
New Developments in T Cell Immunometabolism and Implications for Cancer Immunotherapy


Abstract
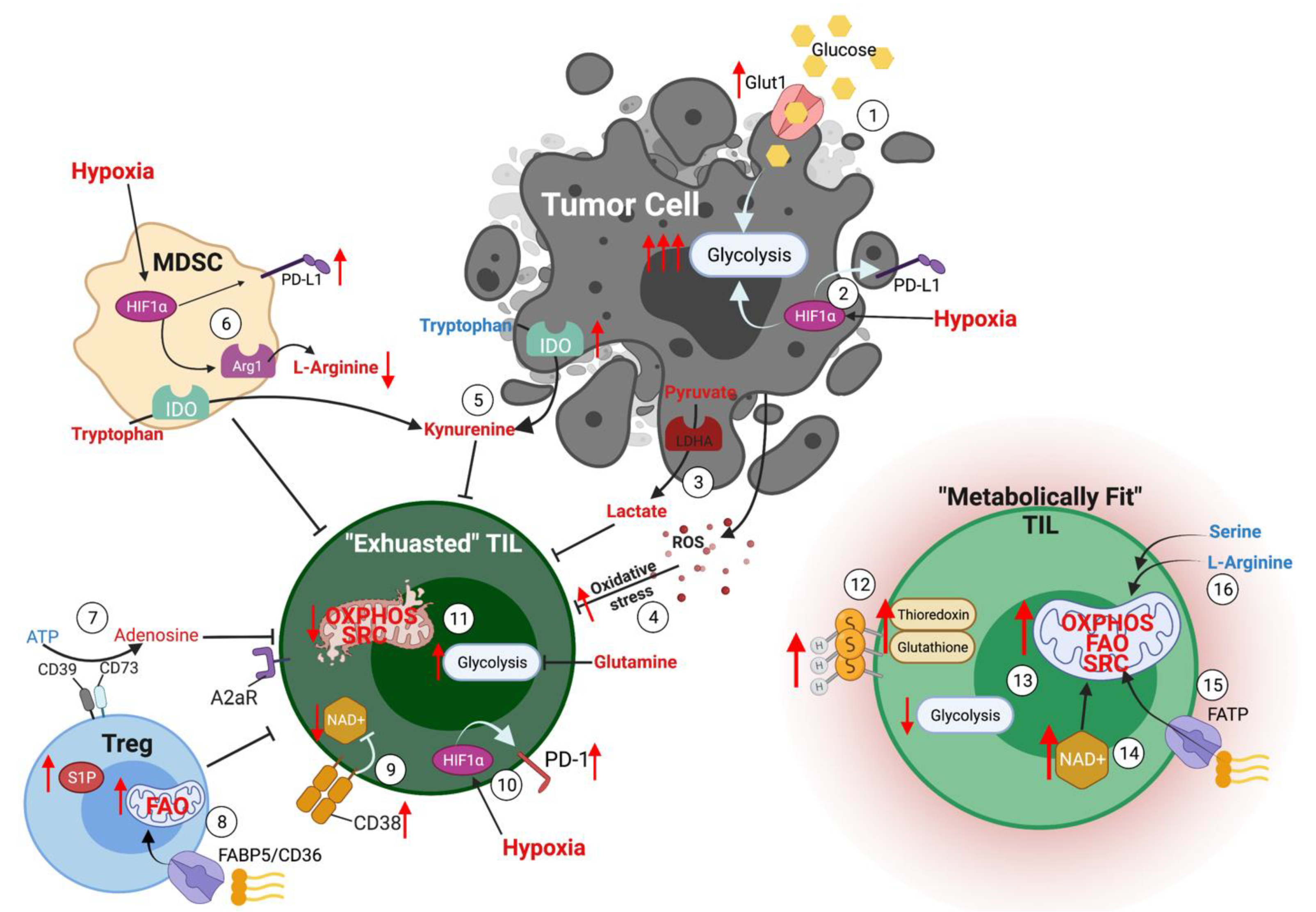
:1. Introduction
2. Metabolic Pathways Influencing Antitumor Immune Function
2.1. Fuel Sources
2.2. Oxidative Phosphorylation and Glycolysis
2.3. Non-Metabolic Functions of Glycolysis-Associated Enzymes
3. Alterations of Metabolic Pathways in the Tumor Microenvironment
3.1. Hypoxia in the TME
3.2. Immunosuppressive Metabolites in the TME
3.3. Metabolic Competition in the TME
3.4. Organoid Methods for Studying the TME
3.5. Strategies to Restore Metabolic Pathways
4. Metabolic Preferences of T Cell Subsets
4.1. Th17 T Cells
4.2. Th1/17 Hybrid T Cells
4.3. IL-9-Secreting T Cells
4.4. Regulatory T Cells
5. Influence of Co-Stimulation on T Cell Metabolism
6. Cytokine Signaling in Dictating T Cell Metabolism
6.1. IL-15
6.2. IL-7
6.3. IL-12
6.4. IL-21
6.5. IL-10
7. Epigenetics
7.1. Histone Acetylation
7.2. Histone Methylation
7.3. Metabolic Intermediates Involved in Epigenetics
8. Clinical Trials Targeting Immunometabolism
9. Conclusions and Future Directions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Old, L.J. Cancer immunology: The search for specificity. Natl. Cancer Inst. Monogr. 1982, 60, 193–209. [Google Scholar] [PubMed]
- Boon, T.; Gajewski, T.F.; Coulie, P.G. From defined human tumor antigens to effective immunization? Immunol. Today 1995, 16, 334–336. [Google Scholar] [CrossRef]
- Cheever, M.A.; Greenberg, P.D.; Irle, C.; Thompson, J.A.; Urdal, D.L.; Mochizuki, D.Y.; Henney, C.S.; Gillis, S. Interleukin 2 administered in vivo induces the growth of cultured T cells in vivo. J. Immunol. 1984, 132, 2259–2265. [Google Scholar] [PubMed]
- Greenberg, P.D.; Cheever, M.A.; Fefer, A. Eradication of disseminated murine leukemia by chemoimmunotherapy with cyclophosphamide and adoptively transferred immune syngeneic Lyt-1+2- lymphocytes. J. Exp. Med. 1981, 154, 952–963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawakami, Y.; Rosenberg, S.A. T-cell recognition of self peptides as tumor rejection antigens. Immunol. Res. 1996, 15, 179–190. [Google Scholar] [CrossRef] [PubMed]
- Mukherji, B.; Chakraborty, N.G. Immunobiology and immunotherapy of melanoma. Curr. Opin. Oncol. 1995, 7, 175–184. [Google Scholar] [CrossRef]
- Rosenberg, S.A. Adoptive immunotherapy of cancer using lymphokine activated killer cells and recombinant interleukin-2. Important Adv. Oncol. 1986, 11, 55–91. [Google Scholar]
- Topalian, S.L.; Hom, S.S.; Kawakami, Y.; Mancini, M.; Schwartzentruber, D.J.; Zakut, R.; Rosenberg, S.A. Recognition of shared melanoma antigens by human tumor-infiltrating lymphocytes. J. Immunother 1992, 12, 203–206. [Google Scholar] [CrossRef]
- Clay, T.M.; Custer, M.C.; Sachs, J.; Hwu, P.; Rosenberg, S.A.; Nishimura, M.I. Efficient transfer of a tumor antigen-reactive TCR to human peripheral blood lymphocytes confers anti-tumor reactivity. J. Immunol. 1999, 163, 507–513. [Google Scholar]
- Duval, L.; Schmidt, H.; Kaltoft, K.; Fode, K.; Jensen, J.J.; Sorensen, S.M.; Nishimura, M.I.; von der Maase, H. Adoptive transfer of allogeneic cytotoxic T lymphocytes equipped with a HLA-A2 restricted MART-1 T-cell receptor: A phase I trial in metastatic melanoma. Clin. Cancer Res. 2006, 12, 1229–1236. [Google Scholar] [CrossRef] [Green Version]
- Morgan, R.A.; Dudley, M.E.; Wunderlich, J.R.; Hughes, M.S.; Yang, J.C.; Sherry, R.M.; Royal, R.E.; Topalian, S.L.; Kammula, U.S.; Restifo, N.P.; et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science 2006, 314, 126–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gross, G.; Eshhar, Z. Therapeutic Potential of T Cell Chimeric Antigen Receptors (CARs) in Cancer Treatment: Counteracting Off-Tumor Toxicities for Safe CAR T Cell Therapy. Annu. Rev. Pharm. Toxicol. 2016, 56, 59–83. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Allison, J.P. Immune checkpoint targeting in cancer therapy: Toward combination strategies with curative potential. Cell 2015, 161, 205–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mellman, I.; Coukos, G.; Dranoff, G. Cancer immunotherapy comes of age. Nature 2011, 480, 480–489. [Google Scholar] [CrossRef] [PubMed]
- Anderson, K.G.; Stromnes, I.M.; Greenberg, P.D. Obstacles Posed by the Tumor Microenvironment to T cell Activity: A Case for Synergistic Therapies. Cancer Cell 2017, 31, 311–325. [Google Scholar] [CrossRef] [Green Version]
- Klein Geltink, R.I.; O’Sullivan, D.; Corrado, M.; Bremser, A.; Buck, M.D.; Buescher, J.M.; Firat, E.; Zhu, X.; Niedermann, G.; Caputa, G.; et al. Mitochondrial Priming by CD28. Cell 2017, 171, 385–397.e311. [Google Scholar] [CrossRef] [Green Version]
- Menk, A.V.; Scharping, N.E.; Rivadeneira, D.B.; Calderon, M.J.; Watson, M.J.; Dunstane, D.; Watkins, S.C.; Delgoffe, G.M. 4-1BB costimulation induces T cell mitochondrial function and biogenesis enabling cancer immunotherapeutic responses. J. Exp. Med. 2018, 215, 1091–1100. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Kurupati, R.; Liu, L.; Zhou, X.Y.; Zhang, G.; Hudaihed, A.; Filisio, F.; Giles-Davis, W.; Xu, X.; Karakousis, G.C.; et al. Enhancing CD8(+) T Cell Fatty Acid Catabolism within a Metabolically Challenging Tumor Microenvironment Increases the Efficacy of Melanoma Immunotherapy. Cancer Cell 2017, 32, 377–391.e379. [Google Scholar] [CrossRef] [Green Version]
- Chowdhury, P.S.; Chamoto, K.; Kumar, A.; Honjo, T. PPAR-Induced Fatty Acid Oxidation in T Cells Increases the Number of Tumor-Reactive CD8(+) T Cells and Facilitates Anti-PD-1 Therapy. Cancer Immunol. Res. 2018, 6, 1375–1387. [Google Scholar] [CrossRef]
- Pilipow, K.; Scamardella, E.; Puccio, S.; Gautam, S.; De Paoli, F.; Mazza, E.M.; De Simone, G.; Polletti, S.; Buccilli, M.; Zanon, V.; et al. Antioxidant metabolism regulates CD8+ T memory stem cell formation and antitumor immunity. JCI Insight 2018, 3, e122299. [Google Scholar] [CrossRef] [Green Version]
- Ma, R.; Ji, T.; Zhang, H.; Dong, W.; Chen, X.; Xu, P.; Chen, D.; Liang, X.; Yin, X.; Liu, Y.; et al. A Pck1-directed glycogen metabolic program regulates formation and maintenance of memory CD8(+) T cells. Nat. Cell Biol. 2018, 20, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Sukumar, M.; Liu, J.; Ji, Y.; Subramanian, M.; Crompton, J.G.; Yu, Z.; Roychoudhuri, R.; Palmer, D.C.; Muranski, P.; Karoly, E.D.; et al. Inhibiting glycolytic metabolism enhances CD8+ T cell memory and antitumor function. J. Clin. Invest. 2013, 123, 4479–4488. [Google Scholar] [CrossRef] [PubMed]
- Ho, P.C.; Bihuniak, J.D.; Macintyre, A.N.; Staron, M.; Liu, X.; Amezquita, R.; Tsui, Y.C.; Cui, G.; Micevic, G.; Perales, J.C.; et al. Phosphoenolpyruvate Is a Metabolic Checkpoint of Anti-tumor T Cell Responses. Cell 2015, 162, 1217–1228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gemta, L.F.; Siska, P.J.; Nelson, M.E.; Gao, X.; Liu, X.; Locasale, J.W.; Yagita, H.; Slingluff, C.L., Jr.; Hoehn, K.L.; Rathmell, J.C.; et al. Impaired enolase 1 glycolytic activity restrains effector functions of tumor-infiltrating CD8(+) T cells. Sci. Immunol. 2019, 4, eaap9520. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.; Chakraborty, P.; Daenthanasanmak, A.; Iamsawat, S.; Andrejeva, G.; Luevano, L.A.; Wolf, M.; Baliga, U.; Krieg, C.; Beeson, C.C.; et al. Targeting PIM Kinase with PD1 Inhibition Improves Immunotherapeutic Antitumor T-cell Response. Clin. Cancer Res. 2019, 25, 1036–1049. [Google Scholar] [CrossRef] [Green Version]
- Van der Windt, G.J.; Everts, B.; Chang, C.H.; Curtis, J.D.; Freitas, T.C.; Amiel, E.; Pearce, E.J.; Pearce, E.L. Mitochondrial respiratory capacity is a critical regulator of CD8+ T cell memory development. Immunity 2012, 36, 68–78. [Google Scholar] [CrossRef] [Green Version]
- O’Sullivan, D.; van der Windt, G.J.; Huang, S.C.; Curtis, J.D.; Chang, C.H.; Buck, M.D.; Qiu, J.; Smith, A.M.; Lam, W.Y.; DiPlato, L.M.; et al. Memory CD8(+) T cells use cell-intrinsic lipolysis to support the metabolic programming necessary for development. Immunity 2014, 41, 75–88. [Google Scholar] [CrossRef] [Green Version]
- Frauwirth, K.A.; Riley, J.L.; Harris, M.H.; Parry, R.V.; Rathmell, J.C.; Plas, D.R.; Elstrom, R.L.; June, C.H.; Thompson, C.B. The CD28 signaling pathway regulates glucose metabolism. Immunity 2002, 16, 769–777. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.; Dillon, C.P.; Shi, L.Z.; Milasta, S.; Carter, R.; Finkelstein, D.; McCormick, L.L.; Fitzgerald, P.; Chi, H.; Munger, J.; et al. The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity 2011, 35, 871–882. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.H.; Qiu, J.; O’Sullivan, D.; Buck, M.D.; Noguchi, T.; Curtis, J.D.; Chen, Q.; Gindin, M.; Gubin, M.M.; van der Windt, G.J.; et al. Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression. Cell 2015, 162, 1229–1241. [Google Scholar] [CrossRef] [Green Version]
- Macintyre, A.N.; Gerriets, V.A.; Nichols, A.G.; Michalek, R.D.; Rudolph, M.C.; Deoliveira, D.; Anderson, S.M.; Abel, E.D.; Chen, B.J.; Hale, L.P.; et al. The glucose transporter Glut1 is selectively essential for CD4 T cell activation and effector function. Cell Metab. 2014, 20, 61–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Araki, K.; Turner, A.P.; Shaffer, V.O.; Gangappa, S.; Keller, S.A.; Bachmann, M.F.; Larsen, C.P.; Ahmed, R. mTOR regulates memory CD8 T-cell differentiation. Nature 2009, 460, 108–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, P.; Chi, H. AGK Unleashes CD8(+) T Cell Glycolysis to Combat Tumor Growth. Cell Metab. 2019, 30, 233–234. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, P.; Vaena, S.G.; Thyagarajan, K.; Chatterjee, S.; Al-Khami, A.; Selvam, S.P.; Nguyen, H.; Kang, I.; Wyatt, M.W.; Baliga, U.; et al. Pro-Survival Lipid Sphingosine-1-Phosphate Metabolically Programs T Cells to Limit Anti-tumor Activity. Cell Rep. 2019, 28, 1879–1893.e1877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Field, C.S.; Baixauli, F.; Kyle, R.L.; Puleston, D.J.; Cameron, A.M.; Sanin, D.E.; Hippen, K.L.; Loschi, M.; Thangavelu, G.; Corrado, M.; et al. Mitochondrial Integrity Regulated by Lipid Metabolism Is a Cell-Intrinsic Checkpoint for Treg Suppressive Function. Cell Metab. 2020, 31, 422–437.e425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geiger, R.; Rieckmann, J.C.; Wolf, T.; Basso, C.; Feng, Y.; Fuhrer, T.; Kogadeeva, M.; Picotti, P.; Meissner, F.; Mann, M.; et al. L-Arginine Modulates T Cell Metabolism and Enhances Survival and Anti-tumor Activity. Cell 2016, 167, 829–842.e813. [Google Scholar] [CrossRef] [Green Version]
- Ma, E.H.; Bantug, G.; Griss, T.; Condotta, S.; Johnson, R.M.; Samborska, B.; Mainolfi, N.; Suri, V.; Guak, H.; Balmer, M.L.; et al. Serine Is an Essential Metabolite for Effector T Cell Expansion. Cell Metab. 2017, 25, 345–357. [Google Scholar] [CrossRef]
- Sukumar, M.; Liu, J.; Mehta, G.U.; Patel, S.J.; Roychoudhuri, R.; Crompton, J.G.; Klebanoff, C.A.; Ji, Y.; Li, P.; Yu, Z.; et al. Mitochondrial Membrane Potential Identifies Cells with Enhanced Stemness for Cellular Therapy. Cell Metab. 2016, 23, 63–76. [Google Scholar] [CrossRef] [Green Version]
- Kesarwani, P.; Al-Khami, A.A.; Scurti, G.; Thyagarajan, K.; Kaur, N.; Husain, S.; Fang, Q.; Naga, O.S.; Simms, P.; Beeson, G.; et al. Promoting thiol expression increases the durability of antitumor T-cell functions. Cancer Res. 2014, 74, 6036–6047. [Google Scholar] [CrossRef] [Green Version]
- Mak, T.W.; Grusdat, M.; Duncan, G.S.; Dostert, C.; Nonnenmacher, Y.; Cox, M.; Binsfeld, C.; Hao, Z.; Brüstle, A.; Itsumi, M.; et al. Glutathione Primes T Cell Metabolism for Inflammation. Immunity 2017, 46, 675–689. [Google Scholar] [CrossRef] [Green Version]
- Chakraborty, P.; Chatterjee, S.; Kesarwani, P.; Thyagarajan, K.; Iamsawat, S.; Dalheim, A.; Nguyen, H.; Selvam, S.P.; Nasarre, P.; Scurti, G.; et al. Thioredoxin-1 improves the immunometabolic phenotype of antitumor T cells. J. Biol. Chem. 2019, 294, 9198–9212. [Google Scholar] [CrossRef] [PubMed]
- Doedens, A.L.; Phan, A.T.; Stradner, M.H.; Fujimoto, J.K.; Nguyen, J.V.; Yang, E.; Johnson, R.S.; Goldrath, A.W. Hypoxia-inducible factors enhance the effector responses of CD8(+) T cells to persistent antigen. Nat. Immunol. 2013, 14, 1173–1182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banerjee, A.; Thyagarajan, K.; Chatterjee, S.; Chakraborty, P.; Kesarwani, P.; Soloshchenko, M.; Al-Hommrani, M.; Andrijauskaite, K.; Moxley, K.; Janakiraman, H.; et al. Lack of p53 Augments Antitumor Functions in Cytolytic T Cells. Cancer Res. 2016, 76, 5229–5240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.; Liu, J.; Wu, R.; Liang, Y.; Lin, M.; Liu, J.; Chan, C.S.; Hu, W.; Feng, Z. Tumor suppressor p53 negatively regulates glycolysis stimulated by hypoxia through its target RRAD. Oncotarget 2014, 5, 5535–5546. [Google Scholar] [CrossRef] [Green Version]
- Roe, J.S.; Kim, H.; Lee, S.M.; Kim, S.T.; Cho, E.J.; Youn, H.D. p53 stabilization and transactivation by a von Hippel-Lindau protein. Mol. Cell 2006, 22, 395–405. [Google Scholar] [CrossRef]
- Chang, C.-H.; Curtis, J.D.; Maggi, L.B., Jr.; Faubert, B.; Villarino, A.V.; O’Sullivan, D.; Huang, S.C.-C.; van der Windt, G.J.W.; Blagih, J.; Qiu, J.; et al. Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell 2013, 153, 1239–1251. [Google Scholar] [CrossRef] [Green Version]
- Peng, M.; Yin, N.; Chhangawala, S.; Xu, K.; Leslie, C.S.; Li, M.O. Aerobic glycolysis promotes T helper 1 cell differentiation through an epigenetic mechanism. Science 2016, 354, 481–484. [Google Scholar] [CrossRef]
- Angiari, S.; Runtsch, M.C.; Sutton, C.E.; Palsson-McDermott, E.M.; Kelly, B.; Rana, N.; Kane, H.; Papadopoulou, G.; Pearce, E.L.; Mills, K.H.G.; et al. Pharmacological Activation of Pyruvate Kinase M2 Inhibits CD4(+) T Cell Pathogenicity and Suppresses Autoimmunity. Cell Metab. 2020, 31, 391–405.e398. [Google Scholar] [CrossRef] [Green Version]
- Brand, A.; Singer, K.; Koehl, G.E.; Kolitzus, M.; Schoenhammer, G.; Thiel, A.; Matos, C.; Bruss, C.; Klobuch, S.; Peter, K.; et al. LDHA-Associated Lactic Acid Production Blunts Tumor Immunosurveillance by T and NK Cells. Cell Metab. 2016, 24, 657–671. [Google Scholar] [CrossRef] [Green Version]
- Eales, K.L.; Hollinshead, K.E.; Tennant, D.A. Hypoxia and metabolic adaptation of cancer cells. Oncogenesis 2016, 5, e190. [Google Scholar] [CrossRef] [Green Version]
- Haas, R.; Smith, J.; Rocher-Ros, V.; Nadkarni, S.; Montero-Melendez, T.; D’Acquisto, F.; Bland, E.J.; Bombardieri, M.; Pitzalis, C.; Perretti, M.; et al. Lactate Regulates Metabolic and Pro-inflammatory Circuits in Control of T Cell Migration and Effector Functions. PLoS Biol. 2015, 13, e1002202. [Google Scholar] [CrossRef] [PubMed]
- Vuillefroy de Silly, R.; Dietrich, P.Y.; Walker, P.R. Hypoxia and antitumor CD8(+) T cells: An incompatible alliance? Oncoimmunology 2016, 5, e1232236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finlay, D.K.; Rosenzweig, E.; Sinclair, L.V.; Feijoo-Carnero, C.; Hukelmann, J.L.; Rolf, J.; Panteleyev, A.A.; Okkenhaug, K.; Cantrell, D.A. PDK1 regulation of mTOR and hypoxia-inducible factor 1 integrate metabolism and migration of CD8+ T cells. J. Exp. Med. 2012, 209, 2441–2453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noman, M.Z.; Hasmim, M.; Lequeux, A.; Xiao, M.; Duhem, C.; Chouaib, S.; Berchem, G.; Janji, B. Improving Cancer Immunotherapy by Targeting the Hypoxic Tumor Microenvironment: New Opportunities and Challenges. Cells 2019, 8, 83. [Google Scholar] [CrossRef] [Green Version]
- Chouaib, S.; Messai, Y.; Couve, S.; Escudier, B.; Hasmim, M.; Noman, M.Z. Hypoxia promotes tumor growth in linking angiogenesis to immune escape. Front. Immunol. 2012, 3, 21. [Google Scholar] [CrossRef] [Green Version]
- Barsoum, I.B.; Smallwood, C.A.; Siemens, D.R.; Graham, C.H. A mechanism of hypoxia-mediated escape from adaptive immunity in cancer cells. Cancer Res. 2014, 74, 665–674. [Google Scholar] [CrossRef] [Green Version]
- Noman, M.Z.; Desantis, G.; Janji, B.; Hasmim, M.; Karray, S.; Dessen, P.; Bronte, V.; Chouaib, S. PD-L1 is a novel direct target of HIF-1α, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J. Exp. Med. 2014, 211, 781–790. [Google Scholar] [CrossRef]
- Messai, Y.; Gad, S.; Noman, M.Z.; Le Teuff, G.; Couve, S.; Janji, B.; Kammerer, S.F.; Rioux-Leclerc, N.; Hasmim, M.; Ferlicot, S.; et al. Renal Cell Carcinoma Programmed Death-ligand 1, a New Direct Target of Hypoxia-inducible Factor-2 Alpha, is Regulated by von Hippel-Lindau Gene Mutation Status. Eur. Urol. 2016, 70, 623–632. [Google Scholar] [CrossRef]
- Hatfield, S.M.; Kjaergaard, J.; Lukashev, D.; Schreiber, T.H.; Belikoff, B.; Abbott, R.; Sethumadhavan, S.; Philbrook, P.; Ko, K.; Cannici, R.; et al. Immunological mechanisms of the antitumor effects of supplemental oxygenation. Sci. Transl. Med. 2015, 7, 277ra230. [Google Scholar] [CrossRef] [Green Version]
- Scharping, N.E.; Menk, A.V.; Whetstone, R.D.; Zeng, X.; Delgoffe, G.M. Efficacy of PD-1 Blockade Is Potentiated by Metformin-Induced Reduction of Tumor Hypoxia. Cancer Immunol. Res. 2017, 5, 9–16. [Google Scholar] [CrossRef] [Green Version]
- Opitz, C.A.; Litzenburger, U.M.; Sahm, F.; Ott, M.; Tritschler, I.; Trump, S.; Schumacher, T.; Jestaedt, L.; Schrenk, D.; Weller, M.; et al. An endogenous tumour-promoting ligand of the human aryl hydrocarbon receptor. Nature 2011, 478, 197–203. [Google Scholar] [CrossRef] [PubMed]
- Mondanelli, G.; Ugel, S.; Grohmann, U.; Bronte, V. The immune regulation in cancer by the amino acid metabolizing enzymes ARG and IDO. Curr. Opin. Pharm. 2017, 35, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Munn, D.H.; Mellor, A.L. IDO in the Tumor Microenvironment: Inflammation, Counter-Regulation, and Tolerance. Trends Immunol. 2016, 37, 193–207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Shen, Z.; Wang, Z.; Wang, X.; Zhang, H.; Qin, J.; Qin, X.; Xu, J.; Sun, Y. Increased expression of IDO associates with poor postoperative clinical outcome of patients with gastric adenocarcinoma. Sci. Rep. 2016, 6, 21319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mezrich, J.D.; Fechner, J.H.; Zhang, X.; Johnson, B.P.; Burlingham, W.J.; Bradfield, C.A. An interaction between kynurenine and the aryl hydrocarbon receptor can generate regulatory T cells. J. Immunol. 2010, 185, 3190–3198. [Google Scholar] [CrossRef] [Green Version]
- Sharma, M.D.; Hou, D.Y.; Liu, Y.; Koni, P.A.; Metz, R.; Chandler, P.; Mellor, A.L.; He, Y.; Munn, D.H. Indoleamine 2,3-dioxygenase controls conversion of Foxp3+ Tregs to TH17-like cells in tumor-draining lymph nodes. Blood 2009, 113, 6102–6111. [Google Scholar] [CrossRef] [Green Version]
- Munn, D.H.; Sharma, M.D.; Baban, B.; Harding, H.P.; Zhang, Y.; Ron, D.; Mellor, A.L. GCN2 kinase in T cells mediates proliferative arrest and anergy induction in response to indoleamine 2,3-dioxygenase. Immunity 2005, 22, 633–642. [Google Scholar] [CrossRef] [Green Version]
- Tang, K.; Wu, Y.-H.; Song, Y.; Yu, B. Indoleamine 2,3-dioxygenase 1 (IDO1) inhibitors in clinical trials for cancer immunotherapy. J. Hematol. Oncol. 2021, 14, 68. [Google Scholar] [CrossRef]
- Prendergast, G.C.; Malachowski, W.P.; DuHadaway, J.B.; Muller, A.J. Discovery of IDO1 Inhibitors: From Bench to Bedside. Cancer Res. 2017, 77, 6795–6811. [Google Scholar] [CrossRef] [Green Version]
- Leone, R.D.; Emens, L.A. Targeting adenosine for cancer immunotherapy. J. Immunother. Cancer 2018, 6, 57. [Google Scholar] [CrossRef] [Green Version]
- Mastelic-Gavillet, B.; Navarro Rodrigo, B.; Décombaz, L.; Wang, H.; Ercolano, G.; Ahmed, R.; Lozano, L.E.; Ianaro, A.; Derré, L.; Valerio, M.; et al. Adenosine mediates functional and metabolic suppression of peripheral and tumor-infiltrating CD8+ T cells. J. ImmunoTher. Cancer 2019, 7, 257. [Google Scholar] [CrossRef] [PubMed]
- Roy, D.G.; Kaymak, I.; Williams, K.S.; Ma, E.H.; Jones, R.G. Immunometabolism in the Tumor Microenvironment. Annu. Rev. Cancer Biol. 2021, 5, 137–159. [Google Scholar] [CrossRef]
- Cascone, T.; McKenzie, J.A.; Mbofung, R.M.; Punt, S.; Wang, Z.; Xu, C.; Williams, L.J.; Wang, Z.; Bristow, C.A.; Carugo, A.; et al. Increased Tumor Glycolysis Characterizes Immune Resistance to Adoptive T Cell Therapy. Cell Metab. 2018, 27, 977–987.e974. [Google Scholar] [CrossRef] [PubMed]
- Reinfeld, B.I.; Madden, M.Z.; Wolf, M.M.; Chytil, A.; Bader, J.E.; Patterson, A.R.; Sugiura, A.; Cohen, A.S.; Ali, A.; Do, B.T.; et al. Cell-programmed nutrient partitioning in the tumour microenvironment. Nature 2021, 593, 282–288. [Google Scholar] [CrossRef] [PubMed]
- Yuki, K.; Cheng, N.; Nakano, M.; Kuo, C.J. Organoid Models of Tumor Immunology. Trends Immunol. 2020, 41, 652–664. [Google Scholar] [CrossRef] [PubMed]
- Cattaneo, C.M.; Dijkstra, K.K.; Fanchi, L.F.; Kelderman, S.; Kaing, S.; van Rooij, N.; van den Brink, S.; Schumacher, T.N.; Voest, E.E. Tumor organoid-T-cell coculture systems. Nat. Protoc. 2020, 15, s019-41596. [Google Scholar] [CrossRef]
- Votanopoulos, K.I.; Forsythe, S.; Sivakumar, H.; Mazzocchi, A.; Aleman, J.; Miller, L.; Levine, E.; Triozzi, P.; Skardal, A. Model of Patient-Specific Immune-Enhanced Organoids for Immunotherapy Screening: Feasibility Study. Ann. Surg. Oncol. 2020, 27, s10434-019. [Google Scholar] [CrossRef]
- Zhang, S.; Iyer, S.; Ran, H.; Dolgalev, I.; Gu, S.; Wei, W.; Foster, C.J.; Loomis, C.A.; Olvera, N.; Dao, F.; et al. Genetically Defined, Syngeneic Organoid Platform for Developing Combination Therapies for Ovarian Cancer. Cancer Discov. 2021, 11, 362–383. [Google Scholar] [CrossRef]
- Vazquez, F.; Lim, J.H.; Chim, H.; Bhalla, K.; Girnun, G.; Pierce, K.; Clish, C.B.; Granter, S.R.; Widlund, H.R.; Spiegelman, B.M.; et al. PGC1α expression defines a subset of human melanoma tumors with increased mitochondrial capacity and resistance to oxidative stress. Cancer Cell 2013, 23, 287–301. [Google Scholar] [CrossRef] [Green Version]
- Scharping, N.E.; Menk, A.V.; Moreci, R.S.; Whetstone, R.D.; Dadey, R.E.; Watkins, S.C.; Ferris, R.L.; Delgoffe, G.M. The Tumor Microenvironment Represses T Cell Mitochondrial Biogenesis to Drive Intratumoral T Cell Metabolic Insufficiency and Dysfunction. Immunity 2016, 45, 374–388. [Google Scholar] [CrossRef] [Green Version]
- Rivadeneira, D.B.; DePeaux, K.; Wang, Y.; Kulkarni, A.; Tabib, T.; Menk, A.V.; Sampath, P.; Lafyatis, R.; Ferris, R.L.; Sarkar, S.N.; et al. Oncolytic Viruses Engineered to Enforce Leptin Expression Reprogram Tumor-Infiltrating T Cell Metabolism and Promote Tumor Clearance. Immunity 2019, 51, 548–560.e544. [Google Scholar] [CrossRef] [PubMed]
- Qiu, J.; Villa, M.; Sanin, D.E.; Buck, M.D.; O’Sullivan, D.; Ching, R.; Matsushita, M.; Grzes, K.M.; Winkler, F.; Chang, C.H.; et al. Acetate Promotes T Cell Effector Function during Glucose Restriction. Cell Rep. 2019, 27, 2063–2074.e2065. [Google Scholar] [CrossRef] [Green Version]
- Muranski, P.; Boni, A.; Antony, P.A.; Cassard, L.; Irvine, K.R.; Kaiser, A.; Paulos, C.M.; Palmer, D.C.; Touloukian, C.E.; Ptak, K.; et al. Tumor-specific Th17-polarized cells eradicate large established melanoma. Blood 2008, 112, 362–373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greenberg, P.D.; Cheever, M.A.; Fefer, A. Pillars article: Eradication of disseminated murine leukemia by chemoimmunotherapy with cyclophosphamide and adoptively transferred immune syngeneic Lyt-1+2- lymphocytes. J. Exp. Med. 1981. 154: 952-963. J. Immunol. 2013, 190, 1899–1910. [Google Scholar] [PubMed]
- Perez-Diez, A.; Joncker, N.T.; Choi, K.; Chan, W.F.; Anderson, C.C.; Lantz, O.; Matzinger, P. CD4 cells can be more efficient at tumor rejection than CD8 cells. Blood 2007, 109, 5346–5354. [Google Scholar] [CrossRef] [PubMed]
- Overwijk, W.W.; Theoret, M.R.; Finkelstein, S.E.; Surman, D.R.; de Jong, L.A.; Vyth-Dreese, F.A.; Dellemijn, T.A.; Antony, P.A.; Spiess, P.J.; Palmer, D.C.; et al. Tumor regression and autoimmunity after reversal of a functionally tolerant state of self-reactive CD8+ T cells. J. Exp. Med. 2003, 198, 569–580. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.; Thyagarajan, K.; Kesarwani, P.; Song, J.H.; Soloshchenko, M.; Fu, J.; Bailey, S.R.; Vasu, C.; Kraft, A.S.; Paulos, C.M.; et al. Reducing CD73 expression by IL1beta-Programmed Th17 cells improves immunotherapeutic control of tumors. Cancer Res. 2014, 74, 6048–6059. [Google Scholar] [CrossRef] [Green Version]
- Muranski, P.; Borman, Z.A.; Kerkar, S.P.; Klebanoff, C.A.; Ji, Y.; Sanchez-Perez, L.; Sukumar, M.; Reger, R.N.; Yu, Z.; Kern, S.J.; et al. Th17 cells are long lived and retain a stem cell-like molecular signature. Immunity 2011, 35, 972–985. [Google Scholar] [CrossRef] [Green Version]
- Wei, S.; Zhao, E.; Kryczek, I.; Zou, W. Th17 cells have stem cell-like features and promote long-term immunity. Oncoimmunology 2012, 1, 516–519. [Google Scholar] [CrossRef] [Green Version]
- Kryczek, I.; Zhao, E.; Liu, Y.; Wang, Y.; Vatan, L.; Szeliga, W.; Moyer, J.; Klimczak, A.; Lange, A.; Zou, W. Human TH17 cells are long-lived effector memory cells. Sci. Transl. Med. 2011, 3, 104ra100. [Google Scholar] [CrossRef] [Green Version]
- Lee, P.H.; Yamamoto, T.N.; Gurusamy, D.; Sukumar, M.; Yu, Z.; Hu-Li, J.; Kawabe, T.; Gangaplara, A.; Kishton, R.J.; Henning, A.N.; et al. Host conditioning with IL-1β improves the antitumor function of adoptively transferred T cells. J. Exp. Med. 2019, 216, 2619–2634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, B.; Benavides, G.A.; Geng, J.; Koralov, S.B.; Hu, H.; Darley-Usmar, V.M.; Harrington, L.E. Mitochondrial Oxidative Phosphorylation Regulates the Fate Decision between Pathogenic Th17 and Regulatory T Cells. Cell Rep. 2020, 30, 1898–1909.e1894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chatterjee, S.; Daenthanasanmak, A.; Chakraborty, P.; Wyatt, M.W.; Dhar, P.; Selvam, S.P.; Fu, J.; Zhang, J.; Nguyen, H.; Kang, I.; et al. CD38-NAD(+)Axis Regulates Immunotherapeutic Anti-Tumor T Cell Response. Cell Metab. 2018, 27, 85–100.e108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chini, E.N. CD38 as a regulator of cellular NAD: A novel potential pharmacological target for metabolic conditions. Curr. Pharm. Des. 2009, 15, 57–63. [Google Scholar] [CrossRef] [Green Version]
- Hogan, K.A.; Chini, C.C.S.; Chini, E.N. The Multi-faceted Ecto-enzyme CD38: Roles in Immunomodulation, Cancer, Aging, and Metabolic Diseases. Front. Immunol. 2019, 10, 1187. [Google Scholar] [CrossRef] [Green Version]
- Escande, C.; Nin, V.; Price, N.L.; Capellini, V.; Gomes, A.P.; Barbosa, M.T.; O’Neil, L.; White, T.A.; Sinclair, D.A.; Chini, E.N. Flavonoid apigenin is an inhibitor of the NAD+ ase CD38: Implications for cellular NAD+ metabolism, protein acetylation, and treatment of metabolic syndrome. Diabetes 2013, 62, 1084–1093. [Google Scholar] [CrossRef] [Green Version]
- Carey, B.W.; Finley, L.W.; Cross, J.R.; Allis, C.D.; Thompson, C.B. Intracellular alpha-ketoglutarate maintains the pluripotency of embryonic stem cells. Nature 2015, 518, 413–416. [Google Scholar] [CrossRef] [Green Version]
- Xu, T.; Stewart, K.M.; Wang, X.; Liu, K.; Xie, M.; Ryu, J.K.; Li, K.; Ma, T.; Wang, H.; Ni, L.; et al. Metabolic control of TH17 and induced Treg cell balance by an epigenetic mechanism. Nature 2017, 548, 228–233. [Google Scholar] [CrossRef]
- Heo, J.; Lim, J.; Lee, S.; Jeong, J.; Kang, H.; Kim, Y.; Kang, J.W.; Yu, H.Y.; Jeong, E.M.; Kim, K.; et al. Sirt1 Regulates DNA Methylation and Differentiation Potential of Embryonic Stem Cells by Antagonizing Dnmt3l. Cell Rep. 2017, 18, 1930–1945. [Google Scholar] [CrossRef] [Green Version]
- Peng, L.; Yuan, Z.; Ling, H.; Fukasawa, K.; Robertson, K.; Olashaw, N.; Koomen, J.; Chen, J.; Lane, W.S.; Seto, E. SIRT1 deacetylates the DNA methyltransferase 1 (DNMT1) protein and alters its activities. Mol. Cell. Biol. 2011, 31, 4720–4734. [Google Scholar] [CrossRef] [Green Version]
- Karmaus, P.W.F.; Chen, X.; Lim, S.A.; Herrada, A.A.; Nguyen, T.M.; Xu, B.; Dhungana, Y.; Rankin, S.; Chen, W.; Rosencrance, C.; et al. Metabolic heterogeneity underlies reciprocal fates of T(H)17 cell stemness and plasticity. Nature 2019, 565, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Wang, Q.; Xue, G.; Bi, E.; Ma, X.; Wang, A.; Qian, J.; Dong, C.; Yi, Q. Th9 Cells Represent a Unique Subset of CD4(+) T Cells Endowed with the Ability to Eradicate Advanced Tumors. Cancer Cell 2018, 33, 1048–1060.e1047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaplan, M.H. Th9 cells: Differentiation and disease. Immunol. Rev. 2013, 252, 104–115. [Google Scholar] [CrossRef]
- Mumm, J.B.; Emmerich, J.; Zhang, X.; Chan, I.; Wu, L.; Mauze, S.; Blaisdell, S.; Basham, B.; Dai, J.; Grein, J.; et al. IL-10 elicits IFNγ-dependent tumor immune surveillance. Cancer Cell 2011, 20, 781–796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ku, C.J.; Hosoya, T.; Maillard, I.; Engel, J.D. GATA-3 regulates hematopoietic stem cell maintenance and cell-cycle entry. Blood 2012, 119, 2242–2251. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Brunner, T.; Carter, L.; Dutton, R.W.; Rogers, P.; Bradley, L.; Sato, T.; Reed, J.C.; Green, D.; Swain, S.L. Unequal death in T helper cell (Th)1 and Th2 effectors: Th1, but not Th2, effectors undergo rapid Fas/FasL-mediated apoptosis. J. Exp. Med. 1997, 185, 1837–1849. [Google Scholar] [CrossRef]
- Dumauthioz, N.; Tschumi, B.; Wenes, M.; Marti, B.; Wang, H.; Franco, F.; Li, W.; Lopez-Mejia, I.C.; Fajas, L.; Ho, P.C.; et al. Enforced PGC-1α expression promotes CD8 T cell fitness, memory formation and antitumor immunity. Cell Mol. Immunol. 2021, 18, 1761–1771. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Bi, Y.; Chen, X.; Li, C.; Li, Y.; Zhang, Z.; Wang, J.; Lu, Y.; Yu, Q.; Su, H.; et al. Histone Deacetylase SIRT1 Negatively Regulates the Differentiation of Interleukin-9-Producing CD4+ T Cells. Immunity 2016, 44, 1337–1349. [Google Scholar] [CrossRef] [Green Version]
- Oberholtzer, N.; Atkinson, C.; Nadig, S.N. Adoptive Transfer of Regulatory Immune Cells in Organ Transplantation. Front. Immunol. 2021, 12, 631365. [Google Scholar] [CrossRef]
- Wang, H.; Franco, F.; Ho, P.C. Metabolic Regulation of Tregs in Cancer: Opportunities for Immunotherapy. Trends Cancer 2017, 3, 583–592. [Google Scholar] [CrossRef] [PubMed]
- Howie, D.; Cobbold, S.P.; Adams, E.; Ten Bokum, A.; Necula, A.S.; Zhang, W.; Huang, H.; Roberts, D.J.; Thomas, B.; Hester, S.S.; et al. Foxp3 drives oxidative phosphorylation and protection from lipotoxicity. JCI Insight 2017, 2, e89160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kempkes, R.W.M.; Joosten, I.; Koenen, H.J.P.M.; He, X. Metabolic Pathways Involved in Regulatory T Cell Functionality. Front. Immunol. 2019, 10, 2839. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Chi, H. Metabolic Control of Treg Cell Stability, Plasticity, and Tissue-Specific Heterogeneity. Front. Immunol. 2019, 10, 2716. [Google Scholar] [CrossRef] [Green Version]
- Beier, U.H.; Angelin, A.; Akimova, T.; Wang, L.; Liu, Y.; Xiao, H.; Koike, M.A.; Hancock, S.A.; Bhatti, T.R.; Han, R.; et al. Essential role of mitochondrial energy metabolism in Foxp3+ T-regulatory cell function and allograft survival. Faseb. J. 2015, 29, 2315–2326. [Google Scholar] [CrossRef] [Green Version]
- Shi, L.Z.; Wang, R.; Huang, G.; Vogel, P.; Neale, G.; Green, D.R.; Chi, H. HIF1alpha-dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. J. Exp. Med. 2011, 208, 1367–1376. [Google Scholar] [CrossRef] [Green Version]
- Pavlova, N.N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef] [Green Version]
- Sugiura, A.; Rathmell, J.C. Metabolic Barriers to T Cell Function in Tumors. J. Immunol. 2018, 200, 400–407. [Google Scholar] [CrossRef]
- Klysz, D.; Tai, X.; Robert, P.A.; Craveiro, M.; Cretenet, G.; Oburoglu, L.; Mongellaz, C.; Floess, S.; Fritz, V.; Matias, M.I.; et al. Glutamine-dependent α-ketoglutarate production regulates the balance between T helper 1 cell and regulatory T cell generation. Sci. Signal. 2015, 8, ra97. [Google Scholar] [CrossRef] [Green Version]
- Lurie, K.G.; Zielinski, T.; McKnite, S.; Aufderheide, T.; Voelckel, W. Use of an Inspiratory Impedance Valve Improves Neurologically Intact Survival in a Porcine Model of Ventricular Fibrillation. Circulation 2002, 105, 124–129. [Google Scholar] [CrossRef] [Green Version]
- Nakaya, M.; Xiao, Y.; Zhou, X.; Chang, J.H.; Chang, M.; Cheng, X.; Blonska, M.; Lin, X.; Sun, S.C. Inflammatory T cell responses rely on amino acid transporter ASCT2 facilitation of glutamine uptake and mTORC1 kinase activation. Immunity 2014, 40, 692–705. [Google Scholar] [CrossRef] [Green Version]
- Sinclair, L.V.; Rolf, J.; Emslie, E.; Shi, Y.B.; Taylor, P.M.; Cantrell, D.A. Control of amino-acid transport by antigen receptors coordinates the metabolic reprogramming essential for T cell differentiation. Nat. Immunol. 2013, 14, 500–508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, J.; Raynor, J.; Nguyen, T.L.; Chi, H. Nutrient and Metabolic Sensing in T Cell Responses. Front. Immunol. 2017, 8, 247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pacella, I.; Procaccini, C.; Focaccetti, C.; Miacci, S.; Timperi, E.; Faicchia, D.; Severa, M.; Rizzo, F.; Coccia, E.M.; Bonacina, F.; et al. Fatty acid metabolism complements glycolysis in the selective regulatory T cell expansion during tumor growth. Proc. Natl. Acad. Sci. USA 2018, 115, e6546–e6555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miska, J.; Lee-Chang, C.; Rashidi, A.; Muroski, M.E.; Chang, A.L.; Lopez-Rosas, A.; Zhang, P.; Panek, W.K.; Cordero, A.; Han, Y.; et al. HIF-1α Is a Metabolic Switch between Glycolytic-Driven Migration and Oxidative Phosphorylation-Driven Immunosuppression of Tregs in Glioblastoma. Cell Rep. 2019, 27, 226–237.e224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Franco, F.; Tsui, Y.-C.; Xie, X.; Trefny, M.P.; Zappasodi, R.; Mohmood, S.R.; Fernández-García, J.; Tsai, C.-H.; Schulze, I.; et al. CD36-mediated metabolic adaptation supports regulatory T cell survival and function in tumors. Nat. Immunol. 2020, 21, 298–308. [Google Scholar] [CrossRef]
- Chang, H.C.; Guarente, L. SIRT1 and other sirtuins in metabolism. Trends Endocrinol. Metab. 2014, 25, 138–145. [Google Scholar] [CrossRef]
- Jeng, M.Y.; Hull, P.A.; Fei, M.; Kwon, H.-S.; Tsou, C.-L.; Kasler, H.; Ng, C.-P.; Gordon, D.E.; Johnson, J.; Krogan, N.; et al. Metabolic reprogramming of human CD8+ memory T cells through loss of SIRT1. J. Exp. Med. 2017, 215, 51–62. [Google Scholar] [CrossRef]
- Kar, A.; Mehrotra, S.; Chatterjee, S. CD38: T Cell Immuno-Metabolic Modulator. Cells 2020, 9, 1716. [Google Scholar] [CrossRef]
- Tyrakis, P.A.; Palazon, A.; Macias, D.; Lee, K.L.; Phan, A.T.; Veliça, P.; You, J.; Chia, G.S.; Sim, J.; Doedens, A.; et al. S-2-hydroxyglutarate regulates CD8+ T-lymphocyte fate. Nature 2016, 540, 236–241. [Google Scholar] [CrossRef]
- Foskolou, I.P.; Barbieri, L.; Vernet, A.; Bargiela, D.; Cunha, P.P.; Velica, P.; Suh, E.; Pietsch, S.; Matuleviciute, R.; Rundqvist, H.; et al. The S enantiomer of 2-hydroxyglutarate increases central memory CD8 populations and improves CAR-T therapy outcome. Blood Adv. 2020, 4, 4483–4493. [Google Scholar] [CrossRef]
- Shyer, J.A.; Flavell, R.A.; Bailis, W. Metabolic signaling in T cells. Cell Res. 2020, 30, 649–659. [Google Scholar] [CrossRef] [PubMed]
- Palmer, C.S.; Ostrowski, M.; Balderson, B.; Christian, N.; Crowe, S.M. Glucose Metabolism Regulates T Cell Activation, Differentiation, and Functions. Front. Immunol. 2015, 6, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, Y.; Delgoffe, G.M.; Meyer, C.F.; Chan, W.; Powell, J.D. Anergic T cells are metabolically anergic. J. Immunol. 2009, 183, 6095–6101. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, S.R.; Herman, C.E.; Maciver, N.J.; Wofford, J.A.; Wieman, H.L.; Hammen, J.J.; Rathmell, J.C. Glucose uptake is limiting in T cell activation and requires CD28-mediated Akt-dependent and independent pathways. J. Immunol. 2008, 180, 4476–4486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marko, A.J.; Miller, R.A.; Kelman, A.; Frauwirth, K.A. Induction of glucose metabolism in stimulated T lymphocytes is regulated by mitogen-activated protein kinase signaling. PLoS ONE 2010, 5, e15425. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Y.; Collins, S.L.; Lutz, M.A.; Allen, A.N.; Kole, T.P.; Zarek, P.E.; Powell, J.D. A role for mammalian target of rapamycin in regulating T cell activation versus anergy. J. Immunol. 2007, 178, 2163–2170. [Google Scholar] [CrossRef] [Green Version]
- Kawalekar, O.U.; O’Connor, R.S.; Fraietta, J.A.; Guo, L.; McGettigan, S.E.; Posey, A.D., Jr.; Patel, P.R.; Guedan, S.; Scholler, J.; Keith, B.; et al. Distinct Signaling of Coreceptors Regulates Specific Metabolism Pathways and Impacts Memory Development in CAR T Cells. Immunity 2016, 44, 380–390. [Google Scholar] [CrossRef] [Green Version]
- Long, A.H.; Haso, W.M.; Shern, J.F.; Wanhainen, K.M.; Murgai, M.; Ingaramo, M.; Smith, J.P.; Walker, A.J.; Kohler, M.E.; Venkateshwara, V.R.; et al. 4-1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat. Med. 2015, 21, 581–590. [Google Scholar] [CrossRef] [Green Version]
- Teijeira, A.; Garasa, S.; Etxeberria, I.; Gato-Cañas, M.; Melero, I.; Delgoffe, G.M. Metabolic Consequences of T-cell Costimulation in Anticancer Immunity. Cancer Immunol. Res. 2019, 7, 1564. [Google Scholar] [CrossRef]
- Guedan, S.; Posey, A.D., Jr.; Shaw, C.; Wing, A.; Da, T.; Patel, P.R.; McGettigan, S.E.; Casado-Medrano, V.; Kawalekar, O.U.; Uribe-Herranz, M.; et al. Enhancing CAR T cell persistence through ICOS and 4-1BB costimulation. JCI Insight 2018, 3, e96976. [Google Scholar] [CrossRef] [Green Version]
- Sabharwal, S.S.; Rosen, D.B.; Grein, J.; Tedesco, D.; Joyce-Shaikh, B.; Ueda, R.; Semana, M.; Bauer, M.; Bang, K.; Stevenson, C.; et al. GITR Agonism Enhances Cellular Metabolism to Support CD8(+) T-cell Proliferation and Effector Cytokine Production in a Mouse Tumor Model. Cancer Immunol. Res. 2018, 6, 1199–1211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bengsch, B.; Johnson, A.L.; Kurachi, M.; Odorizzi, P.M.; Pauken, K.E.; Attanasio, J.; Stelekati, E.; McLane, L.M.; Paley, M.A.; Delgoffe, G.M.; et al. Bioenergetic Insufficiencies Due to Metabolic Alterations Regulated by the Inhibitory Receptor PD-1 Are an Early Driver of CD8(+) T Cell Exhaustion. Immunity 2016, 45, 358–373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toomer, K.H.; Malek, T.R. Cytokine Signaling in the Development and Homeostasis of Regulatory T cells. Cold Spring Harb. Perspect. Biol. 2018, 10, a028597. [Google Scholar] [CrossRef]
- Malek, T.R.; Castro, I. Interleukin-2 receptor signaling: At the interface between tolerance and immunity. Immunity 2010, 33, 153–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, J.X.; Leonard, W.J. The Common Cytokine Receptor γ Chain Family of Cytokines. Cold Spring Harb. Perspect. Biol. 2018, 10, a028449. [Google Scholar] [CrossRef] [PubMed]
- Leonard, W.J.; Lin, J.X.; O’Shea, J.J. The γ(c) Family of Cytokines: Basic Biology to Therapeutic Ramifications. Immunity 2019, 50, 832–850. [Google Scholar] [CrossRef] [Green Version]
- Dwyer, C.J.; Knochelmann, H.M.; Smith, A.S.; Wyatt, M.M.; Rangel Rivera, G.O.; Arhontoulis, D.C.; Bartee, E.; Li, Z.; Rubinstein, M.P.; Paulos, C.M. Fueling Cancer Immunotherapy With Common Gamma Chain Cytokines. Front. Immunol. 2019, 10, 263. [Google Scholar] [CrossRef] [Green Version]
- Robertson, C.N.; Linehan, W.M.; Pass, H.I.; Gomella, L.G.; Haas, G.P.; Berman, A.; Merino, M.; Rosenberg, S.A. Preparative cytoreductive surgery in patients with metastatic renal cell carcinoma treated with adoptive immunotherapy with interleukin-2 or interleukin-2 plus lymphokine activated killer cells. J. Urol. 1990, 144, 614–617, discussion 617–618. [Google Scholar] [CrossRef]
- Yang, J.C.; Shlasko, E.; Ritchey, J.L.; Landry, J.G.; White, D.E.; Rosenberg, S.A. Combination chemoimmunotherapy for metastatic colorectal cancer using 5-fluorouracil, leucovorin and interleukin-2. Eur. J. Cancer 1993, 29, 355–359. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Lotze, M.T.; Yang, J.C.; Topalian, S.L.; Chang, A.E.; Schwartzentruber, D.J.; Aebersold, P.; Leitman, S.; Linehan, W.M.; Seipp, C.A.; et al. Prospective randomized trial of high-dose interleukin-2 alone or in conjunction with lymphokine-activated killer cells for the treatment of patients with advanced cancer. J. Natl. Cancer Inst. 1993, 85, 622–632. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Yang, J.C.; Topalian, S.L.; Schwartzentruber, D.J.; Weber, J.S.; Parkinson, D.R.; Seipp, C.A.; Einhorn, J.H.; White, D.E. Treatment of 283 consecutive patients with metastatic melanoma or renal cell cancer using high-dose bolus interleukin 2. JAMA 1994, 271, 907–913. [Google Scholar] [CrossRef] [PubMed]
- Fyfe, G.; Fisher, R.I.; Rosenberg, S.A.; Sznol, M.; Parkinson, D.R.; Louie, A.C. Results of treatment of 255 patients with metastatic renal cell carcinoma who received high-dose recombinant interleukin-2 therapy. J. Clin. Oncol. 1995, 13, 688–696. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Yang, J.C.; White, D.E.; Steinberg, S.M. Durability of complete responses in patients with metastatic cancer treated with high-dose interleukin-2: Identification of the antigens mediating response. Ann. Surg 1998, 228, 307–319. [Google Scholar] [CrossRef] [PubMed]
- Dudley, M.E.; Wunderlich, J.; Nishimura, M.I.; Yu, D.; Yang, J.C.; Topalian, S.L.; Schwartzentruber, D.J.; Hwu, P.; Marincola, F.M.; Sherry, R.; et al. Adoptive transfer of cloned melanoma-reactive T lymphocytes for the treatment of patients with metastatic melanoma. J. Immunother. 2001, 24, 363–373. [Google Scholar] [CrossRef] [PubMed]
- Weber, J.S.; Yang, J.C.; Topalian, S.L.; Schwartzentruber, D.J.; White, D.E.; Rosenberg, S.A. The use of interleukin-2 and lymphokine-activated killer cells for the treatment of patients with non-Hodgkin’s lymphoma. J. Clin. Oncol. 1992, 10, 33–40. [Google Scholar] [CrossRef] [PubMed]
- MacFarlane, M.P.; Yang, J.C.; Guleria, A.S.; White, R.L., Jr.; Seipp, C.A.; Einhorn, J.H.; White, D.E.; Rosenberg, S.A. The hematologic toxicity of interleukin-2 in patients with metastatic melanoma and renal cell carcinoma. Cancer 1995, 75, 1030–1037. [Google Scholar] [CrossRef]
- Kammula, U.S.; White, D.E.; Rosenberg, S.A. Trends in the safety of high dose bolus interleukin-2 administration in patients with metastatic cancer. Cancer 1998, 83, 797–805. [Google Scholar] [CrossRef]
- Marroquin, C.E.; White, D.E.; Steinberg, S.M.; Rosenberg, S.A.; Schwartzentruber, D.J. Decreased tolerance to interleukin-2 with repeated courses of therapy in patients with metastatic melanoma or renal cell cancer. J. Immunother. 2000, 23, 387–392. [Google Scholar] [CrossRef] [PubMed]
- Fox, C.J.; Hammerman, P.S.; Thompson, C.B. Fuel feeds function: Energy metabolism and the T-cell response. Nat. Rev. Immunol. 2005, 5, 844–852. [Google Scholar] [CrossRef]
- Jones, R.G.; Thompson, C.B. Revving the engine: Signal transduction fuels T cell activation. Immunity 2007, 27, 173–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carrio, R.; Bathe, O.F.; Malek, T.R. Initial Antigen Encounter Programs CD8+ T Cells Competent to Develop into Memory Cells That Are Activated in an Antigen-Free, IL-7- and IL-15-Rich Environment. J. Immunol. 2004, 172, 7315–7323. [Google Scholar] [CrossRef] [Green Version]
- Mengus, C.; Le Magnen, C.; Trella, E.; Yousef, K.; Bubendorf, L.; Provenzano, M.; Bachmann, A.; Heberer, M.; Spagnoli, G.C.; Wyler, S. Elevated levels of circulating IL-7 and IL-15 in patients with early stage prostate cancer. J. Transl. Med. 2011, 9, 162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cieri, N.; Camisa, B.; Cocchiarella, F.; Forcato, M.; Oliveira, G.; Provasi, E.; Bondanza, A.; Bordignon, C.; Peccatori, J.; Ciceri, F.; et al. IL-7 and IL-15 instruct the generation of human memory stem T cells from naive precursors. Blood 2013, 121, 573–584. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Liang, Y.; Xue, D.; Shen, J.; Cai, Y.; Zhu, J.; Fu, Y.-X.; Peng, H. Tumor-conditional IL-15 pro-cytokine reactivates anti-tumor immunity with limited toxicity. Cell Res. 2021, 31, 1190–1198. [Google Scholar] [CrossRef] [PubMed]
- Buck, M.D.; O’Sullivan, D.; Klein Geltink, R.I.; Curtis, J.D.; Chang, C.H.; Sanin, D.E.; Qiu, J.; Kretz, O.; Braas, D.; van der Windt, G.J.; et al. Mitochondrial Dynamics Controls T Cell Fate through Metabolic Programming. Cell 2016, 166, 63–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peschon, J.J.; Morrissey, P.J.; Grabstein, K.H.; Ramsdell, F.J.; Maraskovsky, E.; Gliniak, B.C.; Park, L.S.; Ziegler, S.F.; Williams, D.E.; Ware, C.B.; et al. Early lymphocyte expansion is severely impaired in interleukin 7 receptor-deficient mice. J. Exp. Med. 1994, 180, 1955–1960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, K.; Lee, C.K.; Sayers, T.J.; Muegge, K.; Durum, S.K. The trophic action of IL-7 on pro-T cells: Inhibition of apoptosis of pro-T1, -T2, and -T3 cells correlates with Bcl-2 and Bax levels and is independent of Fas and p53 pathways. J. Immunol. 1998, 160, 5735–5741. [Google Scholar]
- Muegge, K.; Vila, M.P.; Durum, S.K. Interleukin-7: A cofactor for V(D)J rearrangement of the T cell receptor beta gene. Science 1993, 261, 93–95. [Google Scholar] [CrossRef] [Green Version]
- Vivien, L.; Benoist, C.; Mathis, D. T lymphocytes need IL-7 but not IL-4 or IL-6 to survive in vivo. Int. Immunol. 2001, 13, 763–768. [Google Scholar] [CrossRef] [Green Version]
- Rathmell, J.C.; Farkash, E.A.; Gao, W.; Thompson, C.B. IL-7 enhances the survival and maintains the size of naive T cells. J. Immunol. 2001, 167, 6869–6876. [Google Scholar] [CrossRef]
- Hong, C.; Luckey, M.A.; Park, J.H. Intrathymic IL-7: The where, when, and why of IL-7 signaling during T cell development. Semin. Immunol. 2012, 24, 151–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wofford, J.A.; Wieman, H.L.; Jacobs, S.R.; Zhao, Y.; Rathmell, J.C. IL-7 promotes Glut1 trafficking and glucose uptake via STAT5-mediated activation of Akt to support T-cell survival. Blood 2008, 111, 2101–2111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacobs, S.R.; Michalek, R.D.; Rathmell, J.C. IL-7 is essential for homeostatic control of T cell metabolism in vivo. J. Immunol. 2010, 184, 3461–3469. [Google Scholar] [CrossRef] [PubMed]
- Shum, T.; Omer, B.; Tashiro, H.; Kruse, R.L.; Wagner, D.L.; Parikh, K.; Yi, Z.; Sauer, T.; Liu, D.; Parihar, R.; et al. Constitutive Signaling from an Engineered IL7 Receptor Promotes Durable Tumor Elimination by Tumor-Redirected T Cells. Cancer Discov. 2017, 7, 1238–1247. [Google Scholar] [CrossRef] [Green Version]
- Cui, G.; Staron, M.M.; Gray, S.M.; Ho, P.C.; Amezquita, R.A.; Wu, J.; Kaech, S.M. IL-7-Induced Glycerol Transport and TAG Synthesis Promotes Memory CD8+ T Cell Longevity. Cell 2015, 161, 750–761. [Google Scholar] [CrossRef] [Green Version]
- Rubinstein, M.P.; Su, E.W.; Suriano, S.; Cloud, C.A.; Andrijauskaite, K.; Kesarwani, P.; Schwartz, K.M.; Williams, K.M.; Johnson, C.B.; Li, M.; et al. Interleukin-12 enhances the function and anti-tumor activity in murine and human CD8(+) T cells. Cancer Immunol. Immunother. 2015, 64, 539–549. [Google Scholar] [CrossRef] [Green Version]
- Andrijauskaite, K.; Suriano, S.; Cloud, C.A.; Li, M.; Kesarwani, P.; Stefanik, L.S.; Moxley, K.M.; Salem, M.L.; Garrett-Mayer, E.; Paulos, C.M.; et al. IL-12 conditioning improves retrovirally mediated transduction efficiency of CD8+ T cells. Cancer Gene 2015, 22, 360–367. [Google Scholar] [CrossRef] [Green Version]
- Johnson, C.B.; Wrangle, J.; Mehrotra, S.; Li, Z.; Paulos, C.M.; Cole, D.J.; Surh, C.D.; Rubinstein, M.P. Harnessing the IL-7/IL-7Ralpha axis to improve tumor immunotherapy. Oncoimmunology 2016, 5, e1122865. [Google Scholar] [CrossRef] [Green Version]
- Vacaflores, A.; Chapman, N.M.; Harty, J.T.; Richer, M.J.; Houtman, J.C. Exposure of Human CD4 T Cells to IL-12 Results in Enhanced TCR-Induced Cytokine Production, Altered TCR Signaling, and Increased Oxidative Metabolism. PLoS ONE 2016, 11, e0157175. [Google Scholar] [CrossRef] [Green Version]
- Schurich, A.; Pallett, L.J.; Jajbhay, D.; Wijngaarden, J.; Otano, I.; Gill, U.S.; Hansi, N.; Kennedy, P.T.; Nastouli, E.; Gilson, R.; et al. Distinct Metabolic Requirements of Exhausted and Functional Virus-Specific CD8 T Cells in the Same Host. Cell Rep. 2016, 16, 1243–1252. [Google Scholar] [CrossRef] [Green Version]
- Hinrichs, C.S.; Spolski, R.; Paulos, C.M.; Gattinoni, L.; Kerstann, K.W.; Palmer, D.C.; Klebanoff, C.A.; Rosenberg, S.A.; Leonard, W.J.; Restifo, N.P. IL-2 and IL-21 confer opposing differentiation programs to CD8+ T cells for adoptive immunotherapy. Blood 2008, 111, 5326–5333. [Google Scholar] [CrossRef] [Green Version]
- Chapuis, A.G.; Lee, S.M.; Thompson, J.A.; Roberts, I.M.; Margolin, K.A.; Bhatia, S.; Sloan, H.L.; Lai, I.; Wagener, F.; Shibuya, K.; et al. Combined IL-21-primed polyclonal CTL plus CTLA4 blockade controls refractory metastatic melanoma in a patient. J. Exp. Med. 2016, 213, 1133–1139. [Google Scholar] [CrossRef]
- Chapuis, A.G.; Roberts, I.M.; Thompson, J.A.; Margolin, K.A.; Bhatia, S.; Lee, S.M.; Sloan, H.L.; Lai, I.P.; Farrar, E.A.; Wagener, F.; et al. T-Cell Therapy Using Interleukin-21-Primed Cytotoxic T-Cell Lymphocytes Combined With Cytotoxic T-Cell Lymphocyte Antigen-4 Blockade Results in Long-Term Cell Persistence and Durable Tumor Regression. J. Clin. Oncol. 2016, 34, 3787–3795. [Google Scholar] [CrossRef] [PubMed]
- Loschinski, R.; Böttcher, M.; Stoll, A.; Bruns, H.; Mackensen, A.; Mougiakakos, D. IL-21 modulates memory and exhaustion phenotype of T-cells in a fatty acid oxidation-dependent manner. Oncotarget 2018, 9, 13125–13138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Y.; Xie, Y.-Q.; Gao, M.; Zhao, Y.; Franco, F.; Wenes, M.; Siddiqui, I.; Bevilacqua, A.; Wang, H.; Yang, H.; et al. Metabolic reprogramming of terminally exhausted CD8+ T cells by IL-10 enhances anti-tumor immunity. Nat. Immunol. 2021, 22, 746–756. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Qu, G.; Yu, X.; Jiang, H.; Teng, X.-L.; Ding, L.; Hu, Q.; Guo, X.; Zhou, Y.; Wang, F.; et al. Acylglycerol Kinase Maintains Metabolic State and Immune Responses of CD8+ T Cells. Cell Metab. 2019, 30, 290–302.e295. [Google Scholar] [CrossRef]
- Monjazeb, A.M.; Kent, M.S.; Grossenbacher, S.K.; Mall, C.; Zamora, A.E.; Mirsoian, A.; Chen, M.; Kol, A.; Shiao, S.L.; Reddy, A.; et al. Blocking Indolamine-2,3-Dioxygenase Rebound Immune Suppression Boosts Antitumor Effects of Radio-Immunotherapy in Murine Models and Spontaneous Canine Malignancies. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2016, 22, 4328–4340. [Google Scholar] [CrossRef] [Green Version]
- Stagg, J.; Divisekera, U.; McLaughlin, N.; Sharkey, J.; Pommey, S.; Denoyer, D.; Dwyer, K.M.; Smyth, M.J. Anti-CD73 antibody therapy inhibits breast tumor growth and metastasis. Proc. Natl. Acad. Sci. USA 2010, 107, 1547–1552. [Google Scholar] [CrossRef] [Green Version]
- Allard, B.; Pommey, S.; Smyth, M.J.; Stagg, J. Targeting CD73 enhances the antitumor activity of anti-PD-1 and anti-CTLA-4 mAbs. Clin. Cancer Res. 2013, 19, 5626–5635. [Google Scholar] [CrossRef] [Green Version]
- Hay, C.M.; Sult, E.; Huang, Q.; Mulgrew, K.; Fuhrmann, S.R.; McGlinchey, K.A.; Hammond, S.A.; Rothstein, R.; Rios-Doria, J.; Poon, E.; et al. Targeting CD73 in the tumor microenvironment with MEDI9447. Oncoimmunology 2016, 5, e1208875. [Google Scholar] [CrossRef]
- Hayes, G.M.; Cairns, B.; Levashova, Z.; Chinn, L.; Perez, M.; Theunissen, J.W.; Liao-Chan, S.; Bermudez, A.; Flory, M.R.; Schweighofer, K.J.; et al. CD39 is a promising therapeutic antibody target for the treatment of soft tissue sarcoma. Am. J. Transl. Res. 2015, 7, 1181–1188. [Google Scholar] [PubMed]
- Sun, X.; Wu, Y.; Gao, W.; Enjyoji, K.; Csizmadia, E.; Müller, C.E.; Murakami, T.; Robson, S.C. CD39/ENTPD1 expression by CD4+Foxp3+ regulatory T cells promotes hepatic metastatic tumor growth in mice. Gastroenterology 2010, 139, 1030–1040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, S.R.; Deng, W.W.; Liu, J.F.; Mao, L.; Yu, G.T.; Bu, L.L.; Kulkarni, A.B.; Zhang, W.F.; Sun, Z.J. Blockade of adenosine A2A receptor enhances CD8(+) T cells response and decreases regulatory T cells in head and neck squamous cell carcinoma. Mol. Cancer 2017, 16, 99. [Google Scholar] [CrossRef]
- Mittal, D.; Young, A.; Stannard, K.; Yong, M.; Teng, M.W.; Allard, B.; Stagg, J.; Smyth, M.J. Antimetastatic effects of blocking PD-1 and the adenosine A2A receptor. Cancer Res. 2014, 74, 3652–3658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leone, R.D.; Sun, I.M.; Oh, M.H.; Sun, I.H.; Wen, J.; Englert, J.; Powell, J.D. Inhibition of the adenosine A2a receptor modulates expression of T cell coinhibitory receptors and improves effector function for enhanced checkpoint blockade and ACT in murine cancer models. Cancer Immunol. Immunother. 2018, 67, 1271–1284. [Google Scholar] [CrossRef]
- Chung, K.Y.; Park, H.; Abdul-Karim, R.M.; Doroshow, D.B.; Chaves, J.; Coleman, T.A.; Nakai, K.; Patel, P.; Wang, J.; Zhang, H.; et al. Phase I study of BJ-001, a tumor-targeting interleukin-15 fusion protein, in patients with solid tumor. J. Clin. Oncol. 2021, 39, e14545. [Google Scholar] [CrossRef]
- Hamilton, E.; Nikiforow, S.; Bardwell, P.; McInnis, C.; Zhang, J.; Blumenschein, G.; Cristea, M.; Osman, K.; Shields, A.; Motta, M.; et al. 801 PRIME™ IL-15 (RPTR-147): Preliminary clinical results and biomarker analysis from a first-in-human Phase 1 study of IL-15 loaded peripherally-derived autologous T cell therapy in solid tumor patients. J. ImmunoTher. Cancer 2020, 8, A479. [Google Scholar] [CrossRef]
- Liu, Y.; Zhu, B.; Jia, Q.; He, F.; Fang, H.; Chen, J.; Shi, Z.; Jin, T.; Wei, B.; Xie, R.; et al. Durable clinical responses observed from non-Hodgkin lymphoma patients treated with autologous CAR-T cells targeting CD19. J. Clin. Oncol. 2018, 36, 3045. [Google Scholar] [CrossRef]
- Conlon, K.; Leidner, R.; McNeel, D.; Gupta, S.; Wang-Gillam, A.; Waldmann, T.A.; Pavlakis, G.; Dostalek, M.; Kurtulus, S.; Chowdhury, N.R.; et al. Abstract CT082: Phase (Ph) I/Ib study of NIZ985 with and without spartalizumab (PDR001) in patients (pts) with metastatic/unresectable solid tumors. Cancer Res. 2019, 79, CT082. [Google Scholar] [CrossRef]
- Rosser, C.J.; Nix, J.; Ferguson, L.; Hernandez, L.; Wong, H.C. Phase Ib trial of ALT-803, an IL-15 superagonist, plus BCG for the treatment of BCG-naïve patients with non-muscle-invasive bladder cancer. J. Clin. Oncol. 2018, 36, 510. [Google Scholar] [CrossRef]
- Miller, J.S.; Morishima, C.; McNeel, D.G.; Patel, M.R.; Kohrt, H.E.K.; Thompson, J.A.; Sondel, P.M.; Wakelee, H.A.; Disis, M.L.; Kaiser, J.C.; et al. A First-in-Human Phase I Study of Subcutaneous Outpatient Recombinant Human IL15 (rhIL15) in Adults with Advanced Solid Tumors. Clin. Cancer Res. 2018, 24, 1525–1535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conlon, K.C.; Potter, E.L.; Pittaluga, S.; Lee, C.R.; Miljkovic, M.D.; Fleisher, T.A.; Dubois, S.; Bryant, B.R.; Petrus, M.; Perera, L.P.; et al. IL15 by Continuous Intravenous Infusion to Adult Patients with Solid Tumors in a Phase I Trial Induced Dramatic NK-Cell Subset Expansion. Clin. Cancer Res. 2019, 25, 4945–4954. [Google Scholar] [CrossRef] [PubMed]
- Conlon, K.C.; Lugli, E.; Welles, H.C.; Rosenberg, S.A.; Fojo, A.T.; Morris, J.C.; Fleisher, T.A.; Dubois, S.P.; Perera, L.P.; Stewart, D.M.; et al. Redistribution, hyperproliferation, activation of natural killer cells and CD8 T cells, and cytokine production during first-in-human clinical trial of recombinant human interleukin-15 in patients with cancer. J. Clin. Oncol. 2015, 33, 74–82. [Google Scholar] [CrossRef]
- Grünwald, V.; Weikert, S.; Schmidt-Wolf, I.G.H.; Hauser, S.; Magheli, A.; Schroff, M.; Schmidt, M.; Wittig, B. Final results of patients with metastatic renal cell carcinoma treated with MGN1601 in the ASET study. J. Clin. Oncol. 2014, 32, e15590. [Google Scholar] [CrossRef]
- Pang, N.; Shi, J.; Qin, L.; Chen, A.; Tang, Y.; Yang, H.; Huang, Y.; Wu, Q.; Li, X.; He, B.; et al. IL-7 and CCL19-secreting CAR-T cell therapy for tumors with positive glypican-3 or mesothelin. J. Hematol. Oncol. 2021, 14, 118. [Google Scholar] [CrossRef] [PubMed]
- Timmerman, J.M.; Byrd, J.C.; Andorsky, D.J.; Yamada, R.E.; Kramer, J.; Muthusamy, N.; Hunder, N.; Pagel, J.M. A phase I dose-finding trial of recombinant interleukin-21 and rituximab in relapsed and refractory low grade B-cell lymphoproliferative disorders. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2012, 18, 5752–5760. [Google Scholar] [CrossRef] [Green Version]
- Bhatia, S.; Curti, B.; Ernstoff, M.S.; Gordon, M.; Heath, E.I.; Miller, W.H., Jr.; Puzanov, I.; Quinn, D.I.; Flaig, T.W.; VanVeldhuizen, P.; et al. Recombinant interleukin-21 plus sorafenib for metastatic renal cell carcinoma: A phase 1/2 study. J. Immunother. Cancer 2014, 2, 2. [Google Scholar] [CrossRef] [Green Version]
- Youngblood, B.; Hale, J.S.; Kissick, H.T.; Ahn, E.; Xu, X.; Wieland, A.; Araki, K.; West, E.E.; Ghoneim, H.E.; Fan, Y.; et al. Effector CD8 T cells dedifferentiate into long-lived memory cells. Nature 2017, 552, 404–409. [Google Scholar] [CrossRef]
- Abdelsamed, H.A.; Zebley, C.C.; Youngblood, B. Epigenetic Maintenance of Acquired Gene Expression Programs during Memory CD8 T Cell Homeostasis. Front. Immunol. 2018, 9, 6. [Google Scholar] [CrossRef]
- Sen, D.R.; Kaminski, J.; Barnitz, R.A.; Kurachi, M.; Gerdemann, U.; Yates, K.B.; Tsao, H.W.; Godec, J.; LaFleur, M.W.; Brown, F.D.; et al. The epigenetic landscape of T cell exhaustion. Science 2016, 354, 1165–1169. [Google Scholar] [CrossRef] [Green Version]
- Schietinger, A.; Philip, M.; Krisnawan, V.E.; Chiu, E.Y.; Delrow, J.J.; Basom, R.S.; Lauer, P.; Brockstedt, D.G.; Knoblaugh, S.E.; Hammerling, G.J.; et al. Tumor-Specific T Cell Dysfunction Is a Dynamic Antigen-Driven Differentiation Program Initiated Early during Tumorigenesis. Immunity 2016, 45, 389–401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pauken, K.E.; Sammons, M.A.; Odorizzi, P.M.; Manne, S.; Godec, J.; Khan, O.; Drake, A.M.; Chen, Z.; Sen, D.R.; Kurachi, M.; et al. Epigenetic stability of exhausted T cells limits durability of reinvigoration by PD-1 blockade. Science 2016, 354, 1160–1165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghoneim, H.E.; Zamora, A.E.; Thomas, P.G.; Youngblood, B.A. Cell-Intrinsic Barriers of T Cell-Based Immunotherapy. Trends Mol. Med. 2016, 22, 1000–1011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, L.; Orillion, A.; Pili, R. Histone deacetylase inhibitors as immunomodulators in cancer therapeutics. Epigenomics 2016, 8, 415–428. [Google Scholar] [CrossRef]
- Grunstein, M. Histone acetylation in chromatin structure and transcription. Nature 1997, 389, 349–352. [Google Scholar] [CrossRef]
- Pietrocola, F.; Galluzzi, L.; José, M.; Madeo, F.; Kroemer, G. Acetyl Coenzyme A: A Central Metabolite and Second Messenger. Cell Metab. 2015, 21, 805–821. [Google Scholar] [CrossRef] [Green Version]
- Xiao, H.; Jiao, J.; Wang, L.; O’Brien, S.; Newick, K.; Wang, L.-C.S.; Falkensammer, E.; Liu, Y.; Han, R.; Kapoor, V.; et al. HDAC5 controls the functions of Foxp3+ T-regulatory and CD8+ T cells. Int. J. Cancer 2016, 138, 2477–2486. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Wang, L.; Predina, J.; Han, R.; Beier, U.H.; Wang, L.C.; Kapoor, V.; Bhatti, T.R.; Akimova, T.; Singhal, S.; et al. Inhibition of p300 impairs Foxp3+ T regulatory cell function and promotes antitumor immunity. Nat. Med. 2013, 19, 1173–1177. [Google Scholar] [CrossRef]
- Barski, A.; Cuddapah, S.; Cui, K.; Roh, T.-Y.; Schones, D.E.; Wang, Z.; Wei, G.; Chepelev, I.; Zhao, K. High-Resolution Profiling of Histone Methylations in the Human Genome. Cell 2007, 129, 823–837. [Google Scholar] [CrossRef] [Green Version]
- Guenther, M.G.; Levine, S.S.; Boyer, L.A.; Jaenisch, R.; Young, R.A. A chromatin landmark and transcription initiation at most promoters in human cells. Cell 2007, 130, 77–88. [Google Scholar] [CrossRef] [Green Version]
- Chisolm, D.A.; Weinmann, A.S. Connections Between Metabolism and Epigenetics in Programming Cellular Differentiation. Annu. Rev. Immunol. 2018, 36, 221–246. [Google Scholar] [CrossRef]
- Sinclair, L.V.; Howden, A.J.; Brenes, A.; Spinelli, L.; Hukelmann, J.L.; Macintyre, A.N.; Liu, X.; Thomson, S.; Taylor, P.M.; Rathmell, J.C.; et al. Antigen receptor control of methionine metabolism in T cells. Elife 2019, 8, e44210. [Google Scholar] [CrossRef] [PubMed]
- Roy, D.G.; Chen, J.; Mamane, V.; Ma, E.H.; Muhire, B.M.; Sheldon, R.D.; Shorstova, T.; Koning, R.; Johnson, R.M.; Esaulova, E.; et al. Methionine Metabolism Shapes T Helper Cell Responses through Regulation of Epigenetic Reprogramming. Cell Metab. 2020, 31, 250–266.e259. [Google Scholar] [CrossRef]
- Youngblood, B.A.; Ghoniem, H.; Moustaki, A.; Abdelsamed, H.A.; Fan, Y.; Thomas, P.G.; Federico, S.; Stewart, E. De novo DNA methylation programs restrain T cell rejuvenation during immune checkpoint blockade therapy. J. Immunol. 2018, 200, 57.21. [Google Scholar]
- Scott, A.C.; Dündar, F.; Zumbo, P.; Chandran, S.S.; Klebanoff, C.A.; Shakiba, M.; Trivedi, P.; Menocal, L.; Appleby, H.; Camara, S.; et al. TOX is a critical regulator of tumour-specific T cell differentiation. Nature 2019, 571, 270–274. [Google Scholar] [CrossRef]
- Seo, H.; Chen, J.; González-Avalos, E.; Samaniego-Castruita, D.; Das, A.; Wang, Y.H.; López-Moyado, I.F.; Georges, R.O.; Zhang, W.; Onodera, A.; et al. TOX and TOX2 transcription factors cooperate with NR4A transcription factors to impose CD8+ T cell exhaustion. Proc. Natl. Acad. Sci. USA 2019, 116, 12410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alfei, F.; Kanev, K.; Hofmann, M.; Wu, M.; Ghoneim, H.E.; Roelli, P.; Utzschneider, D.T.; von Hoesslin, M.; Cullen, J.G.; Fan, Y.; et al. TOX reinforces the phenotype and longevity of exhausted T cells in chronic viral infection. Nature 2019, 571, 265–269. [Google Scholar] [CrossRef]
- Wang, X.; He, Q.; Shen, H.; Xia, A.; Tian, W.; Yu, W.; Sun, B. TOX promotes the exhaustion of antitumor CD8+ T cells by preventing PD1 degradation in hepatocellular carcinoma. J. Hepatol. 2019, 71, 731–741. [Google Scholar] [CrossRef]
- Khan, O.; Giles, J.R.; McDonald, S.; Manne, S.; Ngiow, S.F.; Patel, K.P.; Werner, M.T.; Huang, A.C.; Alexander, K.A.; Wu, J.E.; et al. TOX transcriptionally and epigenetically programs CD8+ T cell exhaustion. Nature 2019, 571, 211–218. [Google Scholar] [CrossRef]
- He, S.; Liu, Y.; Meng, L.; Sun, H.; Wang, Y.; Ji, Y.; Purushe, J.; Chen, P.; Li, C.; Madzo, J.; et al. Ezh2 phosphorylation state determines its capacity to maintain CD8+ T memory precursors for antitumor immunity. Nat. Commun. 2017, 8, 2125. [Google Scholar] [CrossRef] [Green Version]
- Araki, Y.; Wang, Z.; Zang, C.; Wood, W.H., 3rd; Schones, D.; Cui, K.; Roh, T.Y.; Lhotsky, B.; Wersto, R.P.; Peng, W.; et al. Genome-wide analysis of histone methylation reveals chromatin state-based regulation of gene transcription and function of memory CD8+ T cells. Immunity 2009, 30, 912–925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russ, B.E.; Olshanksy, M.; Smallwood, H.S.; Li, J.; Denton, A.E.; Prier, J.E.; Stock, A.T.; Croom, H.A.; Cullen, J.G.; Nguyen, M.L.; et al. Distinct epigenetic signatures delineate transcriptional programs during virus-specific CD8(+) T cell differentiation. Immunity 2014, 41, 853–865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, G.; Wei, L.; Zhu, J.; Zang, C.; Hu-Li, J.; Yao, Z.; Cui, K.; Kanno, Y.; Roh, T.Y.; Watford, W.T.; et al. Global mapping of H3K4me3 and H3K27me3 reveals specificity and plasticity in lineage fate determination of differentiating CD4+ T cells. Immunity 2009, 30, 155–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Navas, L.E.; Carnero, A. NAD+ metabolism, stemness, the immune response, and cancer. Signal. Transduct. Target. Ther. 2021, 6, 2. [Google Scholar] [CrossRef]
- Philip, M.; Fairchild, L.; Sun, L.; Horste, E.L.; Camara, S.; Shakiba, M.; Scott, A.C.; Viale, A.; Lauer, P.; Merghoub, T.; et al. Chromatin states define tumour-specific T cell dysfunction and reprogramming. Nature 2017, 545, 452–456. [Google Scholar] [CrossRef] [PubMed]
- Pan, D.; Kobayashi, A.; Jiang, P.; Ferrari de Andrade, L.; Tay, R.E.; Luoma, A.M.; Tsoucas, D.; Qiu, X.; Lim, K.; Rao, P.; et al. A major chromatin regulator determines resistance of tumor cells to T cell-mediated killing. Science 2018, 359, 770–775. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Diao, L.; Yang, Y.; Yi, X.; Rodriguez, B.L.; Li, Y.; Villalobos, P.A.; Cascone, T.; Liu, X.; Tan, L.; et al. CD38-mediated immunosuppression as a mechanism of tumor cell escape from PD-1/PD-L1 blockade. Cancer Discov 2018, 8, 1156–1175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verma, V.; Shrimali, R.K.; Ahmad, S.; Dai, W.; Wang, H.; Lu, S.; Nandre, R.; Gaur, P.; Lopez, J.; Sade-Feldman, M.; et al. PD-1 blockade in subprimed CD8 cells induces dysfunctional PD-1(+)CD38(hi) cells and anti-PD-1 resistance. Nat. Immunol. 2019, 20, 1231–1243. [Google Scholar] [CrossRef]
- Iversen, T.Z.; Engell-Noerregaard, L.; Ellebaek, E.; Andersen, R.; Larsen, S.K.; Bjoern, J.; Zeyher, C.; Gouttefangeas, C.; Thomsen, B.M.; Holm, B.; et al. Long-lasting disease stabilization in the absence of toxicity in metastatic lung cancer patients vaccinated with an epitope derived from indoleamine 2,3 dioxygenase. Clin. Cancer Res. 2014, 20, 221–232. [Google Scholar] [CrossRef] [Green Version]
- Kristeleit, R.; Davidenko, I.; Shirinkin, V.; El-Khouly, F.; Bondarenko, I.; Goodheart, M.J.; Gorbunova, V.; Penning, C.A.; Shi, J.G.; Liu, X.; et al. A randomised, open-label, phase 2 study of the IDO1 inhibitor epacadostat (INCB024360) versus tamoxifen as therapy for biochemically recurrent (CA-125 relapse)-only epithelial ovarian cancer, primary peritoneal carcinoma, or fallopian tube cancer. Gynecol. Oncol. 2017, 146, 484–490. [Google Scholar] [CrossRef]
- Mariotti, V.; Han, H.; Ismail-Khan, R.; Tang, S.C.; Dillon, P.; Montero, A.J.; Poklepovic, A.; Melin, S.; Ibrahim, N.K.; Kennedy, E.; et al. Effect of Taxane Chemotherapy With or Without Indoximod in Metastatic Breast Cancer: A Randomized Clinical Trial. JAMA Oncol. 2021, 7, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Kotecki, N.; O’Neil, B.; Jalal, S.; Massard, C.; Wallin, J.; Szpurka, A.; Wang, D.; Galvao, V.R.; Xia, M.S.; Crowe, K.; et al. A Phase I Study of an Anti-IDO1 Inhibitor (LY3381916) as Monotherapy and in Combination with an Anti-PD-L1 Antibody (LY3300054) in Patients with Advanced Cancer. Ann. Oncol. 2019, 30, xi41–xi42. [Google Scholar] [CrossRef]
- Zakharia, Y.; Rixe, O.; Ward, J.H.; Drabick, J.J.; Shaheen, M.F.; Milhem, M.M.; Munn, D.; Kennedy, E.P.; Vahanian, N.N.; Link, C.J.; et al. Phase 2 trial of the IDO pathway inhibitor indoximod plus checkpoint inhibition for the treatment of patients with advanced melanoma. J. Clin. Oncol. 2018, 36, 9512. [Google Scholar] [CrossRef]
- Nayak-Kapoor, A.; Hao, Z.; Sadek, R.; Dobbins, R.; Marshall, L.; Vahanian, N.N.; Jay Ramsey, W.; Kennedy, E.; Mautino, M.R.; Link, C.J.; et al. Phase Ia study of the indoleamine 2,3-dioxygenase 1 (IDO1) inhibitor navoximod (GDC-0919) in patients with recurrent advanced solid tumors. J. Immunother. Cancer 2018, 6, 61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bahary, N.; Wang-Gillam, A.; Haraldsdottir, S.; Somer, B.G.; Lee, J.S.; O’Rourke, M.A.; Nayak-Kapoor, A.; Beatty, G.L.; Liu, M.; Delman, D.; et al. Phase 2 trial of the IDO pathway inhibitor indoximod plus gemcitabine/nab-paclitaxel for the treatment of patients with metastatic pancreas cancer. J. Clin. Oncol. 2018, 36, 4015. [Google Scholar] [CrossRef]
- Johnson, T.S.; Aguilera, D.; Al-Basheer, A.; Berrong, Z.; Castellino, R.C.; Eaton, B.R.; Esiashvili, N.; Foreman, N.; Heger, I.M.; Kennedy, E.P.; et al. Results of the NLG2105 Phase I Trial Using the IDO Pathway Inhibitor Indoximod, in Combination with Radiation and Chemotherapy, for Children with Newly Diagnosed DIPG. Ann. Oncol. 2019, 30, xi38. [Google Scholar] [CrossRef]
- Kelly, C.M.; Chi, P.; Dickson, M.A.; Gounder, M.M.; Keohan, M.L.; Qin, L.-X.; Adamson, T.; Condy, M.M.; Biniakewitz, M.; Phelan, H.; et al. A phase II study of epacadostat and pembrolizumab in patients with advanced sarcoma. J. Clin. Oncol. 2019, 37, 11049. [Google Scholar] [CrossRef]
- Cheng, Y.; Liu, Y.; Xu, J.; Zhu, J.; Wang, Y.; Xin, Y.; Wang, Y.; Wu, C.; Cui, H.; Liu, X.; et al. A phase I study of an IDO inhibitor (SHR9146) plus camrelizumab and in combination with/without apatinib in patients with advanced solid tumors: Safety and efficacy analysis. J. Clin. Oncol. 2021, 39, 3101. [Google Scholar] [CrossRef]
- Chiappori, A.; Williams, C.C.; Creelan, B.C.; Tanvetyanon, T.; Gray, J.E.; Haura, E.B.; Thapa, R.; Chen, D.-T.; Beg, A.A.; Boyle, T.A.; et al. Phase I/II study of the A2AR antagonist NIR178 (PBF-509), an oral immunotherapy, in patients (pts) with advanced NSCLC. J. Clin. Oncol. 2018, 36, 9089. [Google Scholar] [CrossRef]
- Lim, E.A.; Bauer, T.M.; Patel, M.R.; Falchook, G.S.; Karlix, J.L.; Choe, J.H.; George, D.J.; Mugundu, G.M.; Pilling, E.; Chen, H.; et al. A phase I, open-label, multicenter study to assess the safety, pharmacokinetics, and preliminary antitumor activity of AZD4635 both as monotherapy and in combination in patients with advanced solid malignancies: Results from prostate cancer patients (NCT02740985). J. Clin. Oncol. 2020, 38, 5518. [Google Scholar] [CrossRef]
- Siu, L.L.; Burris, H.; Le, D.T.; Hollebecque, A.; Steeghs, N.; Delord, J.-P.; Hilton, J.; Barnhart, B.; Sega, E.; Sanghavi, K.; et al. Abstract CT180: Preliminary phase 1 profile of BMS-986179, an anti-CD73 antibody, in combination with nivolumab in patients with advanced solid tumors. Cancer Res. 2018, 78, CT180. [Google Scholar] [CrossRef]
- Luke, J.J.; Powderly, J.D.; Merchan, J.R.; Barve, M.A.; Hotson, A.N.; Mobasher, M.; Kwei, L.; Luciano, G.; Buggy, J.J.; Piccione, E.; et al. Immunobiology, preliminary safety, and efficacy of CPI-006, an anti-CD73 antibody with immune modulating activity, in a phase 1 trial in advanced cancers. J. Clin. Oncol. 2019, 37, 2505. [Google Scholar] [CrossRef]
- Bendell, J.C.; LoRusso, P.; Overman, M.J.; Noonan, A.M.; Kim, D.-W.; Strickler, J.; Kim, S.-W.; Clarke, S.J.; George, T.J.; Grimison, P.S.; et al. Safety and efficacy of the anti-CD73 monoclonal antibody (mAb) oleclumab ± durvalumab in patients (pts) with advanced colorectal cancer (CRC), pancreatic ductal adenocarcinoma (PDAC), or EGFR-mutant non-small cell lung cancer (EGFRm NSCLC). J. Clin. Oncol. 2021, 39, 9047. [Google Scholar] [CrossRef]
- Villar, M.V.; Simonelli, M.; Eskens, F.A.L.M.; Gil-Martin, M.; Yen, C.-J.; Obermannova, R.; Chao, Y.; Zagonel, V.; Melichar, B.; Moreno, V.; et al. Abstract CT154: Isatuximab (Isa) plus atezolizumab (Atezo) in patients (pts) with advanced malignancies: Results from a Phase 1/2 ½n-label multicenter study. Cancer Res. 2021, 81, CT154. [Google Scholar] [CrossRef]
- Lin, C.-C.; Zucali, P.; Carthon, B.; Bauer, T.M.; Tucci, M.; Italiano, A.; Iacovelli, R.; Su, W.-C.; Massard, C.; Saleh, M.; et al. Abstract LB040: Targeting CD38 and PD-1 with isatuximab (Isa) plus cemiplimab (Cemi) in patients (pts) with advanced malignancies: Results from a Phase 1/2 ½n-label, multicenter study. Cancer Res. 2021, 81, LB040. [Google Scholar] [CrossRef]


{kind=link}
| Metabolite | Role in Antitumor Immune Response | Potential Therapeutic Strategies | Ref. |
|---|---|---|---|
| Fatty acids | Fatty acid oxidation promotes and sustains memory T cell development while also supporting the immunosuppressive function of Tregs | Upregulate or overexpress fatty acid transporters and enzymes involved in FAO in antitumor T cells while inhibiting fatty acid uptake in Tregs | [119] |
| S1P | S1P promotes Treg development via PPARγ and contributes to limited antitumor immune response | Inhibit sphingosine kinase-1 activity in antitumor T cells and Tregs | [34] |
| L-arginine | Increased L-arginine in activated T cells shifts metabolism from glycolysis to OXPHOS, increasing central memory T cells and promoting antitumor activity | Increase extracellular L-arginine levels and increase L-arginine cellular transporters | [36] |
| Serine | Extracellular serine is necessary to support optimal T cell activation and proliferation | Increase extracellular serine levels and increase L-arginine cellular transporters | [37] |
| Lactate | Lactate impairs T cell activation and function, especially blunting activation of NFAT and production of IFNγ | Reduce lactate production by targeting lactate dehydrogenase A (LDHA) | [49,51] |
| Kynurenine | Kynurenine is produced by tumors cells, TAMs, and MDSCs via the enzyme IDO, inhibiting proliferation and effector function of effector T cells and inducing the expansion of Tregs | Block the production of kynurenine by inhibiting IDO activity | [61,63,64,65,68] |
| Tryptophan | Tryptophan is a key nutrient supporting antitumor T cell expansion and effector function. Depletion of tryptophan by IDO deprives T cells of this crucial nutrient and induces a decrease in global protein synthesis, and ultimately leads to T cell anergy | Block the consumption of tryptophan by tumor cells, TAMs, and MDSCs by inhibiting IDO activity | [67] |
| Adenosine | Adenosine interacts with the A2a receptor on effector T cells, inhibiting TCR signaling and expression of the IL-2 receptor while also upregulating the expression of immune checkpoint molecules | Decrease adenosine signaling or overall adenosine levels in the TME by targeting the A2a receptor or inhibiting CD39/CD73, the enzymes responsible for converting ATP to adenosine | [70,71] |
| Glutamine | Glutamine supports cancer cell growth while restricting glucose utilization and glycolysis in antitumor T cells, leading to metabolic dysfunction | Restrict glutamine uptake in the TME using inhibitors of glutamine transport | [74,116] |
| Acetate | Acetate promotes histone acetylation and chromatin accessibility in TILs and enhances IFNγ gene transcription and cytokine production in an acetyl-CoA synthetase (ACSS)-dependent manner | Supplement TILs with acetate and/or overexpress acetyl-CoA synthetase (ACSS) | [82] |
| NAD+ | NAD+ serves as a key substrate for the histone deacetylase Sirt1 and contributes to post-translational modifications and epigenetic stability that lead to fit antitumor memory T cells metabolically | Increase intracellular levels of NAD+ in antitumor T cells, for example, by targeting CD38 expression or by programming T cells to have a hybrid Th1/17 phenotype | [93,126,127,128] |
| Acetyl-CoA | Acetyl-CoA is transferred by HATs for histone acetylation, epigenetically programming both antitumor T cells and Tregs | Determine how acetyl-CoA-producing metabolic processes can be altered to optimize epigenetic programming of antitumor T cells | [119] |
| S-2HG | S-2-hydroxyglutarate drives epigenetic remodeling to enhance IL-2 production in antitumor T cells | Culture antitumor T cells with S-2HG ex vivo to increase the central memory CD8+ population and enhance their antitumor efficacy | [129,130] |
| Target | Model | Strategy | Outcome | Ref. |
|---|---|---|---|---|
| Glycolysis | Murine melanoma | Activate adoptively transferred T cells in the presence of 2-deoxyglucose (glycolysis inhibitor) | Inhibition of glycolysis enhanced generation of memory and antitumor efficacy | [22] |
| Glycolysis/ FAO | Murine viral infection | Systemic administration of rapamycin to inhibit glycolysis and promote FAO | Systemic treatment with rapamycin enhanced memory T cell development and antitumor efficacy | [32] |
| Lipids (AGK) | Murine melanoma and colon cancer | Increase AGK activity in antitumor T cells | AKG-triggered PTEN inactivation promote glycolytic fitness and antitumor efficacy in CD8+ T cells | [186] |
| Lipids (S1P) | Murine melanoma | Pharmacological inhibition of sphingosine kinase 1 to decrease levels of S1P | Inhibition of sphingosine kinase 1 improved metabolic fitness and efficacy of antitumor T cells | [34] |
| L-arginine | Murine melanoma | Supplement antitumor T cells with L-arginine prior to ACT | L-arginine promoted a shift from glycolysis to OXPHOS and enhanced memory formation and antitumor efficacy | [36] |
| Mitochondrial metabolism | Murine melanoma | Selective transfer antitumor T cells with low mitochondrial membrane potential for ACT | ACT using T cells with low mitochondrial membrane potential resulted in superior antitumor efficacy | [38] |
| Mitochondrial metabolism | Murine melanoma | Enforce expression of PGC1a in T cells prior to ACT to enhance mitochondrial fitness | Enforced expression of PGC1a enhanced the ability of T cells to control tumor control | [80] |
| Thiols | Murine melanoma | Selective transfer antitumor T cells with high expression of surface thiols or treatment of T cells with thiol donors for ACT | Antitumor T cells expressing high levels of surface thiols demonstrated superior persistence and tumor control | [39] |
| Thioredoxin | Murine melanoma | Treatment of antitumor T cells with recombinant thioredoxin for ACT | Pre-treatment of T cells with thioredoxin resulted in enhanced persistence and tumor control upon ACT | [41] |
| Lactate | Murine melanoma | Reduce the activity of LDHA in tumor cells to decrease intratumoral levels of lactate | Reducing tumor cell production of lactate resulted in enhanced T cell infiltration and tumor control | [49] |
| Kynurenine | Mouse and canine melanoma, sarcoma, and breast cancer | Inhibit IDO enzyme activity in tumors to decrease levels of kynurenine and increase levels of tryptophan | Inhibition of IDO resulted in enhanced antitumor activity of CD8+ T cells and reduced frequencies of Tregs in the TME | [187] |
| Adenosine | Murine melanoma, colon, sarcoma, and breast cancer | Reduce intratumoral levels of adenosine by pharmacological inhibition of CD39/CD73 activity | Inhibition of CD39/CD73 ectonucleotidase activity decreased intratumoral levels of adenosine and enhanced the antitumor T cells response across multiple tumor models | [188,189,190,191,192] |
| Adenosine | Murine melanoma and head and neck cancer | Antagonize the A2AR receptor to enhance antitumor efficacy | Blockade of the A2AR resulted in enhanced tumor control alone and in combination with anti-PD-1 blockade | [193,194,195] |
| Leptin | Murine melanoma | Enhance the metabolic fitness of TILs using leptin-expressing oncolytic virus | Leptin expression in the TME reprogrammed TIL metabolism to promote tumor control | [81] |
| Acetate | Murine melanoma and lymphoma | Treat exhausted TILs with acetate to rescue effector function | Treatment of TILs ex vivo with acetate rescued their effector function and improved tumor control upon ACT | [82] |
| NAD+ | Murine melanoma | Increased intracellular NAD+ levels in antitumor T cells by blocking CD38 | Combining anti-CD38 antibody therapy with ACT resulted in enhanced tumor control and prolonged T cell survival | [93] |
| S-2HG | Murine lymphoma | Culture T cells with S-2HG to enhance IL-2 production and antitumor efficacy | Treatment of CAR-T ex vivo in the presence of S-2HG enhanced memory formation and antitumor efficacy | [130] |
| IL-15 | Murine melanoma and lymphoma | Selectively increase intratumoral levels of IL-15 using tumor-conditional pro-IL-15 | Use of tumor-conditional pro-IL-15 resulted in enhanced memory development and antitumor efficacy while minimizing systemic toxicity | [164] |
| IL-7 | Murine neuroblastoma and glioblastoma | Engineer CAR-T cells to express a constitutively active IL-7 receptor | Expression of constitutively active IL-7 receptor enhanced the persistence and antitumor efficacy of CAR-T cells | [174] |
| IL-12 | Murine melanoma | Condition antitumor T cells ex vivo with IL-12 prior to ACT | Ex vivo IL-12 conditioning enhanced in vivo expansion, proliferation, and antitumor efficacy of CD8+ T cells | [176] |
| IL-21 | Murine melanoma | Condition antitumor T cells ex vivo with IL-21 prior to ACT | Ex vivo IL-21 conditioning enhanced the capacity of T cells to mediate tumor regression upon ACT | [181] |
| IL-10 | Murine melanoma and colon cancer | Peritumoral administration of half-life-extended interleukin-10–Fc fusion protein, alone and in combination with anti-PD-1 therapy | IL-10–Fc fusion protein induced expansion of terminally exhausted TILs, upregulating OXPHOS and enhancing antitumor capacity of TILs | [185] |
| Clinical Trial | Phase | Intervention | Cancer Type | Outcomes | Status |
|---|---|---|---|---|---|
| NCT05103631 | I | IL-15 + GPC3-CAR-T cells | Hepatocellular carcinoma | N/A | Not yet recruiting (2021) |
| NCT04715191 | I | IL-15 + IL-21 + GPC3-CAR-T cells | Hepatocellular carcinoma | N/A | Not yet recruiting (2021) |
| NCT04628780 | I | anti-PD-1 targeting IL-15 fusion protein (PF07209960) | Advanced solid tumors | N/A | Recruiting (2021) |
| NCT04377932 | I | IL-15 + GPC3-CAR-T cells | Hepatocellular carcinoma | N/A | Recruiting (2021) |
| NCT04294576 | I | IL-15 fusion protein (BJ-001) subcutaneous injection + anti-PD-1 or anti-PD-L1 antibodies | Advanced solid tumors | BJ-001 is well tolerated up to 6 µg/kg [196]. | Recruiting (2020) |
| NCT04261439 | I | IL-15 (NIZ985) subcutaneous injection + anti-PD-1 antibody (spartalizumab) | Melanoma, advanced solid tumors | N/A | Recruiting (2021) |
| NCT03815682 | I | Autologous multi-clonal T cell product loaded with IL15-Fc nanogel (RPTR-147) +/− pembrolizumab | Solid tumors, lymphoma | In a cohort of 17 patients, no dose-limiting toxicities were observed. A dose-dependent increase in inflammatory cytokines and CD8+ TILs was observed [197]. | Recruiting (2021) |
| NCT03721068 | I | Autologous iC9.GD2.CAR.IL-15 T-cells + chemotherapy | Neuroblastoma, osteosarcoma | N/A | Recruiting (2021) |
| NCT03388632 | I | Recombinant IL-15 subcutaneous injection + nivolumab (anti-PD-1) or ipilimumab (anti-CTLA-4) | Metastatic solid tumors | N/A | Recruiting (2021) |
| NCT03127098 | I/II | IL-15 (ALT-803) + ETBX-011 vaccine | Advanced CEA-expressing cancers | N/A | Completed (2019) |
| NCT02652910 | I/II | IL-7/IL-15-programmed anti-CD19 CAR-T cells | Lymphoma | In a cohort of 18 patients, IL-7/IL-15-programmed anti-CD19 CAR-T cells offered substantial clinical benefit for NHL patients with manageable toxicities. ORR was 72.2% with complete remission of 36.7%. One case of CRS over grade 3 was observed [198]. | Unknown (2019) |
| NCT02559674 | I | IL-15 (ALT-803) + chemotherapy | Pancreatic cancer | N/A | Completed (2020) |
| NCT02452268 | I | IL-15 (NIZ985) subcutaneous injection + anti-PD-1 antibody (PDR001) | Metastatic and advanced solid tumors | Combination therapy was well tolerated in pts with advanced solid tumors. No DLTs were observed [199]. | Active, not recruiting (2021) |
| NCT02138734 | I/II | Intravesical IL-15 (ALT-803) + BCG cancer vaccine | Non-muscle invasive bladder cancer | Combination therapy was well-tolerated. All patients were disease-free 24 months following combination therapy [200]. | Recruiting (2021) |
| NCT01946789 | I | IL-15 (ALT-803) | Advanced solid tumors | N/A | Completed (2019) |
| NCT01727076 | I | Recombinant IL-15 subcutaneous injection | Advanced melanoma, kidney cancer, NSCLC, squamous cell head and neck cancer | Treatment was well tolerated. Substantial increases in circulating NK and CD8+ T cells was observed. A total of 2 SAEs were observed out of 19 [201]. | Completed (2017) |
| NCT01572493 | I | Recombinant IL-15 subcutaneous injection | Advanced cancers | 8 SAEs observed out of 27 patients. Significant expansion of circulating CD8+ T cells and NK cells was observed [202]. | Completed (2021) |
| NCT01369888 | I/II | IV IL-15 + TIL infusion | Metastatic melanoma | Study was terminated due to autoimmune toxicity. | Terminated (2015) |
| NCT01189383 | I/II | Autologous dendritic cells manufactured with GM-CSF and IL15 and loaded with melanoma/HIV peptides and KLH. | Metastatic melanoma | N/A | Completed (2016) |
| NCT01021059 | I | Recombinant IL-15 IV injection | Melanoma, renal cell carcinoma | Treatment could be safely administered (0.3 μg/kg). Dose limiting toxicities were observed in patients receiving higher doses (3.0 and 1.0 μg/kg). Clearance of lung lesions was observed in two patients. Significant expansion of NK and memory CD8+ T cells was observed [203] | Completed (2019) |
| NCT01265368 | I/II | Allogenic tumor cell vaccine expressing IL-7, GM-CSF, CD80, and CD154 | Renal cell cancer | MGN1601 treatment was safe and feasible and resulted in improved cellular immune function with preliminary clinical efficacy [204] | Completed (2018) |
| NCT03198546 | I | IL-7- and CCL19-secreting GPC3-specific CAR-T cells | GPC3+ HCC | IL-7 and CCL19-secreting CAR-T cells significantly enhanced antitumor activity, and the therapy was well-tolerated in a cohort of six patients [205] | Recruiting (2020) |
| NCT04099797 | I | GD2-specific CAR with constitutively active IL-7 receptors ((C7R)-GD2.CART) | High grade glioma, diffuse intrinsic pontine glioma | N/A | Recruiting (2021) |
| NCT03635632 | I | GD2-specific CAR with constitutively active IL-7 receptors ((C7R)-GD2.CART) | Refractory neuroblastoma and other GD2+ cancers | N/A | Recruiting (2021) |
| NCT02960594 | I | IL-12 DNA plasmid (INO-9012) + hTERT vaccine (INO-1400 or INO-1401) | Advanced solid tumors | N/A | Completed (2018) |
| NCT04911166 | I | Adenoviral-mediated interleukin-12 gene therapy +/− atezolizumab | NSCLC | N/A | Recruiting (2021) |
| NCT04756505 | I | NHS-IL-12 + bispecific anti-PD-1/TGFβ antibody (bintrafusp alfa) | Hormone receptor positive HER2 negative breast cancer | N/A | Recruiting (2021) |
| NCT01236573 | I/II | IL-12 gen-transduced TILs + chemotherapy | Metastatic melanoma | Terminated due to unexpected toxicities, likely due to TIL product and the low percentage of durable responses. | Terminated (2019) |
| NCT02062827 | I | Genetically engineered IL-12-expressing oncolytic herpes simplex virus (M032) | Glioblastoma multiforme | N/A | Recruiting (2021) |
| NCT00347971 | I | Recombinant IL-21 + rituximab | Non-Hodgkin lymphoma | Clinical responses were seen in 8 of 19 evaluable patients. Durable complete remission was observed in a small subset of patients. Therapy was well-tolerated [206] | Completed (2008) |
| NCT00389285 | I/II | Recombinant IL-21 +/− sorafenib | Renal cell carcinoma | ORR was 21% and disease control rate was 82% with combination therapy. In phase II, the therapy was well-tolerated with toxicities being mostly graded 1 or 2 [207] | Completed (2009) |
| Clinical Trial | Phase | Intervention | Cancer Type | Outcomes | Status |
|---|---|---|---|---|---|
| NCT01219348 | I | IDO peptide vaccination + imiquimod | NSCLC | Long-lasting disease stabilization without toxicity [239] | Completed 2015 |
| NCT03047928 | I/II | IDO peptide vaccination + anti-PD-1 antibody | Metastatic melanoma | Systemic toxicity profile was comparable to nivolumab monotherapy; objective response rate of 80% was reached. | Recruiting (2020) |
| NCT01685255 | II | IDO inhibitor, (epacadostat, INCB024360) | Ovarian cancer | Study terminated due to slow accrual and lack of evidence of superiority [240] | Terminated |
| NCT01792050 | II | IDO inhibitor (indoximod, NLG-8186) + Taxane chemotherapy | Metastatic breast cancer | Addition of indoximod to a taxane did not improve PFS compared with a taxane alone [241] | Completed (2020) |
| NCT03343613 | I | IDO-1 inhibitor (LY3381916) + anti-PD-L1 antibody | NSCLC, renal cell carcinoma, breast cancer | LY3381916 is safely administered as monotherapy and in combination with anti-PD-L1 therapy [242] | Terminated (2020) |
| NCT04106414 | II | IDO inhibitor (BMS-986205) + anti-PD-1 antibody | Endometrial Adenocarcinoma | N/A | Active, not recruiting (2021) |
| NCT03915405 | I | IDO inhibitor (KHK2455) + anti-PD-L1 antibody | Urothelial carcinoma | N/A | Recruiting (2021) |
| NCT02052648 | I/II | IDO inhibitor (indoximod, NLG-8186) + chemotherapy or Bevacizumab | Recurrent glioma | N/A | Completed (2020) |
| NCT03164603 | I | IDO inhibitor (NLG802) | Solid tumors | N/A | Completed (2020) |
| NCT02073123 | I/II | IDO inhibitor (indoximod, NLG-8186) + immune checkpoint inhibition | Metastatic Melanoma | Combination of indoximod and pembrolizumab demonstrated an ORR of 55.7%, CR 18.6%, which compares favorably with reported ORR for pembrolizumab alone [243] | Completed (2020) |
| NCT02048709 | I | IDO1 inhibitor (Navoximod, GDC-0919) | Solid tumors | Navoximod was well-tolerated and decreased plasma kynurenine levels. Stable disease responses were observed [244] | Completed (2017) |
| NCT02077881 | I/II | IDO inhibitor (indoximod, NLG-8186) + chemotherapy | Metastatic pancreatic cancer | Generally well-tolerated. OS of 10.9 months and ORR of 46.2%. Increased intra-tumoral CD8 density was observed [245] | Completed (2020) |
| NCT02502708 | I | IDO inhibitor (indoximod, NLG-8186) + chemotherapy | Pediatric brain tumors | Combining indoximod with chemotherapy was well tolerated with improved outcomes [246] | Completed (2020) |
| NCT02460367 | I | IDO inhibitor (indoximod, NLG-8186) + Tergenpumatucel-L | NSCLC | N/A | Active, not recruiting (2020) |
| NCT03414229 | II | IDO inhibitor (epacadostat, INCB024360) + anti-PD-1 antibody | Sarcoma | Epacadostat + pembrolizumab was well tolerated but had limited anti-tumor activity [247] | Active, not recruiting (2021) |
| NCT02166905 | I/II | IDO inhibitor (epacadostat, INCB024360) + DEC-205/NY-ESO-1 fusion protein CDX-1401 + poly ICLC | Fallopian tube carcinoma, ovarian carcinoma, peritoneal carcinoma | N/A | Completed (2021) |
| NCT03896113 | II | Celecoxib | Endometrium cancer | N/A | Recruiting (2020) |
| NCT01961115 | II | IDO inhibitor (epacadostat, INCB024360) | Melanoma | Epacadostat therapy was considered safe with transient DLTs in only two patients. Pacadostat normalized serum Kyn/Trp ratios in 91% of patients. Clinical activity was observed. Enhanced CD8 T cell infiltration was observed. | Completed (2018) |
| NCT03491631 | I | IDO inhibitor (SHR9146) + apatinib | Solid tumors | SHR9146 plus apatinib demonstrated promising anti-tumor activity with acceptable safety profile [248] | Completed (2018) |
| NCT02118285 | I | IDO inhibitor (epacadostat, INCB024360) + IP NK cells + IL-2 | Fallopian tube carcinoma, ovarian carcinoma, peritoneal carcinoma | N/A | Completed (2017) |
| NCT03459222 | I/II | IDO inhibitor (BMS-986205) + Relatlimab + Nivolumab | Advanced solid cancers | N/A | Recruiting (2021) |
| NCT04047706 | I | IDO inhibitor (BMS-986205) + radiation + nivolumab +/− temozolomide | Glioblastoma | N/A | Recruiting (2020) |
| NCT03361865 | III | IDO inhibitor (epacadostat, INCB024360) + pembrolizumab | Urothelial cancer | ORR of 31.8% in treatment group compared to 24.5% in placebo group. Serious adverse events detected in 30.23% of treatment group compared to 26.53% of placebo group. | Completed (2020) |
| NCT03358472 | III | IDO inhibitor (epacadostat, INCB024360) + pembrolizumab or EXTREME regimen | Head and neck cancer | ORR of 31.4% in combination group compared to 21.1% in pembrolizumab and 34.3% in EXTREME regimen. Similar rates of serious adverse events in all groups. | Active, not recruiting (2021) |
| NCT03322540 | II | IDO inhibitor (epacadostat, INCB024360) + pembrolizumab | Lung cancer | ORR of 32.5% in combination group compared to 39.0% with pembrolizumab alone. Similar rates of serious adverse events in all groups. | Completed (2021) |
| NCT03260894 | III | IDO inhibitor (epacadostat, INCB024360) + pembrolizumab | Renal cell carcinoma | N/A | Active, not recruiting (2020) |
| NCT03322566 | II | IDO inhibitor (epacadostat, INCB024360) + pembrolizumab + chemotherapy | NSCLC | ORR of 26.4% in combination group compared to 44.8% in group receiving pembrolizumab + chemotherapy + placebo. | Completed (2021) |
| NCT03006302 | II | IDO inhibitor (epacadostat) + pembrolizumab + cyclophosphamide +/− GVAX pancreas vaccine | Metastatic pancreatic adenocarcinoma | N/A | Recruiting (2021) |
| NCT05106296 | I | IDO inhibitor (indoximod, NLG-8186) + chemotherapy + ibrutinib | Ependymoma, medulloblastoma, glioblastoma, primitive neuroectodermal tumor (PNET) | N/A | Not yet recruiting (2021) |
| NCT04049669 | II | IDO inhibitor (indoximod, NLG-8186) + chemotherapy + radiation | Ependymoma, medulloblastoma, glioblastoma, diffuse intrinsic pontine glioma | N/A | Recruiting (2021) |
| NCT03347123 | I/II | IDO inhibitor (epacadostat) + nivolumab + ipilimumab or lirilumab | Solid tumors | N/A | Completed (2021) |
| NCT03085914 | I/II | IDO inhibitor (epacadostat) + pubmrolizumab + chemotherapy | Solid tumors | N/A | Completed (2021) |
| NCT03207867 | II | A2a receptor antagonist (NIR178) + anti-PD-1 antibody | Solid tumors, diffuse large B-cell lymphoma | N/A | Recruiting (2021) |
| NCT03381274 | I/II | A2a receptor antagonist (AZD4635) + anti-CD73 antibody (MEDI9447) or Osimertinib | NSCLC | N/A | Active, not recruiting (2021) |
| NCT02403193 | I/II | A2a receptor antagonist (PBF-509) +/− anti-PD-1 antibody | NSCLC | NIR178 was well tolerated. Clinical benefit was observed in patients irrespective of PD-L1 status [249] | Active, not recruiting (2020) |
| NCT04089553 | II | A2a receptor antagonist (AZD4635) + anti-CD73 antibody (oleclumab) or anti-PD-L1 antibody (Durvalumab) | Prostate cancer | N/A | Active, not recruiting (2021) |
| NCT02740985 | I | A2a receptor antagonist (AZD4635) +/− anti-CD73 antibody (oleclumab), anti-PD-L1 antibody (durvalumab), or chemotherapy | NSCLC, prostate cancer, colorectal carcinoma | AZD4635 monotherapy or in combination with durvalumab displayed a tolerable safety profile and was associated with clinical benefit [250] | Active, not recruiting (2021) |
| NCT03267589 | II | anti-CD73 antibody (MEDI9447) +/− anti-PD-1 antibody or anti-CTLA4 antibody | Ovarian cancer | N/A | Recruiting (2021) |
| NCT02754141 | I/II | anti-CD73 antibody (BMS-986179) +/− anti-PD-1 antibody (nivolumab) or rHuPH20 | Advanced solid tumors | BMS-986179 + nivolumab combination therapy was well tolerated with similar safety profile compared to nivolumab alone. Combination therapy demonstrated preliminary antitumor efficacy [251] | Active, not recruiting (2021) |
| NCT03549000 | I | anti-CD73 antibody (NZV930) +/− A2a receptor antagonist (NIR178) or anti-PD-1 antibody | Advanced cancers | N/A | Recruiting (2021) |
| NCT04969315 | I/II | A2a receptor antagonist (TT-10) | Renal cell cancer, castrate-resistant prostate cancer, NSCLC | N/A | Not yet recruiting (2021) |
| NCT02655822 | I | A2a receptor antagonist (ciforadenant) +/− anti-PD-L1 antibody (atezolizumab) | Renal cell cancer, Prostate cancer, NSCLC | Ciforadenant is well tolerated and showed anti-tumor activity as monotherapy and in combination atezolizumab for patients with renal cell cancer and NSCLC | Completed (2021) |
| NCT04797468 | I | CD73 inhibitor (HLX23) | Advanced solid tumors | N/A | Not yet recruiting (2021) |
| NCT05143970 | I | anti-CD73 antibody (IPH5301) +/− chemotherapy and trastuzumab | HER2+ cancers | N/A | Not yet recruiting (2021) |
| NCT04148937 | I | CD73 inhibitor (LY3475070) | Advanced cancers | N/A | Active, not recruiting (2021) |
| NCT04572152 | I | anti-CD73 antibody (AK119) + PD-1/CTLA-4 bispecific antibody | Advanced or metastatic solid tumors | N/A | Recruiting (2021) |
| NCT04672434 | I | anti-CD73 antibody (Sym024) +/− anti-PD-1 antibody (Sym021) | Metastatic solid tumors | N/A | Recruiting (2021) |
| NCT05119998 | I | anti-CD73 antibody (IBI325) +/− anti-PD-1 antibody (sintilima) | Solid tumors | N/A | Not yet recruiting (2021) |
| NCT04940286 | II | anti-CD73 antibody (Oleclumab) + chemotherapy + anti-PD-L1 antibody (Durvalumab) | Pancreatic cancer | N/A | Recruiting (2021) |
| NCT03454451 | I | anti-CD73 antibody (CPI-006) +/− A2a receptor antagonist (ciforadenant) or anti-PD-1 antibody (pembrolizumab) | Advanced cancers | Treatment is well-tolerated with early evidence of anti-tumor activity of CPI-006 monotherapy. Increased CD4+:CD8+ T cell ratio with CPI-006 therapy [252] | Recruiting (2021) |
| NCT02503774 | I | anti-CD73 antibody (MEDI9447) +/− anti-PD-L1 antibody (durvalumab) | Solid tumors | Combination therapy demonstrated a tolerable safety profile with promising antitumor activity in EGFRm NSCLC [253] | Active, not recruiting (2021) |
| NCT05075564 | I | anti-CD39 antibody (ES002023) | Advanced solid tumors | N/A | Not yet recruiting (2021) |
| NCT04306900 | I | anti-CD39 antibody (TTX-030) +/− anti-PD-1 antibody and chemotherapy | Adult solid tumors | N/A | Recruiting (2021) |
| NCT04336098 | I | anti-CD39 antibody (SRF617) +/− chemotherapy or anti-PD-1 antibody | Advanced solid tumors | N/A | Recruiting (2021) |
| NCT03884556 | I | anti-CD39 antibody (TTX-030) +/− anti-PD-1 antibody or chemotherapy | Solid tumors, lymphoma | N/A | Recruiting (2021) |
| NCT03473730 | I | anti-CD38 antibody (daratumumab) | Metastatic renal cell carcinoma, invasive bladder cancer | N/A | Active, not recruiting (2021) |
| NCT03177460 | I | anti-CD38 antibody (daratumumab) +/− FMS Inhibitor | Prostate cancer | N/A | Active, not recruiting (2020) |
| NCT03637764 | I/II | anti-CD38 antibody (isatuximab) +/− anti-PD-L1 (atezolizumab) | HCC, SCCHN, EOC | CD38 inhibition does not seem to influence response to anti-PD-L1 agents in these patients with HCC, SCCHN, or EOC [254] | Active, not recruiting (2021) |
| NCT03367819 | I/II | anti-CD38 antibody (isatuximab) +/− anti-PD-1 (cemiplimab) | Prostate cancer, NSCLC | Combination therapy was associated with a tolerable safety profile, reduction of CD38+ T cells in the TME, and activation of peripheral T cells, but no significant antitumor activity was observed in these small cohorts [255] | Completed |
| NCT04265534 | II | Glutaminase inhibitor (telaglenastat) + anti-PD-1 antibody (pembrolizumab) + chemotherapy | KEAP1/NRF2-mutated NSCLC | N/A | Active, not recruiting (2021) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oberholtzer, N.; Quinn, K.M.; Chakraborty, P.; Mehrotra, S. New Developments in T Cell Immunometabolism and Implications for Cancer Immunotherapy. Cells 2022, 11, 708. https://doi.org/10.3390/cells11040708
Oberholtzer N, Quinn KM, Chakraborty P, Mehrotra S. New Developments in T Cell Immunometabolism and Implications for Cancer Immunotherapy. Cells. 2022; 11(4):708. https://doi.org/10.3390/cells11040708
Chicago/Turabian StyleOberholtzer, Nathaniel, Kristen M. Quinn, Paramita Chakraborty, and Shikhar Mehrotra. 2022. "New Developments in T Cell Immunometabolism and Implications for Cancer Immunotherapy" Cells 11, no. 4: 708. https://doi.org/10.3390/cells11040708