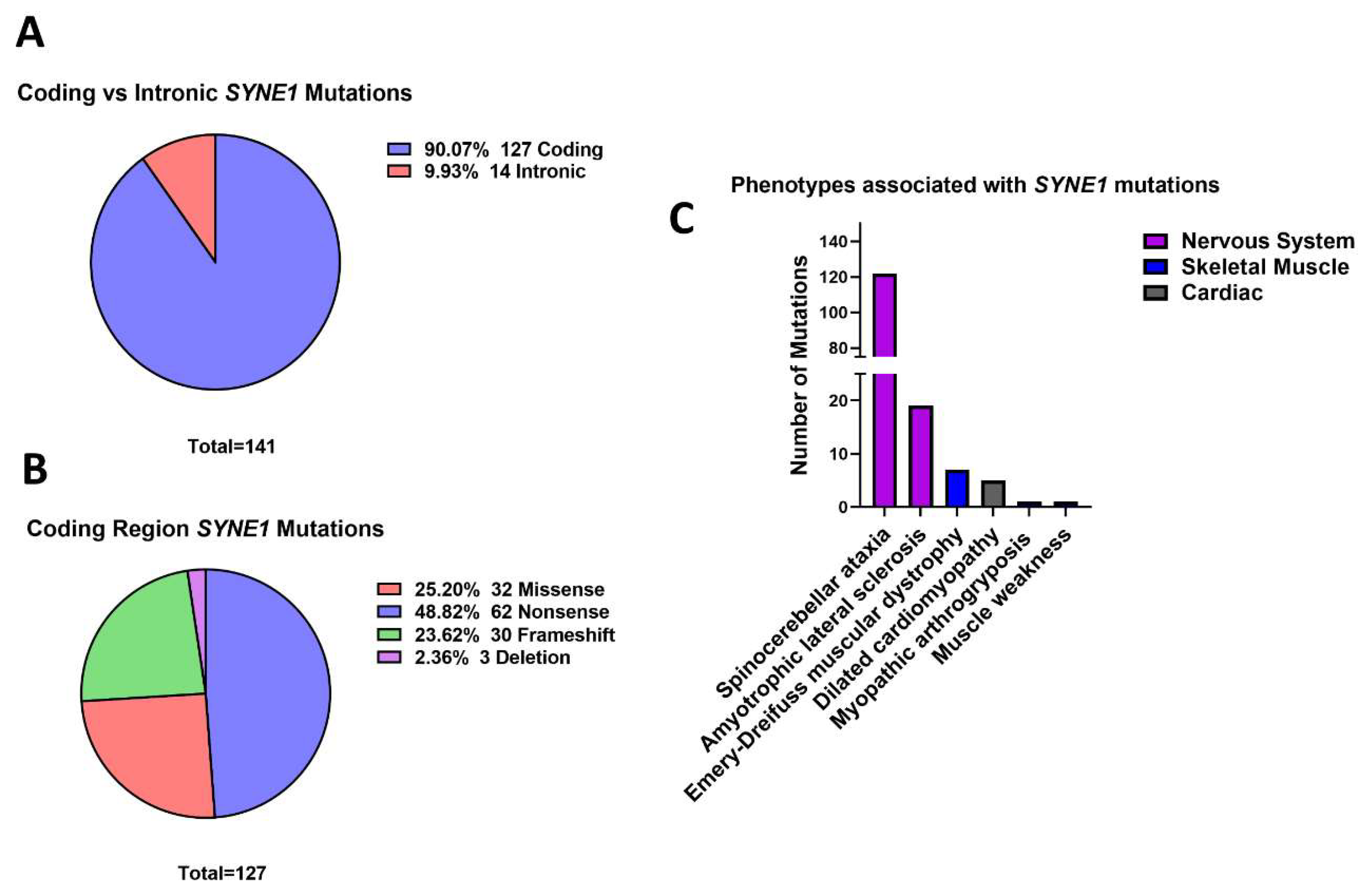
3.5. Outer Nuclear Membrane
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Nagano, A.; Arahata, K. Nuclear Envelope Proteins and Associated Diseases. Curr. Opin. Neurol. 2000, 13, 533–539. [Google Scholar] [CrossRef] [PubMed]
- Chi, Y.H.; Chen, Z.J.; Jeang, K.T. The Nuclear Envelopathies and Human Diseases. J. Biomed. Sci. 2009, 16, 96. [Google Scholar] [CrossRef] [PubMed]
- Somech, R.; Shaklai, S.; Amariglio, N.; Rechavi, G.; Simon, A.J. Nuclear Envelopathies--Raising the Nuclear Veil. Pediatr. Res. 2005, 57, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Worman, H.J.; Bonne, G. “Laminopathies”: A Wide Spectrum of Human Diseases. Exp. Cell Res. 2007, 313, 2121–2133. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Zhang, Y.; Zhong, N. Laminopathies--One Gene, Multiple Diseases. Beijing Da Xue Xue Bao. 2005, 37, 96–99. [Google Scholar] [PubMed]
- Gerace, L.; Burke, B. Functional Organization of the Nuclear Envelope. Annu. Rev. Cell Biol. 2003, 4, 335–374. [Google Scholar] [CrossRef]
- Watson, M.L. The Nuclear Envelope; Its Structure and Relation to Cytoplasmic Membranes. J. Biophys. Biochem. Cytol. 1955, 1, 257–270. [Google Scholar] [CrossRef]
- Linde, N.; Stick, R. Intranuclear Membranes Induced by Lipidated Proteins Are Derived from the Nuclear Envelope. Nucleus 2010, 1, 343–353. [Google Scholar] [CrossRef]
- Güttinger, S.; Laurell, E.; Kutay, U. Orchestrating Nuclear Envelope Disassembly and Reassembly during Mitosis. Nat. Rev. Mol. Cell Biol. 2009, 10, 178–191. [Google Scholar] [CrossRef]
- Taddei, A.; Hediger, F.; Neumann, F.R.; Gasser, S.M. The Function of Nuclear Architecture: A Genetic Approach. Annu. Rev. Genet. 2004, 38, 305–345. [Google Scholar] [CrossRef]
- Stuurman, N.; Heins, S.; Aebi, U. Nuclear Lamins: Their Structure, Assembly, and Interactions. J. Struct. Biol. 1998, 122, 42–66. [Google Scholar] [CrossRef] [PubMed]
- Crisp, M.; Liu, Q.; Roux, K.; Rattner, J.B.; Shanahan, C.; Burke, B.; Stahl, P.D.; Hodzic, D. Coupling of the Nucleus and Cytoplasm: Role of the LINC Complex. J. Cell Biol. 2006, 172, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Skepper, J.N.; Yang, F.; Davies, J.D.; Hegyi, L.; Roberts, R.G.; Weissberg, P.L.; Ellis, J.A.; Shanahan, C.M. Nesprins: A Novel Family of Spectrin-Repeat-Containing Proteins That Localize to the Nuclear Membrane in Multiple Tissues. J. Cell Sci. 2001, 114, 4485–4498. [Google Scholar] [CrossRef] [PubMed]
- Cain, N.E.; Jahed, Z.; Schoenhofen, A.; Valdez, V.A.; Elkin, B.; Hao, H.; Harris, N.J.; Herrera, L.A.; Woolums, B.M.; Mofrad, M.R.K.; et al. Conserved SUN-KASH Interfaces Mediate LINC Complex-Dependent Nuclear Movement and Positioning. Curr. Biol. 2018, 28, 3086–3097.e4. [Google Scholar] [CrossRef] [PubMed]
- Tapley, E.C.; Starr, D.A. Connecting the Nucleus to the Cytoskeleton by SUN-KASH Bridges across the Nuclear Envelope. Curr. Opin. Cell Biol. 2013, 25, 57–62. [Google Scholar] [CrossRef]
- Zhou, Z.; Du, X.; Cai, Z.; Song, X.; Zhang, H.; Mizuno, T.; Suzuki, E.; Yee, M.R.; Berezov, A.; Murali, R.; et al. Structure of Sad1-UNC84 Homology (SUN) Domain Defines Features of Molecular Bridge in Nuclear Envelope. J. Biol. Chem. 2012, 287, 5317–5326. [Google Scholar] [CrossRef]
- Wilhelmsen, K.; Litjens, S.H.M.; Kuikman, I.; Tshimbalanga, N.; Janssen, H.; Van Bout, I.D.; Raymond, K.; Sonnenberg, A. Nesprin-3, a Novel Outer Nuclear Membrane Protein, Associates with the Cytoskeletal Linker Protein Plectin. J. Cell Biol. 2005, 171, 799. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Ragnauth, C.; Greener, M.J.; Shanahan, C.M.; Roberts, R.G. The Nesprins Are Giant Actin-Binding Proteins, Orthologous to Drosophila Melanogaster Muscle Protein MSP-300. Genomics 2002, 80, 473–481. [Google Scholar] [CrossRef]
- Meinke, P.; Nguyen, T.D.; Wehnert, M.S. The LINC Complex and Human Disease. Biochem. Soc. Trans. 2011, 39, 1693–1697. [Google Scholar] [CrossRef] [PubMed]
- Storey, E.C.; Holt, I.; Morris, G.E.; Fuller, H.R. Muscle Cell Differentiation and Development Pathway Defects in Emery-Dreifuss Muscular Dystrophy. Neuromuscul. Disord. 2020, 30, 443–456. [Google Scholar] [CrossRef]
- Gueneau, L.; Bertrand, A.T.; Jais, J.-P.; Salih, M.A.; Stojkovic, T.; Wehnert, M.; Hoeltzenbein, M.; Spuler, S.; Saitoh, S.; Verschueren, A.; et al. Mutations of the FHL1 Gene Cause Emery-Dreifuss Muscular Dystrophy. Am. J. Hum. Genet. 2009, 85, 338–353. [Google Scholar] [CrossRef] [PubMed]
- Bonne, G.; Di Barletta, M.R.; Varnous, S.; Bécane, H.-M.; Hammouda, E.-H.; Merlini, L.; Muntoni, F.; Greenberg, C.R.; Gary, F.; Urtizberea, J.-A.; et al. Mutations in the Gene Encoding Lamin A/C Cause Autosomal Dominant Emery-Dreifuss Muscular Dystrophy. Nat. Genet. 1999, 21, 285–288. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Bethmann, C.; Worth, N.F.; Davies, J.D.; Wasner, C.; Feuer, A.; Ragnauth, C.D.; Yi, Q.; Mellad, J.A.; Warren, D.T.; et al. Nesprin-1 and -2 Are Involved in the Pathogenesis of Emery Dreifuss Muscular Dystrophy and Are Critical for Nuclear Envelope Integrity. Hum. Mol. Genet. 2007, 16, 2816–2833. [Google Scholar] [CrossRef] [PubMed]
- Liang, W.-C.; Mitsuhashi, H.; Keduka, E.; Nonaka, I.; Noguchi, S.; Nishino, I.; Hayashi, Y.K. TMEM43 Mutations in Emery-Dreifuss Muscular Dystrophy-Related Myopathy. Ann. Neurol. 2011, 69, 1005–1013. [Google Scholar] [CrossRef] [PubMed]
- Bione, S.; Maestrini, E.; Rivella, S.; Mancini, M.; Regis, S.; Romeo, G.; Toniolo, D. Identification of a Novel X-Linked Gene Responsible for Emery-Dreifuss Muscular Dystrophy. Nat. Genet. 1994, 8, 323–327. [Google Scholar] [CrossRef] [PubMed]
- Crasto, S.; My, I.; Di Pasquale, E. The Broad Spectrum of LMNA Cardiac Diseases: From Molecular Mechanisms to Clinical Phenotype. Front. Physiol. 2020, 11, 761. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, T.; Escalante-Alcalde, D.; Bhatt, H.; Anver, M.; Bhat, N.; Nagashima, K.; Stewart, C.L.; Burke, B. Loss of A-Type Lamin Expression Compromises Nuclear Envelope Integrity Leading to Muscular Dystrophy. J. Cell Biol. 1999, 147, 913–919. [Google Scholar] [CrossRef]
- Simon, D.N.; Wilson, K.L. Partners and Post-Translational Modifications of Nuclear Lamins. Chromosoma 2013, 122, 13–31. [Google Scholar] [CrossRef]
- Mercuri, E.; Poppe, M.; Quinlivan, R.; Messina, S.; Kinali, M.; Demay, L.; Bourke, J.; Richard, P.; Sewry, C.; Pike, M.; et al. Extreme Variability of Phenotype in Patients with an Identical Missense Mutation in the Lamin A/C Gene: From Congenital Onset with Severe Phenotype to Milder Classic Emery-Dreifuss Variant. Arch. Neurol. 2004, 61, 690–694. [Google Scholar] [CrossRef]
- Rankin, J.; Auer-Grumbach, M.; Bagg, W.; Colclough, K.; Nguyen, T.D.; Fenton-May, J.; Hattersley, A.; Hudson, J.; Jardine, P.; Josifova, D.; et al. Extreme Phenotypic Diversity and Nonpenetrance in Families with the LMNA Gene Mutation R644C. Am. J. Med. Genet. A 2008, 146A, 1530–1542. [Google Scholar] [CrossRef]
- Liberati, A.; Altman, D.G.; Tetzlaff, J.; Mulrow, C.; Gøtzsche, P.C.; Ioannidis, J.P.A.; Clarke, M.; Devereaux, P.J.; Kleijnen, J.; Moher, D. The PRISMA Statement for Reporting Systematic Reviews and Meta-Analyses of Studies That Evaluate Healthcare Interventions: Explanation and Elaboration. BMJ 2009, 339, b2700. [Google Scholar] [CrossRef] [PubMed]
- Bonello-Palot, N.; Simoncini, S.; Robert, S.; Bourgeois, P.; Sabatier, F.; Levy, N.; Dignat-George, F.; Badens, C. Prelamin A Accumulation in Endothelial Cells Induces Premature Senescence and Functional Impairment. Atherosclerosis 2014, 237, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Galant, D.; Gaborit, B.; Desgrouas, C.; Abdesselam, I.; Bernard, M.; Levy, N.; Merono, F.; Coirault, C.; Roll, P.; Lagarde, A.; et al. A Heterozygous ZMPSTE24 Mutation Associated with Severe Metabolic Syndrome, Ectopic Fat Accumulation, and Dilated Cardiomyopathy. Cells 2016, 5, 21. [Google Scholar] [CrossRef] [PubMed]
- Pendás, A.M.; Zhou, Z.; Cadiñanos, J.; Freije, J.M.P.; Wang, J.; Hultenby, K.; Astudillo, A.; Wernerson, A.; Rodríguez, F.; Tryggvason, K.; et al. Defective Prelamin A Processing and Muscular and Adipocyte Alterations in Zmpste24 Metalloproteinase–Deficient Mice. Nat. Genet. 2002, 31, 94–99. [Google Scholar] [CrossRef] [PubMed]
- Mutesa, L.; Pierquin, G.; Cwiny-Ay, N.; Buzizi, P.; Bours, V. Hutchinson-Gilford Progeria Syndrome: Clinical and Molecular Analysis in an African Patient. Rev. Med. Liege 2007, 62, 155–158. [Google Scholar]
- Ouzzani, M.; Hammady, H.; Fedorowicz, Z.; Elmagarmid, A. Rayyan—A Web and Mobile App for Systematic Reviews. Syst. Rev. 2016, 5, 210. [Google Scholar] [CrossRef]
- Lin, F.; Worman, H.J. Structural Organization of the Human Gene Encoding Nuclear Lamin A and Nuclear Lamin C. J. Biol. Chem. 1993, 268, 16321–16326. [Google Scholar] [CrossRef]
- Avila, G.M.; González, A.P.; Abad, A.; Fournier, B.G.; León, S.R.; Corral, J.A.M.; Fernández, C.P. Is the Next Generation Sequencing the Essential Tool for the Early Diagnostic Approach in Congenital Muscular Dystrophy? New Mutation in the Gen LMNA Associated with Serious Phenotype. Neurol. India 2021, 69, 1835–1837. [Google Scholar] [CrossRef]
- Mercuri, E.; Brown, S.C.; Nihoyannopoulos, P.; Poulton, J.; Kinali, M.; Richard, P.; Piercy, R.J.; Messina, S.; Sewry, C.; Burke, M.M.; et al. Extreme Variability of Skeletal and Cardiac Muscle Involvement in Patients with Mutations in Exon 11 of the Lamin A/C Gene. Muscle Nerve 2005, 31, 602–609. [Google Scholar] [CrossRef]
- Perrot, A.; Hussein, S.; Ruppert, V.; Schmidt, H.H.J.; Wehnert, M.S.; Duong, N.T.; Posch, M.G.; Panek, A.; Dietz, R.; Kindermann, I.; et al. Identification of Mutational Hot Spots in LMNA Encoding Lamin A/C in Patients with Familial Dilated Cardiomyopathy. Basic Res. Cardiol. 2009, 104, 90–99. [Google Scholar] [CrossRef]
- Quarta, G.; Syrris, P.; Ashworth, M.; Jenkins, S.; Zuborne Alapi, K.; Morgan, J.; Muir, A.; Pantazis, A.; McKenna, W.J.; Elliott, P.M. Mutations in the Lamin A/C Gene Mimic Arrhythmogenic Right Ventricular Cardiomyopathy. Eur. Heart J. 2012, 33, 1128–1136. [Google Scholar] [CrossRef] [PubMed]
- Parent, J.J.; Towbin, J.A.; Jefferies, J.L. Left Ventricular Noncompaction in a Family with Lamin A/C Gene Mutation. Texas Heart Inst. J. 2015, 42, 73–76. [Google Scholar] [CrossRef] [PubMed]
- Dohrn, M.F.; Glöckle, N.; Mulahasanovic, L.; Heller, C.; Mohr, J.; Bauer, C.; Riesch, E.; Becker, A.; Battke, F.; Hörtnagel, K.; et al. Frequent Genes in Rare Diseases: Panel-Based next Generation Sequencing to Disclose Causal Mutations in Hereditary Neuropathies. J. Neurochem. 2017, 143, 507–522. [Google Scholar] [CrossRef] [PubMed]
- Lin, E.W.; Brady, G.F.; Kwan, R.; Nesvizhskii, A.I.; Omary, M.B. Genotype-Phenotype Analysis of LMNA-Related Diseases Predicts Phenotype-Selective Alterations in Lamin Phosphorylation. FASEB J. 2020, 34, 9051–9073. [Google Scholar] [CrossRef]
- Pasotti, M.; Klersy, C.; Pilotto, A.; Marziliano, N.; Rapezzi, C.; Serio, A.; Mannarino, S.; Gambarin, F.; Favalli, V.; Grasso, M.; et al. Long-Term Outcome and Risk Stratification in Dilated Cardiolaminopathies. J. Am. Coll. Cardiol. 2008, 52, 1250–1260. [Google Scholar] [CrossRef]
- Ditaranto, R.; Boriani, G.; Biffi, M.; Lorenzini, M.; Graziosi, M.; Ziacchi, M.; Pasquale, F.; Vitale, G.; Berardini, A.; Rinaldi, R.; et al. Differences in Cardiac Phenotype and Natural History of Laminopathies with and without Neuromuscular Onset. Orphanet J. Rare Dis. 2019, 14, 263. [Google Scholar] [CrossRef]
- Møller, D.V.; Pham, T.T.; Gustafsson, F.; Hedley, P.; Ersbøll, M.K.; Bundgaard, H.; Andersen, C.B.; Torp-Pedersen, C.; Køber, L.; Christiansen, M. The Role of Lamin A/C Mutations in Danish Patients with Idiopathic Dilated Cardiomyopathy. Eur. J. Heart Fail. 2009, 11, 1031–1035. [Google Scholar] [CrossRef]
- Refaat, M.M.; Hassanieh, S.; Ballout, J.A.; Zakka, P.; Hotait, M.; Khalil, A.; Bitar, F.; Arabi, M.; Arnaout, S.; Skouri, H.; et al. Non-Familial Cardiomyopathies in Lebanon: Exome Sequencing Results for Five Idiopathic Cases. BMC Med. Genom. 2019, 12, 33. [Google Scholar] [CrossRef]
- Sanna, T.; Dello Russo, A.; Toniolo, D.; Vytopil, M.; Pelargonio, G.; De Martino, G.; Ricci, E.; Silvestri, G.; Giglio, V.; Messano, L.; et al. Cardiac Features of Emery-Dreifuss Muscular Dystrophy Caused by Lamin A/C Gene Mutations. Eur. Heart J. 2003, 24, 2227–2236. [Google Scholar] [CrossRef]
- Kandert, S.; Wehnert, M.; Müller, C.R.; Buendia, B.; Dabauvalle, M.-C. Impaired Nuclear Functions Lead to Increased Senescence and Inefficient Differentiation in Human Myoblasts with a Dominant p.R545C Mutation in the LMNA Gene. Eur. J. Cell Biol. 2009, 88, 593–608. [Google Scholar] [CrossRef]
- Favreau, C.; Dubosclard, E.; Östlund, C.; Vigouroux, C.; Capeau, J.; Wehnert, M.; Higuet, D.; Worman, H.J.; Courvalin, J.C.; Buendia, B. Expression of Lamin A Mutated in the Carboxyl-Terminal Tail Generates an Aberrant Nuclear Phenotype Similar to That Observed in Cells from Patients with Dunnigan-Type Partial Lipodystrophy and Emery-Dreifuss Muscular Dystrophy. Exp. Cell Res. 2003, 282, 14–23. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.-E.; Hayashi, Y.K.; Goto, K.; Komaki, H.; Hayashi, Y.; Inuzuka, T.; Noguchi, S.; Nonaka, I.; Nishino, I. Nuclear Changes in Skeletal Muscle Extend to Satellite Cells in Autosomal Dominant Emery-Dreifuss Muscular Dystrophy/Limb-Girdle Muscular Dystrophy 1B. Neuromuscul. Disord. 2009, 19, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Díaz-Manera, J.; Alejaldre, A.; González, L.; Olivé, M.; Gómez-Andrés, D.; Muelas, N.; Vílchez, J.J.; Llauger, J.; Carbonell, P.; Márquez-Infante, C.; et al. Muscle Imaging in Muscle Dystrophies Produced by Mutations in the EMD and LMNA Genes. Neuromuscul. Disord. 2016, 26, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Sewry, C.A.; Brown, S.C.; Mercuri, E.; Bonne, G.; Feng, L.; Camici, G.; Morris, G.E.; Muntoni, F. Skeletal Muscle Pathology in Autosomal Dominant Emery-Dreifuss Muscular Dystrophy with Lamin A/C Mutations. Neuropathol. Appl. Neurobiol. 2001, 27, 281–290. [Google Scholar] [CrossRef] [PubMed]
- Vytopil, M.; Benedetti, S.; Ricci, E.; Galluzzi, G.; Dello Russo, A.; Merlini, L.; Boriani, G.; Gallina, M.; Morandi, L.; Politano, L.; et al. Mutation Analysis of the Lamin A/C Gene (LMNA) among Patients with Different Cardiomuscular Phenotypes. J. Med. Genet. 2003, 40, e132. [Google Scholar] [CrossRef] [PubMed]
- Astejada, M.N.; Goto, K.; Nagano, A.; Ura, S.; Noguchi, S.; Nonaka, I.; Nishino, I.; Hayashi, Y.K. Emerinopathy and Laminopathy Clinical, Pathological and Molecular Features of Muscular Dystrophy with Nuclear Envelopathy in Japan. Acta Myol. Myopathies Cardiomyopathies Off. J. Mediterr. Soc. Myol. 2007, 26, 159–164. [Google Scholar]
- Colomer, J.; Iturriaga, C.; Bonne, G.; Schwartz, K.; Manilal, S.; Morris, G.E.; Puche, M.; Fernández-Alvarez, E. Autosomal Dominant Emery-Dreifuss Muscular Dystrophy: A New Family with Late Diagnosis. Neuromuscul. Disord. 2002, 12, 19–25. [Google Scholar] [CrossRef]
- Bonne, G.; Mercuri, E.; Muchir, A.; Urtizberea, A.; Bécane, H.M.; Recan, D.; Merlini, L.; Wehnert, M.; Boor, R.; Reuner, U.; et al. Clinical and Molecular Genetic Spectrum of Autosomal Dominant Emery-Dreifuss Muscular Dystrophy Due to Mutations of the Lamin A/C Gene. Ann. Neurol. 2000, 48, 170–180. [Google Scholar] [CrossRef]
- Kajino, S.; Ishihara, K.; Goto, K.; Ishigaki, K.; Noguchi, S.; Nonaka, I.; Osawa, M.; Nishino, I.; Hayashi, Y.K. Congenital Fiber Type Disproportion Myopathy Caused by LMNA Mutations. J. Neurol. Sci. 2014, 340, 94–98. [Google Scholar] [CrossRef]
- Ge, L.; Zhang, C.; Wang, Z.; Chan, S.H.S.; Zhu, W.; Han, C.; Zhang, X.; Zheng, H.; Wu, L.; Jin, B.; et al. Congenital Muscular Dystrophies in China. Clin. Genet. 2019, 96, 207–215. [Google Scholar] [CrossRef]
- Ebert, M.; Wijnmaalen, A.P.; de Riva, M.; Trines, S.A.; Androulakis, A.F.A.; Glashan, C.A.; Schalij, M.J.; van Tintelen, J.; Jongbloed, J.D.H.; Zeppenfeld, K. Prevalence and Prognostic Impact of Pathogenic Variants in Patients With Dilated Cardiomyopathy Referred for Ventricular Tachycardia Ablation. JACC. Clin. Electrophysiol. 2020, 6, 1103–1114. [Google Scholar] [CrossRef] [PubMed]
- Vytopil, M.; Vohanka, S.; Vlasinova, J.; Toman, J.; Novak, M.; Toniolo, D.; Ricotti, R.; Lukas, Z. The Screening for X-Linked Emery-Dreifuss Muscular Dystrophy amongst Young Patients with Idiopathic Heart Conduction System Disease Treated by a Pacemaker Implant. Eur. J. Neurol. 2004, 11, 531–534. [Google Scholar] [CrossRef] [PubMed]
- Liang, W.-C.; Jong, Y.-J.; Wang, C.-H.; Wang, C.-H.; Tian, X.; Chen, W.-Z.; Kan, T.-M.; Minami, N.; Nishino, I.; Wong, L.-J.C. Clinical, Pathological, Imaging, and Genetic Characterization in a Taiwanese Cohort with Limb-Girdle Muscular Dystrophy. Orphanet J. Rare Dis. 2020, 15, 160. [Google Scholar] [CrossRef] [PubMed]
- Sanga, S.; Ghosh, A.; Kumar, K.; Polavarapu, K.; Preethish-Kumar, V.; Vengalil, S.; Nashi, S.; Bardhan, M.; Arunachal, G.; Raju, S.; et al. Whole-Exome Analyses of Congenital Muscular Dystrophy and Congenital Myopathy Patients from India Reveal a Wide Spectrum of Known and Novel Mutations. Eur. J. Neurol. 2021, 28, 992–1003. [Google Scholar] [CrossRef]
- Muchir, A.; Medioni, J.; Laluc, M.; Massart, C.; Arimura, T.; van der Kooi, A.J.; Desguerre, I.; Mayer, M.; Ferrer, X.; Briault, S.; et al. Nuclear Envelope Alterations in Fibroblasts from Patients with Muscular Dystrophy, Cardiomyopathy, and Partial Lipodystrophy Carrying Lamin A/C Gene Mutations. Muscle Nerve 2004, 30, 444–450. [Google Scholar] [CrossRef]
- Fan, Y.; Tan, D.; Song, D.; Zhang, X.; Chang, X.; Wang, Z.; Zhang, C.; Chan, S.H.-S.; Wu, Q.; Wu, L.; et al. Clinical Spectrum and Genetic Variations of LMNA -Related Muscular Dystrophies in a Large Cohort of Chinese Patients. J. Med. Genet. 2020, 58, 326–333. [Google Scholar] [CrossRef]
- Magagnotti, C.; Bachi, A.; Zerbini, G.; Fattore, E.; Fermo, I.; Riba, M.; Previtali, S.C.; Ferrari, M.; Andolfo, A.; Benedetti, S. Protein Profiling Reveals Energy Metabolism and Cytoskeletal Protein Alterations in LMNA Mutation Carriers. Biochim. Biophys. Acta 2012, 1822, 970–979. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Choi, S.A.; Cho, A.; Kim, S.Y.; Kim, W.J.; Shim, Y.K.; Lee, J.S.; Jang, S.S.; Lim, B.C.; Kim, H.; Hwang, H.; et al. Importance of Early Diagnosis in LMNA-Related Muscular Dystrophy for Cardiac Surveillance. Muscle Nerve 2019, 60, 668–672. [Google Scholar] [CrossRef]
- Makri, S.; Clarke, N.F.; Richard, P.; Maugenre, S.; Demay, L.; Bonne, G.; Guicheney, P. Germinal Mosaicism for LMNA Mimics Autosomal Recessive Congenital Muscular Dystrophy. Neuromuscul. Disord. 2009, 19, 26–28. [Google Scholar] [CrossRef]
- Tan, D.; Yang, H.; Yuan, Y.; Bonnemann, C.; Chang, X.; Wang, S.; Wu, Y.; Wu, X.; Xiong, H. Phenotype-Genotype Analysis of Chinese Patients with Early-Onset LMNA-Related Muscular Dystrophy. PLoS ONE 2015, 10, e0129699. [Google Scholar] [CrossRef]
- Di Barletta, M.R.; Ricci, E.; Galluzzi, G.; Tonali, P.; Mora, M.; Morandi, L.; Romorini, A.; Voit, T.; Orstavik, K.H.; Merlini, L.; et al. Different Mutations in the LMNA Gene Cause Autosomal Dominant and Autosomal Recessive Emery-Dreifuss Muscular Dystrophy. Am. J. Hum. Genet. 2000, 66, 1407–1412. [Google Scholar] [CrossRef] [PubMed]
- Rudnik-Schöneborn, S.; Botzenhart, E.; Eggermann, T.; Senderek, J.; Schoser, B.G.H.; Schröder, R.; Wehnert, M.; Wirth, B.; Zerres, K. Mutations of the LMNA Gene Can Mimic Autosomal Dominant Proximal Spinal Muscular Atrophy. Neurogenetics 2007, 8, 137–142. [Google Scholar] [CrossRef] [PubMed]
- Francisco, A.R.G.; Santos Gonçalves, I.; Veiga, F.; Mendes Pedro, M.; Pinto, F.J.; Brito, D. Complex Phenotype Linked to a Mutation in Exon 11 of the Lamin A/C Gene: Hypertrophic Cardiomyopathy, Atrioventricular Block, Severe Dyslipidemia and Diabetes. Rev. Port. Cardiol. 2017, 36, 669.e1–669.e4. [Google Scholar] [CrossRef] [PubMed]
- Gigli, M.; Merlo, M.; Graw, S.L.; Barbati, G.; Rowland, T.J.; Slavov, D.B.; Stolfo, D.; Haywood, M.E.; Dal Ferro, M.; Altinier, A.; et al. Genetic Risk of Arrhythmic Phenotypes in Patients With Dilated Cardiomyopathy. J. Am. Coll. Cardiol. 2019, 74, 1480–1490. [Google Scholar] [CrossRef]
- Meinke, P.; Mattioli, E.; Haque, F.; Antoku, S.; Columbaro, M.; Straatman, K.R.; Worman, H.J.; Gundersen, G.G.; Lattanzi, G.; Wehnert, M.; et al. Muscular Dystrophy-Associated SUN1 and SUN2 Variants Disrupt Nuclear-Cytoskeletal Connections and Myonuclear Organization. PLoS Genet. 2014, 10, e1004605. [Google Scholar] [CrossRef]
- Ben Yaou, R.; Toutain, A.; Arimura, T.; Demay, L.; Massart, C.; Peccate, C.; Muchir, A.; Llense, S.; Deburgrave, N.; Leturcq, F.; et al. Multitissular Involvement in a Family with LMNA and EMD Mutations: Role of Digenic Mechanism? Neurology 2007, 68, 1883–1894. [Google Scholar] [CrossRef]
- Muntoni, F.; Bonne, G.; Goldfarb, L.; Mercuri, E.; Piercy, R.; Burke, M.; Yaou, R.; Richard, P.; Récan, D.; Shatunov, A.; et al. Disease Severity in Dominant Emery Dreifuss Is Increased by Mutations in Both Emerin and Desmin Proteins. Brain A J. Neurol. 2006, 129, 1260–1268. [Google Scholar] [CrossRef]
- Granger, B.; Gueneau, L.; Drouin-Garraud, V.; Pedergnana, V.; Gagnon, F.; Ben Yaou, R.; du Montcel, S.; Bonne, G. Modifier Locus of the Skeletal Muscle Involvement in Emery-Dreifuss Muscular Dystrophy. Hum. Genet. 2011, 129, 149–159. [Google Scholar] [CrossRef]
- Riordan, J.D.; Nadeau, J.H. From Peas to Disease: Modifier Genes, Network Resilience, and the Genetics of Health. Am. J. Hum. Genet. 2017, 101, 177. [Google Scholar] [CrossRef]
- Cerrone, M.; Remme, C.A.; Tadros, R.; Bezzina, C.R.; Delmar, M. Beyond the One Gene–One Disease Paradigm. Circulation 2019, 140, 595–610. [Google Scholar] [CrossRef]
- Leitch, C.C.; Zaghloul, N.A.; Davis, E.E.; Stoetzel, C.; Diaz-Font, A.; Rix, S.; Al-Fadhel, M.; Lewis, R.A.; Eyaid, W.; Banin, E.; et al. Hypomorphic Mutations in Syndromic Encephalocele Genes Are Associated with Bardet-Biedl Syndrome. Nat. Genet. 2008, 40, 443–448. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Soave, D.; Miller, M.R.; Keenan, K.; Lin, F.; Gong, J.; Chiang, T.; Stephenson, A.L.; Durie, P.; Rommens, J.; et al. Unraveling the Complex Genetic Model for Cystic Fibrosis: Pleiotropic Effects of Modifier Genes on Early Cystic Fibrosis-Related Morbidities. Hum. Genet. 2014, 133, 151–161. [Google Scholar] [CrossRef] [PubMed]
- Harhouri, K.; Frankel, D.; Bartoli, C.; Roll, P.; De Sandre-Giovannoli, A.; Lévy, N. An Overview of Treatment Strategies for Hutchinson-Gilford Progeria Syndrome. Nucleus 2018, 9, 246–257. [Google Scholar] [CrossRef] [PubMed]
- Coffinier, C.; Chang, S.Y.; Nobumori, C.; Tu, Y.; Farber, E.A.; Toth, J.I.; Fong, L.G.; Young, S.G. Abnormal Development of the Cerebral Cortex and Cerebellum in the Setting of Lamin B2 Deficiency. Proc. Natl. Acad. Sci. USA 2010, 107, 5076–5081. [Google Scholar] [CrossRef] [PubMed]
- Coffinier, C.; Jung, H.J.; Nobumori, C.; Chang, S.; Tu, Y.; Barnes, R.H.; Yoshinaga, Y.; De Jong, P.J.; Vergnes, L.; Reue, K.; et al. Deficiencies in Lamin B1 and Lamin B2 Cause Neurodevelopmental Defects and Distinct Nuclear Shape Abnormalities in Neurons. Mol. Biol. Cell 2011, 22, 4683–4693. [Google Scholar] [CrossRef] [PubMed]
- Coffinier, C.; Fong, L.G.; Young, S.G. LINCing Lamin B2 to Neuronal Migration: Growing Evidence for Cell-Specific Roles of B-Type Lamins. Nucleus 2010, 1, 407–411. [Google Scholar] [CrossRef]
- Vergnes, L.; Péterfy, M.; Bergo, M.O.; Young, S.G.; Reue, K. Lamin B1 Is Required for Mouse Development and Nuclear Integrity. Proc. Natl. Acad. Sci. USA 2004, 101, 10428–10433. [Google Scholar] [CrossRef]
- Lee, J.M.; Tu, Y.; Tatar, A.; Wu, D.; Nobumori, C.; Jung, H.J.; Yoshinaga, Y.; Coffinier, C.; De Jong, P.J.; Fong, L.G.; et al. Reciprocal Knock-in Mice to Investigate the Functional Redundancy of Lamin B1 and Lamin B2. Mol. Biol. Cell 2014, 25, 1666. [Google Scholar] [CrossRef]
- Padiath, Q.S.; Saigoh, K.; Schiffmann, R.; Asahara, H.; Yamada, T.; Koeppen, A.; Hogan, K.; Ptácek, L.J.; Fu, Y.-H. Lamin B1 Duplications Cause Autosomal Dominant Leukodystrophy. Nat. Genet. 2006, 38, 1114–1123. [Google Scholar] [CrossRef]
- Emery, A.E.H. Emery-Dreifuss Syndrome. Syndr. Mon. J. Med. Genet. 1989, 26, 637–641. [Google Scholar] [CrossRef]
- Boriani, G.; Gallina, M.; Merlini, L.; Bonne, G.; Toniolo, D.; Amati, S.; Biffi, M.; Martignani, C.; Frabetti, L.; Bonvicini, M.; et al. Clinical Relevance of Atrial Fibrillation/Flutter, Stroke, Pacemaker Implant, and Heart Failure in Emery-Dreifuss Muscular Dystrophy: A Long-Term Longitudinal Study. Stroke 2003, 34, 901–908. [Google Scholar] [CrossRef] [PubMed]
- Broers, J.L.V.; Peeters, E.A.G.; Kuijpers, H.J.H.; Endert, J.; Bouten, C.V.C.; Oomens, C.W.J.; Baaijens, F.P.T.; Ramaekers, F.C.S. Decreased Mechanical Stiffness in LMNA-/- Cells Is Caused by Defective Nucleo-Cytoskeletal Integrity: Implications for the Development of Laminopathies. Hum. Mol. Genet. 2004, 13, 2567–2580. [Google Scholar] [CrossRef] [PubMed]
- Lammerding, J.; Kamm, R.D.; Lee, R.T.; Schulze, P.C.; Takahashi, T.; Kozlov, S.; Sullivan, T.; Stewart, C.L. Lamin A/C Deficiency Causes Defective Nuclear Mechanics and Mechanotransduction. J. Clin. Investig. 2004, 113, 370–378. [Google Scholar] [CrossRef] [PubMed]
- Hutchison, C.J. Lamins: Building Blocks or Regulators of Gene Expression? Nat. Rev. Mol. Cell Biol. 2002, 3, 848–858. [Google Scholar] [CrossRef] [PubMed]
- Zastrow, M.S.; Vlcek, S.; Wilson, K.L. Proteins That Bind A-Type Lamins: Integrating Isolated Clues. J. Cell Sci. 2004, 117, 979–987. [Google Scholar] [CrossRef]
- Ellis, D.; Jenkins, H.; Whitfield, W.; Hutchinson, C. GST-Lamin Fusion Proteins Act as Dominant Negative Mutants in Xenopus Egg Extract and Reveal the Function of the Lamina in DNA Replication. J. Cell Sci. 1997, 110, 2507–2518. [Google Scholar] [CrossRef]
- Schirmer, E.C.; Gerace, L. The Nuclear Membrane Proteome: Extending the Envelope. Trends Biochem. Sci. 2005, 30, 551–558. [Google Scholar] [CrossRef]
- Dittmer, T.A.; Sahni, N.; Kubben, N.; Hill, D.E.; Vidal, M.; Burgess, R.C.; Roukos, V.; Misteli, T. Systematic Identification of Pathological Lamin A Interactors. Mol. Biol. Cell 2014, 25, 1493–1510. [Google Scholar] [CrossRef]
- Kubben, N.; Voncken, J.W.; Demmers, J.; Calis, C.; Van Almen, G.; Pinto, Y.M.; Misteli, T. Identification of Differential Protein Interactors of Lamin A and Progerin. Nucleus 2010, 1, 513–525. [Google Scholar] [CrossRef]
- Vitkup, D.; Sander, C.; Church, G.M. The Amino-Acid Mutational Spectrum of Human Genetic Disease. Genome Biol. 2003, 4, R72. [Google Scholar] [CrossRef]
- Cooper, D.N.; Youssoufian, H. The CpG Dinucleotide and Human Genetic Disease. Hum. Genet. 1988, 78, 151–155. [Google Scholar] [CrossRef]
- Betts, M.J.; Russell, R.B. Amino Acid Properties and Consequences of Substitutions. In Bioinformatics for Geneticists; Barnes, M.R., Gray, I.C., Eds.; John Wiley & Sons, Ltd.: New York, NY, USA, 2003; pp. 289–316. [Google Scholar]
- Stuurman, N.; Sasse, B.; Fisher, P.A. Intermediate Filament Protein Polymerization: Molecular Analysis of Drosophila Nuclear Lamin Head-to-Tail Binding. J. Struct. Biol. 1996, 117, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Ahn, J.; Jo, I.; Kang, S.; Hong, S.; Kim, S.; Jeong, S.; Kim, Y.H.; Park, B.J.; Ha, N.C. Structural Basis for Lamin Assembly at the Molecular Level. Nat. Commun. 2019, 10, 1–12. [Google Scholar] [CrossRef]
- Bhattacharjee, P.; Dasgupta, D.; Sengupta, K. DCM Associated LMNA Mutations Cause Distortions in Lamina Structure and Assembly. Biochim. Biophys. Acta—Gen. Subj. 2017, 1861, 2598–2608. [Google Scholar] [CrossRef] [PubMed]
- Ostlund, C.; Bonne, G.; Schwartz, K.; Worman, H.J. Properties of Lamin A Mutants Found in Emery-Dreifuss Muscular Dystrophy, Cardiomyopathy and Dunnigan-Type Partial Lipodystrophy. J. Cell Sci. 2001, 114, 4435–4445. [Google Scholar] [CrossRef] [PubMed]
- Vigouroux, C.; Auclair, M.; Dubosclard, E.; Pouchelet, M.; Capeau, J.; Courvalin, J.C.; Buendia, B. Nuclear Envelope Disorganization in Fibroblasts from Lipodystrophic Patients with Heterozygous R482Q/W Mutations in the Lamin A/C Gene. J. Cell Sci. 2001, 114, 4459–4468. [Google Scholar] [CrossRef] [PubMed]
- Muchir, A.; Van Engelen, B.G.; Lammens, M.; Mislow, J.M.; McNally, E.; Schwartz, K.; Bonne, G. Nuclear Envelope Alterations in Fibroblasts from LGMD1B Patients Carrying Nonsense Y259X Heterozygous or Homozygous Mutation in Lamin A/C Gene. Exp. Cell Res. 2003, 291, 352–362. [Google Scholar] [CrossRef]
- Osmanagic-Myers, S.; Foisner, R. The Structural and Gene Expression Hypotheses in Laminopathic Diseases—Not so Different after All. Mol. Biol. Cell 2019, 30, 1786–1790. [Google Scholar] [CrossRef]
- Clements, L.; Manilal, S.; Love, D.R.; Morris, G.E. Direct Interaction between Emerin and Lamin A. Biochem. Biophys. Res. Commun. 2000, 267, 709–714. [Google Scholar] [CrossRef]
- Lee, K.K.; Haraguchi, T.; Lee, R.S.; Koujin, T.; Hiraoka, Y.; Wilson, K.L. Distinct Functional Domains in Emerin Bind Lamin A and DNA-Bridging Protein BAF. J. Cell Sci. 2001, 114, 4567–4573. [Google Scholar] [CrossRef]
- Sakaki, M.; Koike, H.; Takahashi, N.; Sasagawa, N.; Tomioka, S.; Arahata, K.; Ishiura, S. Interaction between Emerin and Nuclear Lamins. J. Biochem. 2001, 129, 321–327. [Google Scholar] [CrossRef] [PubMed]
- Holt, I.; Östlund, C.; Stewart, C.L.; thi Man, N.; Worman, H.J.; Morris, G.E. Effect of Pathogenic Mis-Sense Mutations in Lamin A on Its Interaction with Emerin in Vivo. J. Cell Sci. 2003, 116, 3027–3035. [Google Scholar] [CrossRef] [PubMed]
- Mziaut, H.; Korza, G.; Elkahloun, A.G.; Ozols, J. Induction of Stearoyl CoA Desaturase Is Associated with High-Level Induction of Emerin RNA. Biochem. Biophys. Res. Commun. 2001, 282, 910–915. [Google Scholar] [CrossRef] [PubMed]
- Dharmaraj, T.; Guan, Y.; Liu, J.; Badens, C.; Gaborit, B.; Wilson, K.L. Rare BANF1 Alleles and Relatively Frequent EMD Alleles Including “Healthy Lipid” Emerin p.D149H in the ExAC Cohort. Front. Cell Dev. Biol. 2019, 7, 48. [Google Scholar] [CrossRef]
- Tang, K.; Finley, R.L.; Nie, D.; Honn, K.V. Identification of 12-Lipoxygenase Interaction with Cellular Proteins by Yeast Two-Hybrid Screening. Biochemistry 2000, 39, 3185–3191. [Google Scholar] [CrossRef]
- Dobrian, A.D.; Morris, M.A.; Taylor-Fishwick, D.A.; Holman, T.R.; Imai, Y.; Mirmira, R.G.; Nadler, J.L. Role of the 12-Lipoxygenase Pathway in Diabetes Pathogenesis and Complications. Pharmacol. Ther. 2019, 195, 100–110. [Google Scholar] [CrossRef]
- Dong, C.; Liu, S.; Cui, Y.; Guo, Q. 12-Lipoxygenase as a Key Pharmacological Target in the Pathogenesis of Diabetic Nephropathy. Eur. J. Pharmacol. 2020, 879, 173122. [Google Scholar] [CrossRef]
- Grzesik, W.J.; Nadler, J.L.; Machida, Y.; Nadler, J.L.; Imai, Y.; Morris, M.A. Expression Pattern of 12-Lipoxygenase in Human Islets With Type 1 Diabetes and Type 2 Diabetes. J. Clin. Endocrinol. Metab. 2015, 100, E387–E395. [Google Scholar] [CrossRef]
- Prasad, K.M.R.; Thimmalapura, P.R.R.; Woode, E.A.A.; Nadler, J.L. Evidence That Increased 12-Lipoxygenase Expression Impairs Pancreatic β Cell Function and Viability. Biochem. Biophys. Res. Commun. 2003, 308, 427–432. [Google Scholar] [CrossRef]
- Sasseville, A.M.-J.; Langelier, Y. In Vitro Interaction of the Carboxy-Terminal Domain of Lamin A with Actin. FEBS Lett. 1998, 425, 485–489. [Google Scholar] [CrossRef]
- Stierlé, V.; Couprie, J.; Ostlund, C.; Krimm, I.; Zinn-Justin, S.; Hossenlopp, P.; Worman, H.J.; Courvalin, J.-C.; Duband-Goulet, I. The Carboxyl-Terminal Region Common to Lamins A and C Contains a DNA Binding Domain. Biochemistry 2003, 42, 4819–4828. [Google Scholar] [CrossRef] [PubMed]
- Koch, A.J.; Holaska, J.M. Emerin in Health and Disease. Semin. Cell Dev. Biol. 2014, 29, 95–106. [Google Scholar] [CrossRef] [PubMed]
- Manilal, S.; Recan, D.; Sewry, C.A.; Hoeltzenbein, M.; Llense, S.; Leturcq, F.; Deburgrave, N.; Barbot, J.; Man, N.; Muntoni, F.; et al. Mutations in Emery-Dreifuss Muscular Dystrophy and Their Effects on Emerin Protein Expression. Hum. Mol. Genet. 1998, 7, 855–864. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Li, C.; Zhou, B.; Sun, H.; Koullourou, V.; Holt, I.; Puckelwartz, M.J.; Warren, D.T.; Hayward, R.; Lin, Z.; et al. Novel Nesprin-1 Mutations Associated with Dilated Cardiomyopathy Cause Nuclear Envelope Disruption and Defects in Myogenesis. Hum. Mol. Genet. 2017, 26, 2258–2276. [Google Scholar] [CrossRef]
- Zhang, X.; Lei, K.; Yuan, X.; Wu, X.; Zhuang, Y.; Xu, T.; Xu, R.; Han, M. SUN1/2 and Syne/Nesprin-1/2 Complexes Connect Centrosome to the Nucleus during Neurogenesis and Neuronal Migration in Mice. Neuron 2009, 64, 173–187. [Google Scholar] [CrossRef]
- Clancy, J.P.; Bebök, Z.; Ruiz, F.; King, C.; Jones, J.; Walker, L.; Greer, H.; Hong, J.; Wing, L.; Macaluso, M.; et al. Evidence That Systemic Gentamicin Suppresses Premature Stop Mutations in Patients with Cystic Fibrosis. Am. J. Respir. Crit. Care Med. 2001, 163, 1683–1692. [Google Scholar] [CrossRef]
- Wilschanski, M.; Famini, C.; Blau, H.; Rivlin, J.; Augarten, A.; Avital, A.; Kerem, B.; Kerem, E. A Pilot Study of the Effect of Gentamicin on Nasal Potential Difference Measurements in Cystic Fibrosis Patients Carrying Stop Mutations. Am. J. Respir. Crit. Care Med. 2000, 161, 860–865. [Google Scholar] [CrossRef]
- Malik, V.; Rodino Klapac, L.R.; Viollet, L.; Mendell, J.R. Aminoglycoside-Induced Mutation Suppression (Stop Codon Readthrough) as a Therapeutic Strategy for Duchenne Muscular Dystrophy. Ther. Adv. Neurol. Disord. 2010, 3, 379–389. [Google Scholar] [CrossRef]
- Politano, L.; Nigro, G.; Nigro, V.; Piluso, G.; Papparella, S.; Paciello, O.; Comi, L. Gentamicin Administration in Duchenne Patients with Premature Stop Codon. Preliminary Results. Off. J. Mediterr. Soc. Myol. 2003, 22, 15–21. [Google Scholar]
- James, P.D.; Raut, S.; Rivard, G.E.; Poon, M.C.; Warner, M.; McKenna, S.; Leggo, J.; Lillicrap, D. Aminoglycoside Suppression of Nonsense Mutations in Severe Hemophilia. Blood 2005, 106, 3043–3048. [Google Scholar] [CrossRef]
- Taguchi, A.; Hamada, K.; Shiozuka, M.; Kobayashi, M.; Murakami, S.; Takayama, K.; Taniguchi, A.; Usui, T.; Matsuda, R.; Hayashi, Y. Structure-Activity Relationship Study of Leucyl-3-Epi-Deoxynegamycin for Potent Premature Termination Codon Readthrough. ACS Med. Chem. Lett. 2017, 8, 1060–1065. [Google Scholar] [CrossRef] [PubMed]
- Hamada, K.; Omura, N.; Taguchi, A.; Baradaran-Heravi, A.; Kotake, M.; Arai, M.; Takayama, K.; Taniguchi, A.; Roberge, M.; Hayashi, Y. New Negamycin-Based Potent Readthrough Derivative Effective against TGA-Type Nonsense Mutations. ACS Med. Chem. Lett. 2019, 10, 1450–1456. [Google Scholar] [CrossRef] [PubMed]
- Wilschanski, M.; Yahav, Y.; Yaacov, Y.; Blau, H.; Bentur, L.; Rivlin, J.; Aviram, M.; Bdolah-Abram, T.; Bebok, Z.; Shushi, L.; et al. Gentamicin-Induced Correction of CFTR Function in Patients with Cystic Fibrosis and CFTR Stop Mutations. N. Engl. J. Med. 2009, 349, 1433–1441. [Google Scholar] [CrossRef] [PubMed]
- Kellermayer, R.; Szigeti, R.; Keeling, K.M.; Bedekovics, T.; Bedwell, D.M. Aminoglycosides as Potential Pharmacogenetic Agents in the Treatment of Hailey-Hailey Disease. J. Investig. Dermatol. 2006, 126, 229–231. [Google Scholar] [CrossRef]
- PTC Therapeutics, Inc. PTC Therapeutics Receives Conditional Approval in the European Union for TranslarnaTM for the Treatment of Nonsense Mutation Duchenne Muscular Dystrophy; PTC Therapeutics, Inc.: South Plainfield, NJ, USA, 2014. [Google Scholar]
- McDonald, C.M.; Campbell, C.; Torricelli, R.E.; Finkel, R.S.; Flanigan, K.M.; Goemans, N.; Heydemann, P.; Kaminska, A.; Kirschner, J.; Muntoni, F.; et al. Ataluren in Patients with Nonsense Mutation Duchenne Muscular Dystrophy (ACT DMD): A Multicentre, Randomised, Double-Blind, Placebo-Controlled, Phase 3 Trial. Lancet 2017, 390, 1489–1498. [Google Scholar] [CrossRef]
- Folker, E.S.; Östlund, C.; Luxton, G.W.G.; Worman, H.J.; Gundersen, G.G. Lamin A Variants That Cause Striated Muscle Disease Are Defective in Anchoring Transmembrane Actin-Associated Nuclear Lines for Nuclear Movement. Proc. Natl. Acad. Sci. USA 2011, 108, 131–136. [Google Scholar] [CrossRef]
- Chang, W.; Antoku, S.; Östlund, C.; Worman, H.J.; Gundersen, G.G. Linker of Nucleoskeleton and Cytoskeleton (LINC) Complex-Mediated Actin-Dependent Nuclear Positioning Orients Centrosomes in Migrating Myoblasts. Nucleus 2015, 6, 77–88. [Google Scholar] [CrossRef]
- Kirby, T.J.; Lammerding, J. Emerging Views of the Nucleus as a Cellular Mechanosensor. Nat. Cell Biol. 2018, 20, 373–381. [Google Scholar] [CrossRef]
- Lombardi, M.L.; Jaalouk, D.E.; Shanahan, C.M.; Burke, B.; Roux, K.J.; Lammerding, J. The Interaction between Nesprins and Sun Proteins at the Nuclear Envelope Is Critical for Force Transmission between the Nucleus and Cytoskeleton. J. Biol. Chem. 2011, 286, 26743–26753. [Google Scholar] [CrossRef]
- Swift, J.; Ivanovska, I.L.; Buxboim, A.; Harada, T.; Dingal, P.C.D.P.; Pinter, J.; Pajerowski, J.D.; Spinler, K.R.; Shin, J.W.; Tewari, M.; et al. Nuclear Lamin-A Scales with Tissue Stiffness and Enhances Matrix-Directed Differentiation. Science 2013, 341, 1240104. [Google Scholar] [CrossRef]
- Brosig, M.; Ferralli, J.; Gelman, L.; Chiquet, M.; Chiquet-Ehrismann, R. Interfering with the Connection between the Nucleus and the Cytoskeleton Affects Nuclear Rotation, Mechanotransduction and Myogenesis. Int. J. Biochem. Cell Biol. 2010, 42, 1717–1728. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, A.; Hieda, M.; Yokoyama, Y.; Nishioka, Y.; Yoshidome, K.; Tsujimoto, M.; Matsuura, N. Global Loss of a Nuclear Lamina Component, Lamin A/C, and LINC Complex Components SUN1, SUN2, and Nesprin-2 in Breast Cancer. Cancer Med. 2015, 4, 1547–1557. [Google Scholar] [CrossRef] [PubMed]
- Masica, D.L.; Karchin, R. Correlation of Somatic Mutation and Expression Identifies Genes Important in Human Glioblastoma Progression and Survival. Cancer Res. 2011, 71, 4561. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Luo, H.; Hu, Q.; Zhu, H. Identification of Potential Driver Genes Based on Multi-Genomic Data in Cervical Cancer. Front. Genet. 2021, 12, 598304. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Narayanan, V.; Mui, K.L.; O’Bryan, C.S.; Anderson, R.H.; KC, B.; Cabe, J.I.; Denis, K.B.; Antoku, S.; Roux, K.J.; et al. Mechanical Stabilization of the Glandular Acinus by Linker of Nucleoskeleton and Cytoskeleton Complex. Curr. Biol. 2019, 29, 2839.e4. [Google Scholar] [CrossRef]
- De Sandre-Giovannoli, A.; Bernard, R.; Cau, P.; Navarro, C.; Amiel, J.; Boccaccio, I.; Lyonnet, S.; Stewart, C.L.; Munnich, A.; Le Merrer, M.; et al. Lamin A Truncation in Hutchinson-Gilford Progeria. Science 2003, 300, 2055. [Google Scholar] [CrossRef]
- Chang, W.; Wang, Y.; Gant Luxton, G.W.; Östlund, C.; Worman, H.J.; Gundersen, G.G. Imbalanced Nucleocytoskeletal Connections Create Common Polarity Defects in Progeria and Physiological Aging. Proc. Natl. Acad. Sci. USA 2019, 116, 3578–3583. [Google Scholar] [CrossRef]
- Cann, F.; Corbett, M.; O’Sullivan, D.; Tennant, S.; Hailey, H.; Grieve, J.H.K.; Broadhurst, P.; Rankin, R.; Dean, J.C.S. Phenotype-Driven Molecular Autopsy for Sudden Cardiac Death. Clin. Genet. 2017, 91, 22–29. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein Nomenclature | c.DNA Nomenclature | Associated Diseases | Min. Number of Reports of Variant | References | |||
|---|---|---|---|---|---|---|---|
| Skeletal Muscle | Cardiac | Metabolic | Nervous System | ||||
| p.Arg644Cys | 1930C > T | EDMD, muscular dystrophy, LGMD, L-CMD | AVB, DCM, ARVC, LVNC | IR, FPLD | CMT | 23 | [24,30,39,40,41,42,43,44,45,46,47] |
| p.Arg453Trp | 1357C > T | EDMD, LGMD, L-CMD, CFTD | DCM | FPLD | 37 | [44,48,49,50,51,52,53,54,55,56,57,58,59,60,61,62,63,64,65,66,67,68,69] | |
| p.Arg527Pro | 1580G > C | EDMD, LGMD, L-CMD | DCM | FPLD | 21 | [44,46,56,22,58,60,65,66,68,69,70,71] | |
| p.Ser573Leu | 1718C > T | LGMD | DCM, AVB | Diabetes, FPLD | 14 | [44,45,72,73,74] | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Storey, E.C.; Fuller, H.R. Genotype-Phenotype Correlations in Human Diseases Caused by Mutations of LINC Complex-Associated Genes: A Systematic Review and Meta-Summary. Cells 2022, 11, 4065. https://doi.org/10.3390/cells11244065
Storey EC, Fuller HR. Genotype-Phenotype Correlations in Human Diseases Caused by Mutations of LINC Complex-Associated Genes: A Systematic Review and Meta-Summary. Cells. 2022; 11(24):4065. https://doi.org/10.3390/cells11244065
Chicago/Turabian StyleStorey, Emily C., and Heidi R. Fuller. 2022. "Genotype-Phenotype Correlations in Human Diseases Caused by Mutations of LINC Complex-Associated Genes: A Systematic Review and Meta-Summary" Cells 11, no. 24: 4065. https://doi.org/10.3390/cells11244065
APA StyleStorey, E. C., & Fuller, H. R. (2022). Genotype-Phenotype Correlations in Human Diseases Caused by Mutations of LINC Complex-Associated Genes: A Systematic Review and Meta-Summary. Cells, 11(24), 4065. https://doi.org/10.3390/cells11244065