
Sympathetic Nerves and Innate Immune System in the Spleen: Implications of Impairment in HIV-1 and Relevant Models

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. MAIDS and Simian AIDs (SAIDS) as Models for HIV-1
2.1. MAIDS Model
2.2. SAIDS
3. Splenic Sympathetic Nerves Influence HIV-1 Cell Entry and Replication in Acute Disease
4. HIV-1 Establishes Cellular Reservoirs and Induces Splenocyte Dysfunction
4.1. HIV-1-Induced Pathogenesis in the Spleen and Establishing the Viral Reservoir
4.2. Spleen Dysfunction in SIV and HIV-1 Infection
4.3. Activation of Type I Interferon Gene Expression in the SIV-Infected Spleens
5. Functional Consequences of SIV-Induced Pathology in the Spleen
6. HIV-1 Infection and Consequences for Sympathetic Innervation of the Spleen
7. HIV-1 and Inflammation in Autonomic Ganglia
Author Contributions
Funding
Conflicts of Interest
References
- CDC HIV Website. Available online: https://www.hiv.gov/hiv-basics/overview/data-and-trends/global-statistics (accessed on 7 February 2022).
- UNAIDS. Global HIV & AIDS Statistics—2020 Fact Sheet. Available online: https://www.unaids.org/en/resources/fact-sheet (accessed on 28 December 2021).
- Sagaon-Tevssier, L.; Singh, S.; Dongmo-Nguimfack, B.; Moati, J.-P. Affordability of adult HIV/AIDS treatment in developing countries: Modelling price determinants for a better insight of the market functioning. J. Int. AIDS Soc. 2016, 19, 20619. [Google Scholar]
- Ndung’u, T.; McCune, J.M.; Deeks, S.G. Why and where an HIV cure is needed and how it might be achieved. Nature 2019, 576, 397–405. [Google Scholar] [CrossRef] [PubMed]
- Kityo, C.; Makamdop, K.N.; Rothenberger, M.; Chipman, J.G.; Hoskuldsson, T.; Beilman, G.J.; Grzywacz, B.; Mugyenyi, P.; Ssali, F.; Akondy, R.S.; et al. Lymphoid tissue fibrosis is associated with impaired vaccine responses. J. Clin. Investig. 2018, 128, 2763–2773. [Google Scholar] [CrossRef] [PubMed]
- Dias, A.S.; Bester, M.J.; Britz, R.F.; Apostolides, Z. Animal models used for the evaluation of antiretroviral therapies. Curr. HIV Res. 2006, 4, 431–446. [Google Scholar] [CrossRef] [PubMed]
- Bartholomew, O.I.; Ashano, E. (5 November 2018) Experimental Animal Models of HIV/AIDS for Vaccine Trials, Experimental Animal Models of Human Diseases—An Effective Therapeutic Strategy, Ibeh Bartholomew, IntechOpen. Available online: https://www.intechopen.com/chapters/60960 (accessed on 5 January 2022).
- Nolan, D.J.; Rose, R.; Rodriguez, P.H.; Salemi, M.; Singer, E.J.; Lamers, S.L.; McGrath, M.S. The Spleen Is an HIV-1 Sanctuary During Combined Antiretroviral Therapy. AIDS Res. Hum. Retrovir. 2018, 34, 123–125. [Google Scholar] [CrossRef] [PubMed]
- Remor, E.; Penedo, F.J.; Shen, B.J.; Schneiderman, N. Perceived stress is associated with CD4+ cell decline in men and women living with HIV/AIDS in Spain. AIDS Care 2007, 19, 215–219. [Google Scholar] [CrossRef]
- Farinpour, R.; Miller, E.N.; Satz, P.; Selnes, O.A.; Cohen, B.A.; Becker, J.T.; Skolasky, R.L., Jr.; Visscher, B.R. Psychosocial risk factors of HIV morbidity and mortality: Findings from the Multicenter AIDS Cohort Study (MACS). J. Clin. Exp. Neuropsychol. 2003, 25, 654–670. [Google Scholar] [CrossRef]
- Robinson-Papp, J.; Astha, V.; Nmashie, A.; Sharma, S.K.; Kim-Schulze, S.; Murray, J.; George, M.C.; Morgello, S.; Mueller, B.R.; Lawrence, S.A.; et al. Sympathetic function and markers of inflammation in well-controlled HIV. Brain Behav. Immun. Health 2020, 3, 100112. [Google Scholar] [CrossRef]
- Leserman, J.; Petitto, J.M.; Gu, H.; Gaynes, B.N.; Barroso, J.; Golden, R.N.; Perkins, D.O.; Folds, J.D.; Evans, D.L. Progression to AIDS, a clinical AIDS condition and mortality: Psychosocial and physiological predictors. Psychol. Med. 2002, 32, 1059–1073. [Google Scholar] [CrossRef]
- Ironson, G.; Solomon, G.F.; Balbin, E.G.; O’Cleirigh, C.; George, A.; Kumar, M.; Larson, D.; Woods, T.E. The Ironson-woods Spirituality/Religiousness Index is associated with long survival, health behaviors, less distress, and low cortisol in people with HIV/AIDS. Ann. Behav. Med. 2002, 24, 34–48. [Google Scholar] [CrossRef]
- Cole, S.W.; Naliboff, B.D.; Kemeny, M.E.; Griswold, M.P.; Fahey, J.L.; Zack, J.A. Impaired response to HAART in HIV-infected individuals with high autonomic nervous system activity. Proc. Natl. Acad. Sci. USA 2001, 98, 12695–12700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cole, S.W.; Kemeny, M.E.; Fahey, J.L.; Zack, J.A.; Naliboff, B.D. Psychological risk factors for HIV pathogenesis: Mediation by the autonomic nervous system. Biol. Psychiatry 2003, 54, 1444–1456. [Google Scholar] [CrossRef]
- Ironson, G.; Balbin, E.; Stieren, E.; Detz, K.; Fletcher, M.A.; Schneiderman, N.; Kumar, M. Perceived stress and norepinephrine predict the effectiveness of response to protease inhibitors in HIV. Int. J. Behav. Med. 2008, 15, 221–226. [Google Scholar] [CrossRef]
- Capitanio, J.P.; Mendoza, S.P.; Lerche, N.W.; Mason, W.A. Social stress results in altered glucocorticoid regulation and shorter survival in simian acquired immune deficiency syndrome. Proc. Natl. Acad. Sci. USA 1998, 95, 4714–4719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sloan, E.K.; Capitanio, J.P.; Tarara, R.P.; Mendoza, S.P.; Mason, W.A.; Cole, S.W. Social stress enhances sympathetic innervation of primate lymph nodes: Mechanisms and implications for viral pathogenesis. J. Neurosci. 2007, 27, 8857–8865. [Google Scholar] [CrossRef] [PubMed]
- Fulcher, J.A.; Javanbakht, M.; Shover, C.L.; Ragsdale, A.; Brookmeyer, R.; Shoptaw, S.; Gorbach, P.M. Comparative impact of methamphetamine and other drug use on viral suppression among sexual minority men on antiretroviral therapy. Drug Alcohol Depend. 2021, 221, 108622. [Google Scholar] [CrossRef]
- Jiang, J.; Wang, M.; Liang, B.; Shi, Y.; Su, Q.; Chen, H.; Huang, J.; Su, J.; Pan, P.; Li, Y.; et al. In vivo effects of methamphetamine on HIV-1 replication: A population-based study. Drug Alcohol Depend. 2016, 159, 246–254. [Google Scholar] [CrossRef] [PubMed]
- Sloan, E.K.; Cox, B.F.; Tarara, R.P.; Capitanio, J.P.; Mason, W.A.; Cole, S.W. Beta-blockers for SIV infection: Impact on lymph node innervation and viral replication. Brain Behav. Immun. 2007, 21, e37. [Google Scholar]
- Sloan, E.K.; Tarara, R.P.; Capitanio, J.P.; Cole, S.W. Enhanced replication of Simian Immunodeficiency Virus adjacent to catecholaminergic varicosities in primate lymph nodes. J. Virol. 2006, 80, 4326–4335. [Google Scholar] [CrossRef] [Green Version]
- Bellinger, D.L.; Millar, B.A.; Perez, S.; Carter, J.; Wood, C.; ThyagaRajan, S.; Molinaro, C.; Lubahn, C.; Lorton, D. Sympathetic modulation of immunity: Relevance to disease. Cell Immunol. 2008, 252, 27–56. [Google Scholar] [CrossRef] [Green Version]
- Reid, W.; Sadowska, M.; Denaro, F.; Rao, S.; Foulke, J., Jr.; Hayes, N.; Jones, O.; Doodnauth, D.; Davis, H.; Sill, A.; et al. An HIV-1 transgenic rat that develops HIV-related pathology and immunologic dysfunction. Proc Natl. Acad. Sci. USA 2001, 98, 9271–9276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bucsek, M.J.; Giridharan, T.; MacDonald, C.R.; Hylander, B.L.; Repasky, E.A. An overview of the role of sympathetic regulation of immune responses in infectious disease and autoimmunity. Int. J. Hyperth. Off. J. Eur. Soc. Hyperthermic Oncol. N. Am. Hyperth. Group 2018, 34, 135–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pahar, B.; Kuebler, D.; Rasmussen, T.; Wang, X.; Srivastav, S.K.; Das, A.; Veazey, R.S. Quantification of Viral RNA and DNA Positive Cells in Tissues from Simian Immunodeficiency Virus/Simian Human Immunodeficiency Virus Infected Controller and Progressor Rhesus Macaques. Front. Microbiol. 2019, 10, 2933. [Google Scholar] [CrossRef] [PubMed]
- Gulzar, N.; Copeland, K.F. CD8+ T-cells: Function and response to HIV infection. Curr. HIV Res. 2004, 2, 23–37. [Google Scholar] [CrossRef] [PubMed]
- Merino, K.M.; Allers, C.; Didier, E.S.; Kuroda, M.J. Role of Monocyte/Macrophages during HIV/SIV Infection in Adult and Pediatric Acquired Immune Deficiency Syndrome. Front. Immunol. 2017, 8, 1693. [Google Scholar] [CrossRef] [Green Version]
- Kelley, S.P.; Moynihan, J.A.; Stevens, S.Y.; Grota, L.J.; Felten, D.L. Sympathetic nerve destruction in spleen in murine AIDS. Brain Behav. Immun. 2003, 17, 94–109. [Google Scholar] [CrossRef]
- Sloan, E.K.; Nguyen, C.T.; Cox, B.F.; Tarara, R.P.; Capitanio, J.P.; Cole, S.W. SIV infection decreases sympathetic innervation of primate lymph nodes: The role of neurotrophins. Brain Behav. Immun. 2008, 22, 185–194. [Google Scholar] [CrossRef] [Green Version]
- Marincowitz, C.; Genis, A.; Goswami, N.; De Boever, P.; Nawrot, T.S.; Strijdom, H. Vascular endothelial dysfunction in the wake of HIV and ART. FEBS J. 2019, 286, 1256–1270. [Google Scholar] [CrossRef] [Green Version]
- Fitzgerald, P.J. Serious infection may systemically increase noradrenergic signaling and produce psychological effects. Med. Hypotheses 2020, 139, 109692. [Google Scholar] [CrossRef]
- Bloemker, D.; Mollerus, S.; Gibbert, K.; Dittmer, U.; Del Rey, A.; Schedlowski, M.; Engler, H. Inhibition of catecholamine degradation ameliorates while chemical sympathectomy aggravates the severity of acute Friend retrovirus infection in mice. Brain Behav. Immun. 2016, 54, 252–259. [Google Scholar] [CrossRef]
- Gonzalez-Duarte, A.; Cikurel, K.; Simpson, D.M. Managing HIV peripheral neuropathy. Curr. HIV/AIDS Rep. 2007, 4, 114–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aziz, D.C.; Hanna, Z.; Jolicoeur, P. Severe immunodeficiency disease induced by a defective murine leukaemia virus. Nature 1989, 338, 505–508. [Google Scholar] [CrossRef] [PubMed]
- Jolicoeur, P. Murine acquired immunodeficiency syndrome (MAIDS): An animal model to study the AIDS pathogenesis. FASEB J. 1991, 5, 2398–2405. [Google Scholar] [CrossRef] [Green Version]
- Cao, L.; Butler, M.B.; Tan, L.; Draleau, K.S.; Koh, W.Y. Murine immunodeficiency virus-induced peripheral neuropathy and the associated cytokine responses. J. Immunol. 2012, 189, 3724–3733. [Google Scholar] [CrossRef] [PubMed]
- Liang, B.; Wang, J.Y.; Watson, R.R. Murine AIDS, a key to understanding retrovirus-induced immunodeficiency. Viral Immunol. 1996, 9, 225–239. [Google Scholar] [CrossRef] [PubMed]
- Morse, H.C., III; Chattopadhyay, S.K.; Makino, M.; Fredrickson, T.N.; Hügin, A.W.; Hartley, J.W. Retrovirus-induced immunodeficiency in the mouse: MAIDS as a model for AIDS. AIDS 1992, 6, 607–621. [Google Scholar] [CrossRef] [PubMed]
- Mosier, D.E.; Yetter, R.A.; Morse, H.C., III. Retroviral induction of acute lymphoproliferative disease and profound immunosuppression in adult C57BL/6 mice. J. Exp. Med. 1985, 161, 766–784. [Google Scholar] [CrossRef] [Green Version]
- Hartley, J.W.; Fredrickson, T.N.; Yetter, R.A.; Makino, M.; Morse, H.C., III. Retrovirus-induced murine acquired immunodeficiency syndrome: Natural history of infection and differing susceptibility of inbred mouse strains. J. Virol. 1989, 63, 1223–1231. [Google Scholar] [CrossRef] [Green Version]
- Umemura, M.; Nishimura, H.; Yajima, T.; Wajjwalk, W.; Matsuguchi, T.; Takahashi, M.; Nishiyama, Y.; Makino, M.; Nagai, Y.; Yoshikai, Y. Overexpression of interleukin-15 prevents the development of murine retrovirus-induced acquired immunodeficiency syndrome. FASEB J. 2002, 16, 1755–1763. [Google Scholar] [CrossRef] [Green Version]
- Fredrickson, T.N.; Morse, H.C., III; Yetter, R.A.; Rowe, W.P.; Hartley, J.W.; Pattengale, P.K. Multiparameter analyses of spontaneous nonthymic lymphomas occurring in NFS/N mice congenic for ecotropic murine leukemia viruses. Am. J. Pathol. 1985, 121, 349–360. [Google Scholar]
- Klinken, S.P.; Fredrickson, T.N.; Hartley, J.W.; Yetter, R.A.; Morse, H.C., III. Evolution of B cell lineage lymphomas in mice with a retrovirus-induced immunodeficiency syndrome, MAIDS. J. Immunol. 1988, 140, 1123–1131. [Google Scholar] [PubMed]
- Li, W.; Green, W.R. Murine AIDS requires CD154/CD40L expression by the CD4 T cells that mediate retrovirus-induced disease: Is CD4 T cell receptor ligation needed? Virology 2007, 360, 58–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mosier, D.E.; Yetter, R.A.; Morse, I.H.C., III. Functional T lymphocytes are required for a murine retrovirus-induced immunodeficiency disease (MAIDS). J. Exp. Med. 1987, 165, 1737–1742. [Google Scholar] [CrossRef] [PubMed]
- Klinman, D.M.; Morse, H.C., III. Characteristic of B cell proliferation and action in murine AIDS. J. Immunol. 1989, 142, 1144–1149. [Google Scholar]
- Basham, T.; Rios, C.D.; Holdener, T.; Merigan, T.C. Zidovudine (AZT) reduces virus titer, retards immune dysfunction, and prolongs survival in the LP-BM5 murine induced immunodeficiency model. J. Infect. Dis. 1990, 161, 1006–1009. [Google Scholar] [CrossRef]
- Chattopadhyay, S.K.; Morse, H.C.; Makino, M.; Ruscetti, S.K.; Hartley, J.W. A defective virus is associated with induction of a murine retrovirus-induced immunodeficiency syndrome, MAIDS. Proc. Natl. Acad. Sci. USA 1989, 86, 3862–3866. [Google Scholar] [CrossRef] [Green Version]
- Huang, M.; Simard, C.; Kay, D.G.; Jolicoeur, P. The majority of cells infected with the defective murine AIDS virus belong to the B-cell lineage. J. Virol. 1991, 65, 6562–6571. [Google Scholar] [CrossRef] [Green Version]
- Kim, W.K.; Tang, Y.; Kenny, J.J.; Longo, D.L.; Morse, H.C., III. In murine AIDS, B cells are early targets of defective virus and are required for efficient infection and expression of defective virus in T cells and macrophages. J. Virol. 1994, 68, 6767–6769. [Google Scholar] [CrossRef] [Green Version]
- Itoh, N.; Yonehara, S.; Ishii, A.; Yonehara, M.; Mizushima, S.; Sameshima, M.; Hase, A.; Seto, Y.; Nagata, S. The polypeptide encoded by the cDNA for human cell surface antigen Fas can mediate apoptosis. Cell 1991, 66, 233–243. [Google Scholar] [CrossRef]
- Muralidhar, G.; Koch, S.; Haas, M.; Swain, S.L. CD4 T cells in murine acquired immunodeficiency syndrome: Polyclonal progression to anergy. J. Exp. Med. 1992, 175, 1589–1599. [Google Scholar] [CrossRef] [Green Version]
- Debatin, K.M.; Fahrig, A.F.; Enenkel, S.S.; Kreuz, W.; Brenner, A.; Krammer, P.H. High expression of APO1 (CD95) on T lymphocytes from human immunodeficiency virus-1-infected children. Blood 1994, 83, 3101–3103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobayashi, N.; Hamamoto, Y.; Yamamoto, N.; Ishii, A.; Yonehara, M.; Yonehara, S. Anti-Fas monoclonal antibody is cytocidal to human immunodeficiency virus-infected cells without augmenting viral replication. Proc. Natl. Acad. Sci. USA 1990, 87, 9620–9624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Westendorp, M.O.; Frank, R.; Ochsenbauer, C.; Stricker, K.; Dhein, J.; Walczak, H.; Debatin, K.-M.; Krammer, P.H. Sensitization of T cells to CD95-mediated apoptosis by HIV-1 Tat and gp120. Nature 1995, 375, 497–500. [Google Scholar] [CrossRef] [PubMed]
- Hiromatsu, K.; Aoki, Y.; Makino, M.; Matsumoto, Y.; Mizuochi, T.; Gotoh, Y.; Nomoto, K.; Ogasawara, J.; Nagata, S.; Yoshikai, Y. Increased Fas antigen expression in murine retrovirus-induced immunodeficiency syndrome, MAIDS. Eur. J. Immunol. 1994, 24, 2446–2451. [Google Scholar] [CrossRef]
- Poonia, B.; Pauza, C.D.; Salvato, M.S. Role of the Fas/FasL pathway in HIV or SIV disease. Retrovirology 2009, 6, 91. [Google Scholar] [CrossRef] [Green Version]
- Fraternale, A.; Casabianca, A.; Tonelli, A.; Chiarantini, L.; Brandi, G.; Magnani, M. New drug combinations for the treatment of murine AIDS and macrophage protection. Eur. J. Clin. Investig. 2001, 31, 248–252. [Google Scholar] [CrossRef]
- Rahmouni, S.; Aandahl, E.M.; Trebak, M.; Boniver, J.; Taskén, K.; Moutschen, M. Increased cAMP levels and protein kinase (PKA) type I activation in CD4+ T cells and B cells contribute to retrovirus-induced immunodeficiency of mice (MAIDS): A useful in vivo model for drug testing. FASEB J. 2001, 15, 1466–1468. [Google Scholar] [CrossRef]
- Furukawa, K.; Sasaki, H.; Pollard, R.B.; Suzuki, F. Lanoconazole, a new imidazole antimycotic compound, protects MAIDS mice against encephalitis caused by Cryptococcus neoformans. J. Antimicrob. Chemother. 2000, 46, 443–450. [Google Scholar] [CrossRef] [Green Version]
- Van Rompay, K.K. The use of nonhuman primate models of HIV infection for the evaluation of antiviral strategies. AIDS Res. Hum. Retrovir. 2012, 28, 16–35. [Google Scholar] [CrossRef]
- Thippeshappa, R.; Kimata, J.T.; Kaushal, D. Toward a macaque model of HIV-1 infection: Roadblocks, progress, and future strategies. Front. Microbiol. 2020, 11, 882. [Google Scholar] [CrossRef]
- Sharer, L.R.; Baskin, G.B.; Cho, E.S.; Murphey-Corb, M.; Blumberg, B.M.; Epstein, L.G. Comparison of simian immunodeficiency virus and human immunodeficiency virus encephalitides in the immature host. Ann. Neurol. 1988, 23, S108–S112. [Google Scholar] [CrossRef] [PubMed]
- Williams, K.; Lackner, A.; Mallard, J. Non-human primate models of SIV infection and CNS neuropathology. Curr. Opin. Virol. 2016, 19, 92–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beck, S.E.; Queen, S.E.; Metcalf Pate, K.A.; Mangus, L.M.; Abreu, C.M.; Gama, L.; Witwer, K.W.; Adams, R.J.; Zink, M.C.; Clements, J.E.; et al. An SIV/macaque model targeted to study HIV-associated neurocognitive disorders. J. Neurovirol. 2018, 24, 204–212. [Google Scholar] [CrossRef] [PubMed]
- Mallard, J.; Williams, K.C. Animal models of HIV-associated disease of the central nervous system. Handb. Clin. Neurol. 2018, 152, 41–53. [Google Scholar] [PubMed]
- Bellinger, D.L.; Lorton, D. Autonomic regulation of cellular immune function. Auton. Neurosci. 2014, 182, 15–41. [Google Scholar] [CrossRef]
- Lorton, D.; Bellinger, D.L. Molecular mechanisms underlying β-adrenergic receptor-mediated cross-talk between sympathetic neurons and immune cells. Int. J. Mol. Sci. 2015, 16, 5635–5665. [Google Scholar] [CrossRef] [Green Version]
- Cole, S.W.; Korin, Y.D.; Fahey, J.L.; Zack, J.A. Norepinephrine accelerates HIV replication via protein kinase A-dependent effects on cytokine production. J. Immunol. 1998, 161, 610–616. [Google Scholar]
- Cole, S.W. Psychosocial influences on HIV-1 disease progression: Neural, endocrine, and virologic mechanisms. Psychosom. Med. 2008, 70, 562–568. [Google Scholar] [CrossRef] [Green Version]
- Cole, S.W.; Jamieson, B.D.; Zack, J.A. cAMP externalizes lymphocyte CXCR4: Implications for chemotaxis and HIV infection. J. Immunol. 1999, 162, 1392–1400. [Google Scholar]
- Collado-Hidalgo, A.; Sung, C.; Cole, S. Adrenergic inhibition of innate anti-viral response: PKA blockade of Type I interferon gene transcription mediates catecholamine support for HIV-1 replication. Brain Behav. Immun. 2006, 20, 552–563. [Google Scholar] [CrossRef]
- Cheng, L.; Yu, H.; Li, G.; Li, F.; Ma, J.; Li, J.; Chi, L.; Zhang, L.; Su, L. Type I interferons suppress viral replication but contribute to T cell depletion and dysfunction during chronic HIV-1 infection. JCI Insight 2017, 2, e94366. [Google Scholar] [CrossRef] [Green Version]
- Ivashkiv, L.B.; Donlin, L.T. Regulation of type I interferon responses. Nat. Rev. Immunol. 2014, 14, 36–49. [Google Scholar] [CrossRef] [Green Version]
- Mikulak, J.; Oriolo, R.; Zaghi, E.; Di Vito, C.; Mavilio, D. Natural killer cells in HIV-1 Infection and therapy. AIDS 2017, 31, 2317–2330. [Google Scholar] [CrossRef]
- Flórez-Álvarez, L.; Hernandez, J.C.; Zapata, W. NK Cells in HIV-1 Infection: From Basic Science to Vaccine Strategies. Front. Immunol. 2018, 9, 2290. [Google Scholar] [CrossRef] [PubMed]
- Williams, D.W.; Engle, E.L.; Shirk, E.N.; Queen, S.E.; Gama, L.; Mankowski, J.L.; Zink, M.C.; Clements, J.E. Splenic Damage during SIV Infection: Role of T-Cell Depletion and Macrophage Polarization and Infection. Am. J. Pathol. 2016, 186, 2068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thaney, V.E.; Kaul, M. Type I Interferons in NeuroHIV. Viral Immunol. 2019, 32, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Zang, Y.C.; Halder, J.B.; Samanta, A.K.; Hong, J.; Rivera, V.M.; Zhang, J.Z. Regulation of chemokine receptor CCR5 and production of RANTES and MIP-1alpha by interferon-beta. J. Neuroimmunol. 2001, 112, 174–180. [Google Scholar] [CrossRef]
- Shirazi, Y.; Pitha, P.M. Interferon downregulates CXCR4 (fusin) gene expression in peripheral blood mononuclear cells. J. Hum. Virol. 1998, 1, 69–76. [Google Scholar]
- Cremer, I.; Vieillard, V.; De Maeyer, E. Interferon-beta-induced human immunodeficiency virus resistance in CD34+ human hematopoietic progenitor cells: Correlation with a down-regulation of CCR-5 expression. Virology 1999, 253, 241–249. [Google Scholar] [CrossRef] [Green Version]
- Cremer, I.; Vieillard, V.; De Maeyer, E. Retrovirally mediated IFN-beta transduction of macrophages induces resistance to HIV, correlated with up-regulation of RANTES production and down-regulation of C-C chemokine receptor-5 expression. J. Immunol. 2000, 164, 1582–1587. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.-F.; Tomura, M.; Iwasaki, M.; Ono, S.; Zou, J.-P.; Uno, K.; Shearer, G.M.; Fujiwara, H.; Hamaoka, T. IFN-alpha acts on T-cell receptor-triggered human peripheral leukocytes to up-regulate CCR5 expression on CD4 and CD8 T cells. J. Clin. Immunol. 2001, 21, 402–409. [Google Scholar] [CrossRef] [PubMed]
- Araínga, M.; Edagwa, B.; Mosley, R.L.; Poluektova, L.Y.; Gorantla, S.; Gendelman, H.E. A mature macrophage is a principal HIV-1 cellular reservoir in humanized mice after treatment with long- acting antiretroviral therapy. Retrovirology 2017, 14, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fonteneau, J.F.; Larsson, M.; Beignon, A.S.; McKenna, K.; Dasilva, I.; Amara, A.; Liu, Y.J.; Lifson, J.D.; Littman, D.R.; Bhardwaj, N. Human immunodeficiency virus type 1 activates plasmacytoid dendritic cells and concomitantly induces the bystander maturation of myeloid dendritic cells. J. Virol. 2004, 78, 5223–5232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bukrinsky, M.I.; Stanwick, T.L.; Dempsey, M.P.; Stevenson, M. Quiescent T lymphocytes as an inducible virus reservoir in HIV-1 infection. Science 1991, 254, 423–427. [Google Scholar] [CrossRef] [PubMed]
- Levy, J.A. Pathogenesis of human immunodeficiency virus infection. Microbiol. Rev. 1993, 57, 183–289. [Google Scholar] [CrossRef]
- Ho, D.D.; Neumann, A.U.; Perelson, A.S.; Chen, W.; Leonard, J.M.; Markowitz, M. Rapid turnover of plasma virions and CD4 lymphocytes in HIV-1 infection. Nature 1995, 373, 123–126. [Google Scholar] [CrossRef]
- Mellors, J.W.; Rinaldo, C.R., Jr.; Gupta, P.; White, R.M.; Todd, J.A.; Kingsley, L.A. Prognosis in HIV-1 infection predicted by the quantity of virus in plasma. Science 1996, 272, 1167–1170. [Google Scholar] [CrossRef]
- Perelson, A.S.; Neumann, A.U.; Markowitz, M.; Leonard, J.M.; Ho, D.D. HIV-1 dynamics in vivo: Virion clearance rate, infected cell life-span, and viral generation time. Science 1996, 271, 1582–1586. [Google Scholar] [CrossRef] [Green Version]
- Cole, S.W.; Kemeny, M.E.; Taylor, S.E. Social identity and physical health: Accelerated HIV progression in rejection-sensitive gay men. J. Pers. Soc. Psychol. 1997, 72, 320–336. [Google Scholar] [CrossRef]
- Cole, S.W.; Kemeny, M.E.; Taylor, S.E.; Visscher, B.R.; Fahey, J.L. Accelerated course of Human Immunodeficiency Virus infection in gay men who conceal their homosexual identity. Psychosom. Med. 1996, 58, 219–231. [Google Scholar] [CrossRef] [Green Version]
- Kagan, J.; Reznick, J.S.; Snidman, N. Biological bases of childhood shyness. Science 1988, 240, 167–671. [Google Scholar] [CrossRef] [PubMed]
- Nolan, R.; Gaskill, P.J. The role of catecholamines in HIV neuropathogenesis. Brain Res. 2019, 1702, 54–73. [Google Scholar] [CrossRef] [PubMed]
- Woodham, A.W.; Skeate, J.G.; Sanna, A.M.; Taylor, J.R.; Da Silva, D.M.; Cannon, P.M.; Kast, W.M. Human Immunodeficiency Virus Immune Cell Receptors, Coreceptors, and Cofactors: Implications for Prevention and Treatment. AIDS Patient Care STDS 2016, 30, 291–306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basova, L.; Najera, J.A.; Bortell, N.; Wang, D.; Moya, R.; Lindsey, A.; Semenova, S.; Ellis, R.J.; Marcondes, M.C.G. Dopamine and its receptors play a role in the modulation of CCR5 expression in innate immune cells following exposure to methamphetamine: Implications to HIV infection. PLoS ONE 2018, 13, e0199861. [Google Scholar] [CrossRef] [PubMed]
- Najera, J.A.; Bustamante, E.A.; Bortel, L.N.; Morsey, B.; Fox, H.S.; Ravasi, T.; Marcondes, M.C. Methamphetamine abuse affects gene expression in brain-derived microglia of SIV-infected macaques to enhance inflammation and promote virus targets. BMC Immunol. 2016, 17, 7. [Google Scholar] [CrossRef] [Green Version]
- Nyberg, M.; Suni, J.; Haltia, M. Isolation of human immunodeficiency virus (HIV) at autopsy one to six days postmortem. Am. J. Clin. Pathol. 1990, 94, 422–425. [Google Scholar] [CrossRef]
- Bronte, V.; Pittet Mikael, J. The spleen in local and systemic regulation of immunity. Immunity 2013, 39, 806–818. [Google Scholar] [CrossRef] [Green Version]
- Davies, L.C.; Jenkins, S.J.; Allen, J.E.; Taylor, P.R. Tissue-resident macrophages. Nat. Immunol. 2013, 14, 986–995. [Google Scholar] [CrossRef]
- Den Haan, J.M.; Martinez-Pomares, L. Macrophage heterogeneity in lymphoid tissues. Semin. Immunopathol. 2013, 35, 541–552. [Google Scholar] [CrossRef]
- Fitzgerald-Bocarsly, P.; Dai, J.; Singh, S. Plasmacytoid dendritic cells and type I IFN: 50 years of convergent history. Cytokine Growth Factor Rev. 2008, 19, 3–19. [Google Scholar] [CrossRef] [Green Version]
- Bronnimann, M.P.; Skinner, P.J.; Connick, E. The B-Cell Follicle in HIV Infection: Barrier to a Cure. Front. Immunol. 2018, 9, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cesta, M.F. Normal structure, function, and histology of the spleen. Toxicol. Pathol. 2006, 34, 455–465. [Google Scholar] [CrossRef]
- Nascimbeni, M.; Perié, L.; Chorro, L.; Diocou, S.; Kreitmann, L.; Louis, S.; Garderet, L.; Fabiani, B.; Berger, A.; Schmitz, J.; et al. Plasmacytoid dendritic cells accumulate in spleens from chronically HIV-infected patients but barely participate in interferon-alpha expression. Blood 2009, 113, 6112–6119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schnittman, S.M.; Lane, H.C.; Greenhouse, J.; Justement, J.S.; Baseler, M.; Fauci, A.S. Preferential infection of CD4+ memory T cells by human immunodeficiency virus type 1: Evidence for a role in the selective T-cell functional defects observed in infected individuals. Proc. Natl. Acad. Sci. USA 1990, 87, 6058–6062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doitsh, G.; Greene, W.C. Dissecting how CD4 T cells are lost during HIV infection. Cell Host Microbe 2016, 19, 280–291. [Google Scholar] [CrossRef] [Green Version]
- Diaz, L.K.; Murphy, R.L.; Phair, J.P.; Variakojis, D. The AIDS autopsy spleen: A comparison of the pre-anti-retroviral and highly active anti-retroviral therapy eras. Mod. Pathol. 2002, 15, 406–412. [Google Scholar] [CrossRef] [Green Version]
- Costiniuk, C.T.; Jenabian, M.-A. Cell-to-cell transfer of HIV infection: Implications for HIV viral persistence. J. Gen. Virol. 2014, 95, 2346–2355. [Google Scholar] [CrossRef]
- Alexaki, A.; Liu, Y.; Wigdahl, B. Cellular reservoirs of HIV-1 and their role in viral persistence. Curr. HIV Res. 2008, 6, 388. [Google Scholar] [CrossRef] [Green Version]
- Carr, J.; Hocking, H.; Li, P.; Burrell, C. Rapid and efficient cell-to-cell transmission of human immunodeficiency virus infection from monocyte-derived macrophages to peripheral blood lymphocytes. Virology 1999, 265, 319–329. [Google Scholar] [CrossRef]
- Gratton, S.; Cheynler, R.; Dumaurier, M.J.; Oksenhendler, E.; Wain-Hobson, S. Highly restricted spread of HIV-1 and multiply infected cells within splenic germinal centers. Proc. Natl. Acad. Sci. USA 2000, 97, 14566–14571. [Google Scholar] [CrossRef] [Green Version]
- McElrath, M.J.; Steinman, R.M.; Cohn, Z.A. Latent HIV-1 infection in enriched populations of blood monocytes and T cells from seropositive patients. J. Clin. Investig. 1991, 87, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Falk, S.; Stutte, H.J. The spleen in HIV infection: Morphological evidence of HIV-associated macrophage dysfunction. Res. Virol. 1990, 141, 161–169. [Google Scholar] [CrossRef]
- Okoye, A.A.; Picker, L.J. CD4+ T-cell depletion in HIV infection: Mechanisms of immunological failure. Immunol. Rev. 2013, 254, 54–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nie, C.; Sato, K.; Misawa, N.; Kitayama, H.; Fujino, H.; Hiramatsu, H.; Heike, T.; Nakahata, T.; Tanaka, Y.; Ito, M.; et al. Selective infection of CD4+ effector memory T lymphocytes leads to preferential depletion of memory T lymphocytes in R5 HIV-1-infected humanized NOD/SCID/IL-2Rgnull mice. Virology 2009, 394, 64–72. [Google Scholar] [CrossRef] [Green Version]
- Mattapallil, J.J.; Douek, D.C.; Hill, B.; Nishimura, Y.; Martin, M.; Roederer, M. Massive infection and loss of memory CD4+ T cells in multiple tissues during acute SIV infection. Nature 2005, 434, 1093–1097. [Google Scholar] [CrossRef]
- Costiniuk, C.T.; Jenabian, M.-A. HIV reservoir dynamics in the face of highly active antiretroviral therapy. AIDS Patient Care STDs 2015, 29, 55–68. [Google Scholar] [CrossRef]
- Keele, B.F.; Jones, J.H.; Terio, K.A.; Estes, J.D.; Rudicell, R.S.; Wilson, M.L.; Li, Y.; Learn, G.H.; Beasley, T.M.; Schumacher-Stankey, J.; et al. Increased mortality and AIDS-like immunopathology in wild chimpanzees infected with SIVcpz. Nature 2009, 460, 515–519. [Google Scholar] [CrossRef] [Green Version]
- Brandtzaeg, P.; Flavell, D.J.; Fagerhol, M.K. Mac 387 antibody and detection of formalin resistant myelomonocytic L1 antigen. J. Clin. Pathol. 1988, 41, 963–970. [Google Scholar] [CrossRef] [Green Version]
- Soulas, C.; Conerly, C.; Kim, W.-K.; Burdo, T.H.; Alvarez, X.; Lackner, A.A.; Williams, K.C. Recently infiltrating MAC387+ monocytes/macrophages. Am. J. Pathol. 2011, 178, 2121–2135. [Google Scholar] [CrossRef] [Green Version]
- Lichtnekert, J.; Kawakami, T.; Parks, W.C.; Duffield, J.S. Changes in macrophage phenotype as the immune response evolves. Curr. Opin. Pharmacol. 2013, 13, 555–564. [Google Scholar] [CrossRef] [Green Version]
- George, M.P.; Brower, A.; Kling, H.; Shipley, T.; Kristoff, J.; Reinhart, T.A.; Murphey-Corb, M.; Gladwin, M.T.; Champion, H.C.; Morris, A.; et al. Pulmonary vascular lesions are common in SIV- and SHIV-env-infected macaques. AIDS Res. Hum. Retrovir. 2011, 27, 103–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chalifoux, L.V.; Simon, M.A.; Pauley, D.R.; MacKey, J.J.; Wyand, M.S.; Ringler, D.J. Arteriopathy in macaques infected with simian immunodeficiency virus. Lab. Investig. 1992, 67, 338–349. [Google Scholar] [PubMed]
- Varešlija, D.; Tipton, K.F.; Davey, G.P.; McDonald, A.G. 6-Hydroxydopamine: A far from simple neurotoxin. J. Neural. Transm. 2020, 127, 213–230. [Google Scholar] [CrossRef]
- Lorton, D.; Lubahn, C.; Sweeney, S.; Major, A.; Lindquist, C.A.; Schaller, J.; Washington, C.; Bellinger, D.L. Differences in the injury/sprouting response of splenic noradrenergic nerves in Lewis rats with adjuvant-induced arthritis compared with rats treated with 6-hydroxydopamine. Brain Behav. Immun. 2009, 23, 276–285. [Google Scholar] [CrossRef]
- ThyagaRajan, S.; Felten, D.L. Modulation of neuroendocrine--immune signaling by L-deprenyl and L-desmethyldeprenyl in aging and mammary cancer. Mech. Ageing Dev. 2002, 123, 1065–1079. [Google Scholar] [CrossRef]
- Bai, Y.; Wang, T.; Wang, Z.; Cao, J.; Dong, Y.; Chen, Y. Effect of sympathetic nerves on proliferation of splenic lymphocytes and antioxidant function of maternal spleen in early pregnant mice. Anat. Rec. 2011, 294, 875–882. [Google Scholar] [CrossRef] [PubMed]
- Abbas, W.; Tariq, M.; Iqbal, M.; Kumar, A.; Herbein, G. Eradication of HIV-1 from the macrophage reservoir: An uncertain goal? Viruses 2015, 7, 1578–1598. [Google Scholar] [CrossRef] [Green Version]
- Hernandez-Vargas, E.A.; Middleton, R.H. Modeling the three stages in HIV infection. J. Theor. Biol. 2013, 320, 33–40. [Google Scholar] [CrossRef] [Green Version]
- McIntosh, R.C.; Hurwitz, B.E.; Antoni, M.; Gonzalez, A.; Seay, J.; Schneiderman, N. The ABCs of Trait Anger, Psychological Distress, and Disease Severity in HIV. Ann. Behav. Med. 2015, 49, 420–433. [Google Scholar] [CrossRef] [Green Version]
- Guerrero-Martin, S.M.; Rubin, L.H.; McGee, K.M.; Shirk, E.N.; Queen, S.E.; Li, M.; Bullock, B.; Carlson, B.W.; Adams, R.J.; Gama, L.; et al. Psychosocial Stress Alters the Immune Response and Results in Higher Viral Load During Acute SIV Infection in a Pigtailed Macaque Model of HIV. J. Infect. Dis. 2021, 10, jiab252. [Google Scholar]
- Buechler, C.; Eisinger, K.; Krautbauer, S. Diagnostic and prognostic potential of the macrophage specific receptor CD163 in inflammatory diseases. Inflamm. Allergy Drug Targets 2013, 12, 391–402. [Google Scholar] [CrossRef] [PubMed]
- Gordon, S. Homeostasis: A scavenger receptor for haemoglobin. Curr. Biol. 2001, 11, R399–R401. [Google Scholar] [CrossRef] [Green Version]
- Högger, P.; Dreier, J.; Droste, A.; Buck, F.; Sorg, C. Identification of the integral membrane protein RM3/1 on human monocytes as a glucocorticoid-inducible member of the scavenger receptor cysteine-rich family (CD163). J. Immunol. 1998, 161, 1883–1890. [Google Scholar] [PubMed]
- Holness, C.; Simmons, D. Molecular cloning of CD68, a human macrophage marker related to lysosomal glycoproteins. Blood 1993, 81, 1607–1613. [Google Scholar] [CrossRef] [Green Version]
- Ito, S.; Ansari, P.; Sakatsume, M.; Dickensheets, H.; Vazquez, N.; Donnelly, R.P.; Larner, A.C.; Finbloom, D.S. Interleukin-10 inhibits expression of both interferon ¦Á- and interferon ¦Ã-induced genes by suppressing tyrosine phosphorylation of STAT1. Blood 1999, 93, 1456–1463. [Google Scholar] [CrossRef]
- Witwer, K.W.; Gama, L.; Li, M.; Bartizal, C.M.; Queen, S.E.; Varrone, J.J.; Brice, A.K.; Graham, D.R.; Tarwater, P.M.; Mankowski, J.L.; et al. Coordinated regulation of SIV replication and immune responses in the CNS. PLoS ONE 2009, 4, e8129. [Google Scholar] [CrossRef]
- Zaritsky, L.A.; Dery, A.; Leong, W.Y.; Gama, L.; Clements, J.E. Tissue-specific interferon alpha subtype response to SIV infection in brain, spleen, and lung. J. Interferon Cytokine Res. 2013, 33, 24–33. [Google Scholar] [CrossRef] [Green Version]
- Qin, H.; Holdbrooks, A.T.; Liu, Y.; Reynolds, S.L.; Yanagisawa, L.L.; Benveniste, E.N. SOCS3 deficiency promotes M1 macrophage polarization and inflammation. J. Immunol. 2012, 189, 3439–3448. [Google Scholar] [CrossRef] [Green Version]
- Vasilescu, A.; Heath, S.C.; Ivanova, R.; Hendel, H.; Do, H.; Mazoyer, A.; Khadivpour, E.; Goutalier, F.X.; Khalili, K.; Rappaport, J.; et al. Genomic analysis of Th1-Th2 cytokine genes in an AIDS cohort: Identification of IL4 and IL10 haplotypes associated with the disease progression. Genes Immun. 2003, 4, 441–449. [Google Scholar] [CrossRef] [Green Version]
- Becker, Y. The changes in the T helper 1 (Th1) and T helper 2 (Th2) cytokine balance during HIV-1 infection are indicative of an allergic response to viral proteins that may be reversed by Th2 cytokine inhibitors and immune response modifiers—A review and hypothesis. Virus Genes 2004, 28, 5–18. [Google Scholar] [CrossRef]
- Cassol, E.; Cassetta, L.; Rizzi, C.; Alfano, M.; Poli, G. M1 and M2a Polarization of human monocyte-derived macrophages inhibits HIV1 replication by distinct mechanisms. J. Immunol. 2009, 182, 6237–6246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cassol, E.; Cassetta, L.; Alfano, M.; Poli, G. Macrophage polarization and HIV-1 infection. J. Leukoc. Biol. 2010, 87, 599–608. [Google Scholar] [CrossRef] [PubMed]
- Ganor, Y.; Real, F.; Sennepin, A.C.; Dutertre, A.; Prevedel, L.; Xu, L.; Tudor, D.; Charmeteau, B.; Couedel-Courteille, A.; Marion, S.; et al. HIV-1 reservoirs in urethral macrophages of patients under suppressive antiretroviral therapy. Nat. Microbiol. 2019, 1, 633–644. [Google Scholar] [CrossRef] [PubMed]
- Tuluc, F.; Meshki, J.; Spitsin, S.; Douglas, S.D. HIV infection of macrophages is enhanced in the presence of increased expression of CD163 induced by substance P. J. Leukoc. Biol. 2014, 96, 143–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nowlin, B.T.; Burdo, T.H.; Midkiff, C.C.; Salemi, M.; Alvarez, X.; Williams, K. C: SIV encephalitis lesions are composed of CD163+ macrophages present in the central nervous system during early SIV infection and SIV-positive macrophages recruited terminally with AIDS. Am. J. Pathol. 2015, 185, 1649–1665. [Google Scholar] [CrossRef] [PubMed]
- Lim, H.; Kim, K.C.; Son, J.; Shin, Y.; Yoon, C.H.; Kang, C.; Choi, B.S. Synergistic reactivation of latent HIV-1 provirus by PKA activator dibutyryl-cAMP in combination with an HDAC inhibitor. Virus Res. 2017, 227, 1–5. [Google Scholar] [CrossRef]
- Nokta, M.A.; Pollard, R.B. Human immunodeficiency virus replication: Modulation by cellular levels of cAMP. AIDS Res. Hum. Retrovir. 1992, 8, 1255–1261. [Google Scholar] [CrossRef]
- Bellinger, D.L.; Lorton, D. Sympathetic Nerve Hyperactivity in the Spleen: Causal for Nonpathogenic-Driven Chronic Immune-Mediated Inflammatory Diseases (IMIDs)? Int. J. Mol. Sci. 2018, 19, 1188. [Google Scholar] [CrossRef] [Green Version]
- Herbein, G.; Varin, A. The macrophage in HIV-1 infection: From activation to deactivation. Retrovirology 2010, 7, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Swirski, F.K.; Nahrendorf, M.; Etzrodt, M.; Wildgruber, M.; Cortez-Retamozo, V.; Panizzi, P.; Figueiredo, J.-L.; Kohler, R.H.; Chudnovskiy, A.; Waterman, P.; et al. Identification of splenic reservoir monocytes and their deployment to inflammatory sites. Science 2009, 325, 612–616. [Google Scholar] [CrossRef] [Green Version]
- Van der Laan, A.M.; ter Horst, E.N.; Delewi, R.; Begieneman, M.P.V.; Krijnen, P.A.J.; Hirsch, A.; Lavaei, M.; Nahrendorf, M.; Horrevoets, A.J.; Niessen, H.W.M.; et al. Monocyte subset accumulation in the human heart following acute myocardial infarction and the role of the spleen as monocyte reservoir. Eur. Heart J. 2014, 35, 376–385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Otani, I.; Mori, K.; Sata, T.; Terao, K.; Doi, K.; Akari, H.; Yoshikawa, Y. Accumulation of MAC387+ macrophages in paracortical areas of lymph nodes in rhesus monkeys acutely infected with simian immunodeficiency virus. Microbes Infect. 1999, 1, 977–985. [Google Scholar] [CrossRef]
- Lakritz, J.R.; Bodair, A.; Shah, N.; O’Donnell, R.; Polydefkis, M.J.; Miller, A.D.; Burdo, T.H. Monocyte traffic, dorsal root ganglion histopathology, and loss of intraepidermal nerve fiber density in SIV peripheral neuropathy. Am. J. Pathol. 2015, 185, 1912–1923. [Google Scholar] [CrossRef] [PubMed]
- Williams, D.W.; Eugenin, E.A.; Calderon, T.M.; Berman, J.W. Monocyte maturation, HIV susceptibility, and transmigration across the blood brain barrier are critical in HIV neuropathogenesis. J. Leukoc. Biol. 2012, 91, 401–415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Connick, E.; Folkvord, J.M.; Lind, K.T.; Rakasz, E.G.; Miles, B.; Wilson, N.A.; Santiago, M.L.; Schmitt, K.; Stephens, E.B.; Kim, H.O. Compartmentalization of simian immunodeficiency virus replication within secondary lymphoid tissues of rhesus macaques is linked to disease stage and inversely related to localization of virus-specific CTL. J. Immunol. 2014, 193, 5613–5625. [Google Scholar] [CrossRef] [PubMed]
- Rainho, J.N.; Martins, M.A.; Cunyat, F.; Watkins, I.T.; Watkins, D.I.; Stevenson, M. Nef is dispensable for resistance of SIV-infected macrophages to CD8+ T cell killing. J. Virol. 2015, 89, 10625–10636. [Google Scholar] [CrossRef] [Green Version]
- Esiri, M.M.; Morris, C.S.; Millard, P.R. Sensory and sympathetic ganglia in HIV-1 infection: Immunocytochemical demonstration of HIV-1 viral antigens, increased MHC class II antigen expression and mild reactive inflammation. J. Neurol. Sci. 1993, 114, 178–187. [Google Scholar] [CrossRef]
- Ensoli, F.; Fiorelli, V.; DeCristofaro, M.; Santini Muratori, D.; Novi, A.; Vannelli, B.; Thiele, C.J.; Luzi, G.; Aiuti, F. Inflammatory cytokines and HIV-1-associated neurodegeneration: Oncostatin-M produced by mononuclear cells from HIV-1-infected individuals induces apoptosis of primary neurons. J. Immunol. 1999, 162, 6268–6277. [Google Scholar]
- Geffin, R.; Martinez, R.; de Las Pozas, A.; Issac, B.; McCarthy, M. Apolipoprotein E4 Suppresses Neuronal-Specific Gene Expression in Maturing Neuronal Progenitor Cells Exposed to HIV. J. Neuroimmune Pharmacol. 2017, 12, 462–483. [Google Scholar] [CrossRef] [Green Version]
- Muema, D.M.; Akilimali, N.A.; Ndumnego, O.C.; Rasehlo, S.S.; Durgiah, R.; Ojwach, D.; Ismail, N.; Dong, M.; Moodley, A.; Dong, K.L.; et al. Association between the cytokine storm, immune cell dynamics, and viral replicative capacity in hyperacute HIV infection. BMC Med. 2020, 18, 81. [Google Scholar] [CrossRef] [Green Version]
- Kaul, M.; Garden, G.A.; Lipton, S.A. Pathways to neuronal injury and apoptosis in HIV-associated dementia. Nature 2001, 410, 988–994. [Google Scholar] [CrossRef] [PubMed]
- Orandle, M.S.; Veazey, R.S.; Lackner, A.A. Enteric ganglionitis in rhesus macaques infected with simian immunodeficiency virus. J. Virol. 2007, 81, 6265–6275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Triantafilou, K.; Ward, C.J.K.; Czubala, M.; Ferris, R.G.; Koppe, E.; Haffner, C.; Piguet, V.; Patel, V.K.; Amrine-Madsen, H.; Modis, L.K.; et al. Differential recognition of HIV-stimulated IL-1β and IL-18 secretion through NLR and NAIP signalling in monocyte-derived macrophages. PLoS Pathog. 2021, 17, e1009417. [Google Scholar] [CrossRef] [PubMed]
- Robinson-Papp, J.; Nmashie, A.; Pedowitz, E.; Benn, E.K.T.; George, M.C.; Sharma, S.; Murray, J.; Machac, J.; Heiba, S.; Mehandru, S.; et al. Vagal dysfunction and small intestinal bacterial overgrowth: Novel pathways to chronic inflammation in HIV. AIDS 2018, 32, 1147–1156. [Google Scholar] [CrossRef]
- Vida, G.; Peña, G.; Deitch, E.A.; Ulloa, L. α7-cholinergic receptor mediates vagal induction of splenic norepinephrine. J. Immunol. 2011, 186, 4340–4346. [Google Scholar] [CrossRef] [Green Version]
- Enes, J.; Haburčák, M.; Sona, S.; Gerard, N.; Mitchell, A.C.; Fu, W.; Birren, S.J. Satellite glial cells modulate cholinergic transmission between sympathetic neurons. PLoS ONE 2020, 15, e0218643. [Google Scholar] [CrossRef]
- Zauli, G.; Marchisio, M.; Bertagnolo, V.; Celeghini, C.; Capitani, S. Hiv-1 tat protein suppresses the nerve growth-factor (ngf)-mediated differentiation of PC12 rat pheochromocytoma cell-line. Oncol. Rep. 1994, 1, 773–777. [Google Scholar] [CrossRef]
- Quizon, P.M.; Sun, W.L.; Yuan, Y.; Midde, N.M.; Zhan, C.G.; Zhu, J. Molecular mechanism: The human dopamine transporter histidine 547 regulates basal and HIV-1 Tat protein-inhibited dopamine transport. Sci. Rep. 2016, 6, 39–48. [Google Scholar] [CrossRef]
- Prehaud, C.; Megret, F.; Lafage, M.; Lafon, M. Virus infection switches TLR-3-positive human neurons to become strong producers of beta interferon. J. Virol. 2005, 79, 12893–12904. [Google Scholar] [CrossRef] [Green Version]
- Posch, W.; Bermejo-Jambrina, M.; Steger, M.; Witting, C.; Diem, G.; Hörtnagl, P.; Hackl, H.; Lass-Flörl, C.; Huber, L.A.; Geijtenbeek, T.B.H.; et al. Complement Potentiates Immune Sensing of HIV-1 and Early Type I Interferon Responses. mBio 2021, 12, e0240821. [Google Scholar] [CrossRef]
- Wekerle, H. Planting and pruning in the brain: MHC antigens involved in synaptic plasticity? Proc. Natl. Acad. Sci. USA 2005, 102, 3–4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neumann, H.; Schmidt, H.; Cavalie, A.; Jenne, D.; Wekerle, H. Major histocompatibility complex (MHC) class I gene expression in single neurons of the central nervous system: Differential regulation by interferon (IFN)-gamma and tumor necrosis factor (TNF)-alpha. J. Exp. Med. 1997, 185, 305–316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Redwine, J.M.; Buchmeier, M.J.; Evans, C.F. In vivo expression of major histocompatibility complex molecules on oligodendrocytes and neurons during viral infection. Am. J. Pathol. 2001, 159, 1219–1224. [Google Scholar] [CrossRef] [Green Version]
- Petito, C.K.; Torres-Munoz, J.E.; Zielger, F.; McCarthy, M. Brain CD8+ and cytotoxic T lymphocytes are associated with, and may be specific for, human immunodeficiency virus type 1 encephalitis in patients with acquired immunodeficiency syndrome. J. Neurovirol. 2006, 12, 272–283. [Google Scholar] [CrossRef] [PubMed]
- Lorton, D.; Hewitt, D.; Bellinger, D.L.; Felten, S.Y.; Felten, D.L. Noradrenergic reinnervation of the rat spleen following chemical sympathectomy with 6-hydroxydopamine: Pattern and time course of reinnervation. Brain Behav. Immun. 1990, 4, 198–222. [Google Scholar] [CrossRef]
- Peruzzi, F.; Gordon, J.; Darbinian, N.; Amini, S. Tat-induced deregulation of neuronal differentiation and survival by nerve growth factor pathway. J. Neurovirol. 2002, 8 (Suppl. S2), 91–96. [Google Scholar] [CrossRef]
- Schiavoni, I.; Muratori, C.; Piacentini, V.; Giammarioli, A.M.; Federico, M. The HIV-1 Nef protein: How an AIDS pathogenetic factor turns to a tool for combating AIDS. Curr. Drug Targets Immune Endocr. Metabol. Disord. 2004, 4, 19–27. [Google Scholar] [CrossRef]
- Rivera-Ortiz, J.; Pla-Tenorio, J.; Cruz, M.L.; Colon, K.; Perez-Morales, J.; Rodriguez, J.A., Jr.; Martinez-Sicari, J.; Noel, R.J., Jr. Blockade of beta adrenergic receptors protects the blood brain barrier and reduces systemic pathology caused by HIV-1 Nef protein. PLoS ONE 2021, 16, e0259446. [Google Scholar] [CrossRef]


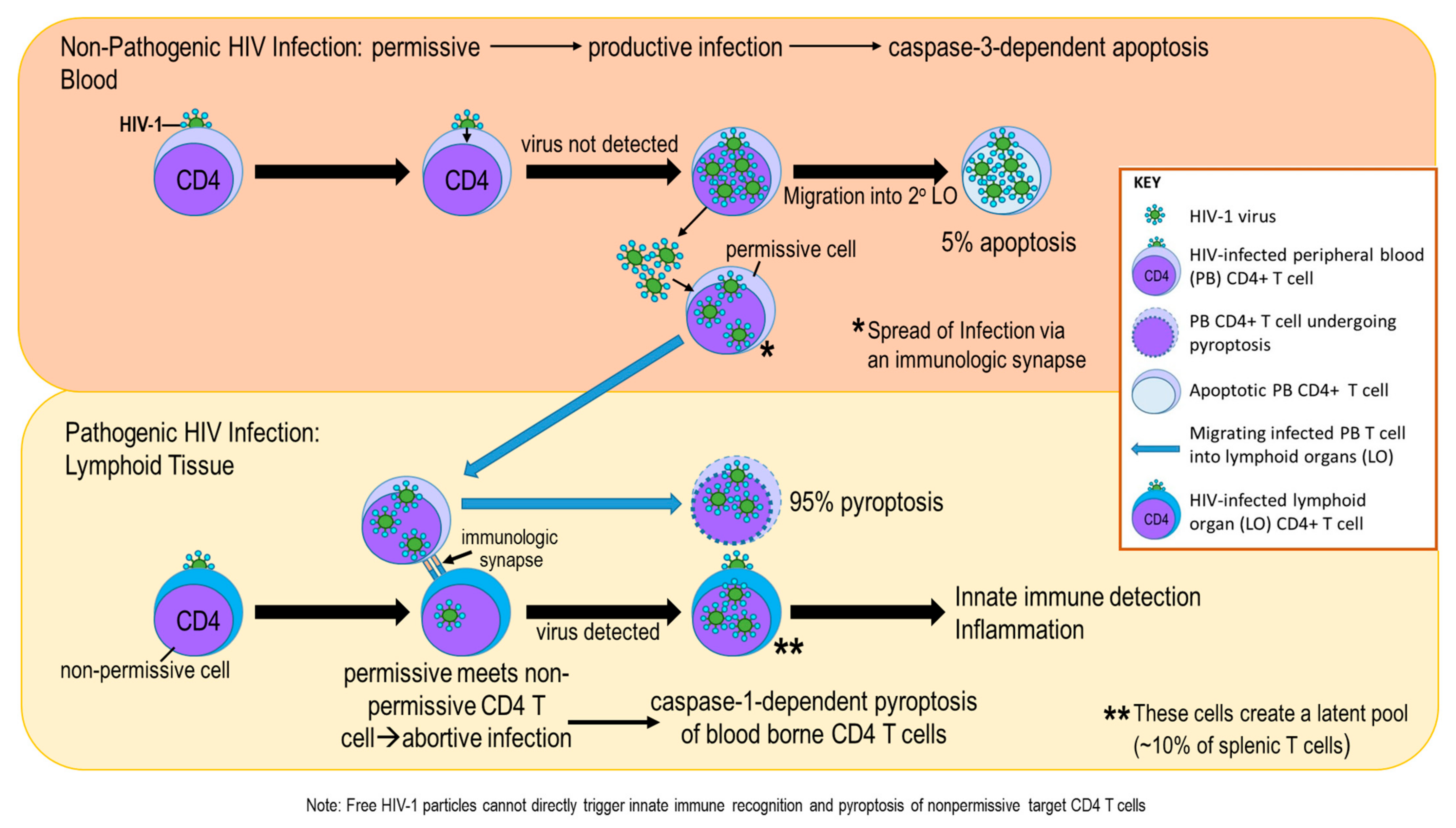
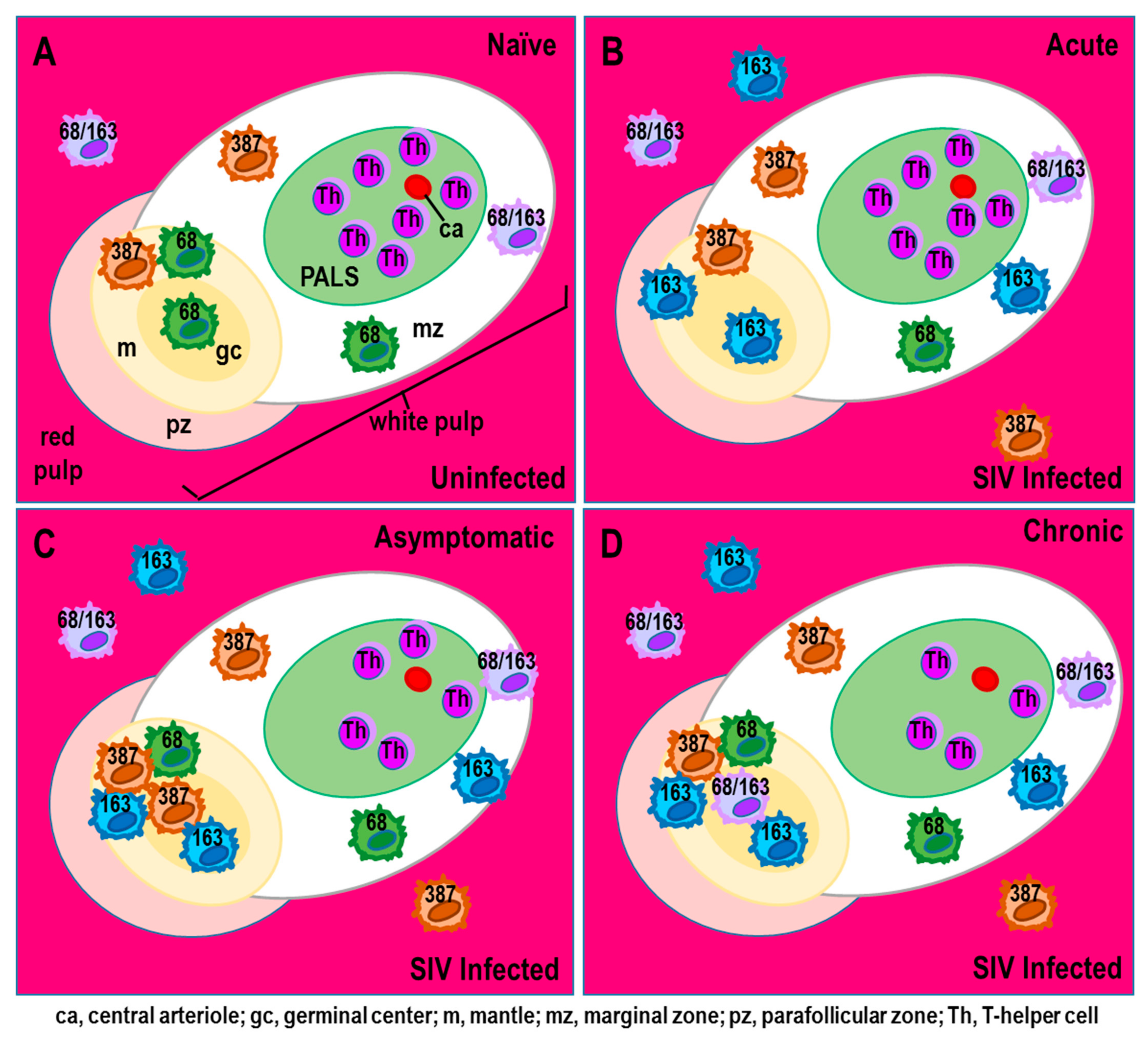
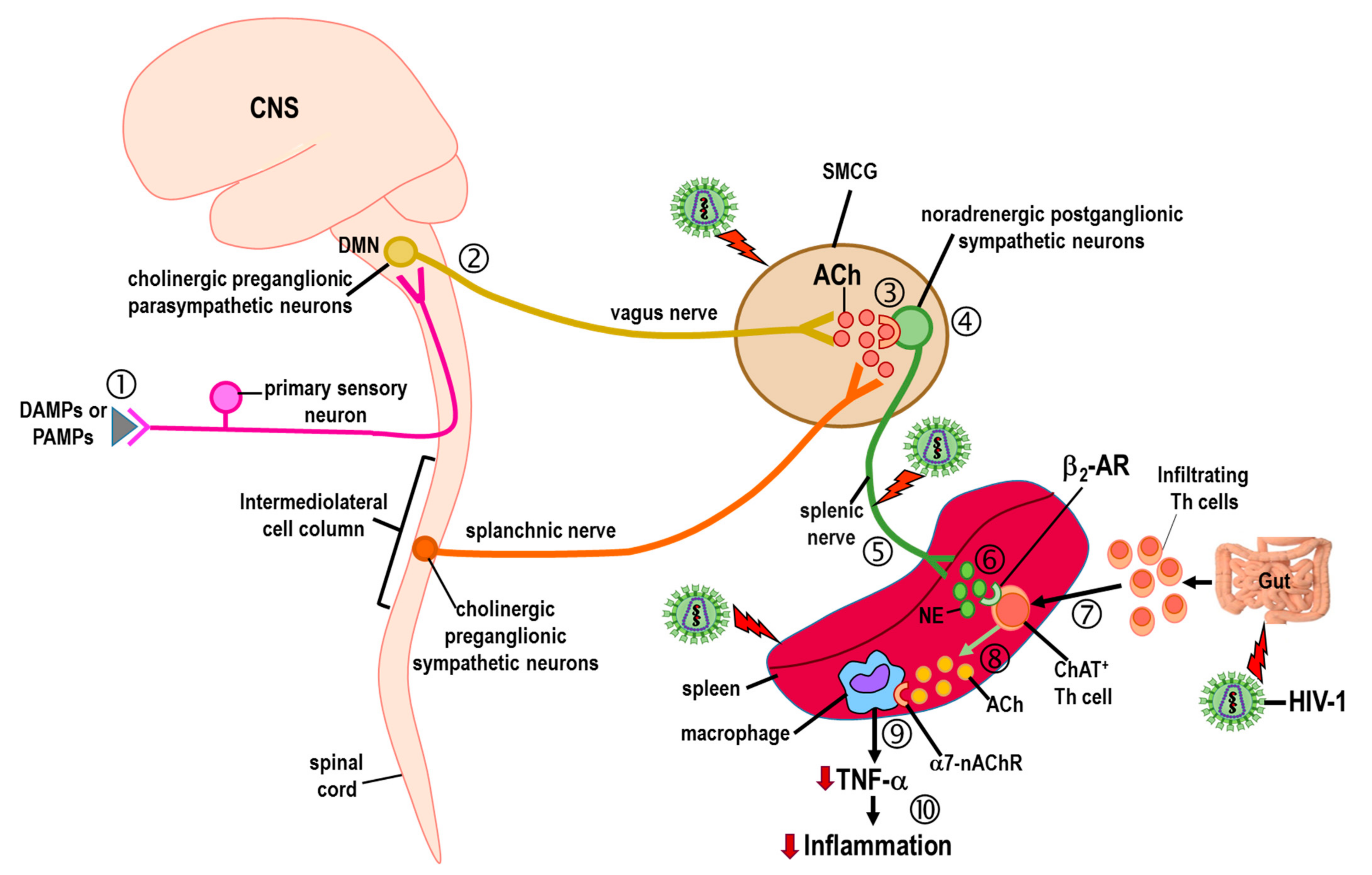
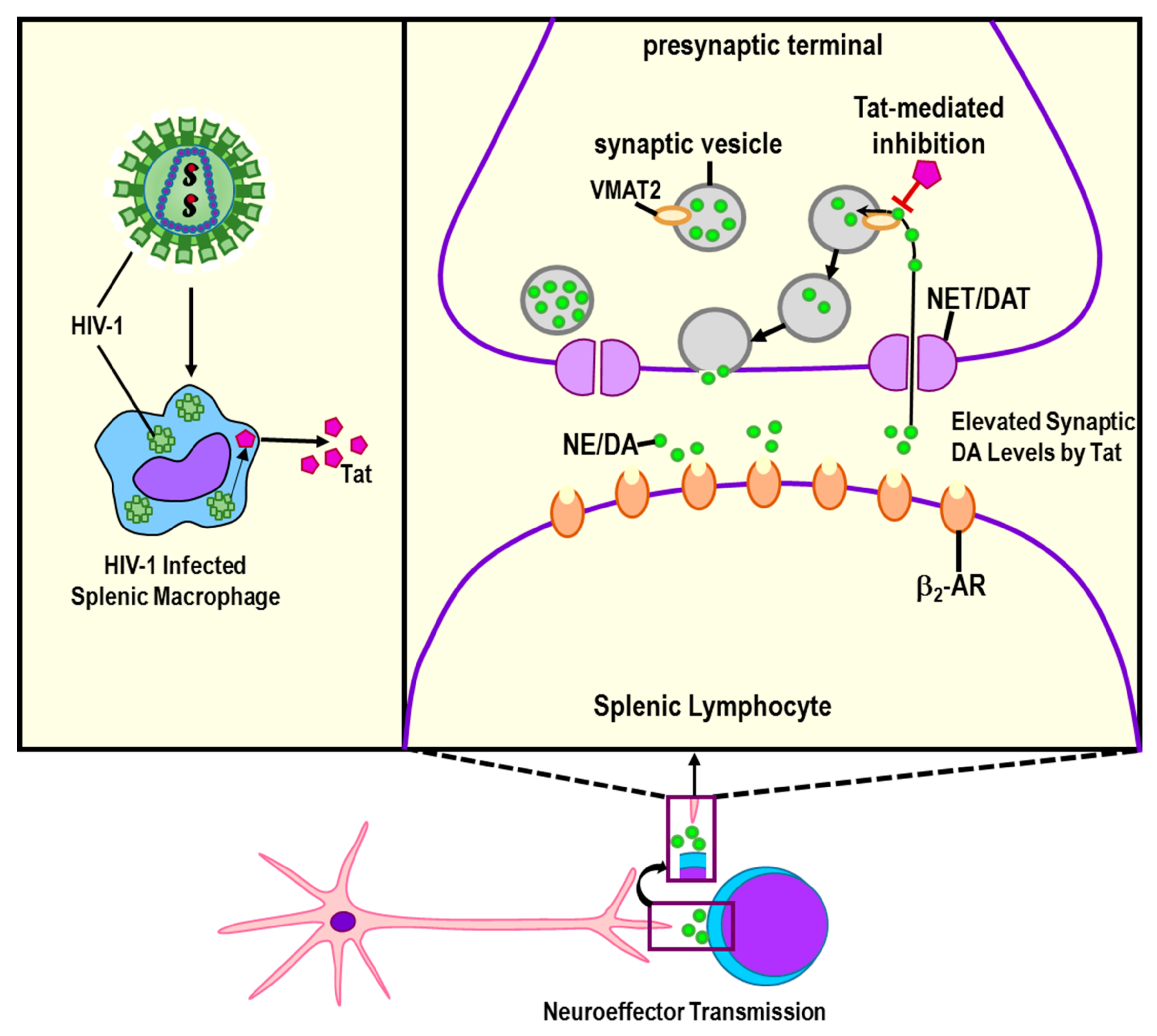
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bellinger, D.L.; Lorton, D. Sympathetic Nerves and Innate Immune System in the Spleen: Implications of Impairment in HIV-1 and Relevant Models. Cells 2022, 11, 673. https://doi.org/10.3390/cells11040673
Bellinger DL, Lorton D. Sympathetic Nerves and Innate Immune System in the Spleen: Implications of Impairment in HIV-1 and Relevant Models. Cells. 2022; 11(4):673. https://doi.org/10.3390/cells11040673
Chicago/Turabian StyleBellinger, Denise L., and Dianne Lorton. 2022. "Sympathetic Nerves and Innate Immune System in the Spleen: Implications of Impairment in HIV-1 and Relevant Models" Cells 11, no. 4: 673. https://doi.org/10.3390/cells11040673
APA StyleBellinger, D. L., & Lorton, D. (2022). Sympathetic Nerves and Innate Immune System in the Spleen: Implications of Impairment in HIV-1 and Relevant Models. Cells, 11(4), 673. https://doi.org/10.3390/cells11040673