
In-Cell Labeling Coupled to Direct Analysis of Extracellular Vesicles in the Conditioned Medium to Study Extracellular Vesicles Secretion with Minimum Sample Processing and Particle Loss

 , , , ,
, , , , 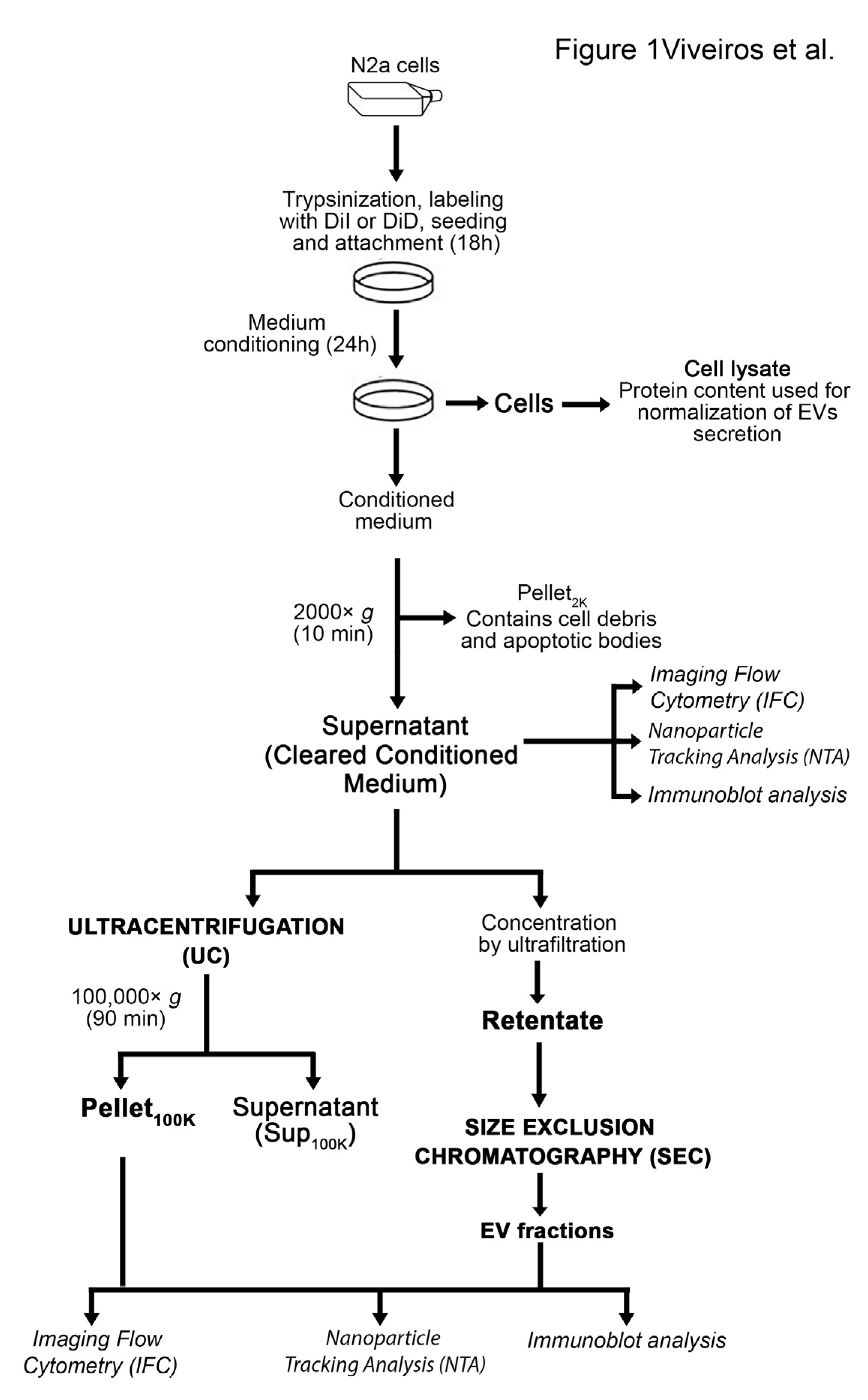{kind=link}
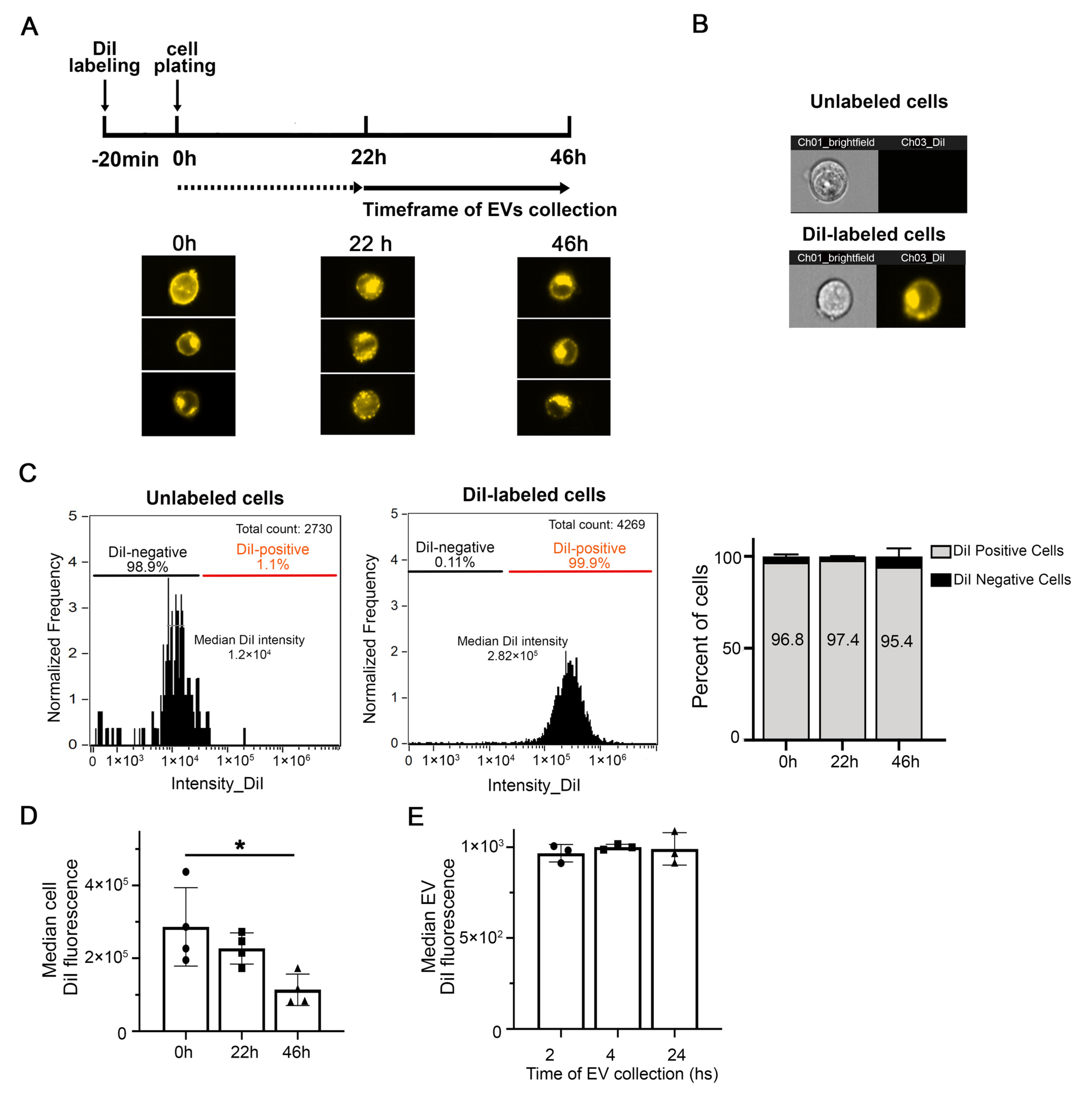{kind=link}
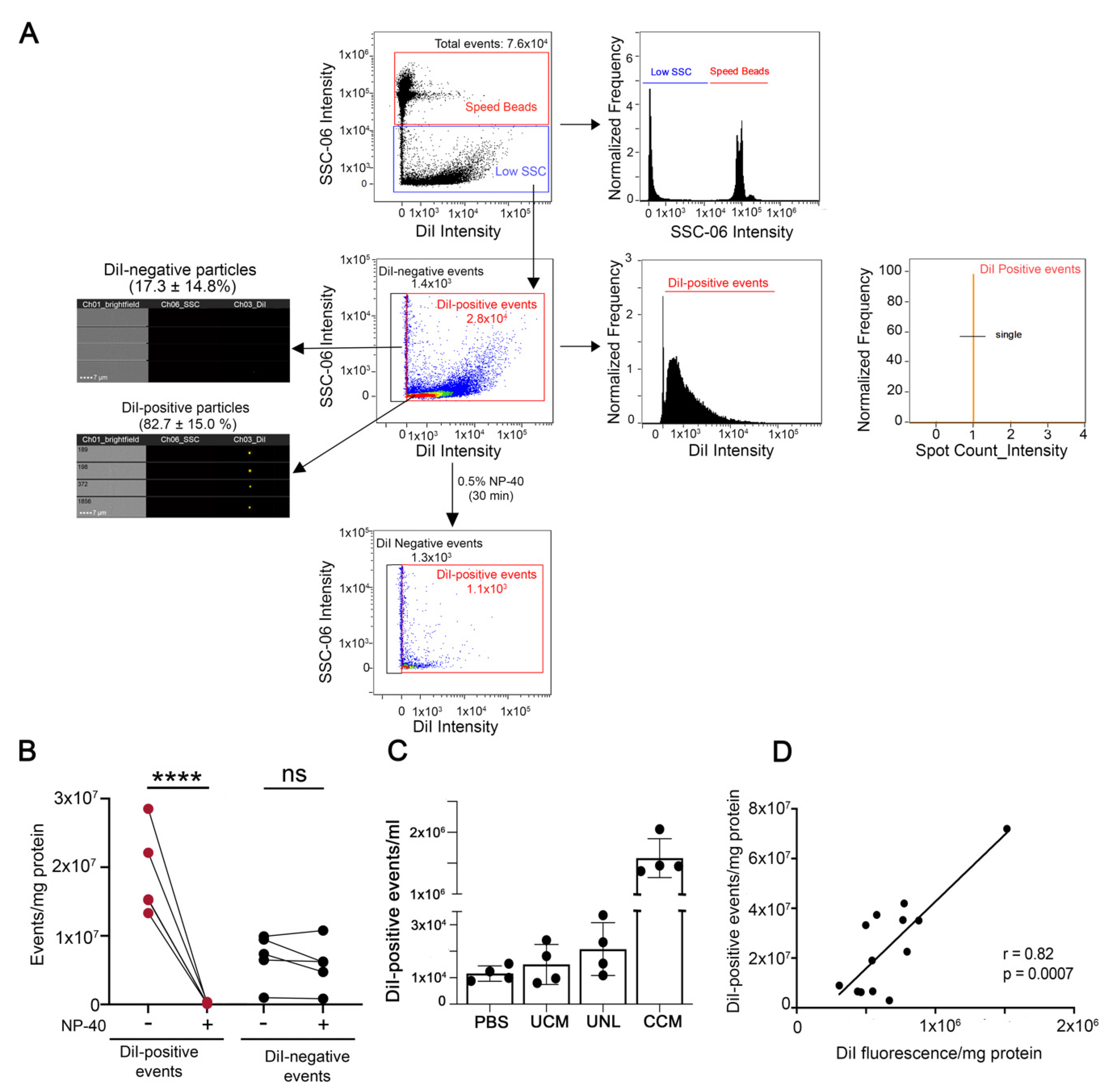{kind=link}
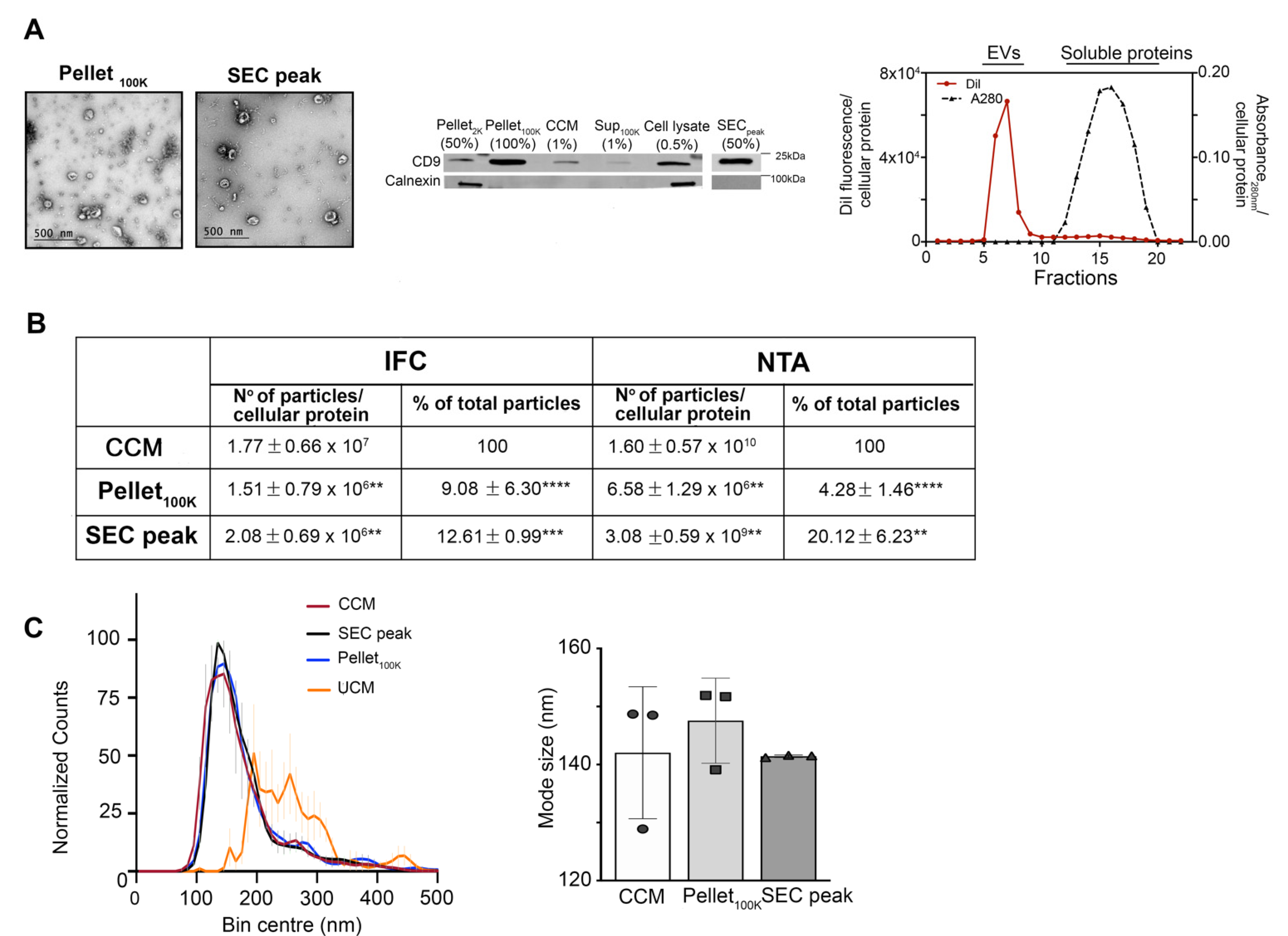{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Cell Culture
2.3. Fluorescent Labeling of Cells for EV Detection
2.4. Collection of EVs in Cell-Conditioned Media
2.5. Fluorometry
2.6. Separation of EVs by Sequential Ultracentrifugation
2.7. Separation of EVs by Ultrafiltration Combined with Size Exclusion Chromatography
2.8. In Vitro Labeling of EVs
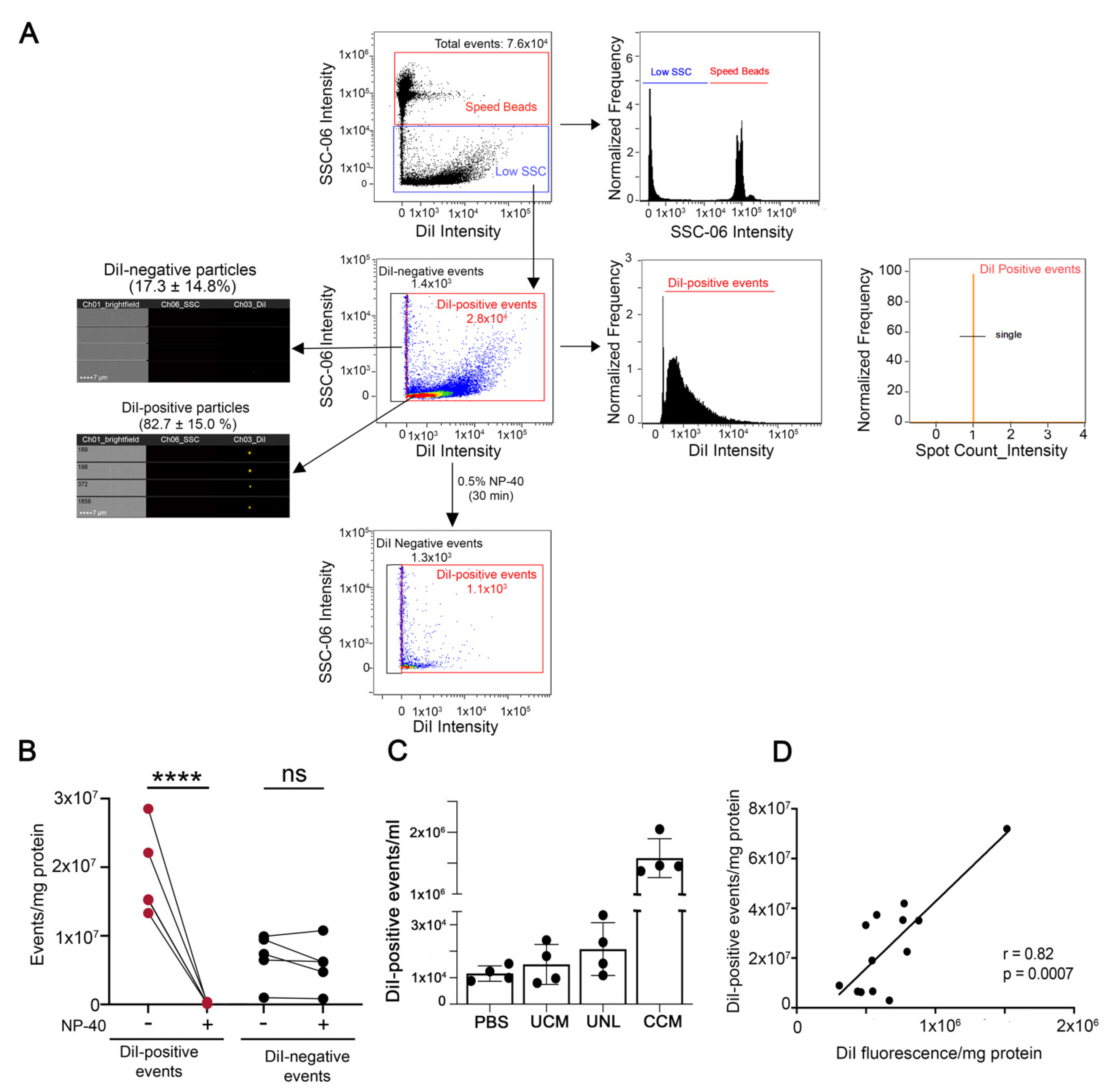
2.9. Analysis of EVs by Image Flow Cytometry (IFC)
2.10. Nanoparticle Tracking Analysis (NTA)
2.11. Transmission Electron Microscopy
2.12. Immunoblotting
2.13. Statistical Analysis
3. Results
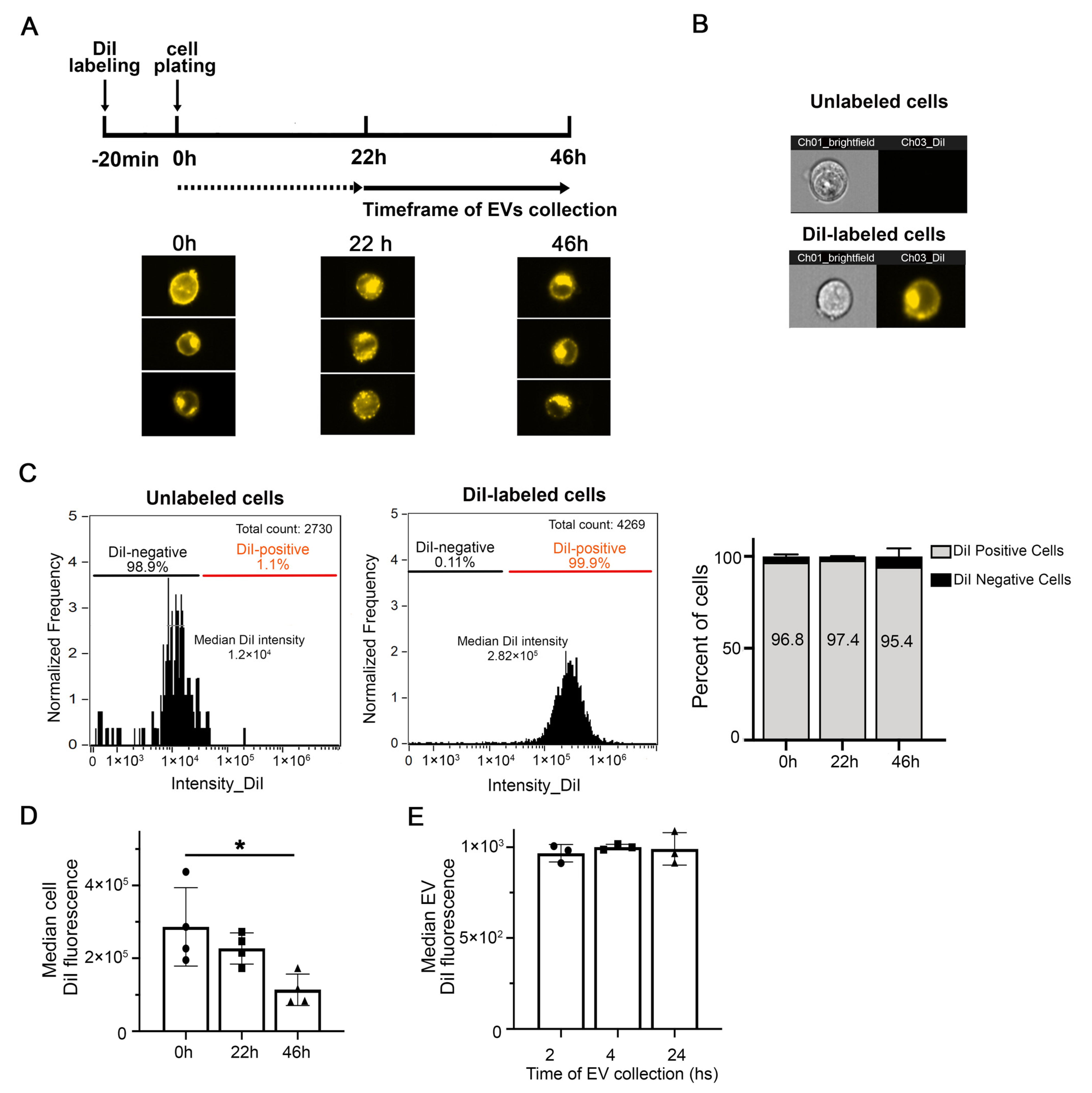
3.1. Cell and EV Labeling
3.2. Cell Culture Conditions
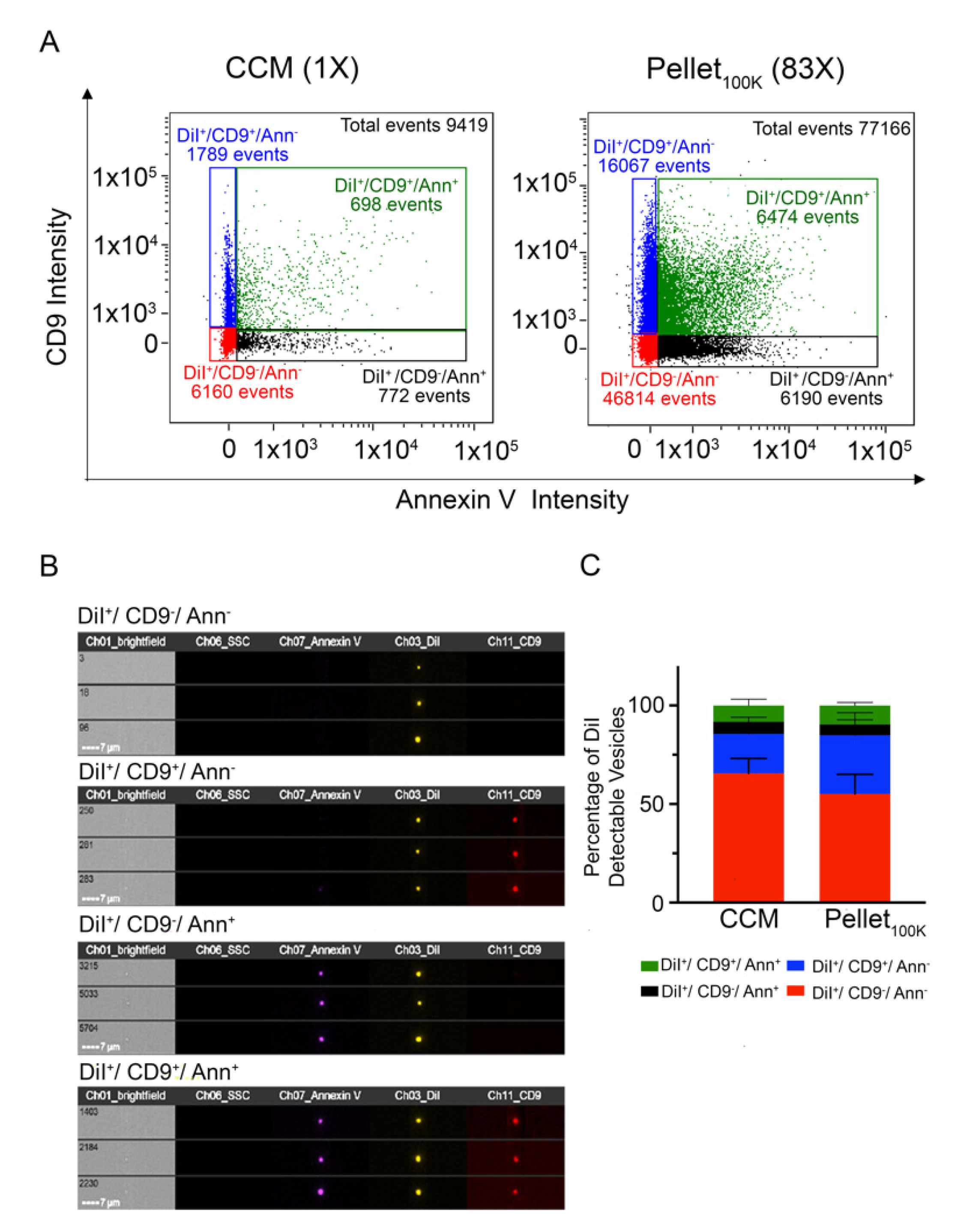
3.3. Comparative Analysis of EVs in the Cleared Conditioned Medium and upon Separation by Ultracentrifugation or Ultrafiltration-Size Exclusion Chromatography
3.4. Direct Analysis of EVs in the Cleared Conditioned Medium after Cell Treatment with Bafilomycin
4. Discussion
4.1. EVs Labeling
4.2. EVs Separation and Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Raposo, G.; Stahl, P.D. Extracellular vesicles: A new communication paradigm? Nat. Rev. Mol. Cell Biol. 2019, 20, 509–510. [Google Scholar] [CrossRef]
- Mathews, P.M.; Levy, E. Exosome Production Is Key to Neuronal Endosomal Pathway Integrity in Neurodegenerative Diseases. Front. Neurosci. 2019, 13, 1347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murao, A.; Brenner, M.; Aziz, M.; Wang, P. Exosomes in Sepsis. Front. Immunol. 2020, 11, 2140. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, A.; Rathore, S.; Munshi, A.; Ramesh, R. Extracellular Vesicles in Oncology: From Immune Suppression to Immunotherapy. AAPS J. 2021, 23, 1–15. [Google Scholar] [CrossRef]
- Kim, I.A.; Hur, J.Y.; Kim, H.J.; Lee, S.E.; Kim, W.S.; Lee, K.Y. Liquid biopsy using extracellular vesicle–derived DNA in lung adenocarcinoma. J. Pathol. Transl. Med. 2020, 54, 453–461. [Google Scholar] [CrossRef]
- Hu, T.; Wolfram, J.; Srivastava, S. Extracellular Vesicles in Cancer Detection: Hopes and Hypes. Trends Cancer 2021, 7, 122–133. [Google Scholar] [CrossRef] [PubMed]
- Sarko, D.K.; McKinney, C.E. Exosomes: Origins and Therapeutic Potential for Neurodegenerative Disease. Front. Neurosci. 2017, 11, 82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nam, G.; Choi, Y.; Kim, G.B.; Kim, S.; A Kim, S.; Kim, I. Emerging Prospects of Exosomes for Cancer Treatment: From Conventional Therapy to Immunotherapy. Adv. Mater. 2020, 32, e2002440. [Google Scholar] [CrossRef]
- Born, L.J.; Harmon, J.W.; Jay, S.M. Therapeutic potential of extracellular vesicle-associated long noncoding RNA. Bioeng. Transl. Med. 2020, 5, 10172. [Google Scholar] [CrossRef] [PubMed]
- Zocchi, M.R.; Tosetti, F.; Benelli, R.; Poggi, A. Cancer Nanomedicine Special Issue Review Anticancer Drug Delivery with Nanoparticles: Extracellular Vesicles or Synthetic Nanobeads as Therapeutic Tools for Conventional Treatment or Immunotherapy. Cancers 2020, 12, 1886. [Google Scholar] [CrossRef]
- Kostyushev, D.; Kostyusheva, A.; Brezgin, S.; Smirnov, V.; Volchkova, E.; Lukashev, A.; Chulanov, V. Gene Editing by Extracellular Vesicles. Int. J. Mol. Sci. 2020, 21, 7362. [Google Scholar] [CrossRef]
- Mehryab, F.; Rabbani, S.; Shahhosseini, S.; Shekari, F.; Fatahi, Y.; Baharvand, H.; Haeri, A. Exosomes as a next-generation drug delivery system: An update on drug loading approaches, characterization, and clinical application challenges. Acta Biomater. 2020, 113, 42–62. [Google Scholar] [CrossRef]
- Yan, H.-C.; Yu, T.-T.; Li, J.; Qiao, Y.-Q.; Wang, L.-C.; Zhang, T.; Li, Q.; Zhou, Y.-H.; Liu, D.-W. The Delivery of Extracellular Vesicles Loaded in Biomaterial Scaffolds for Bone Regeneration. Front. Bioeng. Biotechnol. 2020, 8, 1015. [Google Scholar] [CrossRef]
- Andjus, P.; Kosanović, M.; Milićević, K.; Gautam, M.; Vainio, S.J.; Jagečić, D.; Kozlova, E.N.; Pivoriūnas, A.; Chachques, J.-C.; Sakaj, M.; et al. Extracellular Vesicles as Innovative Tool for Diagnosis, Regeneration and Protection against Neurological Damage. Int. J. Mol. Sci. 2020, 21, 6859. [Google Scholar] [CrossRef]
- Margolis, L.; Sadovsky, Y. The biology of extracellular vesicles: The known unknowns. PLoS Biol. 2019, 17, e3000363. [Google Scholar] [CrossRef]
- Stahl, P.D.; Raposo, G. Extracellular Vesicles: Exosomes and Microvesicles, Integrators of Homeostasis. Physiology 2019, 34, 169–177. [Google Scholar] [CrossRef]
- Gardiner, C.; Di Vizio, D.; Sahoo, S.; Théry, C.; Witwer, K.W.; Wauben, M.; Hill, A.F. Techniques used for the isolation and characterization of extracellular vesicles: Results of a worldwide survey. J. Extracell. Vesicles 2016, 5, 32945. [Google Scholar] [CrossRef] [PubMed]
- Royo, F.; Théry, C.; Falcón-Pérez, J.M.; Nieuwland, R.; Witwer, K.W. Methods for Separation and Characterization of Extracellular Vesicles: Results of a Worldwide Survey Performed by the ISEV Rigor and Standardization Subcommittee. Cells 2020, 9, 1955. [Google Scholar] [CrossRef]
- van der Vlist, E.J.; Nolte-’t Hoen, E.N.; Stoorvogel, W.; Arkesteijn, G.J.; Wauben, M.H. Fluorescent labeling of nano-sized vesicles released by cells and subsequent quantitative and qualitative analysis by high-resolution flow cytometry. Nat. Protoc. 2012, 7, 1311–1326. [Google Scholar] [CrossRef] [PubMed]
- Erdbrügger, U.; Rudy, C.K.; Etter, M.E.; Dryden, K.A.; Yeager, M.; Klibanov, A.L.; Lannigan, J. Imaging flow cytometry elucidates limitations of microparticle analysis by conventional flow cytometry. Cytom. Part A 2014, 85, 756–770. [Google Scholar] [CrossRef] [PubMed]
- Görgens, A.; Bremer, M.; Ferrer-Tur, R.; Murke, F.; Tertel, T.; Horn, P.A.; Thalmann, S.; Welsh, J.A.; Probst, C.; Guerin, C.; et al. Optimisation of imaging flow cytometry for the analysis of single extracellular vesicles by using fluorescence-tagged vesicles as biological reference material. J. Extracell. Vesicles 2019, 8, 1587567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lannigan, J.; Erdbruegger, U. Imaging flow cytometry for the characterization of extracellular vesicles. Methods 2017, 112, 55–67. [Google Scholar] [CrossRef] [PubMed]
- Morales-Kastresana, A.; Telford, B.; Musich, T.A.; McKinnon, K.; Clayborne, C.; Braig, Z.; Rosner, A.; Demberg, T.; Watson, D.C.; Karpova, T.; et al. Labeling Extracellular Vesicles for Nanoscale Flow Cytometry. Sci. Rep. 2017, 7, 1878. [Google Scholar] [CrossRef]
- Dominkuš, P.P.; Stenovec, M.; Sitar, S.; Lasič, E.; Zorec, R.; Plemenitaš, A.; Žagar, E.; Kreft, M.; Lenassi, M. PKH26 labeling of extracellular vesicles: Characterization and cellular internalization of contaminating PKH26 nanoparticles. Biochim. Biophys. Acta Biomembr. 2018, 1860, 1350–1361. [Google Scholar] [CrossRef] [PubMed]
- Lai, C.P.-K.; Kim, E.Y.; Badr, C.E.; Weissleder, R.; Mempel, T.R.; Tannous, B.A.; Breakefield, X.O. Visualization and tracking of tumour extracellular vesicle delivery and RNA translation using multiplexed reporters. Nat. Commun. 2015, 6, 7029. [Google Scholar] [CrossRef] [PubMed]
- Russell, A.E.; Sneider, A.; Witwer, K.W.; Bergese, P.; Bhattacharyya, S.N.; Cocks, A.; Cocucci, E.; Erdbrügger, U.; Falcon-Perez, J.M.; Freeman, D.W.; et al. Biological membranes in EV biogenesis, stability, uptake, and cargo transfer: An ISEV position paper arising from the ISEV membranes and EVs workshop. J. Extracell. Vesicles 2019, 8, 1684862. [Google Scholar] [CrossRef] [Green Version]
- Lobb, R.J.; Becker, M.; Wen, S.W.; Wong, C.S.F.; Wiegmans, A.P.; Leimgruber, A.; Möller, A. Optimized exosome isolation protocol for cell culture supernatant and human plasma. J. Extracell. Vesicles 2015, 4, 27031. [Google Scholar] [CrossRef]
- Lee, K.; Mower, R.; Hollenbeck, T.; Castelo, J.; Johnson, N.; Gordon, P.; Sinko, P.J.; Holme, K.; Lee, Y. Modulation of nonspecific binding in ultrafiltration protein binding studies. Pharm. Res. 2003, 20, 1015–1021. [Google Scholar] [CrossRef] [PubMed]
- Inglis, H.C.; Danesh, A.; Shah, A.; Lacroix, J.; Spinella, P.C.; Norris, P.J. Techniques to improve detection and analysis of extracellular vesicles using flow cytometry. Cytom. Part A 2015, 87, 1052–1063. [Google Scholar] [CrossRef] [Green Version]
- Théry, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef] [Green Version]
- Coumans, F.A.W.; Brisson, A.R.; Buzas, E.I.; Dignat-George, F.; Drees, E.E.E.; El-Andaloussi, S.; Emanueli, C.; Gasecka, A.; Hendrix, A.; Hill, A.F.; et al. Methodological Guidelines to Study Extracellular Vesicles. Circ. Res. 2017, 120, 1632–1648. [Google Scholar] [CrossRef]
- Lehrich, B.M.; Liang, Y.; Fiandaca, M.S. Foetal bovine serum influence on in vitro extracellular vesicle analyses. J. Extracell. Vesicles 2021, 10, e12061. [Google Scholar] [CrossRef]
- Linares, R.; Tan, S.; Gounou, C.; Arraud, N.; Brisson, A.R. High-speed centrifugation induces aggregation of extracellular vesicles. J. Extracell. Vesicles 2015, 4, 29509. [Google Scholar] [CrossRef] [PubMed]
- Baixauli, F.; López-Otín, C.; Mittelbrunn, M. Exosomes and Autophagy: Coordinated Mechanisms for the Maintenance of Cellular Fitness. Front. Immunol. 2014, 5, 403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savina, A.; Furlán, M.; Vidal, M.; Colombo, M.I.; Gamel-Didelon, K.; Kunz, L.; Föhr, K.J.; Gratzl, M.; Mayerhofer, A. Exosome Release Is Regulated by a Calcium-dependent Mechanism in K562 Cells. J. Biol. Chem. 2003, 278, 20083–20090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cashikar, A.G.; Hanson, P.I. A cell-based assay for CD63-containing extracellular vesicles. PLoS ONE 2019, 14, e0220007. [Google Scholar] [CrossRef] [PubMed]
- Pospíchalová, V.; Svoboda, J.; Dave, Z.; Kotrbová, A.; Kaiser, K.; Klemova, D.; Ilkovics, L.; Hampl, A.; Crha, I.; Jandakova, E.; et al. Simplified protocol for flow cytometry analysis of fluorescently labeled exosomes and microvesicles using dedicated flow cytometer. J. Extracell. Vesicles 2015, 4, 25530. [Google Scholar] [CrossRef]
- Wiklander, O.P.B.; Nordin, J.Z.; O’Loughlin, A.; Gustafsson, Y.; Corso, G.; Mäger, I.; Vader, P.; Lee, Y.; Sork, H.; Seow, Y.; et al. Extracellular vesicle in vivo biodistribution is determined by cell source, route of administration and targeting. J. Extracell. Vesicles 2015, 4, 26316. [Google Scholar] [CrossRef] [Green Version]
- Grange, C.; Tapparo, M.; Bruno, S.; Chatterjee, D.; Quesenberry, P.J.; Tetta, C.; Camussi, G. Biodistribution of mesenchymal stem cell-derived extracellular vesicles in a model of acute kidney injury monitored by optical imaging. Int. J. Mol. Med. 2014, 33, 1055–1063. [Google Scholar] [CrossRef] [Green Version]
- Nicola, A.M.; Frases, S.; Casadevall, A. Lipophilic Dye Staining of Cryptococcus neoformans Extracellular Vesicles and Capsule. Eukaryot. Cell 2009, 8, 1373–1380. [Google Scholar] [CrossRef] [Green Version]
- Tamura, R.; Uemoto, S.; Tabata, Y. Immunosuppressive effect of mesenchymal stem cell-derived exosomes on a concanavalin A-induced liver injury model. Inflamm. Regen. 2016, 36, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimomura, T.; Seino, R.; Umezaki, K.; Shimoda, A.; Ezoe, T.; Ishiyama, M.; Akiyoshi, K. New Lipophilic Fluorescent Dyes for Labeling Extracellular Vesicles: Characterization and Monitoring of Cellular Uptake. Bioconjug. Chem. 2021, 32, 680–684. [Google Scholar] [CrossRef] [PubMed]
- Panagopoulou, M.S.; Wark, A.; Birch, D.; Gregory, C.D. Phenotypic analysis of extracellular vesicles: A review on the applications of fluorescence. J. Extracell. Vesicles 2020, 9, 1710020. [Google Scholar] [CrossRef] [PubMed]
- Dehghani, M.; Gulvin, S.M.; Flax, J.; Gaborski, T.R. Systematic Evaluation of PKH Labelling on Extracellular Vesicle Size by Nanoparticle Tracking Analysis. Sci. Rep. 2020, 10, 9533. [Google Scholar] [CrossRef]
- Ender, F.; Zamzow, P.; Von Bubnoff, N.; Gieseler, F. Detection and Quantification of Extracellular Vesicles via FACS: Membrane Labeling Matters! Int. J. Mol. Sci. 2019, 21, 291. [Google Scholar] [CrossRef] [Green Version]
- Dabrowska, S.; Del Fattore, A.; Karnas, E.; Frontczak-Baniewicz, M.; Kozlowska, H.; Muraca, M.; Janowski, M.; Lukomska, B. Imaging of extracellular vesicles derived from human bone marrow mesenchymal stem cells using fluorescent and magnetic labels. Int. J. Nanomed. 2018, 13, 1653–1664. [Google Scholar] [CrossRef] [Green Version]
- Samlowski, W.E.; Robertson, B.A.; Draper, B.K.; Prystas, E.; McGregor, J.R. Effects of supravital fluorochromes used to analyze the in vivo homing of murine lymphocytes on cellular function. J. Immunol. Methods 1991, 144, 101–115. [Google Scholar] [CrossRef]
- Mittelbrunn, M.; Gutiérrez-Vázquez, C.; Villarroya-Beltri, C.; González, S.; Sánchez-Cabo, F.; González, M.Á.; Bernad, A.; Sánchez-Madrid, F. Unidirectional transfer of microRNA-loaded exosomes from T cells to antigen-presenting cells. Nat. Commun. 2011, 2, 282. [Google Scholar] [CrossRef] [Green Version]
- Piao, Y.J.; Kim, H.S.; Hwang, E.H.; Woo, J.; Zhang, M.; Moon, W.K. Breast cancer cell-derived exosomes and macrophage polarization are associated with lymph node metastasis. Oncotarget 2017, 9, 7398–7410. [Google Scholar] [CrossRef]
- Verweij, F.J.; Bebelman, M.P.; Jimenez, C.R.; Garcia-Vallejo, J.J.; Janssen, H.; Neefjes, J.; Knol, J.C.; Haas, R.D.G.-D.; Piersma, S.R.; Baglio, S.R.; et al. Quantifying exosome secretion from single cells reveals a modulatory role for GPCR signaling. J. Cell Biol. 2018, 217, 1129–1142. [Google Scholar] [CrossRef]
- Chuo, S.T.-Y.; Chien, J.C.-Y.; Lai, C.P.-K. Imaging extracellular vesicles: Current and emerging methods. J. Biomed. Sci. 2018, 25, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Lee, T.S.; Kim, Y.; Zhang, W.; Song, I.H.; Tung, C.-H. Facile metabolic glycan labeling strategy for exosome tracking. Biochim. Biophys. Acta Gen. Subj. 2018, 1862, 1091–1100. [Google Scholar] [CrossRef]
- Zhang, P.; Dong, B.; Zeng, E.; Wang, F.; Jiang, Y.; Li, D.; Liu, D. In Vivo Tracking of Multiple Tumor Exosomes Labeled by Phospholipid-Based Bioorthogonal Conjugation. Anal. Chem. 2018, 90, 11273–11279. [Google Scholar] [CrossRef]
- Gardiner, C.; Ferreira, Y.J.; Dragovic, R.A.; Redman, C.W.G.; Sargent, I.L. Extracellular vesicle sizing and enumeration by nanoparticle tracking analysis. J. Extracell. Vesicles 2013, 2, 19671. [Google Scholar] [CrossRef] [PubMed]
- Livshits, M.A.; Khomyakova, E.; Evtushenko, E.; Lazarev, V.N.; Kulemin, N.; Semina, S.E.; Generozov, E.; Govorun, V.M. Isolation of exosomes by differential centrifugation: Theoretical analysis of a commonly used protocol. Sci. Rep. 2015, 5, 17319. [Google Scholar] [CrossRef] [PubMed]
- Brennan, K.; Martin, K.; Fitzgerald, S.P.; O’Sullivan, J.; Wu, Y.; Blanco, A.; Richardson, C.; Mc Gee, M.M. A comparison of methods for the isolation and separation of extracellular vesicles from protein and lipid particles in human serum. Sci. Rep. 2020, 10, 1039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mastoridis, S.; Bertolino, G.M.; Whitehouse, G.; Dazzi, F.; Sanchez-Fueyo, A.; Martinez-Llordella, M. Multiparametric Analysis of Circulating Exosomes and Other Small Extracellular Vesicles by Advanced Imaging Flow Cytometry. Front. Immunol. 2018, 9, 1583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filipe, V.; Hawe, A.; Jiskoot, W. Critical Evaluation of Nanoparticle Tracking Analysis (NTA) by NanoSight for the Measurement of Nanoparticles and Protein Aggregates. Pharm. Res. 2010, 27, 796–810. [Google Scholar] [CrossRef] [Green Version]
- Erdbrügger, U.; Lannigan, J. Analytical challenges of extracellular vesicle detection: A comparison of different techniques. Cytom. Part A 2015, 89, 123–134. [Google Scholar] [CrossRef]
- Lamparski, H.G.; Metha-Damani, A.; Yao, J.-Y.; Patel, S.; Hsu, D.-H.; Ruegg, C.; Le Pecq, J.-B. Production and characterization of clinical grade exosomes derived from dendritic cells. J. Immunol. Methods 2002, 270, 211–226. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Viveiros, A.; Kadam, V.; Monyror, J.; Morales, L.C.; Pink, D.; Rieger, A.M.; Sipione, S.; Posse de Chaves, E. In-Cell Labeling Coupled to Direct Analysis of Extracellular Vesicles in the Conditioned Medium to Study Extracellular Vesicles Secretion with Minimum Sample Processing and Particle Loss. Cells 2022, 11, 351. https://doi.org/10.3390/cells11030351
Viveiros A, Kadam V, Monyror J, Morales LC, Pink D, Rieger AM, Sipione S, Posse de Chaves E. In-Cell Labeling Coupled to Direct Analysis of Extracellular Vesicles in the Conditioned Medium to Study Extracellular Vesicles Secretion with Minimum Sample Processing and Particle Loss. Cells. 2022; 11(3):351. https://doi.org/10.3390/cells11030351
Chicago/Turabian StyleViveiros, Anissa, Vaibhavi Kadam, John Monyror, Luis Carlos Morales, Desmond Pink, Aja M. Rieger, Simonetta Sipione, and Elena Posse de Chaves. 2022. "In-Cell Labeling Coupled to Direct Analysis of Extracellular Vesicles in the Conditioned Medium to Study Extracellular Vesicles Secretion with Minimum Sample Processing and Particle Loss" Cells 11, no. 3: 351. https://doi.org/10.3390/cells11030351
APA StyleViveiros, A., Kadam, V., Monyror, J., Morales, L. C., Pink, D., Rieger, A. M., Sipione, S., & Posse de Chaves, E. (2022). In-Cell Labeling Coupled to Direct Analysis of Extracellular Vesicles in the Conditioned Medium to Study Extracellular Vesicles Secretion with Minimum Sample Processing and Particle Loss. Cells, 11(3), 351. https://doi.org/10.3390/cells11030351