
Secreted Neutrophil Gelatinase-Associated Lipocalin Shows Stronger Ability to Inhibit Cyst Enlargement of ADPKD Cells Compared with Nonsecreted Form

 ,
,  , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. Cell Culture
2.3. Lectin Staining
2.4. DNA Lysate and Genotyping
2.5. Quantitative Reverse Transcription Real-Time Polymerase Chain Reaction (RT–qPCR)
2.6. Cell Viability Assay
2.7. Cell Proliferation Assay
2.8. Apoptosis Assay
2.9. Western Blot Analysis
2.10. Three-Dimensional Cell Culture (3D Culture)
2.11. Immunocytochemistry (ICC)
2.12. Confocal Microscopy
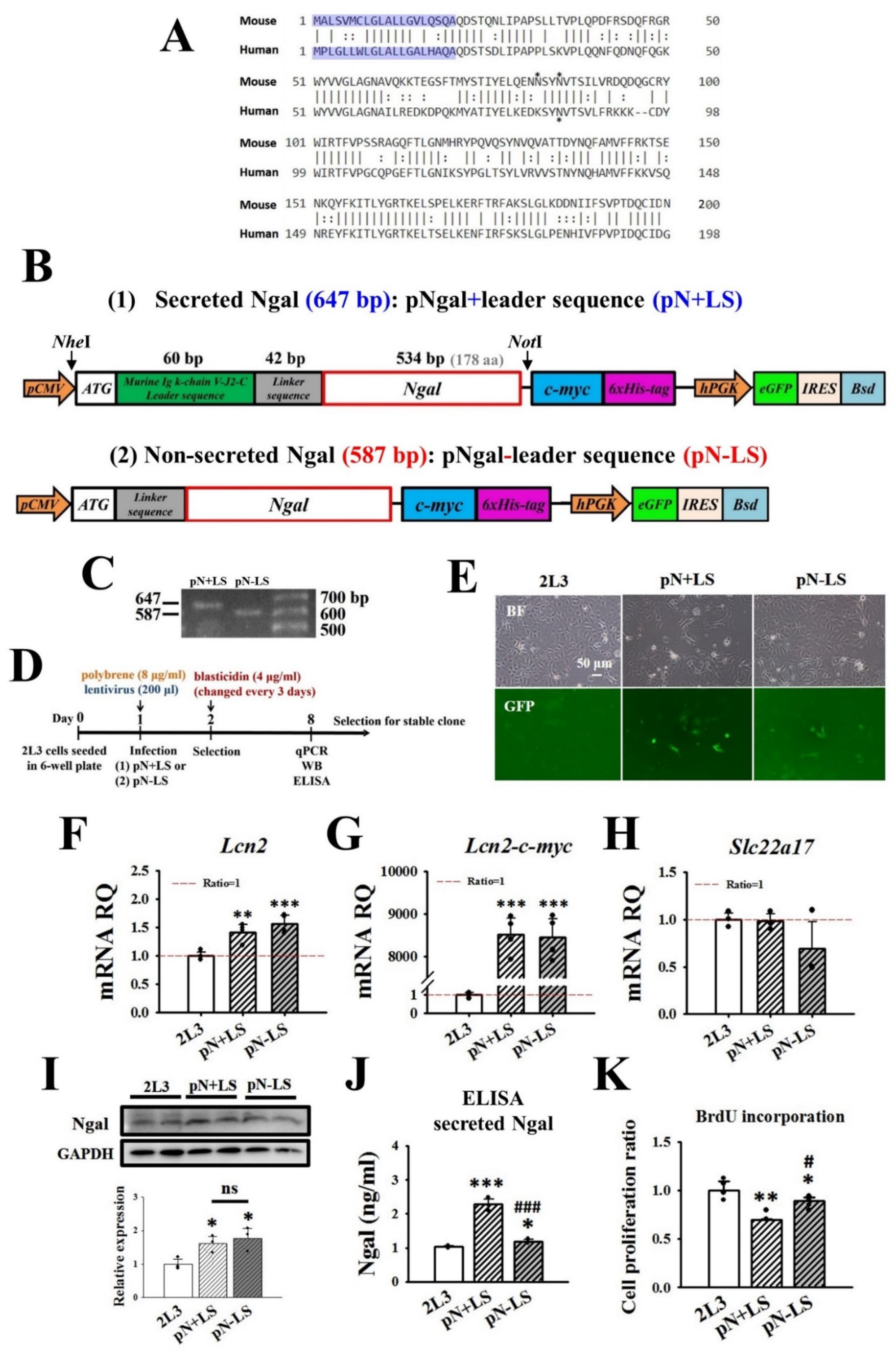
2.13. Plasmid Construction and Virus Infection
2.14. Enzyme-Linked Immunosorbent Assay (ELISA)
2.15. Statistical Analysis
3. Results
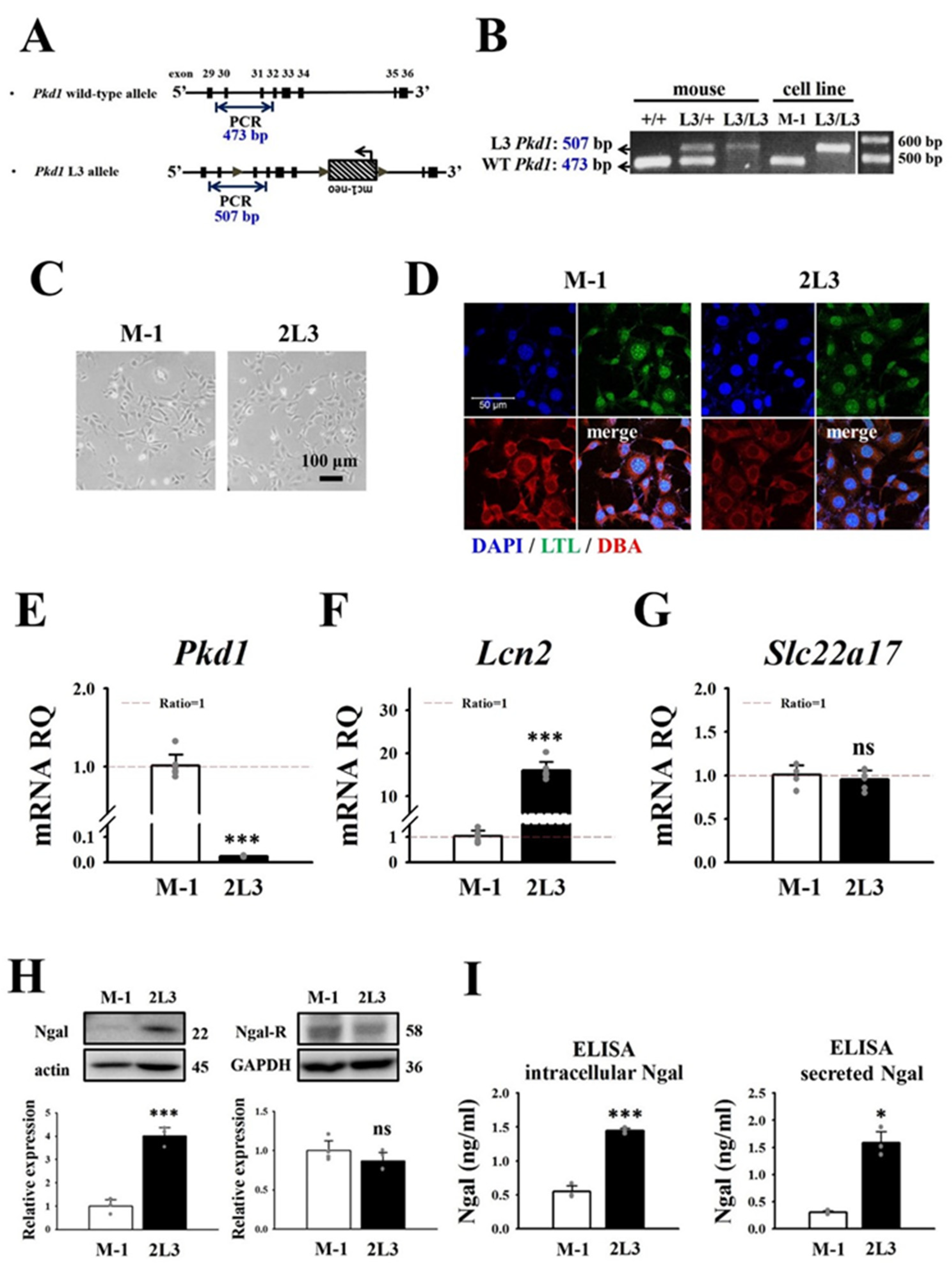
3.1. Comparison of NGAL Expression and Cell Proliferation between M-1 Wild Type and 2L3 ADPKD Cells
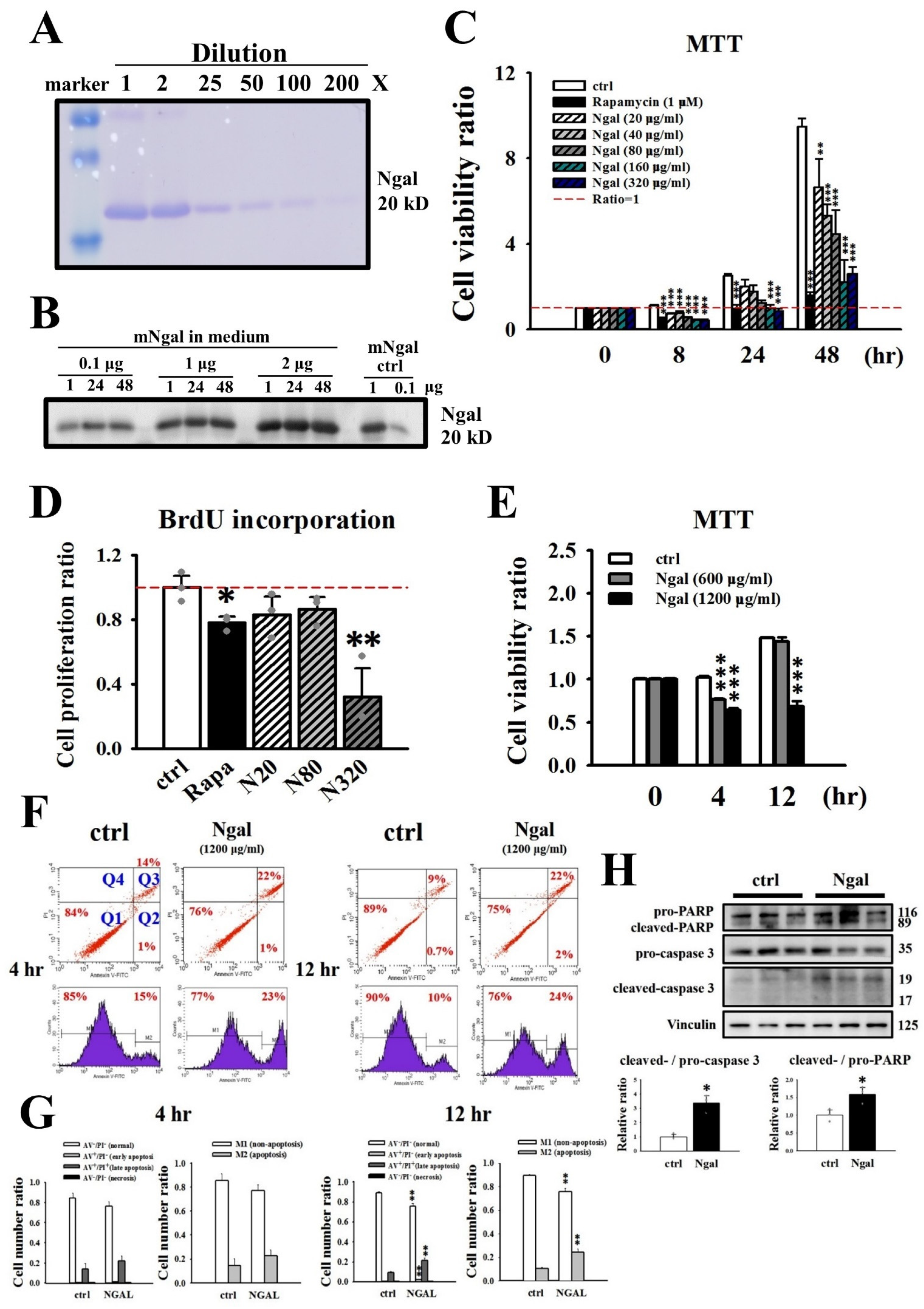
3.2. Effects of the Addition of mNGAL on the Viability and Proliferation of 2L3 Cells

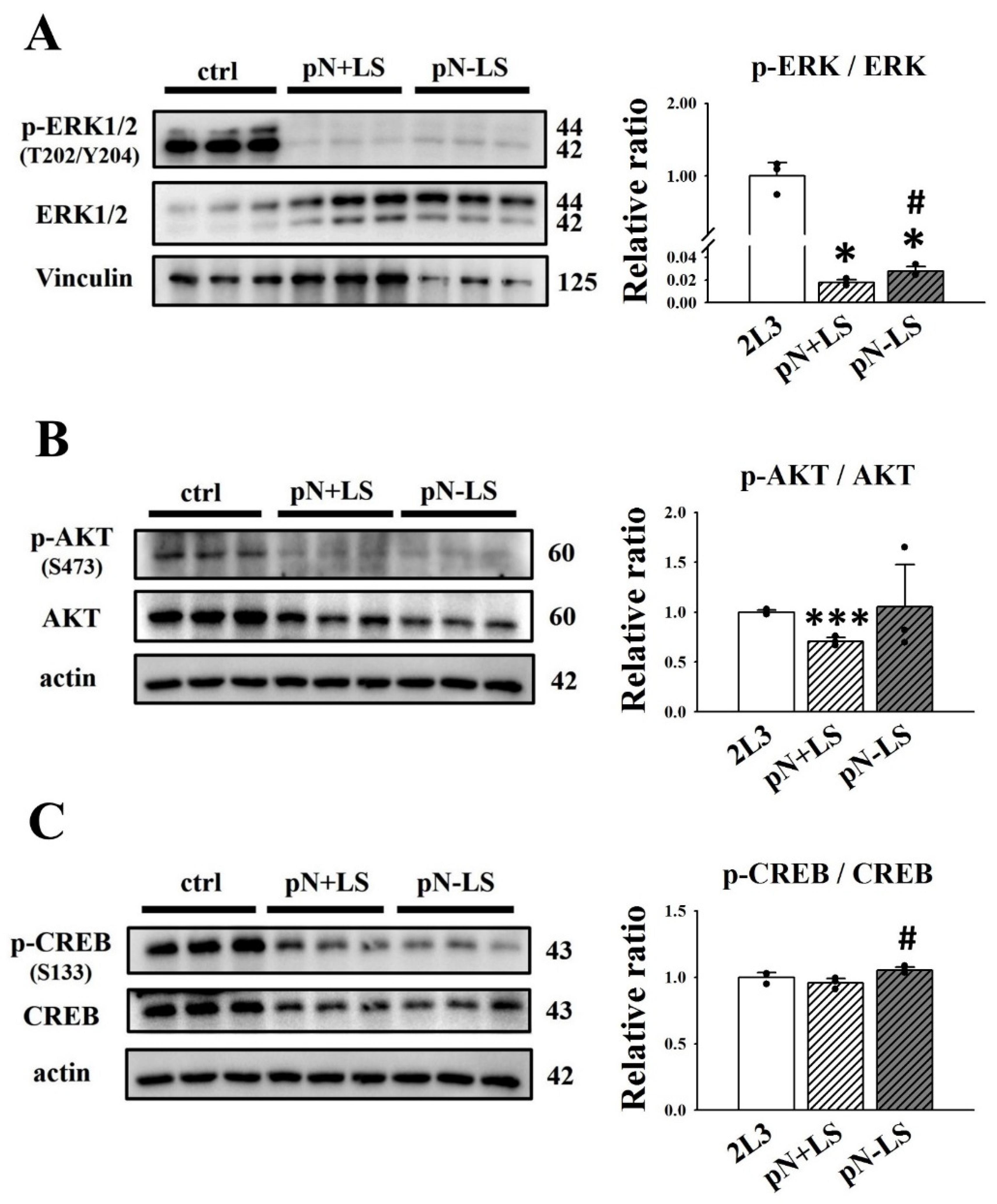
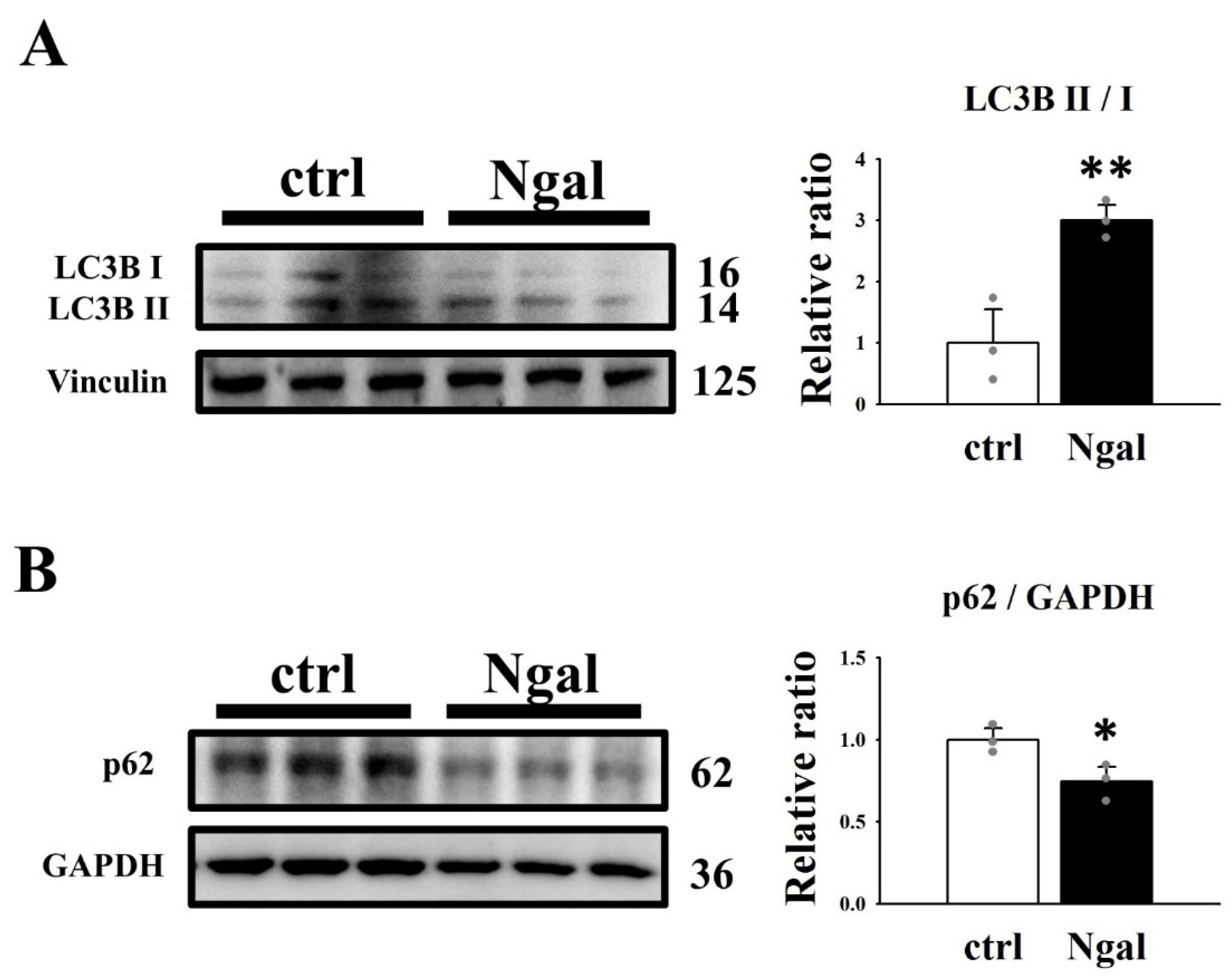
3.3. Signaling Pathways Affected by the Addition of mNGAL to 2L3 Cells
3.4. Effects of mNGAL on Cyst Formation of 2L3 Cells in 3D Culture
3.5. Effects of Transfection with pN + LS and pN − LS on NGAL Expression and Cell Proliferation in 2L3 Cells
3.6. Effects of mNGAL Overexpression Leading to Signaling Pathways in 2L3 Cells
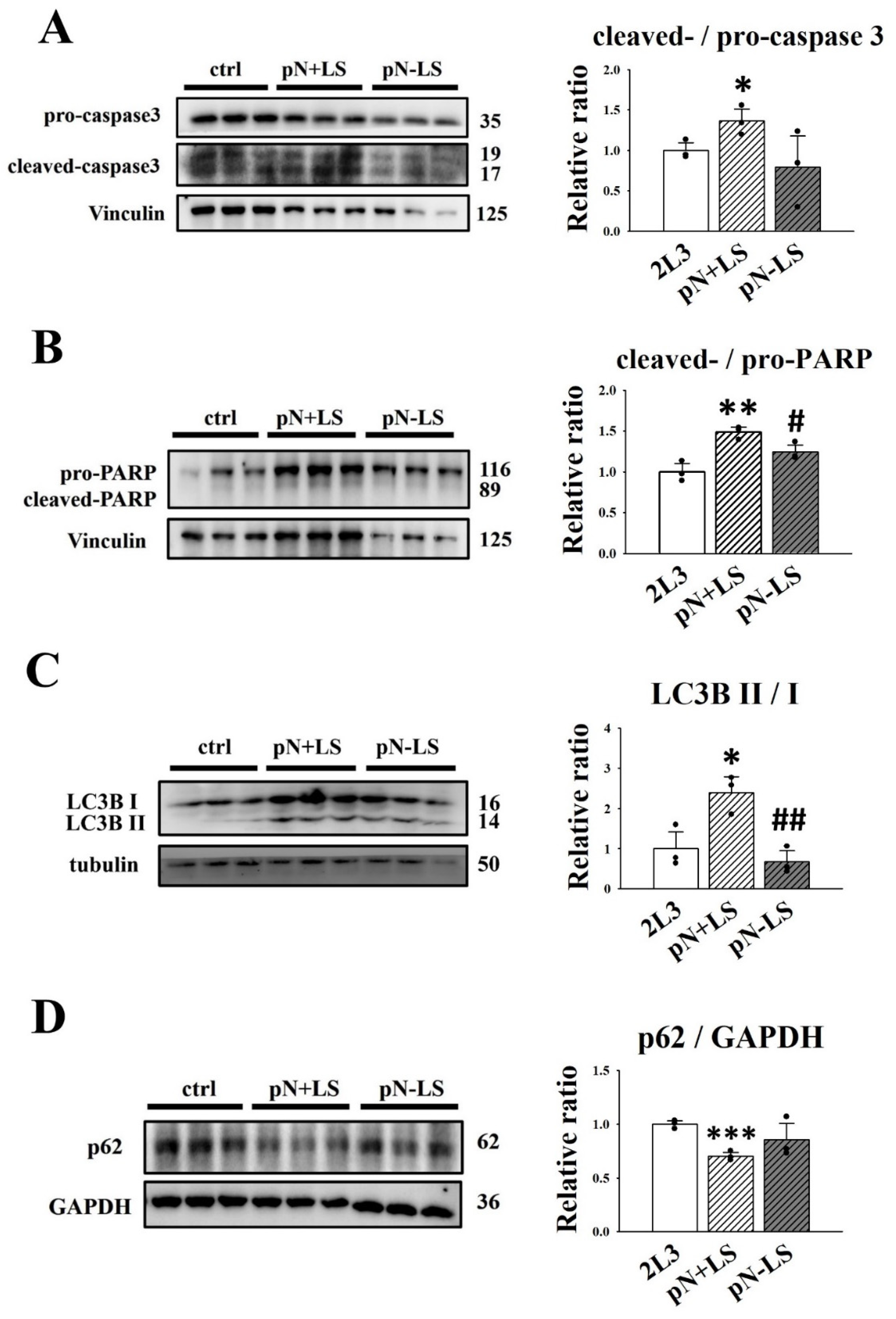
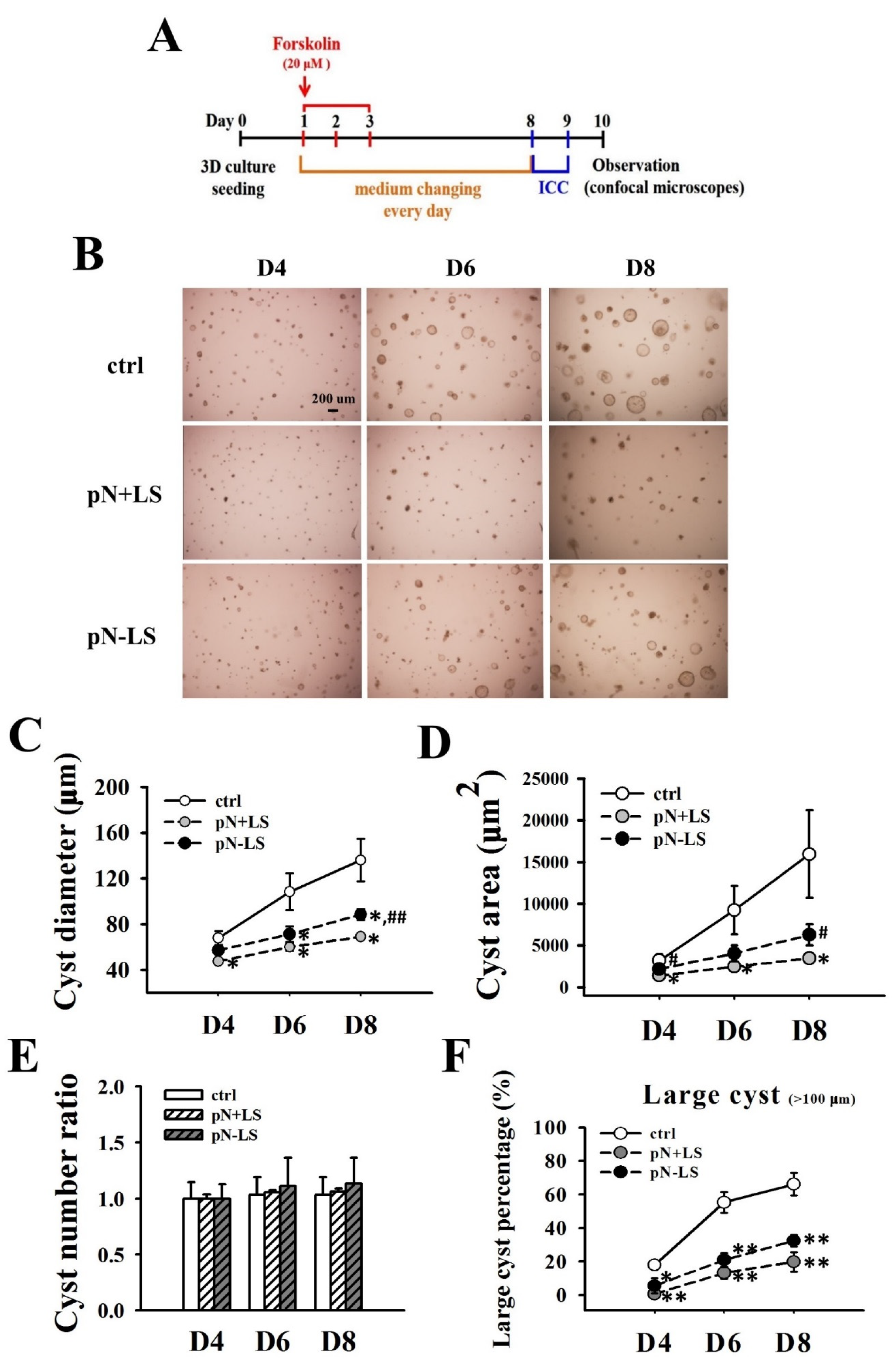
3.7. Effects of Overexpression of mNGAL on Cyst Formation of 2L3 Cells in 3D Culture
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bergmann, C.; Guay-Woodford, L.M.; Harris, P.C.; Horie, S.; Peters, D.J.; Torres, V.E. Polycystic kidney disease. Nat. Rev. Dis. Primers 2018, 4, 50. [Google Scholar] [CrossRef] [PubMed]
- Grantham, J.J. Clinical practice. Autosomal dominant polycystic kidney disease. N. Engl. J. Med. 2008, 359, 1477–1485. [Google Scholar] [CrossRef] [PubMed]
- Chapman, A.B.; Devuyst, O.; Eckardt, K.-U.; Gansevoort, R.T.; Harris, T.; Horie, S.; Kasiske, B.L.; Odland, D.; Pei, Y.; Perrone, R.D.; et al. Autosomal-dominant polycystic kidney disease (ADPKD): Executive summary from a Kidney Disease: Improving Global Outcomes (KDIGO) Controversies Conference. Kidney Int. 2015, 88, 17–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grantham, J.J.; Mulamalla, S.; Swenson-Fields, K.I. Why kidneys fail in autosomal dominant polycystic kidney disease. Nat. Rev. Nephrol. 2011, 7, 556–566. [Google Scholar] [CrossRef] [PubMed]
- Harris, P.C.; Torres, V.E. Genetic mechanisms and signaling pathways in autosomal dominant polycystic kidney disease. J. Clin. Investig. 2014, 124, 2315–2324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ong, A.C.; Harris, P.C. A polycystin-centric view of cyst formation and disease: The polycystins revisited. Kidney Int. 2015, 88, 699–710. [Google Scholar] [CrossRef] [Green Version]
- Weimbs, T. Third-Hit Signaling in Renal Cyst Formation. J. Am. Soc. Nephrol. 2011, 22, 793–795. [Google Scholar] [CrossRef]
- Hanaoka, K.; Guggino, W.B. cAMP regulates cell proliferation and cyst formation in autosomal polycystic kidney disease cells. J. Am. Soc. Nephrol. 2000, 11, 1179–1187. [Google Scholar] [CrossRef]
- Starremans, P.; Li, X.; Finnerty, P.; Guo, L.; Takakura, A.; Neilson, E.; Zhou, J. A mouse model for polycystic kidney disease through a somatic in-frame deletion in the 5’ end of Pkd1. Kidney Int. 2008, 73, 1394–1405. [Google Scholar] [CrossRef] [Green Version]
- Yamaguchi, T.; Nagao, S.; Wallace, D.P.; Belibi, F.A.; Cowley, B.D.; Pelling, J.C.; Grantham, J.J. Cyclic AMP activates B-Raf and ERK in cyst epithelial cells from autosomal-dominant polycystic kidneys. Kidney Int. 2003, 63, 1983–1994. [Google Scholar] [CrossRef] [Green Version]
- YamaguchiJill, T.; Pelling, J.C.; Ramaswamy, N.T.; Eppler, J.W.; Wallace, D.P.; Nagao, S.; Rome, L.A.; Sullivan, L.P.; Grantham, J.J. cAMP stimulates the in vitro proliferation of renal cyst epithelial cells by activating the extracellular signal-regulated kinase pathway. Kidney Int. 2000, 57, 1460–1471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, P.; Sieben, C.J.; Xu, X.; Harris, P.C.; Lin, X. Autophagy activators suppress cystogenesis in an autosomal dominant polycystic kidney disease model. Hum. Mol. Genet. 2016, 26, 158–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belibi, F.; Zafar, I.; Ravichandran, K.; Segvic, A.B.; Jani, A.; Ljubanovic, D.G.; Edelstein, C.L. Hypoxia-inducible factor-1α (HIF-1α) and autophagy in polycystic kidney disease (PKD). Am. J. Physiol.-Ren. Physiol. 2011, 300, F1235–F1243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chun, Y.; Kim, J. Autophagy: An Essential Degradation Program for Cellular Homeostasis and Life. Cells 2018, 7, 278. [Google Scholar] [CrossRef] [Green Version]
- Rubinsztein, D.C.; Shpilka, T.; Elazar, Z. Mechanisms of Autophagosome Biogenesis. Curr. Biol. 2012, 22, R29–R34. [Google Scholar] [CrossRef] [Green Version]
- Mukhopadhyay, S.; Panda, P.K.; Sinha, N.; Das, D.N.; Bhutia, S.K. Autophagy and apoptosis: Where do they meet? Apoptosis 2014, 19, 555–566. [Google Scholar] [CrossRef]
- Jiang, S.-T.; Chiou, Y.-Y.; Wang, E.; Lin, H.-K.; Lin, Y.-T.; Chi, Y.-C.; Wang, C.-K.L.; Tang, M.-J.; Li, H. Defining a Link with Autosomal-Dominant Polycystic Kidney Disease in Mice with Congenitally Low Expression of Pkd1. Am. J. Pathol. 2006, 168, 205–220. [Google Scholar] [CrossRef] [Green Version]
- Tsai, Y.-C.; Teng, I.-L.; Jiang, S.-T.; Lee, Y.-C.; Chiou, Y.-Y.; Cheng, F.-Y. Safe Nanocomposite-Mediated Efficient Delivery of MicroRNA Plasmids for Autosomal Dominant Polycystic Kidney Disease (ADPKD) Therapy. Adv. Healthc. Mater. 2019, 8, e1801358. [Google Scholar] [CrossRef]
- Parikh, C.R.; Dahl, N.K.; Chapman, A.B.; Bost, J.E.; Edelstein, C.L.; Comer, D.M.; Zeltner, R.; Tian, X.; Grantham, J.J.; Somlo, S. Evaluation of urine biomarkers of kidney injury in polycystic kidney disease. Kidney Int. 2012, 81, 784–790. [Google Scholar] [CrossRef] [Green Version]
- Tsingos, M.; Merlini, L.; Solcà, M.; Goischke, A.; Wilhelm-Bals, A.; Parvex, P. Early Urinary Biomarkers in Pediatric Autosomal Dominant Polycystic Kidney Disease (ADPKD): No Evi-dence in the Interest of Urinary Neutrophil Gelatinase-Associated Lipocalin (uNGAL). Front. Pediatrics 2019, 7, 88. [Google Scholar] [CrossRef]
- Shen, S.-J.; Hu, Z.-X.; Li, Q.-H.; Wang, S.-M.; Song, C.-J.; Wu, D.-D.; He, J.-L.; Guan, J.-C.; Shan, J.-P. Implications of the changes in serum neutrophil gelatinase-associated lipocalin and cystatin C in patients with chronic kidney disease. Nephrology 2014, 19, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Mishra, J.; Dent, C.; Tarabishi, R.; Mitsnefes, M.M.; Ma, Q.; Kelly, C.; Ruff, S.M.; Zahedi, K.; Shao, M.; Bean, J.; et al. Neutrophil gelatinase-associated lipocalin (NGAL) as a biomarker for acute renal injury after cardiac surgery. Lancet 2005, 365, 1231–1238. [Google Scholar] [CrossRef]
- Cassidy, H.; Slyne, J.; Higgins, M.; Radford, R.; Conlon, P.J.; Watson, A.J.; Ryan, M.P.; McMorrow, T.; Slattery, C. Neutrophil gelatinase-associated lipocalin (NGAL) is localised to the primary cilium in renal tubular epithelial cells—A novel source of urinary biomarkers of renal injury. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2019, 1865, 165532. [Google Scholar] [CrossRef]
- An, S.; Zang, X.; Yuan, W.; Zhuge, Y.; Yu, Q. Neutrophil Gelatinase-Associated Lipocalin (NGAL) May Play a Protective Role Against Rats Ischemia/Reperfusion Renal Injury via Inhibiting Tubular Epithelial Cell Apoptosis. Ren. Fail. 2012, 35, 143–149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, F.; Karihaloo, A.; Yu, Z.; Marlier, A.; Seth, P.; Shibazaki, S.; Wang, T.; Somlo, S.; Cantley, L.G.; Sukhatme, V.P. Neutrophil gelatinase-associated lipocalin suppresses cyst growth by Pkd1 null cells in vitro and in vivo. Kidney Int. 2008, 74, 1310–1318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolignano, D.; Coppolino, G.; Campo, S.; Aloisi, C.; Nicocia, G.; Frisina, N.; Buemi, M. Neutrophil Gelatinase-Associated Lipocalin in Patients with Autosomal-Dominant Polycystic Kidney Disease. Am. J. Nephrol. 2007, 27, 373–378. [Google Scholar] [CrossRef] [PubMed]
- Bauvois, B.; Susin, S.A. Revisiting Neutrophil Gelatinase-Associated Lipocalin (NGAL) in Cancer: Saint or Sinner? Cancers 2018, 10, 336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langelueddecke, C.; Roussa, E.; Fenton, R.A.; Wolff, N.A.; Lee, W.K.; Thévenod, F. Lipocalin-2 (24p3/neutrophil gelatinase-associated lipocalin (NGAL)) receptor is expressed in distal nephron and mediates protein endocytosis. J. Biol. Chem. 2012, 287, 159–169. [Google Scholar] [CrossRef] [Green Version]
- Candido, S.; Abrams, S.L.; Steelman, L.S.; Lertpiriyapong, K.; Fitzgerald, T.L.; Martelli, A.M.; Cocco, L.; Montalto, G.; Cervello, M.; Polesel, J.; et al. Roles of NGAL and MMP-9 in the tumor microenvironment and sensitivity to targeted therapy. Biochim. Biophys. Acta 2016, 1863, 438–448. [Google Scholar] [CrossRef]
- Goetz, D.H.; Holmes, M.A.; Borregaard, N.; Bluhm, M.E.; Raymond, K.N.; Strong, R.K. The neutrophil lipocalin NGAL is a bacteriostatic agent that interferes with siderophore-mediated iron acquisition. Mol. Cell 2002, 10, 1033–1043. [Google Scholar] [CrossRef]
- Wang, E.; Chiou, Y.-Y.; Jeng, W.-Y.; Lin, H.-K.; Lin, H.-H.; Chin, H.-J.; Wang, C.-K.L.; Yu, S.-S.; Tsai, S.-C.; Chiang, C.-Y.; et al. Overexpression of exogenous kidney-specific Ngal attenuates progressive cyst development and prolongs lifespan in a murine model of polycystic kidney disease. Kidney Int. 2016, 91, 412–422. [Google Scholar] [CrossRef] [PubMed]
- Shillingford, J.M.; Piontek, K.B.; Germino, G.; Weimbs, T. Rapamycin Ameliorates PKD Resulting from Conditional Inactivation of Pkd1. J. Am. Soc. Nephrol. 2010, 21, 489–497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vermes, I.; Haanen, C.; Steffens-Nakken, H.; Reutellingsperger, C. A novel assay for apoptosis Flow cytometric detection of phosphatidylserine expression on early apoptotic cells using fluorescein labelled Annexin V. J. Immunol. Methods 1995, 184, 39–51. [Google Scholar] [CrossRef] [Green Version]
- Majno, G.; Joris, I. Apoptosis, oncosis, and necrosis. An overview of cell death. Am. J. Pathol. 1995, 146, 3–15. [Google Scholar]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef] [PubMed]
- Los, M.J.; Mozoluk, M.; Ferrari, D.; Stepczynska, A.; Stroh, C.; Renz, A.; Herceg, Z.; Wang, Z.-Q.; Schulze-Osthoff, K. Activation and Caspase-mediated Inhibition of PARP: A Molecular Switch between Fibroblast Necrosis and Apoptosis in Death Receptor Signaling. Mol. Biol. Cell 2002, 13, 978–988. [Google Scholar] [CrossRef] [Green Version]
- Shillingford, J.M.; Murcia, N.S.; Larson, C.H.; Low, S.H.; Hedgepeth, R.; Brown, N.; Flask, C.A.; Novick, A.C.; Goldfarb, D.A.; Kramer-Zucker, A.; et al. The mTOR pathway is regulated by polycystin-1, and its inhibition reverses renal cystogenesis in poly-cystic kidney disease. Proc. Natl. Acad. Sci. USA 2006, 103, 5466. [Google Scholar] [CrossRef] [Green Version]
- Torres, V.E.; Harris, P.C. Autosomal dominant polycystic kidney disease: The last 3 years. Kidney Int. 2009, 76, 149–168. [Google Scholar] [CrossRef] [Green Version]
- Torres, V.E.; Harris, P.C. Strategies Targeting cAMP Signaling in the Treatment of Polycystic Kidney Disease. J. Am. Soc. Nephrol. 2013, 25, 18–32. [Google Scholar] [CrossRef]
- Yamamoto, K.K.; Gonzalez, G.A.; Biggs, W.H.; Montminy, M.R. Phosphorylation-induced binding and transcriptional efficacy of nuclear factor CREB. Nature 1988, 334, 494–498. [Google Scholar] [CrossRef]
- Carlezon, W.A.; Duman, R.S.; Nestler, E.J. The many faces of CREB. Trends Neurosci. 2005, 28, 436–445. [Google Scholar] [CrossRef] [PubMed]
- Kabeya, Y.; Mizushima, N.; Ueno, T.; Yamamoto, A.; Kirisako, T.; Noda, T.; Kominami, E.; Ohsumi, Y.; Yoshimori, T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000, 19, 5720–5728. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of Cells and Tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coloma, M.; Hastings, A.; Wims, L.A.; Morrison, S.L. Novel vectors for the expression of antibody molecules using variable regions generated by polymerase chain reaction. J. Immunol. Methods 1992, 152, 89–104. [Google Scholar] [CrossRef]
- Kusaba, T.; Lalli, M.; Kramann, R.; Kobayashi, A.; Humphreys, B.D. Differentiated kidney epithelial cells repair injured proximal tubule. Proc. Natl. Acad. Sci. USA 2014, 111, 1527–1532. [Google Scholar] [CrossRef] [Green Version]
- Woo, Y.M.; Kim, D.Y.; Koo, N.J.; Kim, Y.-M.; Lee, S.; Ko, J.Y.; Shin, Y.; Kim, B.H.; Mun, H.; Choi, S.; et al. Profiling of miRNAs and target genes related to cystogenesis in ADPKD mouse models. Sci. Rep. 2017, 7, 14151. [Google Scholar] [CrossRef] [Green Version]
- Kawano, H.; Muto, S.; Ohmoto, Y.; Iwata, F.; Fujiki, H.; Mori, T.; Yan, L.; Horie, S. Exploring urinary biomarkers in autosomal dominant polycystic kidney disease. Clin. Exp. Nephrol. 2014, 19, 968–973. [Google Scholar] [CrossRef]
- Vareesangthip, K.; Limwongse, C.; Reesukumal, K. Role of Urinary Neutrophil Gelatinase-Associated Lipocalin for Predicting the Severity of Renal Func-tions in Patients With Autosomal-Dominant Polycystic Kidney Disease. Transpl. Proc. 2017, 49, 950–954. [Google Scholar] [CrossRef]
- Messchendorp, A.L.; Meijer, E.; Boertien, W.E.; Engels, G.E.; Casteleijn, N.F.; Spithoven, E.M.; Losekoot, M.; Burgerhof, J.G.; Peters, D.J.; Gansevoort, R.T. Urinary Biomarkers to Identify Autosomal Dominant Polycystic Kidney Disease Patients With a High Likelihood of Disease Progression. Kidney Int. Rep. 2017, 3, 291–301. [Google Scholar] [CrossRef]
- Lefloch, R.; Pouyssegur, J.; Lenormand, P. Single and combined silencing of ERK1 and ERK2 reveals their positive contribu-tion to growth signaling depending on their expression levels. Mol. Cell Biol. 2008, 28, 511–527. [Google Scholar] [CrossRef] [Green Version]
- Roskoski, R., Jr. ERK1/2 MAP kinases: Structure, function, and regulation. Pharmacol. Res. 2012, 66, 105–143. [Google Scholar] [CrossRef] [PubMed]
- Bao, Q.; Hughes, R.C. Galectin-3 expression and effects on cyst enlargement and tubulogenesis in kidney epithelial MDCK cells cultured in three-dimensional matrices in vitro. J. Cell Sci. 1995, 108, 2791–2800. [Google Scholar] [CrossRef] [PubMed]
- DesRochers, T.M.; Suter, L.; Roth, A.; Kaplan, D.L. Bioengineered 3D human kidney tissue, a platform for the determination of nephrotoxicity. PLoS ONE 2013, 8, e59219. [Google Scholar] [CrossRef]
- Lee, G.Y.; A Kenny, P.; Lee, E.H.; Bissell, M.J. Three-dimensional culture models of normal and malignant breast epithelial cells. Nat. Methods 2007, 4, 359–365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Booij, T.H.; Bange, H.; Leonhard, W.N.; Yan, K.; Fokkelman, M.; Kunnen, S.J.; Dauwerse, J.G.; Qin, Y.; van de Water, B.; van Westen, G.J.P.; et al. High-Throughput Phenotypic Screening of Kinase Inhibitors to Identify Drug Targets for Polycystic Kidney Disease. Slas Discov. Adv. Life Sci. RD 2017, 22, 974–984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhatwa, A.; Wang, W.; Hassan, Y.I.; Abraham, N.; Li, X.-Z.; Zhou, T. Challenges Associated With the Formation of Recombinant Protein Inclusion Bodies in Escherichia coli and Strategies to Address Them for Industrial Applications. Front. Bioeng. Biotechnol. 2021, 9, 630551. [Google Scholar] [CrossRef]
- Nothaft, H.; Szymanski, C.M. Bacterial Protein N-Glycosylation: New Perspectives and Applications. J. Biol. Chem. 2013, 288, 6912–6920. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Locus | Primer Sequence (5′-3′) | Product Size (bp) | |
|---|---|---|---|
| Gapdh | Forward | CATCA CTGCC ACCCA GAAGA CTG | 153 |
| Reverse | ATGCC AGTGA GCTTC CCGTT CAG | ||
| Lcn2 | Forward | CCACC ACGGA CTACA ACCAG | 100 |
| Reverse | AGCTC CTTGG TTCTT CCATA CA | ||
| Lcn2-myc | Forward | ATCTT CTCTG TCCCC ACCGA | 98 |
| Reverse | ACGGC GCTAT TCAGA TCCTC | ||
| Pkd1 | Forward | TCAAT TGCTC CGGCC GCTG | 102 |
| Reverse | CCAGC GTCTG AAGTA GGTTG TGGG | ||
| 2Slc22a17 | Forward | CAGCC ACCTC CTAAC CGCTG TG | 86 |
| Reverse | CTCCC ACTAG GCTCA AAGGC TGCT | ||
| Antibody | Manufacture | Catalog | Titer | MW (kDa) | Host |
|---|---|---|---|---|---|
| NGAL | abcam | ab63929 | 1:500 | 20, 22 | Rabbit |
| NGAL-R | NOVUS | NBP1-20975 | 1:1000 | 58 | Goat |
| Caspase-3 | Cell signaling | 9662S | 1:750 | 17, 19, 35 | Rabbit |
| PARP | Cell signaling | 9542S | 1:1000 | 89, 116 | Rabbit |
| p–ERK (T202/Y204) | Cell signaling | 4377S | 1:1000 | 42, 44 | Rabbit |
| ERK | Cell signaling | 9102S | 1:1000 | 42, 44 | Rabbit |
| p-AKT (S473) | Cell signaling | 4060S | 1:1000 | 60 | Rabbit |
| AKT | Cell signaling | 9272S | 1:1000 | 60 | Rabbit |
| p-CREB (S133) | Cell signaling | 9198S | 1:1000 | 43 | Rabbit |
| CREB | Cell signaling | 9197S | 1:1000 | 43 | Rabbit |
| LC3B | Cell signaling | 2775S | 1:1000 | 14, 16 | Rabbit |
| p62 | Millipore | MABC32 | 1:1000 | 62 | Mouse |
| Actin | Millipore | MAB1501 | 1:5000 | 42 | Mouse |
| GAPDH | Arigo | ARG10112 | 1:5000 | 36 | Mouse |
| Vinculin | Millipore | 05-386 | 1:2000 | 125 | Mouse |
| tubulin | Thermo Fisher Scientific | A11126 | 1:2000 | 50 | Mouse |
| Antibody | Manufacture | Catalog | WB | ICC |
|---|---|---|---|---|
| Anti-mouse IgG HRP-linked | GE Healthcare | NA-931 | 1:2000 | |
| Anti-rabbit IgG HRP-linked | GE Healthcare | NA-934 | 1:2000 | |
| Anti-rabbit IgG 647 | Invitrogen | A27040 | 1:200 | |
| Anti-mouse IgG 555 | Invitrogen | A31570 | 1:200 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chuang, H.-Y.; Jeng, W.-Y.; Wang, E.; Jiang, S.-T.; Hsu, C.-M.; Hsieh-Li, H.M.; Chiou, Y.-Y. Secreted Neutrophil Gelatinase-Associated Lipocalin Shows Stronger Ability to Inhibit Cyst Enlargement of ADPKD Cells Compared with Nonsecreted Form. Cells 2022, 11, 483. https://doi.org/10.3390/cells11030483
Chuang H-Y, Jeng W-Y, Wang E, Jiang S-T, Hsu C-M, Hsieh-Li HM, Chiou Y-Y. Secreted Neutrophil Gelatinase-Associated Lipocalin Shows Stronger Ability to Inhibit Cyst Enlargement of ADPKD Cells Compared with Nonsecreted Form. Cells. 2022; 11(3):483. https://doi.org/10.3390/cells11030483
Chicago/Turabian StyleChuang, Hsin-Yin, Wen-Yih Jeng, Ellian Wang, Si-Tse Jiang, Chen-Ming Hsu, Hsiu Mei Hsieh-Li, and Yuan-Yow Chiou. 2022. "Secreted Neutrophil Gelatinase-Associated Lipocalin Shows Stronger Ability to Inhibit Cyst Enlargement of ADPKD Cells Compared with Nonsecreted Form" Cells 11, no. 3: 483. https://doi.org/10.3390/cells11030483
APA StyleChuang, H. -Y., Jeng, W. -Y., Wang, E., Jiang, S. -T., Hsu, C. -M., Hsieh-Li, H. M., & Chiou, Y. -Y. (2022). Secreted Neutrophil Gelatinase-Associated Lipocalin Shows Stronger Ability to Inhibit Cyst Enlargement of ADPKD Cells Compared with Nonsecreted Form. Cells, 11(3), 483. https://doi.org/10.3390/cells11030483