
Epigenetic Regulation of Cellular Senescence

{kind=link}
{kind=link}
Abstract
:1. Introduction
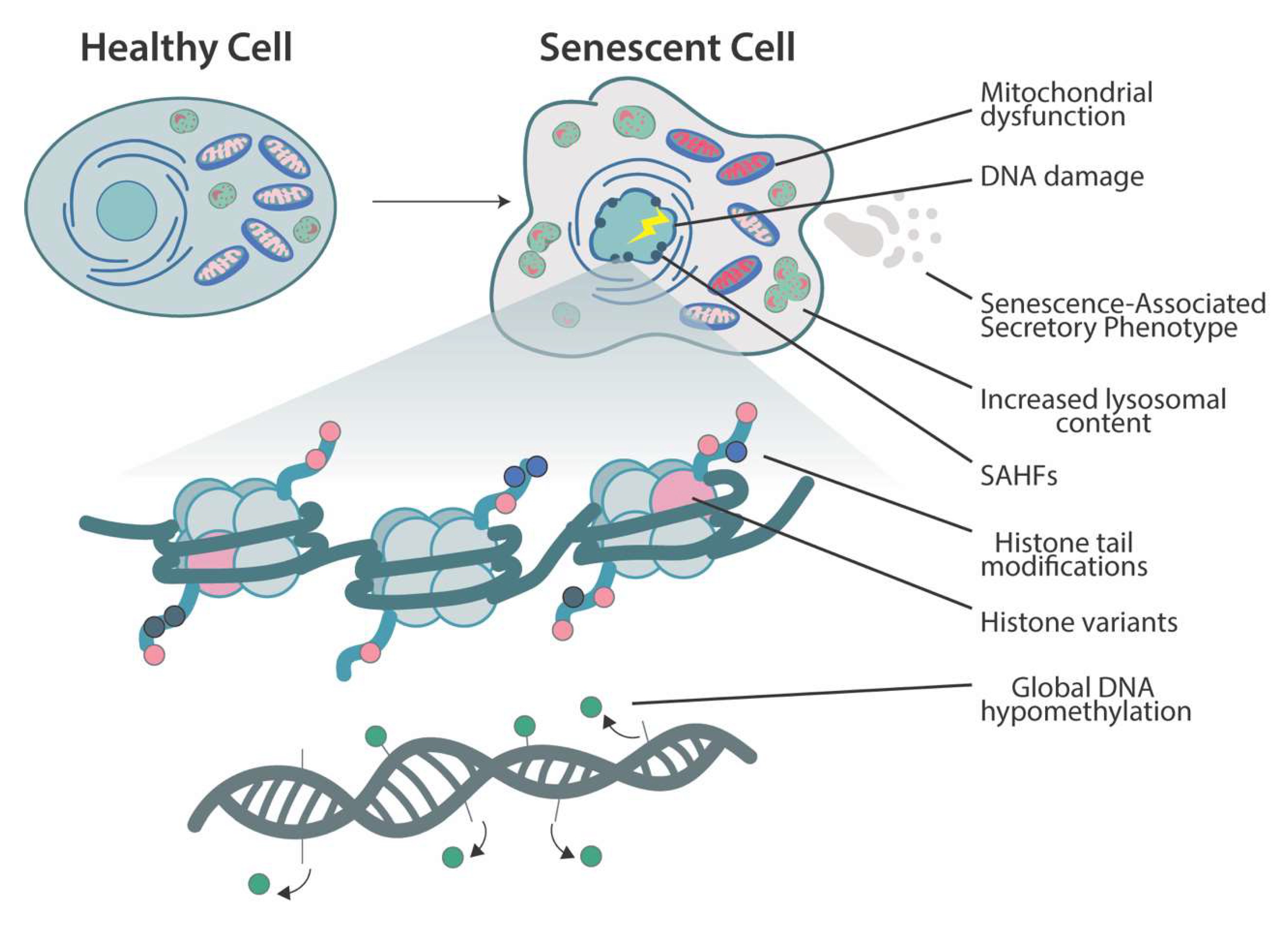
2. Cellular Senescence
3. Chromatin Changes and Histone-Modifying Enzymes in Senescence
3.1. Polycomb-Group Proteins and Other Histone-Modifying Enzymes
3.2. Histone Acetylation
3.3. Senescence-Associated Heterochromatin Foci
3.4. Histone Variants
4. DNA Methylation
5. Epigenetic Regulation of SASP
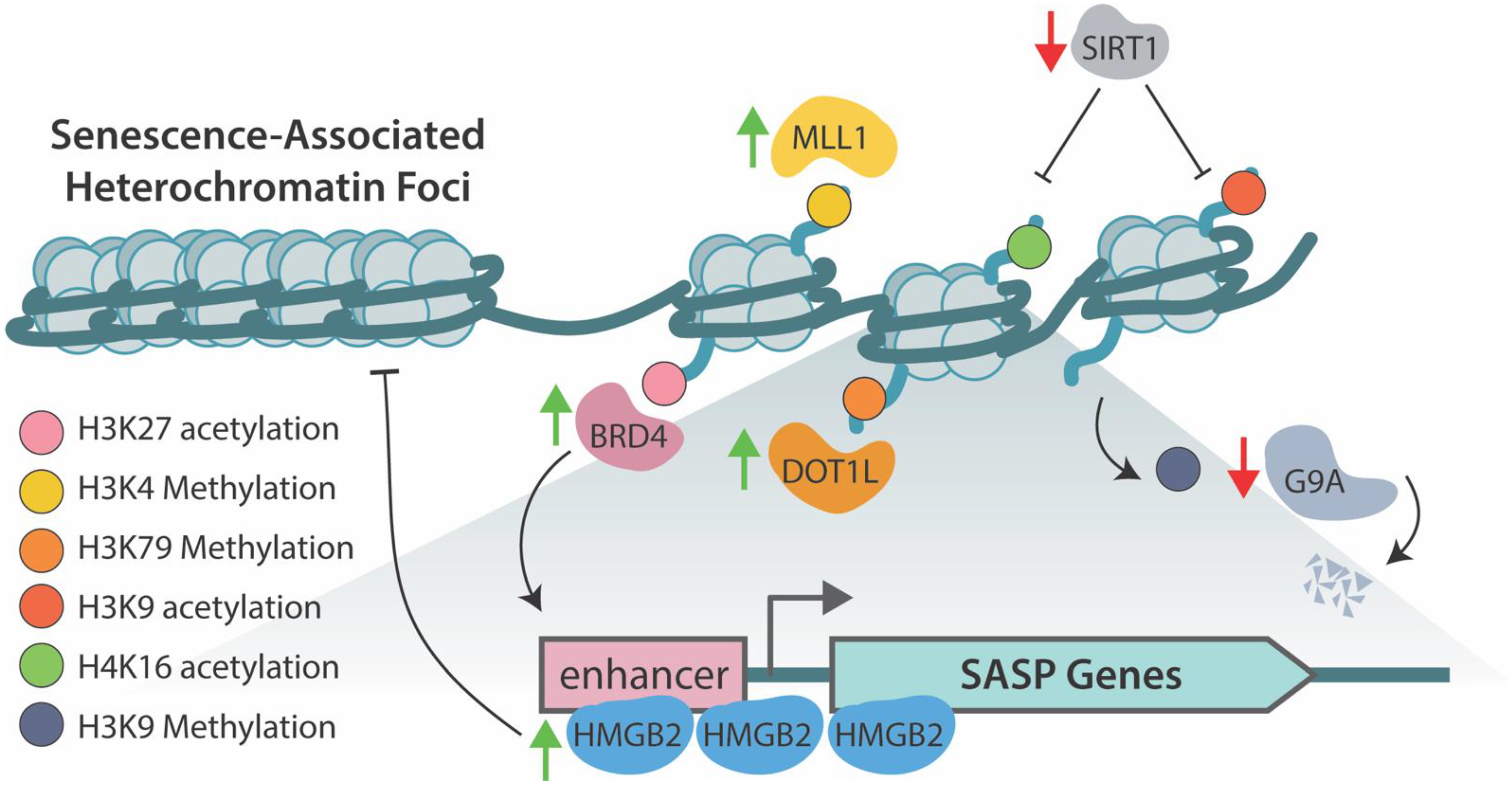
6. Therapeutics Targeting Epigenetic Mechanisms
7. Conclusions and Future Perspectives/Directions
Funding
Conflicts of Interest
References
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campisi, J. Cellular senescence as a tumor-suppressor mechanism. Trends Cell Biol. 2001, 11, S27–S31. [Google Scholar] [CrossRef] [Green Version]
- Demaria, M.; Ohtani, N.; Youssef, S.A.; Rodier, F.; Toussaint, W.; Mitchell, J.R.; Laberge, R.-M.; Vijg, J.; Van Steeg, H.; Dollé, M.E.; et al. An Essential Role for Senescent Cells in Optimal Wound Healing through Secretion of PDGF-AA. Dev. Cell 2014, 31, 722–733. [Google Scholar] [CrossRef] [Green Version]
- Ritschka, B.; Storer, M.; Mas, A.; Heinzmann, F.; Ortells, M.C.; Morton, J.P.; Sansom, O.J.; Zender, L.; Keyes, W.M. The senescence-associated secretory phenotype induces cellular plasticity and tissue regeneration. Genes Dev. 2017, 31, 172–183. [Google Scholar] [CrossRef] [Green Version]
- Baker, D.J.; Childs, B.G.; Durik, M.; Wijers, M.E.; Sieben, C.J.; Zhong, J.; Saltness, R.A.; Jeganathan, K.B.; Verzosa, G.C.; Pezeshki, A.; et al. Naturally occurring p16Ink4a-positive cells shorten healthy lifespan. Nature 2016, 530, 184–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gibaja, A.; Aburto, M.R.; Pulido, S.; Collado, M.; Hurle, J.M.; Varela-Nieto, I.; Magariños, M. TGFβ2-induced senescence during early inner ear development. Sci. Rep. 2019, 9, 5912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muñoz-Espín, D.; Cañamero, M.; Maraver, A.; López, G.G.; Contreras, J.; Murillo-Cuesta, S.; Rodríguez-Baeza, A.; Varela-Nieto, I.; Ruberte, J.; Collado, M.; et al. Programmed Cell Senescence during Mammalian Embryonic Development. Cell 2013, 155, 1104–1118. [Google Scholar] [CrossRef] [Green Version]
- Hayflick, L.; Moorhead, P.S. The serial cultivation of human diploid cell strains. Exp. Cell Res. 1961, 25, 585–621. [Google Scholar] [CrossRef]
- Bernadotte, A.; Mikhelson, V.M.; Spivak, I.M. Markers of cellular senescence. Telomere shortening as a marker of cellular senescence. Aging 2016, 8, 3–11. [Google Scholar] [CrossRef] [Green Version]
- Di Micco, R.; Fumagalli, M.; Cicalese, A.; Piccinin, S.; Gasparini, P.; Luise, C.; Schurra, C.; Garre, M.; Nuciforo, P.; Bensimon, A.; et al. Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature 2006, 444, 638–642. [Google Scholar] [CrossRef]
- Michaloglou, C.; Vredeveld, L.C.; Soengas, M.S.; Denoyelle, C.; Kuilman, T.; Van Der Horst, C.M.; Majoor, D.M.; Shay, J.W.; Mooi, W.J.; Peeper, D.S. BRAF E600-associated senescence-like cell cycle arrest of human naevi. Nature 2005, 436, 720–724. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Durbin, K.R.; Sweet, S.M.; Tipton, J.D.; Zheng, Y.; Kelleher, N.L. Oncogene-induced cellular senescence elicits an anti-W arburg effect. Proteomics 2013, 13, 2585–2596. [Google Scholar] [CrossRef] [PubMed]
- Karimian, A.; Ahmadi, Y.; Yousefi, B. Multiple functions of p21 in cell cycle, apoptosis and transcriptional regulation after DNA damage. DNA Repair 2016, 42, 63–71. [Google Scholar] [CrossRef]
- Yang, N.; Sen, P. The senescent cell epigenome. Aging 2018, 10, 3590–3609. [Google Scholar] [CrossRef] [PubMed]
- Wiley, C.D.; Velarde, M.C.; Lecot, P.; Liu, S.; Sarnoski, E.A.; Freund, A.; Shirakawa, K.; Lim, H.W.; Davis, S.S.; Ramanathan, A.; et al. Mitochondrial Dysfunction Induces Senescence with a Distinct Secretory Phenotype. Cell Metab. 2016, 23, 303–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milanovic, M.; Fan, D.N.Y.; Belenki, D.; Däbritz, J.H.M.; Zhao, Z.; Yu, Y.; Dörr, J.R.; Dimitrova, L.; Lenze, D.; Barbosa, I.A.M.; et al. Senescence-associated reprogramming promotes cancer stemness. Nature 2017, 553, 96–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sage, J.; Miller, A.L.; Pérez-Mancera, P.A.; Wysocki, J.M.; Jacks, T. Acute mutation of retinoblastoma gene function is sufficient for cell cycle re-entry. Nature 2003, 424, 223–228. [Google Scholar] [CrossRef]
- Beauséjour, C.M.; Krtolica, A.; Galimi, F.; Narita, M.; Lowe, S.W.; Yaswen, P.; Campisi, J. Reversal of human cellular senescence: Roles of the p53 and p16 pathways. EMBO J. 2003, 22, 4212–4222. [Google Scholar] [CrossRef]
- Wang, A.S.; Dreesen, O. Biomarkers of cellular senescence and skin aging. Front. Genet. 2018, 9, 247. [Google Scholar] [CrossRef]
- Vasileiou, P.V.; Evangelou, K.; Vlasis, K.; Fildisis, G.; Panayiotidis, M.I.; Chronopoulos, E.; Passias, P.-G.; Kouloukoussa, M.; Gorgoulis, V.G.; Havaki, S. Mitochondrial Homeostasis and Cellular Senescence. Cells 2019, 8, 686. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.M.; Liu, J.; Merrett, J.B. Apoptosis or senescence-like growth arrest: Influence of cell-cycle position, p53, p21 and bax in H2O2 response of normal human fibroblasts. Biochem. J. 2000, 347, 543–551. [Google Scholar] [CrossRef]
- Hampel, B.; Malisan, F.; Niederegger, H.; Testi, R.; Jansen-Dürr, P. Differential regulation of apoptotic cell death in senescent human cells. Exp. Gerontol. 2004, 39, 1713–1721. [Google Scholar] [CrossRef] [PubMed]
- Narita, M.; Nuñez, S.; Heard, E.; Narita, M.; Lin, A.W.; Hearn, S.A.; Spector, D.L.; Hannon, G.J.; Lowe, S.W. Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell 2003, 113, 703–716. [Google Scholar] [CrossRef] [Green Version]
- Narita, M.; Narita, M.; Krizhanovsky, V.; Nuñez, S.; Chicas, A.; Hearn, S.A.; Myers, P.M.; Lowe, S.W. A novel role for high-mobility group a proteins in cellular senescence and heterochromatin formation. Cell 2006, 126, 503–514. [Google Scholar] [CrossRef] [Green Version]
- Shimi, T.; Butin-Israeli, V.; Adam, S.A.; Hamanaka, R.B.; Goldman, A.E.; Lucas, C.A.; Shumaker, D.K.; Kosak, S.T.; Chandel, N.S.; Goldman, R.D. The role of nuclear lamin B1 in cell proliferation and senescence. Genes Dev. 2011, 25, 2579–2593. [Google Scholar] [CrossRef] [Green Version]
- Matias, I.; Diniz, L.P.; Damico, I.V.; Araujo, A.P.B.; Neves, L.D.S.; Vargas, G.; Leite, R.E.P.; Suemoto, C.K.; Nitrini, R.; Jacob-Filho, W.; et al. Loss of lamin-B1 and defective nuclear morphology are hallmarks of astrocyte senescence in vitro and in the aging human hippocampus. Aging Cell 2021, 21, e13521. [Google Scholar] [CrossRef]
- Pathak, R.U.; Soujanya, M.; Mishra, R.K. Deterioration of nuclear morphology and architecture: A hallmark of senescence and aging. Ageing Res. Rev. 2021, 67, 101264. [Google Scholar] [CrossRef]
- Oberdoerffer, P.; Sinclair, D. The role of nuclear architecture in genomic instability and ageing. Nat. Rev. Mol. Cell Biol. 2007, 8, 692–702. [Google Scholar] [CrossRef] [PubMed]
- Lopatina, N.; Haskell, J.F.; Andrews, L.G.; Poole, J.C.; Saldanha, S.; Tollefsbol, T. Differential maintenance and de novo methylating activity by three DNA methyltransferases in aging and immortalized fibroblasts. J. Cell. Biochem. 2002, 84, 324–334. [Google Scholar] [CrossRef] [PubMed]
- Coppé, J.-P.; Desprez, P.-Y.; Krtolica, A.; Campisi, J. The Senescence-Associated Secretory Phenotype: The Dark Side of Tumor Suppression. Annu. Rev. Pathol. Mech. Dis. 2010, 5, 99–118. [Google Scholar] [CrossRef] [Green Version]
- Nacarelli, T.; Liu, P.; Zhang, R. Epigenetic Basis of Cellular Senescence and Its Implications in Aging. Genes 2017, 8, 343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sidler, C.; Kovalchuk, O.; Kovalchuk, I. Epigenetic Regulation of Cellular Senescence and Aging. Front. Genet. 2017, 8, 138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, T.; Notta, F.; Navab, R.; Joseph, J.; Ibrahimov, E.; Xu, J.; Zhu, C.-Q.; Borgida, A.; Gallinger, S.; Tsao, M.-S. Senescent Carcinoma-associated Fibroblasts Upregulate IL8 to Enhance Prometastatic Phenotypes. Mol. Cancer Res. 2016, 15, 3–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kehrl, J.; Wakefield, L.M.; Roberts, A.B.; Jakowlew, S.; Alvarez-Mon, M.; Derynck, R.; Sporn, M.B.; Fauci, A.S. Production of transforming growth factor beta by human T lymphocytes and its potential role in the regulation of T cell growth. J. Exp. Med. 1986, 163, 1037–1050. [Google Scholar] [CrossRef]
- Hoare, M.; Ito, Y.; Kang, T.W.; Weekes, M.P.; Matheson, N.J.; Patten, D.A.; Shetty, S.; Parry, A.J.; Menon, S.; Salama, R.; et al. NOTCH1 mediates a switch between two distinct secretomes during senescence. Nat. Cell Biol. 2016, 18, 979–992. [Google Scholar] [CrossRef] [Green Version]
- Acosta, J.C.; Banito, A.; Wuestefeld, T.; Georgilis, A.; Janich, P.; Morton, J.P.; Athineos, D.; Kang, T.-W.; Lasitschka, F.; Andrulis, M.; et al. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat. Cell Biol. 2013, 15, 978–990. [Google Scholar] [CrossRef]
- van Deursen, J.M. The role of senescent cells in ageing. Nature 2014, 509, 439–446. [Google Scholar] [CrossRef] [Green Version]
- Minamino, T.; Miyauchi, H.; Yoshida, T.; Komuro, I. Endothelial cell senescence in human atherosclerosis: Role of telomeres in endothelial dysfunction. J. Cardiol. 2003, 41, 1541–1544. [Google Scholar] [CrossRef] [Green Version]
- Schroth, J.; Thiemermann, C.; Henson, S.M. Senescence and the Aging Immune System as Major Drivers of Chronic Kidney Disease. Front. Cell Dev. Biol. 2020, 8, 564461. [Google Scholar] [CrossRef]
- Ogrodnik, M.; Miwa, S.; Tchkonia, T.; Tiniakos, D.; Wilson, C.L.; Lahat, A.; Day, C.P.; Burt, A.; Palmer, A.; Anstee, Q.M.; et al. Cellular senescence drives age-dependent hepatic steatosis. Nat. Commun. 2017, 8, 15691. [Google Scholar] [CrossRef]
- Jeyapalan, J.C.; Ferreira, M.; Sedivy, J.M.; Herbig, U. Accumulation of senescent cells in mitotic tissue of aging primates. Mech. Ageing Dev. 2007, 128, 36–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krishnamurthy, J.; Ramsey, M.; Ligon, K.L.; Torrice, C.; Koh, A.; Bonner-Weir, S.; Sharpless, N. p16INK4a induces an age-dependent decline in islet regenerative potential. Nature 2006, 443, 453–457. [Google Scholar] [CrossRef]
- Molofsky, A.V.; Slutsky, S.G.; Joseph, N.M.; He, S.; Pardal, R.; Krishnamurthy, J.; Sharpless, N.; Morrison, S.J. Increasing p16INK4a expression decreases forebrain progenitors and neurogenesis during ageing. Nature 2006, 443, 448–452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, R.; Ramanathan, A. The Aging Metabolome—Biomarkers to Hub Metabolites. Proteomics 2020, 20, e1800407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, W.; Liu, J.-L.; Lu, X.; Yang, Q. Epigenetic regulation of energy metabolism in obesity. J. Mol. Cell Biol. 2021, 13, 480–499. [Google Scholar] [CrossRef]
- Matilainen, O.; Quiros, P.M.; Auwerx, J. Mitochondria and Epigenetics—Crosstalk in Homeostasis and Stress. Trends Cell Biol. 2017, 27, 453–463. [Google Scholar] [CrossRef]
- Schell, J.C.; Rutter, J. Mitochondria link metabolism and epigenetics in haematopoiesis. Nat. Cell Biol. 2017, 19, 589–591. [Google Scholar] [CrossRef]
- Zhu, X.; Chen, Z.; Shen, W.; Huang, G.; Sedivy, J.M.; Wang, H.; Ju, Z. Inflammation, epigenetics, and metabolism converge to cell senescence and ageing: The regulation and intervention. Signal Transduct. Target. Ther. 2021, 6, 1–29. [Google Scholar] [CrossRef]
- Minocherhomji, S.; Tollefsbol, T.O.; Singh, K.K. Mitochondrial regulation of epigenetics and its role in human diseases. Epigenetics 2012, 7, 326–334. [Google Scholar] [CrossRef] [Green Version]
- Grewal, S.I.S.; Moazed, D. Heterochromatin and Epigenetic Control of Gene Expression. Science 2003, 301, 798–802. [Google Scholar] [CrossRef] [Green Version]
- Smeal, T.; Claus, J.; Kennedy, B.; Cole, F.; Guarente, L. Loss of Transcriptional Silencing Causes Sterility in Old Mother Cells of S. cerevisiae. Cell 1996, 84, 633–642. [Google Scholar] [CrossRef] [Green Version]
- Villeponteau, B. The heterochromatin loss model of aging. Exp. Gerontol. 1997, 32, 383–394. [Google Scholar] [CrossRef]
- Haithcock, E.; Dayani, Y.; Neufeld, E.; Zahand, A.J.; Feinstein, N.; Mattout, A.; Gruenbaum, Y.; Liu, J. Age-related changes of nuclear architecture in Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA 2005, 102, 16690–16695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larson, K.; Yan, S.-J.; Tsurumi, A.; Liu, J.; Zhou, J.; Gaur, K.; Guo, D.; Eickbush, T.H.; Li, W.X. Heterochromatin Formation Promotes Longevity and Represses Ribosomal RNA Synthesis. PLoS Genet. 2012, 8, e1002473. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Li, J.; Suzuki, K.; Qu, J.; Wang, P.; Zhou, J.; Liu, X.; Ren, R.; Xu, X.; Ocampo, A.; et al. A Werner syndrome stem cell model unveils heterochromatin alterations as a driver of human aging. Science 2015, 348, 1160–1163. [Google Scholar] [CrossRef] [Green Version]
- Goldman, R.D.; Shumaker, D.K.; Erdos, M.R.; Eriksson, M.; Goldman, A.E.; Gordon, L.B.; Gruenbaum, Y.; Khuon, S.; Mendez, M.; Varga, R.; et al. Accumulation of mutant lamin A causes progressive changes in nuclear architecture in Hutchinson–Gilford progeria syndrome. Proc. Natl. Acad. Sci. USA 2004, 101, 8963–8968. [Google Scholar] [CrossRef] [Green Version]
- O’Sullivan, R.J.; Kubicek, S.; Schreiber, S.L.; Karlseder, J. Reduced histone biosynthesis and chromatin changes arising from a damage signal at telomeres. Nat. Struct. Mol. Biol. 2010, 17, 1218–1225. [Google Scholar] [CrossRef] [Green Version]
- Ivanov, A.; Pawlikowski, J.; Manoharan, I.; Van Tuyn, J.; Nelson, D.M.; Rai, T.S.; Shah, P.P.; Hewitt, G.; Korolchuk, V.; Passos, J.; et al. Lysosome-mediated processing of chromatin in senescence. J. Cell Biol. 2013, 202, 129–143. [Google Scholar] [CrossRef]
- Bannister, A.J.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381–395. [Google Scholar] [CrossRef]
- Jambhekar, A.; Dhall, A.; Shi, Y. Roles and regulation of histone methylation in animal development. Nat. Rev. Mol. Cell Biol. 2019, 20, 625–641. [Google Scholar] [CrossRef]
- Lessard, J.; Sauvageau, G. Bmi-1 determines the proliferative capacity of normal and leukaemic stem cells. Nature 2003, 423, 255–260. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, J.; Kieboom, K.; Marino, S.; DePinho, R.; Van Lohuizen, M. The oncogene and Polycomb-group gene bmi-1 regulates cell proliferation and senescence through the ink4a locus. Nature 1999, 397, 164–168. [Google Scholar] [CrossRef] [PubMed]
- Beà, S.; Tort, F.; Pinyol, M.; Puig, X.; Hernández, L.; Hernández, S.; Fernandez, P.L.; Van Lohuizen, M.; Colomer, D.; Campo, E. BMI-1 gene amplification and overexpression in hematological malignancies occur mainly in mantle cell lymphomas. Cancer Res. 2001, 61, 2409–2412. [Google Scholar] [PubMed]
- Vonlanthen, S.; Heighway, J.; Altermatt, H.J.; Gugger, M.; Kappeler, A.; Borner, M.M.; Van Lohuizen, M.; Betticher, D.C. The bmi-1 oncoprotein is differentially expressed in non-small cell lung cancer and correlates with INK4A-ARF locus expression. Br. J. Cancer 2001, 84, 1372–1376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.H.; Yoon, S.Y.; Kim, C.N.; Joo, J.H.; Moon, S.K.; Choe, I.S.; Choe, Y.K.; Kim, J.W. The Bmi-1 oncoprotein is overexpressed in human colorectal cancer and correlates with the reduced p16INK4a/p14ARF proteins. Cancer Lett. 2004, 203, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Neo, S.Y.; Leow, C.K.; Vega, V.B.; Long, P.M.; Islam, A.F.; Lai, P.B.; Liu, E.T.; Ren, E.C. Identification of discriminators of hepatoma by gene expression profiling using a minimal dataset approach. Hepatology 2004, 39, 944–953. [Google Scholar] [CrossRef]
- Gil, J.; Bernard, D.; Martínez, D.; Beach, D. Polycomb CBX7 has a unifying role in cellular lifespan. Nat. Cell Biol. 2003, 6, 67–72. [Google Scholar] [CrossRef]
- Itahana, K.; Zou, Y.; Itahana, Y.; Martinez-Guitarte, J.-L.; Beausejour, C.; Jacobs, J.; van Lohuizen, M.; Band, V.; Campisi, J.; Dimri, G.P. Control of the Replicative Life Span of Human Fibroblasts by p16 and the Polycomb Protein Bmi-1. Mol. Cell. Biol. 2003, 23, 389–401. [Google Scholar] [CrossRef] [Green Version]
- Guo, W.-J.; Datta, S.; Band, V.; Dimri, G.P. Mel-18, a Polycomb Group Protein, Regulates Cell Proliferation and Senescence via Transcriptional Repression of Bmi-1 and c-Myc Oncoproteins. Mol. Biol. Cell 2007, 18, 536–546. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.K.; Nikodem, V.M. hnRNP U Inhibits Carboxy-Terminal Domain Phosphorylation by TFIIH and Represses RNA Polymerase II Elongation. Mol. Cell. Biol. 1999, 19, 6833–6844. [Google Scholar] [CrossRef] [Green Version]
- Weidensdorfer, D.; Stöhr, N.; Baude, A.; Lederer, M.; Köhn, M.; Schierhorn, A.; Buchmeier, S.; Wahle, E.; Hüttelmaier, S. Control of c-myc mRNA stability by IGF2BP1-associated cytoplasmic RNPs. RNA 2008, 15, 104–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nozawa, R.-S.; Boteva, L.; Soares, D.; Naughton, C.; Dun, A.R.; Buckle, A.; Ramsahoye, B.; Bruton, P.C.; Saleeb, R.S.; Arnedo, M.; et al. SAF-A Regulates Interphase Chromosome Structure through Oligomerization with Chromatin-Associated RNAs. Cell 2017, 169, 1214–1227.e18. [Google Scholar] [CrossRef] [Green Version]
- Puvvula, P.K.; Desetty, R.D.; Pineau, P.; Marchio, A.; Moon, A.; Dejean, A.; Bischof, O. Long noncoding RNA PANDA and scaffold-attachment-factor SAFA control senescence entry and exit. Nat. Commun. 2014, 5, 5323. [Google Scholar] [CrossRef] [PubMed]
- Puvvula, P.K. LncRNAs Regulatory Networks in Cellular Senescence. Int. J. Mol. Sci. 2019, 20, 2615. [Google Scholar] [CrossRef] [Green Version]
- Suh, N. MicroRNA controls of cellular senescence. BMB Rep. 2018, 51, 493–499. [Google Scholar] [CrossRef] [Green Version]
- Kim, E.; Kim, Y.K.; Lee, S.-J.V. Emerging functions of circular RNA in aging. Trends Genet. 2021, 37, 819–829. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, A.; Faust, C.; Magnuson, T. Positional cloning of a global regulator of anterior–posterior patterning in mice. Nature 1996, 383, 250–253. [Google Scholar] [CrossRef]
- Ito, T.; Teo, Y.V.; Evans, S.A.; Neretti, N.; Sedivy, J.M. Regulation of Cellular Senescence by Polycomb Chromatin Modifiers through Distinct DNA Damage- and Histone Methylation-Dependent Pathways. Cell Rep. 2018, 22, 3480–3492. [Google Scholar] [CrossRef] [Green Version]
- Barradas, M.; Anderton, E.; Acosta, J.C.; Li, S.; Banito, A.; Rodriguez-Niedenführ, M.; Maertens, G.; Banck, M.; Zhou, M.-M.; Walsh, M.J.; et al. Histone demethylase JMJD3 contributes to epigenetic control of INK4a/ARF by oncogenic RAS. Genes Dev. 2009, 23, 1177–1182. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Qin, G.; Zhao, T.C. HDAC4: Mechanism of regulation and biological functions. Epigenomics 2014, 6, 139–150. [Google Scholar] [CrossRef] [Green Version]
- Di Giorgio, E.; Dalla, E.; Franforte, E.; Paluvai, H.; Minisini, M.; Trevisanut, M.; Picco, R.; Brancolini, C. Different class IIa HDACs repressive complexes regulate specific epigenetic responses related to cell survival in leiomyosarcoma cells. Nucleic Acids Res. 2020, 48, 646–664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Giorgio, E.; Paluvai, H.; Dalla, E.; Ranzino, L.; Renzini, A.; Moresi, V.; Minisini, M.; Picco, R.; Brancolini, C. HDAC4 degradation during senescence unleashes an epigenetic program driven by AP-1/p300 at selected enhancers and super-enhancers. Genome Biol. 2021, 22, 1–25. [Google Scholar] [CrossRef]
- Sen, P.; Lan, Y.; Li, C.Y.; Sidoli, S.; Donahue, G.; Dou, Z.; Frederick, B.; Chen, Q.; Luense, L.J.; Garcia, B.A.; et al. Histone Acetyltransferase p300 Induces De Novo Super-Enhancers to Drive Cellular Senescence. Mol. Cell 2019, 73, 684–698.e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandra, T.; Kirschner, K.; Thuret, J.-Y.; Pope, B.; Ryba, T.; Newman, S.; Ahmed, K.; Samarajiwa, S.; Salama, R.; Carroll, T.; et al. Independence of Repressive Histone Marks and Chromatin Compaction during Senescent Heterochromatic Layer Formation. Mol. Cell 2012, 47, 203–214. [Google Scholar] [CrossRef] [Green Version]
- Funayama, R.; Saito, M.; Tanobe, H.; Ishikawa, F. Loss of linker histone H1 in cellular senescence. J. Cell Biol. 2006, 175, 869–880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braig, M.; Lee, S.; Loddenkemper, C.; Rudolph, C.; Peters, A.H.; Schlegelberger, B.; Stein, H.; Dörken, B.; Jenuwein, T.; Schmitt, C.A. Oncogene-induced senescence as an initial barrier in lymphoma development. Nature 2005, 436, 660–665. [Google Scholar] [CrossRef]
- Kovatcheva, M.; Liao, W.; Klein, M.E.; Robine, N.; Geiger, H.; Crago, A.M.; Dickson, M.A.; Tap, W.D.; Singer, S.; Koff, A. ATRX is a regulator of therapy induced senescence in human cells. Nat. Commun. 2017, 8, 386. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Gao, Y.; Zhao, L.; Han, L.; Lu, Y.; Hou, P.; Shi, X.; Liu, X.; Tian, B.; Wang, X.; et al. Mitogen-activated protein kinase p38 and retinoblastoma protein signalling is required for DNA damage-mediated formation of senescence-associated heterochromatic foci in tumour cells. FEBS J. 2013, 280, 4625–4639. [Google Scholar] [CrossRef]
- Zhao, L.; Zhang, Y.; Gao, Y.; Geng, P.; Lu, Y.; Liu, X.; Yao, R.; Hou, P.; Liu, D.; Lu, J.; et al. JMJD3 promotes SAHF formation in senescent WI38 cells by triggering an interplay between demethylation and phosphorylation of RB protein. Cell Death Differ. 2015, 22, 1630–1640. [Google Scholar] [CrossRef]
- Košař, M.; Bartkova, J.; Hubackova, S.; Hodny, Z.; Lukas, J.; Bartek, J. Senescence-associated heterochromatin foci are dispensable for cellular senescence, occur in a cell type- and insult-dependent manner and follow expression of p16 ink4a. Cell Cycle 2011, 10, 457–468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henikoff, S.; Smith, M.M. Histone Variants and Epigenetics. Cold Spring Harb. Perspect. Biol. 2015, 7, a019364. [Google Scholar] [CrossRef] [Green Version]
- Duarte, L.F.; Young, A.; Wang, Z.; Wu, H.-A.; Panda, T.; Kou, Y.; Kapoor, A.; Hasson, D.; Mills, N.R.; Ma’Ayan, A.; et al. Histone H3.3 and its proteolytically processed form drive a cellular senescence programme. Nat. Commun. 2014, 5, 5210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rai, T.S.; Cole, J.J.; Nelson, D.M.; Dikovskaya, D.; Faller, W.J.; Vizioli, M.G.; Hewitt, R.N.; Anannya, O.; McBryan, T.; Manoharan, I.; et al. HIRA orchestrates a dynamic chromatin landscape in senescence and is required for suppression of neoplasia. Genes Dev. 2014, 28, 2712–2725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, R.; Poustovoitov, M.V.; Ye, X.; Santos, H.A.; Chen, W.; Daganzo, S.M.; Erzberger, J.P.; Serebriiskii, I.G.; Canutescu, A.A.; Dunbrack, R.; et al. Formation of MacroH2A-Containing Senescence-Associated Heterochromatin Foci and Senescence Driven by ASF1a and HIRA. Dev. Cell 2005, 8, 19–30. [Google Scholar] [CrossRef] [Green Version]
- Contrepois, K.; Coudereau, C.; Benayoun, B.A.; Schuler, N.; Roux, P.-F.; Bischof, O.; Courbeyrette, R.; Carvalho, C.; Thuret, J.-Y.; Ma, Z.; et al. Histone variant H2A.J accumulates in senescent cells and promotes inflammatory gene expression. Nat. Commun. 2017, 8, 14995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gévry, N.; Chan, H.M.; Laflamme, L.; Livingston, D.M.; Gaudreau, L. p21 transcription is regulated by differential localization of histone H2A.Z. Genes Dev. 2007, 21, 1869–1881. [Google Scholar] [CrossRef] [Green Version]
- Hovest, M.G.; Brüggenolte, N.; Hosseini, K.S.; Krieg, T.; Herrmann, G. Senescence of Human Fibroblasts after Psoralen Photoactivation Is Mediated by ATR Kinase and Persistent DNA Damage Foci at Telomeres. Mol. Biol. Cell 2006, 17, 1758–1767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cruickshanks, H.A.; McBryan, T.; Nelson, D.M.; VanderKraats, N.D.; Shah, P.P.; Van Tuyn, J.; Rai, T.S.; Brock, C.; Donahue, G.; Dunican, D.S.; et al. Senescent cells harbour features of the cancer epigenome. Nat. Cell Biol. 2013, 15, 1495–1506. [Google Scholar] [CrossRef]
- Sakaki, M.; Ebihara, Y.; Okamura, K.; Nakabayashi, K.; Igarashi, A.; Matsumoto, K.; Hata, K.; Kobayashi, Y.; Maehara, K. Potential roles of DNA methylation in the initiation and establishment of replicative senescence revealed by array-based methylome and transcriptome analyses. PLoS ONE 2017, 12, e0171431. [Google Scholar] [CrossRef]
- Arand, J.; Spieler, D.; Karius, T.; Branco, M.R.; Meilinger, D.; Meissner, A.; Jenuwein, T.; Xu, G.; Leonhardt, H.; Wolf, V.; et al. In Vivo Control of CpG and Non-CpG DNA Methylation by DNA Methyltransferases. PLoS Genet. 2012, 8, e1002750. [Google Scholar] [CrossRef] [Green Version]
- Scott-Browne, J.P.; Lio, C.-W.J.; Rao, A. TET proteins in natural and induced differentiation. Curr. Opin. Genet. Dev. 2017, 46, 202–208. [Google Scholar] [CrossRef] [PubMed]
- Parry, A.; Rulands, S.; Reik, W. Active turnover of DNA methylation during cell fate decisions. Nat. Rev. Genet. 2021, 22, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.-L.; Bochtler, M. Reversal of nucleobase methylation by dioxygenases. Nat. Chem. Biol. 2020, 16, 1160–1169. [Google Scholar] [CrossRef] [PubMed]
- Lio, C.-W.J.; Yue, X.; Lopez-Moyado, I.F.; Tahiliani, M.; Aravind, L.; Rao, A. TET methylcytosine oxidases: New insights from a decade of research. J. Biosci. 2020, 45, 21. [Google Scholar] [CrossRef]
- Rinaldi, L.; Datta, D.; Serrat, J.; Morey, L.; Solanas, G.; Avgustinova, A.; Blanco, E.; Pons, J.I.; Matallanas, D.; von Kriegsheim, A.; et al. Dnmt3a and Dnmt3b Associate with Enhancers to Regulate Human Epidermal Stem Cell Homeostasis. Cell Stem Cell 2016, 19, 491–501. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Wang, H.; Xu, G.; Liu, L. Tet1 Deficiency Leads to Premature Ovarian Failure. Front. Cell Dev. Biol. 2021, 9, 620. [Google Scholar] [CrossRef]
- Huang, G.; Liu, L.; Wang, H.; Gou, M.; Gong, P.; Tian, C.; Deng, W.; Yang, J.; Zhou, T.-T.; Xu, G.-L.; et al. Tet1 Deficiency Leads to Premature Reproductive Aging by Reducing Spermatogonia Stem Cells and Germ Cell Differentiation. IScience 2020, 23, 100908. [Google Scholar] [CrossRef] [Green Version]
- Ferrone, C.K.; Blydt-Hansen, M.; Rauh, M.J. Age-Associated TET2 Mutations: Common Drivers of Myeloid Dysfunction, Cancer and Cardiovascular Disease. Int. J. Mol. Sci. 2020, 21, 626. [Google Scholar] [CrossRef] [Green Version]
- Weidner, C.I.; Lin, Q.; Koch, C.M.; Eisele, L.; Beier, F.; Ziegler, P.; Bauerschlag, D.O.; Jöckel, K.-H.; Erbel, R.; Mühleisen, T.W.; et al. Aging of blood can be tracked by DNA methylation changes at just three CpG sites. Genome Biol. 2014, 15, R24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meer, M.V.; Podolskiy, D.I.; Tyshkovskiy, A.; Gladyshev, V.N. A whole lifespan mouse multi-tissue DNA methylation clock. Elife 2018, 7, 7. [Google Scholar] [CrossRef]
- Koch, C.M.; Joussen, S.; Schellenberg, A.; Lin, Q.; Zenke, M.; Wagner, W. Monitoring of cellular senescence by DNA-methylation at specific CpG sites. Aging Cell 2012, 11, 366–369. [Google Scholar] [CrossRef] [PubMed]
- Bork, S.; Pfister, S.; Witt, H.; Horn, P.; Korn, B.; Ho, A.D.; Wagner, W. DNA methylation pattern changes upon long-term culture and aging of human mesenchymal stromal cells. Aging Cell 2010, 9, 54–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ting, A.H.; McGarvey, K.M.; Baylin, S.B. The cancer epigenome—components and functional correlates. Genes Dev. 2006, 20, 3215–3231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, W.; Kagiampakis, I.; Pan, L.; Zhang, Y.W.; Murphy, L.; Tao, Y.; Kong, X.; Kang, B.; Xia, L.; Carvalho, F.L.; et al. DNA Methylation Patterns Separate Senescence from Transformation Potential and Indicate Cancer Risk. Cancer Cell 2018, 33, 309–321.e5. [Google Scholar] [CrossRef] [Green Version]
- Sarkar, T.J.; Quarta, M.; Mukherjee, S.; Colville, A.; Paine, P.; Doan, L.; Tran, C.M.; Chu, C.R.; Horvath, S.; Qi, L.S.; et al. Transient non-integrative expression of nuclear reprogramming factors promotes multifaceted amelioration of aging in human cells. Nat. Commun. 2020, 11, 1545. [Google Scholar] [CrossRef] [Green Version]
- Lapasset, L.; Milhavet, O.; Prieur, A.; Besnard, E.; Babled, A.; Aït-Hamou, N.; Leschik, J.; Pellestor, F.; Ramirez, J.-M.; De Vos, J.; et al. Rejuvenating senescent and centenarian human cells by reprogramming through the pluripotent state. Genes Dev. 2011, 25, 2248–2253. [Google Scholar] [CrossRef] [Green Version]
- Ocampo, A.; Reddy, P.; Redondo, P.M.; Luengo, A.P.; Hatanaka, F.; Hishida, T.; Li, M.; Lam, D.; Kurita, M.; Beyret, E.; et al. In Vivo Amelioration of Age-Associated Hallmarks by Partial Reprogramming. Cell 2016, 167, 1719–1733.e12. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.; Brommer, B.; Tian, X.; Krishnan, A.; Meer, M.; Wang, C.; Vera, D.L.; Zeng, Q.; Yu, D.; Bonkowski, M.S.; et al. Reprogramming to recover youthful epigenetic information and restore vision. Nature 2020, 588, 124–129. [Google Scholar] [CrossRef]
- Voong, C.K.; Goodrich, J.A.; Kugel, J.F. Interactions of HMGB Proteins with the Genome and the Impact on Disease. Biomolecules 2021, 11, 1451. [Google Scholar] [CrossRef]
- Aird, K.M.; Iwasaki, O.; Kossenkov, A.V.; Tanizawa, H.; Fatkhutdinov, N.; Bitler, B.; Le, L.; Alicea, G.; Yang, T.-L.; Johnson, F.B.; et al. HMGB2 orchestrates the chromatin landscape of senescence-associated secretory phenotype gene loci. J. Cell Biol. 2016, 215, 325–334. [Google Scholar] [CrossRef]
- Tasdemir, N.; Banito, A.; Roe, J.-S.; Curbelo, D.A.; Camiolo, M.; Tschaharganeh, D.F.; Huang, C.-H.; Aksoy, O.; Bolden, J.E.; Chen, C.-C.; et al. BRD4 Connects Enhancer Remodeling to Senescence Immune Surveillance. Cancer Discov. 2016, 6, 612–629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Capell, B.C.; Drake, A.M.; Zhu, J.; Shah, P.P.; Dou, Z.; Dorsey, J.; Simola, D.F.; Donahue, G.; Sammons, M.; Rai, T.S.; et al. MLL1 is essential for the senescence-associated secretory phenotype. Genes Dev. 2016, 30, 321–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leon, K.E.; Buj, R.; Lesko, E.; Dahl, E.S.; Chen, C.-W.; Tangudu, N.K.; Kawasawa, Y.I.; Kossenkov, A.V.; Hobbs, R.P.; Aird, K.M. DOT1L modulates the senescence-associated secretory phenotype through epigenetic regulation of IL1A. J. Cell Biol. 2021, 220, 202008101. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, A.; Imai, Y.; Yamakoshi, K.; Kuninaka, S.; Ohtani, N.; Yoshimoto, S.; Hori, S.; Tachibana, M.; Anderton, E.; Takeuchi, T.; et al. DNA Damage Signaling Triggers Degradation of Histone Methyltransferases through APC/CCdh1 in Senescent Cells. Mol. Cell 2012, 45, 123–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perrigue, P.M.; Silva, M.E.; Warden, C.D.; Feng, N.L.; Reid, M.; Mota, D.J.; Joseph, L.P.; Tian, Y.I.; Glackin, C.A.; Gutova, M.; et al. The Histone Demethylase Jumonji Coordinates Cellular Senescence Including Secretion of Neural Stem Cell–Attracting Cytokines. Mol. Cancer Res. 2015, 13, 636–650. [Google Scholar] [CrossRef] [Green Version]
- Hayakawa, T.; Iwai, M.; Aoki, S.; Takimoto, K.; Maruyama, M.; Maruyama, W.; Motoyama, N. SIRT1 Suppresses the Senescence-Associated Secretory Phenotype through Epigenetic Gene Regulation. PLoS ONE 2015, 10, e0116480. [Google Scholar] [CrossRef] [PubMed]
- Pospelova, T.V.; Demidenko, Z.N.; Bukreeva, E.I.; Pospelov, V.A.; Gudkov, A.; Blagosklonny, M.V. Pseudo-DNA damage response in senescent cells. Cell Cycle 2009, 8, 4112–4118. [Google Scholar] [CrossRef] [Green Version]
- Pazolli, E.; Alspach, E.; Milczarek, A.; Prior, J.; Piwnica-Worms, D.; Stewart, S.A. Chromatin Remodeling Underlies the Senescence-Associated Secretory Phenotype of Tumor Stromal Fibroblasts That Supports Cancer Progression. Cancer Res. 2012, 72, 2251–2261. [Google Scholar] [CrossRef] [Green Version]
- Barger, J.L.; Kayo, T.; Vann, J.M.; Arias, E.B.; Wang, J.; Hacker, T.A.; Wang, Y.; Raederstorff, D.; Morrow, J.D.; Leeuwenburgh, C.; et al. A low dose of dietary resveratrol partially mimics caloric restriction and retards aging parameters in mice. PLoS ONE 2008, 3, e2264. [Google Scholar] [CrossRef]
- Fischer-Posovszky, P.; Kukulus, V.; Tews, D.; Unterkircher, T.; Debatin, K.-M.; Fulda, S.; Wabitsch, M. Resveratrol regulates human adipocyte number and function in a Sirt1-dependent manner. Am. J. Clin. Nutr. 2010, 92, 5–15. [Google Scholar] [CrossRef]
- Subramanian, L.; Youssef, S.; Bhattacharya, S.; Kenealey, J.; Polans, A.S.; van Ginkel, P.R. Resveratrol: Challenges in translation to the clinic—a critical discussion. Clin. Cancer Res. 2010, 16, 5942–5948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiley, J.C.; Pettan-Brewer, C.; Ladiges, W.C. Phenylbutyric acid reduces amyloid plaques and rescues cognitive behavior in AD transgenic mice. Aging Cell 2011, 10, 418–428. [Google Scholar] [CrossRef]
- Yoo, Y.-E.; Ko, C.-P. Treatment with trichostatin A initiated after disease onset delays disease progression and increases survival in a mouse model of amyotrophic lateral sclerosis. Exp. Neurol. 2011, 231, 147–159. [Google Scholar] [CrossRef] [PubMed]
- Harrison, I.F.; Crum, W.R.; Vernon, A.C.; Dexter, D.T. Neurorestoration induced by the HDAC inhibitor sodium valproate in the lactacystin model of P arkinson’s is associated with histone acetylation and up-regulation of neurotrophic factors. Br. J. Pharmacol. 2015, 172, 4200–4215. [Google Scholar] [CrossRef] [Green Version]
- Gao, Z.; Yin, J.; Zhang, J.; Ward, R.E.; Martin, R.J.; Lefevre, M.; Cefalu, W.T.; Ye, J. Butyrate Improves Insulin Sensitivity and Increases Energy Expenditure in Mice. Diabetes 2009, 58, 1509–1517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henagan, T.M.; Stefanska, B.; Fang, Z.; Navard, A.M.; Ye, J.; Lenard, N.R.; Devarshi, P. Sodium butyrate epigenetically modulates high-fat diet-induced skeletal muscle mitochondrial adaptation, obesity and insulin resistance through nucleosome positioning. J. Cereb. Blood Flow Metab. 2015, 172, 2782–2798. [Google Scholar] [CrossRef]
- Piekarz, R.L.; Frye, R.; Turner, M.; Wright, J.J.; Allen, S.; Kirschbaum, M.H.; Zain, J.; Prince, H.M.; Leonard, J.P.; Geskin, L.J.; et al. Phase II Multi-Institutional Trial of the Histone Deacetylase Inhibitor Romidepsin As Monotherapy for Patients with Cutaneous T-Cell Lymphoma. J. Clin. Oncol. 2009, 27, 5410–5417. [Google Scholar] [CrossRef] [Green Version]
- Whittaker, S.J.; Demierre, M.-F.; Kim, E.J.; Rook, A.H.; Lerner, A.; Duvic, M.; Scarisbrick, J.; Reddy, S.; Robak, T.; Becker, J.C.; et al. Final Results from a Multicenter, International, Pivotal Study of Romidepsin in Refractory Cutaneous T-Cell Lymphoma. J. Clin. Oncol. 2010, 28, 4485–4491. [Google Scholar] [CrossRef]
- Coiffier, B.; Pro, B.; Prince, H.M.; Foss, F.; Sokol, L.; Greenwood, M.; Caballero, D.; Borchmann, P.; Morschhauser, F.; Wilhelm, M.; et al. Results from a Pivotal, Open-Label, Phase II Study of Romidepsin in Relapsed or Refractory Peripheral T-Cell Lymphoma After Prior Systemic Therapy. J. Clin. Oncol. 2012, 30, 631–636. [Google Scholar] [CrossRef]
- O’Connor, O.A.; Horwitz, S.; Masszi, T.; Van Hoof, A.; Brown, P.D.N.; Doorduijn, J.; Hess, G.; Jurczak, W.; Knoblauch, P.; Chawla, S.; et al. Belinostat in Patients with Relapsed or Refractory Peripheral T-Cell Lymphoma: Results of the Pivotal Phase II BELIEF (CLN-19) Study. J. Clin. Oncol. 2015, 33, 2492–2499. [Google Scholar] [CrossRef] [Green Version]
- Derissen, E.J.; Beijnen, J.H.; Schellens, J.H. Concise Drug Review: Azacitidine and Decitabine. Oncologist 2013, 18, 619–624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Italiano, A.; Soria, J.C.; Toulmonde, M.; Michot, J.M.; Lucchesi, C.; Varga, A.; Coindre, J.M.; Blakemore, S.J.; Clawson, A.; Suttle, B.; et al. Tazemetostat, an EZH2 inhibitor, in relapsed or refractory B-cell non-Hodgkin lymphoma and advanced solid tumours: A first-in-human, open-label, phase 1 study. Lancet Oncol. 2018, 19, 649–659. [Google Scholar] [CrossRef]
- Thomas, S.; Thurn, K.T.; Biçaku, E.; Marchion, D.C.; Münster, P.N. Addition of a histone deacetylase inhibitor redirects tamoxifen-treated breast cancer cells into apoptosis, which is opposed by the induction of autophagy. Breast Cancer Res. Treat. 2011, 130, 437–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hodges-Gallagher, L.; Valentine, C.D.; El Bader, S.; Kushner, P.J. Inhibition of histone deacetylase enhances the anti-proliferative action of antiestrogens on breast cancer cells and blocks tamoxifen-induced proliferation of uterine cells. Breast Cancer Res. Treat. 2006, 105, 297–309. [Google Scholar] [CrossRef] [PubMed]
- Morel, D.; Jeffery, D.; Aspeslagh, S.; Almouzni, G.; Postel-Vinay, S. Combining epigenetic drugs with other therapies for solid tumours—Past lessons and future promise. Nat. Rev. Clin. Oncol. 2020, 17, 91–107. [Google Scholar] [CrossRef] [PubMed]
- Pasyukova, E.G.; Vaiserman, A.M. HDAC inhibitors: A new promising drug class in anti-aging research. Mech. Ageing Dev. 2017, 166, 6–15. [Google Scholar] [CrossRef] [PubMed]
- De Lera, A.R.; Ganesan, A. Two-hit wonders: The expanding universe of multitargeting epigenetic agents. Curr. Opin. Chem. Biol. 2020, 57, 135–154. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Crouch, J.; Shvedova, M.; Thanapaul, R.J.R.S.; Botchkarev, V.; Roh, D. Epigenetic Regulation of Cellular Senescence. Cells 2022, 11, 672. https://doi.org/10.3390/cells11040672
Crouch J, Shvedova M, Thanapaul RJRS, Botchkarev V, Roh D. Epigenetic Regulation of Cellular Senescence. Cells. 2022; 11(4):672. https://doi.org/10.3390/cells11040672
Chicago/Turabian StyleCrouch, Jack, Maria Shvedova, Rex Jeya Rajkumar Samdavid Thanapaul, Vladimir Botchkarev, and Daniel Roh. 2022. "Epigenetic Regulation of Cellular Senescence" Cells 11, no. 4: 672. https://doi.org/10.3390/cells11040672
APA StyleCrouch, J., Shvedova, M., Thanapaul, R. J. R. S., Botchkarev, V., & Roh, D. (2022). Epigenetic Regulation of Cellular Senescence. Cells, 11(4), 672. https://doi.org/10.3390/cells11040672