
Prevention of Lipotoxicity in Pancreatic Islets with Gammahydroxybutyrate

 ,
,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Islet Treatment In Vitro
2.3. Glucose-Stimulated Insulin Secretion (GSIS) Assay
2.4. Reactive Oxygen Species (ROS) Measurement
2.5. Oxygen Consumption Rate
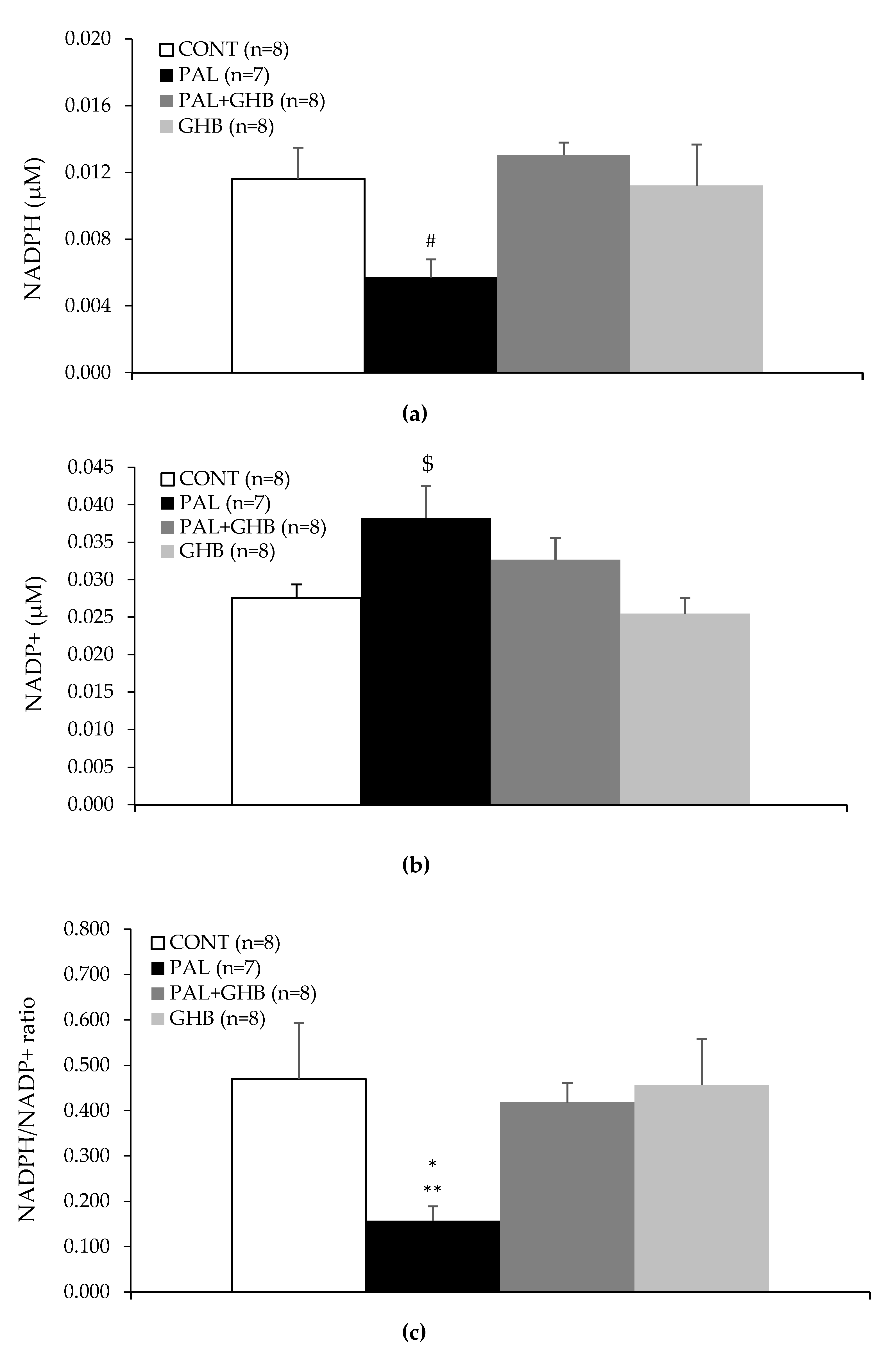
2.6. NADPH and NADP+ Measurement
2.7. Statistical Analysis
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Oprescu, A.I.; Bikopoulos, G.; Naassan, A.; Allister, E.M.; Tang, C.; Park, E.; Uchino, H.; Lewis, G.F.; Fantus, I.G.; Rozakis-Adcock, M.; et al. Free Fatty Acid–Induced Reduction in Glucose-Stimulated Insulin Secretion: Evidence for a Role of Oxidative Stress In Vitro and In Vivo. Diabetes 2007, 56, 2927–2937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koulajian, K.; Desai, T.; Liu, G.C.; Ivovic, A.; Patterson, J.N.; Tang, C.; El-Benna, J.; Joseph, J.W.; Scholey, J.W.; Giacca, A. NADPH oxidase inhibition prevents beta cell dysfunction induced by prolonged elevation of oleate in rodents. Diabetologia 2013, 56, 1078–1087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mamelak, M.; Hyndman, D. Gammahydroxybutyrate and oxidative stress. In Gammahydroxybutyrate: Molecular, Functional and Clinical Aspects; Tunnicliff, G., Cash, C., Eds.; Taylor and Francis: New York, NY, USA, 2002; pp. 218–235. [Google Scholar]
- Mamelak, M. Energy and the Alzheimer brain. Neurosci. Biobehav. Rev. 2017, 75, 297–313. [Google Scholar] [CrossRef] [PubMed]
- Bouix, O.; Reynier, M.; Guintrand-Hugret, R.; Orsetti, A. Protective effect of gamma-hydroxybutyrate and nicotinamide on low-dose streptozotocin-induced diabetes in mice. Horm. Metab. Res. 1995, 27, 216–220. [Google Scholar] [CrossRef] [PubMed]
- Pierrefiche, G.; Topall, G.; Henriet, I.; Laborit, H. Protective effect of gamma-hydroxybutyrate on alloxan induced diabetes in mice. Res. Commun. Chem. Pathol. Pharmacol. 1991, 71, 309–319. [Google Scholar]
- Laborit, H. Sodium 4-hydroxybutyrate. Int. J. Neuropharmacol. 1964, 3, 433-IN8. [Google Scholar] [CrossRef]
- Taberner, P.V.; Rick, J.T.; Kerkut, G.A. The Action of Gamma—Hydroxybutyric Acid on Cerebral Glucose Metabolism. J. Neurochem. 1972, 19, 245–254. [Google Scholar] [CrossRef]
- Abad, V.C. An evaluation of sodium oxybate as a treatment option for narcolepsy. Expert Opin Pharm. 2019, 20, 1189–1199. [Google Scholar] [CrossRef]
- Joseph, J.W.; Koshkin, V.; Saleh, M.C.; Sivitz, W.I.; Zhang, C.-Y.; Lowell, B.B.; Chan, C.B.; Wheeler, M.B. Free Fatty Acid-induced ß-Cell Defects Are Dependent on Uncoupling Protein 2 Expression. J. Biol. Chem. 2004, 279, 51049–51056. [Google Scholar] [CrossRef] [Green Version]
- Lenzen, S.; Drinkgern, J.; Tiedge, M. Low antioxidant enzyme gene expression in pancreatic islets compared with various other mouse tissues. Free. Radic. Biol. Med. 1996, 20, 463–466. [Google Scholar] [CrossRef]
- Ivovic, A.; Oprescu, A.I.; Koulajian, K.; Mori, Y.; Eversley, J.A.; Zhang, L.; Nino-Fong, R.; Lewis, G.F.; Donath, M.Y.; Karin, M.; et al. IKKß inhibition prevents fat-induced beta cell dysfunction in vitro and in vivo in rodents. Diabetologia 2017, 60, 2021–2032. [Google Scholar] [CrossRef] [Green Version]
- Giacca, A.; Xiao, C.; Oprescu, A.I.; Carpentier, A.C.; Lewis, G.F. Lipid-induced pancreatic ß-cell dysfunction: Focus on in vivo studies. Am. J. Physiol. -Endocrinol. Metab. 2011, 300, E255–E262. [Google Scholar] [CrossRef] [Green Version]
- Morgan, D.; Oliveira-Emilio, H.R.; Keane, D.; Hirata, A.E.; Da Rocha, M.S.; Bordin, S.; Curi, R.; Newsholme, P.; Carpinelli, A.R. Glucose, palmitate and pro-inflammatory cytokines modulate production and activity of a phagocyte-like NADPH oxidase in rat pancreatic islets and a clonal beta cell line. Diabetologia 2007, 50, 359–369. [Google Scholar] [CrossRef] [Green Version]
- Newsholme, P.; Haber, E.P.; Hirabara, S.M.; Rebelato, E.L.O.; Procopio, J.; Morgan, D.; Oliveira-Emilio, H.C.; Carpinelli, A.R.; Curi, R. Diabetes associated cell stress and dysfunction: Role of mitochondrial and non-mitochondrial ROS production and activity. J Physiol. 2007, 583, 9–24. [Google Scholar] [CrossRef]
- Fontayne, A.; Dang, P.M.-C.; Gougerot-Pocidalo, A.M.-A.; El Benna, J. Phosphorylation of p47phox sites by PKC alpha, beta II, delta, and zeta: Effect on binding to p22phox and on NADPH oxidase activation. Biochemistry 2002, 41, 7743–7750. [Google Scholar] [CrossRef]
- Macdonald, M.J. Feasibility of a Mitochondrial Pyruvate Malate Shuttle in Pancreatic Islets Further Implication of Cytosolic Nadph in Insulin Secretion. J. Biol. Chem. 1995, 270, 20051–20058. [Google Scholar] [CrossRef]
- Pike, L.S.; Smift, A.L.; Croteau, N.J.; Ferrick, D.A.; Wu, M. Inhibition of fatty acid oxidation by etomoxir impairs NADPH production and increases reactive oxygen species resulting in ATP depletion and cell death in human glioblastoma cells. Biochim. Biophys. Acta (BBA)-Bioenerg. 2011, 1807, 726–734. [Google Scholar] [CrossRef] [Green Version]
- Ronchi, J.A.; Figueira, T.R.; Ravagnani, F.G.; Oliveira, H.C.F.; Vercesi, A.E.; Castilho, R.F. A spontaneous mutation in the nicotinamide nucleotide transhydrogenase gene of C57BL/6J mice results in mitochondrial redox abnormalities. Free. Radic. Biol. Med. 2013, 63, 446–456. [Google Scholar] [CrossRef] [Green Version]
- Iizuka, K.; Nakajima, H.; Namba, M.; Miyagawa, J.-I.; Miyazaki, J.-I.; Hanafusa, T.; Matsuzawa, Y. Metabolic consequence of long-term exposure of pancreatic ß cells to free fatty acid with special reference to glucose insensitivity. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2002, 1586, 23–31. [Google Scholar] [CrossRef] [Green Version]
- Ivarsson, R.; Quintens, R.; Dejonghe, S.; Tsukamoto, K.; Veld, P.I.; Renström, E.; Schuit, F.C. Redox Control of Exocytosis: Regulatory Role of NADPH, Thioredoxin, and Glutaredoxin. Diabetes 2005, 54, 2132–2142. [Google Scholar] [CrossRef] [Green Version]
- MacDonald, P.E.; Wheeler, M.B. Voltage-dependent K+ channels in pancreatic beta cells: Role, regulation and potential as therapeutic targets. Diabetologia 2003, 46, 1046–1062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopatin, A.F.; Riabtseva, E.G.; Riabova, V.V.; TIu, L. Effect of sodium oxybate on metabolic indices in ischemis hypoxia of muscle tissue. Farmakol. Toksikol. 1984, 47, 53–55. [Google Scholar] [PubMed]
- Stanton, R.C. Glucose-6-Phosphate Dehydrogenase, NADPH, and Cell Survival. IUBMB Life 2012, 64, 362–369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaufman, E.E.; Nelson, T. An overview of gamma-hydroxybutyrate catabolism: The role of the cytosolic NADP(+)-dependent oxidoreductase EC 1.1.1.19 and of a mitochondrial hydroxyacid-oxoacid transhydrogenase in the initial, rate-limiting step in this pathway. Neurochem. Res. 1991, 16, 965–974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaufman, E.E. Metabolism and distribution of gammahydroxybutyrate in the brain. In Gammahydroxybutyrate: Molecular, Functional and Clinical Aspects; Hyndman, D., Cash, C., Eds.; Taylor and Francis: New York, NY, USA, 2002; pp. 1–16. [Google Scholar]
- Vogel, R.; Wiesinger, H.; Hamprecht, B.; Dringen, R. The regeneration of reduced glutathione in rat forebrain mitochondria identifies metabolic pathways providing the NADPH required. Neurosci. Lett. 1999, 275, 97–100. [Google Scholar] [CrossRef]
- Förstermann, U.; Xia, N.; Li, H. Roles of Vascular Oxidative Stress and Nitric Oxide in the Pathogenesis of Atherosclerosis. Circ. Res. 2017, 120, 713–735. [Google Scholar] [CrossRef] [PubMed]
- Sena, C.M.; Pereira, A.M.; Seiça, R. Endothelial dysfunction—A major mediator of diabetic vascular disease. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2013, 1832, 2216–2231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Depner, C.M.; Stothard, E.R.; Wright, K.P. Metabolic consequences of sleep and circadian disorders. Curr. Diabetes Rep. 2014, 14, 507. [Google Scholar] [CrossRef]
- Garnock-Jones, K.P. Naloxegol: A Review of Its Use in Patients with Opioid-Induced Constipation. Drugs 2015, 75, 419–425. [Google Scholar] [CrossRef]
- Mahú, I.; Barateiro, A.; Rial-Pensado, E.; Martinez-Sanchez, N.; Vaz, S.; Cal, P.M.; Jenkins, B.; Rodrigues, T.; Cordeiro, C.; Costa, M.F.; et al. Domingos, Brain-Sparing Sympathofacilitators Mitigate Obesity without Adverse Cardiovascular Effects. Cell Metab. 2020, 31, 1120–1135.e7. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yung, J.H.M.; Yeung, L.S.N.; Ivovic, A.; Tan, Y.F.; Jentz, E.M.; Batchuluun, B.; Gohil, H.; Wheeler, M.B.; Joseph, J.W.; Giacca, A.; et al. Prevention of Lipotoxicity in Pancreatic Islets with Gammahydroxybutyrate. Cells 2022, 11, 545. https://doi.org/10.3390/cells11030545
Yung JHM, Yeung LSN, Ivovic A, Tan YF, Jentz EM, Batchuluun B, Gohil H, Wheeler MB, Joseph JW, Giacca A, et al. Prevention of Lipotoxicity in Pancreatic Islets with Gammahydroxybutyrate. Cells. 2022; 11(3):545. https://doi.org/10.3390/cells11030545
Chicago/Turabian StyleYung, Justin Hou Ming, Lucy Shu Nga Yeung, Aleksandar Ivovic, Yao Fang Tan, Emelien Mariella Jentz, Battsetseg Batchuluun, Himaben Gohil, Michael B. Wheeler, Jamie W. Joseph, Adria Giacca, and et al. 2022. "Prevention of Lipotoxicity in Pancreatic Islets with Gammahydroxybutyrate" Cells 11, no. 3: 545. https://doi.org/10.3390/cells11030545