
Normal and Neoplastic Growth Suppression by the Extended Myc Network



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Myc
3. Max
4. Mxd1
5. Mxd2
6. Mxd3
7. Mxd4
8. Mnt
9. Mga
10. ChREBP
11. MondoA
12. Mlx
13. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Alitalo, K.; Bishop, J.M.; Smith, D.H.; Chen, E.Y.; Colby, W.W.; Levinson, A.D. Nucleotide sequence to the v-myc oncogene of avian retrovirus MC29. Proc. Natl. Acad. Sci. USA 1983, 80, 100–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blackwood, E.M.; Eisenman, R.N. Max: A Helix-Loop-Helix Zipper Protein That Forms a Sequence-Specific DNA-Binding Complex with Myc. Science 1991, 251, 1211–1217. [Google Scholar] [CrossRef] [PubMed]
- Reddy, E.P.; Reynolds, R.K.; Watson, D.K.; Schultz, R.A.; Lautenberger, J.; Papas, T.S. Nucleotide sequence analysis of the proviral genome of avian myelocytomatosis virus (MC29). Proc. Natl. Acad. Sci. USA 1983, 80, 2500–2504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vennstrom, B.; Sheiness, D.; Zabielski, J.; Bishop, J.M. Isolation and characterization of c-myc, a cellular homolog of the oncogene (v-myc) of avian myelocytomatosis virus strain 29. J. Virol. 1982, 42, 773–779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watson, D.K.; Reddy, E.P.; Duesberg, P.H.; Papas, T.S. Nucleotide sequence analysis of the chicken c-myc gene reveals homologous and unique coding regions by comparison with the transforming gene of avian myelocytomatosis virus MC29, delta gag-myc. Proc. Natl. Acad. Sci. USA 1983, 80, 2146–2150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amati, B.; Dalton, S.; Brooks, M.W.; Littlewood, T.D.; Evan, G.I.; Land, H. Transcriptional activation by the human c-Myc oncoprotein in yeast requires interaction with Max. Nature 1992, 359, 423–426. [Google Scholar] [CrossRef] [PubMed]
- Amati, B.; Brooks, M.W.; Levy-Strumpf, N.; Littlewood, T.D.; Evan, G.I.; Land, H. Oncogenic activity of the c-Myc protein requires dimerization with Max. Cell 1993, 72, 233–245. [Google Scholar] [CrossRef]
- Amati, B.; Littlewood, T.; Evan, G.; Land, H. The c-Myc protein induces cell cycle progression and apoptosis through dimerization with Max. EMBO J. 1993, 12, 5083–5087. [Google Scholar] [CrossRef]
- Gu, W.; Cechova, K.; Tassi, V.; Dalla-Favera, R. Opposite regulation of gene transcription and cell proliferation by c-Myc and Max. Proc. Natl. Acad. Sci. USA 1993, 90, 2935–2939. [Google Scholar] [CrossRef] [Green Version]
- Kretzner, L.; Blackwood, E.M.; Eisenman, R.N. Myc and Max proteins possess distinct transcriptional activities. Nature 1992, 359, 426–429. [Google Scholar] [CrossRef]
- Kretzner, L.; Blackwood, E.M.; Eisenman, R.N. Transcriptional Activities of the Myc and Max Proteins in Mammalian Cells. Curr. Top. Microbiol. Immunol. 1992, 182, 435–443. [Google Scholar] [CrossRef] [PubMed]
- Min, S.; Taparowsky, E.J. v-Myc, but not Max, possesses domains that function in both transcription activation and cellular transformation. Oncogene 1992, 7, 1531–1540. [Google Scholar] [PubMed]
- Kato, G.J.; Barrett, J.; Villa-Garcia, M.; Dang, C.V. An amino-terminal c-myc domain required for neoplastic transformation activates transcription. Mol. Cell. Biol. 1990, 10, 5914–5920. [Google Scholar] [CrossRef] [PubMed]
- Amente, S.; Bertoni, A.; Morano, A.; Lania, L.; Avvedimento, E.V.; Majello, B. LSD1-mediated demethylation of histone H3 lysine 4 triggers Myc-induced transcription. Oncogene 2010, 29, 3691–3702. [Google Scholar] [CrossRef] [Green Version]
- de Pretis, S.; Kress, T.R.; Morelli, M.J.; Sabò, A.; Locarno, C.; Verrecchia, A.; Doni, M.; Campaner, S.; Amati, B.; Pelizzola, M. Integrative analysis of RNA polymerase II and transcriptional dynamics upon MYC activation. Genome Res. 2017, 27, 1658–1664. [Google Scholar] [CrossRef] [Green Version]
- Lourenco, C.; Resetca, D.; Redel, C.; Lin, P.; MacDonald, A.S.; Ciaccio, R.; Kenney, T.M.G.; Wei, Y.; Andrews, D.W.; Sunnerhagen, M.; et al. MYC protein interactors in gene transcription and cancer. Nat. Rev. Cancer 2021, 21, 579–591. [Google Scholar] [CrossRef]
- Price, D.H. Regulation of RNA Polymerase II Elongation by c-Myc. Cell 2010, 141, 399–400. [Google Scholar] [CrossRef] [Green Version]
- Rahl, P.B.; Lin, C.Y.; Seila, A.C.; Flynn, R.A.; McCuine, S.; Burge, C.B.; Sharp, P.A.; Young, R.A. c-Myc Regulates Transcriptional Pause Release. Cell 2010, 141, 432–445. [Google Scholar] [CrossRef] [Green Version]
- Berberich, S.J.; Cole, M.D. Casein kinase II inhibits the DNA-binding activity of Max homodimers but not Myc/Max heterodimers. Genes Dev. 1992, 6, 166–176. [Google Scholar] [CrossRef] [Green Version]
- Prochownik, E.V.; VanAntwerp, M.E. Differential patterns of DNA binding by myc and max proteins. Proc. Natl. Acad. Sci. USA 1993, 90, 960–964. [Google Scholar] [CrossRef] [Green Version]
- Minn, A.H.; Hafele, C.; Shalev, A. Thioredoxin-Interacting Protein Is Stimulated by Glucose through a Carbohydrate Response Element and Induces β-Cell Apoptosis. Endocrinology 2005, 146, 2397–2405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hurlin, P.J.; Quéva, C.; Koskinen, P.; Steingrimsson, E.; Ayer, D.E.; Copeland, N.G.; Jenkins, N.A.; Eisenman, R.N. Mad3 and Mad4: Novel Max-interacting transcriptional repressors that suppress c-myc dependent transformation and are expressed during neural and epidermal differentiation. EMBO J. 1995, 14, 5646–5659. [Google Scholar] [CrossRef] [PubMed]
- Popov, N.; Wahlström, T.; Hurlin, P.J.; Henriksson, M. Mnt transcriptional repressor is functionally regulated during cell cycle progression. Oncogene 2005, 24, 8326–8337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quéva, C.; Hurlin, P.J.; Foley, K.P.; Eisenman, R.N. Sequential expression of the MAD family of transcriptional repressors during differentiation and development. Oncogene 1998, 16, 967–977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, G.; Hurlin, P.J. MNT and Emerging Concepts of MNT-MYC Antagonism. Genes 2017, 8, 83. [Google Scholar] [CrossRef]
- Yang, L.; Venneti, S.; Nagrath, D. Glutaminolysis: A Hallmark of Cancer Metabolism. Annu. Rev. Biomed. Eng. 2017, 19, 163–194. [Google Scholar] [CrossRef]
- Zhou, Z.-Q.; Hurlin, P.J. The interplay between Mad and Myc in proliferation and differentiation. Trends Cell Biol. 2001, 11, S10–S14. [Google Scholar] [CrossRef]
- Ayer, D.E.; Lawrence, Q.A.; Eisenman, R.N. Mad-max transcriptional repression is mediated by ternary complex formation with mammalian homologs of yeast repressor Sin3. Cell 1995, 80, 767–776. [Google Scholar] [CrossRef] [Green Version]
- Llabata, P.; Mitsuishi, Y.; Choi, P.; Cai, D.; Francis, J.M.; Torres-Diz, M.; Udeshi, N.D.; Golomb, L.; Wu, Z.; Zhou, J.; et al. Multi-Omics Analysis Identifies MGA as a Negative Regulator of the MYC Pathway in Lung Adenocarcinoma. Mol. Cancer Res. 2020, 18, 574–584. [Google Scholar] [CrossRef] [Green Version]
- Mathsyaraja, H.; Catchpole, J.; Freie, B.; Eastwood, E.; Babaeva, E.; Geuenich, M.; Cheng, P.F.; Ayers, J.; Yu, M.; Wu, N.; et al. Loss of MGA repression mediated by an atypical polycomb complex promotes tumor progression and invasiveness. eLife 2021, 10, e64212. [Google Scholar] [CrossRef]
- Billin, A.N.; Ayer, D.E. The Mlx Network: Evidence for a Parallel Max-Like Transcriptional Network That Regulates Energy Metabolism. Curr. Top. Microbiol. Immunol. 2006, 302, 255–278. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.L.-Y.; Merl, D.; Peterson, C.W.; Wu, J.; Liu, P.Y.; Yin, H.; Muoio, D.M.; Ayer, D.E.; West, M.; Chi, J.-T. Lactic Acidosis Triggers Starvation Response with Paradoxical Induction of TXNIP through MondoA. PLoS Genet. 2010, 6, e1001093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davies, M.N.; O’Callaghan, B.L.; Towle, H.C. Glucose Activates ChREBP by Increasing Its Rate of Nuclear Entry and Relieving Repression of Its Transcriptional Activity. J. Biol. Chem. 2008, 283, 24029–24038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davies, M.N.; O’Callaghan, B.L.; Towle, H.C. Activation and repression of glucose-stimulated ChREBP requires the concerted action of multiple domains within the MondoA conserved region. Am. J. Physiol. Endocrinol. Metab. 2010, 299, E665–E674. [Google Scholar] [CrossRef] [Green Version]
- Li, M.V.; Chang, B.; Imamura, M.; Poungvarin, N.; Chan, L. Glucose-Dependent Transcriptional Regulation by an Evolutionarily Conserved Glucose-Sensing Module. Diabetes 2006, 55, 1179–1189. [Google Scholar] [CrossRef] [Green Version]
- O’Shea, J.M.; Ayer, D.E. Coordination of Nutrient Availability and Utilization by MAX- and MLX-Centered Transcription Networks. Cold Spring Harb. Perspect. Med. 2013, 3, a014258. [Google Scholar] [CrossRef]
- Peterson, C.W.; Stoltzman, C.A.; Sighinolfi, M.P.; Han, K.-S.; Ayer, D.E. Glucose Controls Nuclear Accumulation, Promoter Binding, and Transcriptional Activity of the MondoA-Mlx Heterodimer. Mol. Cell. Biol. 2010, 30, 2887–2895. [Google Scholar] [CrossRef] [Green Version]
- Christopher, W.P.; Peterson, C.W.; Ayer, D.E. An extended Myc network contributes to glucose homeostasis in cancer and diabetes. Front. Biosci. 2011, 16, 2206–2223. [Google Scholar] [CrossRef] [Green Version]
- Poungvarin, N.; Chang, B.; Imamura, M.; Chen, J.; Moolsuwan, K.; Sae-Lee, C.; Li, W.; Chanachai, S.-L. Genome-Wide Analysis of ChREBP Binding Sites on Male Mouse Liver and White Adipose Chromatin. Endocrinology 2015, 156, 1982–1994. [Google Scholar] [CrossRef] [Green Version]
- Sans, C.L.; Satterwhite, D.J.; Stoltzman, C.A.; Breen, K.T.; Ayer, D.E. MondoA-Mlx Heterodimers Are Candidate Sensors of Cellular Energy Status: Mitochondrial Localization and Direct Regulation of Glycolysis. Mol. Cell. Biol. 2006, 26, 4863–4871. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Dolezal, J.M.; Kulkarni, S.; Lu, J.; Mandel, J.; Jackson, L.E.; Alencastro, F.; Duncan, A.W.; Prochownik, E.V. Myc and ChREBP transcription factors cooperatively regulate normal and neoplastic hepatocyte proliferation in mice. J. Biol. Chem. 2018, 293, 14740–14757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilde, B.; Ayer, D. Interactions between Myc and MondoA transcription factors in metabolism and tumourigenesis. Br. J. Cancer 2015, 113, 1529–1533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, X.; Grove, L.; Prochownik, E.V. Lack of transcriptional repression by max homodimers. Oncogene 1998, 16, 2629–2637. [Google Scholar] [CrossRef] [Green Version]
- Carroll, P.A.; Diolaiti, D. A novel role for the extended MYC network in cancer cell survival. Mol. Cell. Oncol. 2016, 3, e1026528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diolaiti, D.; McFerrin, L.; Carroll, P.A.; Eisenman, R.N. Functional interactions among members of the MAX and MLX transcriptional network during oncogenesis. Biochim. Biophys. Acta 2015, 1849, 484–500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carroll, P.A.; Freie, B.W.; Mathsyaraja, H.; Eisenman, R.N. The MYC transcription factor network: Balancing metabolism, proliferation and oncogenesis. Front. Med. 2018, 12, 412–425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poole, C.J.; Van Riggelen, J. MYC—Master Regulator of the Cancer Epigenome and Transcriptome. Genes 2017, 8, 142. [Google Scholar] [CrossRef]
- Foley, K.P.; McArthur, G.A.; Quéva, C.; Hurlin, P.J.; Soriano, P.; Eisenman, R.N. Targeted disruption of the MYC antagonist MAD1 inhibits cell cycle exit during granulocyte differentiation. EMBO J. 1998, 17, 774–785. [Google Scholar] [CrossRef] [Green Version]
- Foley, K.P.; Eisenman, R.N. Two MAD tails: What the recent knockouts of Mad1 and Mxi1 tell us about the MYC/MAX/MAD network. Biochim. Biophys. Acta 1999, 1423, M37–M47. [Google Scholar] [CrossRef]
- Grandori, C.; Cowley, S.M.; James, L.P.; Eisenman, R.N. The Myc/Max/Mad Network and the Transcriptional Control of Cell Behavior. Annu. Rev. Cell Dev. Biol. 2000, 16, 653–699. [Google Scholar] [CrossRef]
- Hurlin, P.J.; Quéva, C.; Eisenman, R.N. Mnt, a novel Max-interacting protein is coexpressed with Myc in proliferating cells and mediates repression at Myc binding sites. Genes Dev. 1997, 11, 44–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hurlin, P.J.; Steingrimsson, E.; Copeland, N.G.; Jenkins, N.A.; Eisenman, R.N. Mga, a dual-specificity transcription factor that interacts with Max and contains a T-domain DNA-binding motif. EMBO J. 2000, 19, 3841–3842. [Google Scholar] [CrossRef] [Green Version]
- Hurlin, P.J.; Zhou, Z.; Toyo-Oka, K.; Ota, S.; Walker, W.L.; Hirotsune, S.; Wynshaw-Boris, A. Deletion of Mnt leads to disrupted cell cycle control and tumorigenesis. EMBO J. 2003, 22, 4584–4596. [Google Scholar] [CrossRef] [Green Version]
- Hurlin, P.J.; Zhou, Z.-Q.; Toyooka, K.; Ota, S.; Walker, W.L.; Hirotsune, S.; Wynshaw-Boris, A. Evidence of mnt-myc antagonism revealed by mnt gene deletion. Cell Cycle 2004, 3, 95–97. [Google Scholar] [CrossRef]
- Quéva, C.; McArthur, G.A.; Iritani, B.M.; Eisenman, R.N. Targeted Deletion of the S-Phase-Specific Myc Antagonist Mad3 Sensitizes Neuronal and Lymphoid Cells to Radiation-Induced Apoptosis. Mol. Cell. Biol. 2001, 21, 703–712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Billin, A.; Eilers, A.L.; Coulter, K.L.; Logan, J.S.; Ayer, D.E. MondoA, a Novel Basic Helix-Loop-Helix–Leucine Zipper Transcriptional Activator That Constitutes a Positive Branch of a Max-Like Network. Mol. Cell. Biol. 2000, 20, 8845–8854. [Google Scholar] [CrossRef] [Green Version]
- Billin, A.N.; Eilers, A.L.; Queva, C.; Ayer, D.E. Mlx, a Novel Max-like BHLHZip Protein That Interacts with the Max Network of Transcription Factors. J. Biol. Chem. 1999, 274, 36344–36350. [Google Scholar] [CrossRef] [Green Version]
- Yamashita, H.; Takenoshita, M.; Sakurai, M.; Bruick, R.K.; Henzel, W.; Shillinglaw, W.; Arnot, D.; Uyeda, K. A glucose-responsive transcription factor that regulates carbohydrate metabolism in the liver. Proc. Natl. Acad. Sci. USA 2001, 98, 9116–9121. [Google Scholar] [CrossRef] [Green Version]
- Han, K.-S.; Ayer, D.E. MondoA senses adenine nucleotides: Transcriptional induction of thioredoxin-interacting protein. Biochem. J. 2013, 453, 209–218. [Google Scholar] [CrossRef] [Green Version]
- Li, M.V.; Chen, W.; Harmancey, R.N.; Nuotio-Antar, A.M.; Imamura, M.; Saha, P.; Taegtmeyer, H.; Chan, L. Glucose-6-phosphate mediates activation of the carbohydrate responsive binding protein (ChREBP). Biochem. Biophys. Res. Commun. 2010, 395, 395–400. [Google Scholar] [CrossRef] [Green Version]
- Petrie, J.L.; Al-Oanzi, Z.H.; Arden, C.; Tudhope, S.J.; Mann, J.; Kieswich, J.; Yaqoob, M.M.; Towle, H.C.; Agius, L. Glucose Induces Protein Targeting to Glycogen in Hepatocytes by Fructose 2,6-Bisphosphate-Mediated Recruitment of MondoA to the Promoter. Mol. Cell. Biol. 2013, 33, 725–738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stoltzman, C.A.; Kaadige, M.R.; Peterson, C.W.; Ayer, D. MondoA Senses Non-glucose Sugars: Regulation of thioredoxin-interacting protein (txnip) and the hexose transport curb. J. Biol. Chem. 2011, 286, 38027–38034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stoltzman, C.A.; Peterson, C.W.; Breen, K.T.; Muoio, D.M.; Billin, A.N.; Ayer, D.E. Glucose sensing by MondoA:Mlx complexes: A role for hexokinases and direct regulation of thioredoxin-interacting protein expression. Proc. Natl. Acad. Sci. USA 2008, 105, 6912–6917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilde, B.R.; Kaadige, M.R.; Guillen, K.P.; Butterfield, A.; Welm, B.E.; Ayer, D.E. Protein synthesis inhibitors stimulate MondoA transcriptional activity by driving an accumulation of glucose 6-phosphate. Cancer Metab. 2020, 8, 27. [Google Scholar] [CrossRef]
- Wilde, B.R.; Ye, Z.; Lim, T.-Y.; Ayer, D.E. Cellular acidosis triggers human MondoA transcriptional activity by driving mitochondrial ATP production. eLife 2019, 8, e40199. [Google Scholar] [CrossRef]
- Yu, F.-X.; Chai, T.F.; He, H.; Hagen, T.; Luo, Y. Thioredoxin-interacting Protein (Txnip) Gene Expression: Sensing oxidative phosphorylation status and glycolytic rate. J. Biol. Chem. 2010, 285, 25822–25830. [Google Scholar] [CrossRef] [Green Version]
- Yu, F.-X.; Goh, S.-R.; Dai, R.-P.; Luo, Y. Adenosine-Containing Molecules Amplify Glucose Signaling and Enhance Txnip Expression. Mol. Endocrinol. 2009, 23, 932–942. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Fu, T.; He, Q.; Gao, X.; Luo, Y. Glucose-6-Phosphate Upregulates Txnip Expression by Interacting with MondoA. Front. Mol. Biosci. 2020, 6, 147. [Google Scholar] [CrossRef]
- Mejhert, N.; Kuruvilla, L.; Gabriel, K.R.; Elliott, S.D.; Guie, M.-A.; Wang, H.; Lai, Z.W.; Lane, E.A.; Christiano, R.; Danial, N.N.; et al. Partitioning of MLX-Family Transcription Factors to Lipid Droplets Regulates Metabolic Gene Expression. Mol. Cell 2020, 77, 1251–1264.e9. [Google Scholar] [CrossRef]
- Carroll, P.; Diolaiti, D.; McFerrin, L.; Gu, H.; Djukovic, D.; Du, J.; Cheng, P.F.; Anderson, S.; Ulrich, M.; Hurley, J.B.; et al. Deregulated Myc Requires MondoA/Mlx for Metabolic Reprogramming and Tumorigenesis. Cancer Cell 2015, 27, 271–285. [Google Scholar] [CrossRef] [Green Version]
- Ma, L.; Robinson, L.N.; Towle, H.C. ChREBP•Mlx Is the Principal Mediator of Glucose-induced Gene Expression in the Liver. J. Biol. Chem. 2006, 281, 28721–28730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, L.; Sham, Y.Y.; Walters, K.J.; Towle, H.C. A critical role for the loop region of the basic helix-loop-helix/leucine zipper protein Mlx in DNA binding and glucose-regulated transcription. Nucleic Acids Res. 2007, 35, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Tsatsos, N.G.; Towle, H.C. Direct Role of ChREBP·Mlx in Regulating Hepatic Glucose-responsive Genes. J. Biol. Chem. 2005, 280, 12019–12027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stoeckman, A.; Ma, L.; Towle, H.C. Mlx Is the Functional Heteromeric Partner of the Carbohydrate Response Element-binding Protein in Glucose Regulation of Lipogenic Enzyme Genes. J. Biol. Chem. 2004, 279, 15662–15669. [Google Scholar] [CrossRef] [Green Version]
- Wutthisathapornchai, A.; Vongpipatana, T.; Muangsawat, S.; Boonsaen, T.; Macdonald, M.J.; Jitrapakdee, S. Multiple E-Boxes in the Distal Promoter of the Rat Pyruvate Carboxylase Gene Function as a Glucose-Responsive Element. PLoS ONE 2014, 9, e102730. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Lu, J.; Alencastro, F.; Roberts, A.; Fiedor, J.; Carroll, P.; Eisenman, R.N.; Ranganathan, S.; Torbenson, M.; Duncan, A.W.; et al. Coordinated Cross-Talk Between the Myc and Mlx Networks in Liver Regeneration and Neoplasia. Cell Mol. Gastroenterol. Hepatol. 2022; in press. [Google Scholar]
- Zhang, P.; Metukuri, M.R.; Bindom, S.M.; Prochownik, E.V.; O’Doherty, R.M.; Scott, D.K. c-Myc Is Required for the ChREBP-Dependent Activation of Glucose-Responsive Genes. Mol. Endocrinol. 2010, 24, 1274–1286. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Lu, J.; Edmunds, L.R.; Kulkarni, S.; Dolezal, J.; Tao, J.; Ranganathan, S.; Jackson, L.; Fromherz, M.; Beer-Stolz, D.; et al. Coordinated Activities of Multiple Myc-dependent and Myc-independent Biosynthetic Pathways in Hepatoblastoma. J. Biol. Chem. 2016, 291, 26241–26251. [Google Scholar] [CrossRef] [Green Version]
- Denechaud, P.-D.; Bossard, P.; Lobaccaro, J.-M.A.; Millatt, L.; Staels, B.; Girard, J.; Postic, C. ChREBP, but not LXRs, is required for the induction of glucose-regulated genes in mouse liver. J. Clin. Investig. 2008, 118, 956–964. [Google Scholar] [CrossRef]
- Denechaud, P.-D.; Dentin, R.; Girard, J.; Postic, C. Role of ChREBP in hepatic steatosis and insulin resistance. FEBS Lett. 2007, 582, 68–73. [Google Scholar] [CrossRef] [Green Version]
- Havula, E.; Hietakangas, V. Glucose sensing by ChREBP/MondoA–Mlx transcription factors. Semin. Cell Dev. Biol. 2012, 23, 640–647. [Google Scholar] [CrossRef] [PubMed]
- Havula, E.; Hietakangas, V. Sugar sensing by ChREBP/Mondo-Mlx—New insight into downstream regulatory networks and integration of nutrient-derived signals. Curr. Opin. Cell Biol. 2018, 51, 89–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Havula, E.; Teesalu, M.; Hyötyläinen, T.; Seppälä, H.; Hasygar, K.; Auvinen, P.; Orešič, M.; Sandmann, T.; Hietakangas, V. Mondo/ChREBP-Mlx-Regulated Transcriptional Network Is Essential for Dietary Sugar Tolerance in Drosophila. PLoS Genet. 2013, 9, e1003438. [Google Scholar] [CrossRef] [Green Version]
- Iizuka, K.; Bruick, R.K.; Liang, G.; Horton, J.D.; Uyeda, K. From The Cover: Deficiency of carbohydrate response element-binding protein (ChREBP) reduces lipogenesis as well as glycolysis. Proc. Natl. Acad. Sci. USA 2004, 101, 7281–7286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeong, Y.-S.; Kim, D.; Lee, Y.S.; Kim, H.-J.; Han, J.-Y.; Im, S.-S.; Chong, H.K.; Kwon, J.-K.; Cho, Y.-H.; Kim, W.K.; et al. Integrated Expression Profiling and Genome-Wide Analysis of ChREBP Targets Reveals the Dual Role for ChREBP in Glucose-Regulated Gene Expression. PLoS ONE 2011, 6, e22544. [Google Scholar] [CrossRef] [PubMed]
- Ke, H.; Luan, Y.; Wu, S.; Zhu, Y.; Tong, X. The Role of Mondo Family Transcription Factors in Nutrient-Sensing and Obesity. Front. Endocrinol. 2021, 12, 653972. [Google Scholar] [CrossRef] [PubMed]
- Lane, E.A.; Choi, D.W.; Garcia-Haro, L.; Levine, Z.G.; Tedoldi, M.; Walker, S.; Danial, N.N. HCF-1 Regulates De Novo Lipogenesis through a Nutrient-Sensitive Complex with ChREBP. Mol. Cell 2019, 75, 357–371.e7. [Google Scholar] [CrossRef]
- Richards, P.; Ourabah, S.; Montagne, J.; Burnol, A.-F.; Postic, C.; Guilmeau, S. MondoA/ChREBP: The usual suspects of transcriptional glucose sensing; Implication in pathophysiology. Metabolism 2017, 70, 133–151. [Google Scholar] [CrossRef]
- Richards, P.; Rachdi, L.; Oshima, M.; Marchetti, P.; Bugliani, M.; Armanet, M.; Postic, C.; Guilmeau, S.; Scharfmann, R. MondoA Is an Essential Glucose-Responsive Transcription Factor in Human Pancreatic β-Cells. Diabetes 2017, 67, 461–472. [Google Scholar] [CrossRef] [Green Version]
- Airley, R.E.; McHugh, P.; Evans, A.R.; Harris, B.; Winchester, L.; Buffa, F.; Al-Tameemi, W.; Leek, R.; Harris, A. Role of carbohydrate response element-binding protein (ChREBP) in generating an aerobic metabolic phenotype and in breast cancer progression. Br. J. Cancer 2014, 110, 715–723. [Google Scholar] [CrossRef]
- Buttgereit, F.; Brand, M. A hierarchy of ATP-consuming processes in mammalian cells. Biochem. J. 1995, 312 Pt 1, 163–167. [Google Scholar] [CrossRef] [PubMed]
- Elgort, M.G.; O’Shea, J.M.; Jiang, Y.; Ayer, D.E. Transcriptional and Translational Downregulation of Thioredoxin Interacting Protein Is Required for Metabolic Reprogramming during G1. Genes Cancer 2010, 1, 893–907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaadige, M.R.; Looper, R.E.; Kamalanaadhan, S.; Ayer, D.E. Glutamine-dependent anapleurosis dictates glucose uptake and cell growth by regulating MondoA transcriptional activity. Proc. Natl. Acad. Sci. USA 2009, 106, 14878–14883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaadige, M.R.; Yang, J.; Wilde, B.R.; Ayer, D. MondoA-Mlx Transcriptional Activity Is Limited by mTOR-MondoA Interaction. Mol. Cell. Biol. 2015, 35, 101–110. [Google Scholar] [CrossRef] [Green Version]
- Sipol, A.; Hameister, E.; Xue, B.; Hofstetter, J.; Barenboim, M.; Öllinger, R.; Jain, G.; Prexler, C.; Rubio, R.A.; Baldauf, M.C.; et al. MondoA Drives B-ALL Malignancy through Enhanced Adaptation to Metabolic Stress. Blood 2021, in press. [Google Scholar] [CrossRef]
- Wernicke, C.M.; Richter, G.H.; Beinvogl, B.C.; Plehm, S.; Schlitter, A.M.; Bandapalli, O.R.; da Costa, O.P.; Hattenhorst, U.E.; Volkmer, I.; Staege, M.S.; et al. MondoA is highly overexpressed in acute lymphoblastic leukemia cells and modulates their metabolism, differentiation and survival. Leuk. Res. 2012, 36, 1185–1192. [Google Scholar] [CrossRef] [PubMed]
- Afshar, A.R.; Pekmezci, M.; Bloomer, M.M.; Cadenas, N.J.; Stevers, M.; Banerjee, A.; Roy, R.; Olshen, A.B.; Van Ziffle, J.; Onodera, C.; et al. Next-Generation Sequencing of Retinoblastoma Identifies Pathogenic Alterations beyond RB1 Inactivation That Correlate with Aggressive Histopathologic Features. Ophthalmology 2020, 127, 804–813. [Google Scholar] [CrossRef]
- Berry, J.L.; Polski, A.; Cavenee, W.K.; Dryja, T.P.; Murphree, A.L.; Gallie, B.L. The RB1 Story: Characterization and Cloning of the First Tumor Suppressor Gene. Genes 2019, 10, 879. [Google Scholar] [CrossRef] [Green Version]
- Garber, J.E.; Offit, K. Hereditary Cancer Predisposition Syndromes. J. Clin. Oncol. 2005, 23, 276–292. [Google Scholar] [CrossRef]
- Gargallo, P.; Yáñez, Y.; Segura, V.; Juan, A.; Torres, B.; Balaguer, J.; Oltra, S.; Castel, V.; Cañete, A. Li–Fraumeni syndrome heterogeneity. Clin. Transl. Oncol. 2020, 22, 978–988. [Google Scholar] [CrossRef]
- Guha, T.; Malkin, D. Inherited TP53 Mutations and the Li–Fraumeni Syndrome. Cold Spring Harb. Perspect. Med. 2017, 7, a026187. [Google Scholar] [CrossRef] [Green Version]
- Marmolejo, D.H.; Wong, M.Y.Z.; Bajalica-Lagercrantz, S.; Tischkowitz, M.; Balmaña, J.; Patócs, A.B.; Chappuis, P.; Colas, C.; Genuardi, M.; Haanpää, M.; et al. Overview of hereditary breast and ovarian cancer (HBOC) guidelines across Europe. Eur. J. Med Genet. 2021, 64, 104350. [Google Scholar] [CrossRef] [PubMed]
- Beroukhim, R.; Mermel, C.H.; Porter, D.; Wei, G.; Raychaudhuri, S.; Donovan, J.; Barretina, J.; Boehm, J.S.; Dobson, J.; Urashima, M.; et al. The landscape of somatic copy-number alteration across human cancers. Nature 2010, 463, 899–905. [Google Scholar] [CrossRef] [PubMed]
- Schaub, F.X.; Dhankani, V.; Berger, A.C.; Trivedi, M.; Richardson, A.B.; Shaw, R.; Zhao, W.; Zhang, X.; Ventura, A.; Liu, Y.; et al. Pan-cancer Alterations of the MYC Oncogene and Its Proximal Network across the Cancer Genome Atlas. Cell Syst. 2018, 6, 282–300.e2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zack, T.I.; Schumacher, S.E.; Carter, S.L.; Cherniack, A.D.; Saksena, G.; Tabak, B.; Lawrence, M.S.; Zhang, C.Z.; Wala, J.; Mermel, C.H.; et al. Pan-cancer patterns of somatic copy number alteration. Nat. Genet. 2013, 45, 1134–1140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fallah, Y.; Brundage, J.; Allegakoen, P.; Shajahan-Haq, A.N. MYC-Driven Pathways in Breast Cancer Subtypes. Biomolecules 2017, 7, 53. [Google Scholar] [CrossRef]
- Kawate, S.; Fukusato, T.; Ohwada, S.; Watanuki, A.; Morishita, Y. Amplification of c-myc in Hepatocellular Carcinoma: Correlation with Clinicopathologic Features, Proliferative Activity and p53 Overexpression. Oncology 1999, 57, 157–163. [Google Scholar] [CrossRef]
- Li, C.; Bonazzoli, E.; Bellone, S.; Choi, J.; Dong, W.; Menderes, G.; Altwerger, G.; Han, C.; Manzano, A.; Bianchi, A.; et al. Mutational landscape of primary, metastatic, and recurrent ovarian cancer reveals c-MYC gains as potential target for BET inhibitors. Proc. Natl. Acad. Sci. USA 2019, 116, 619–624. [Google Scholar] [CrossRef] [Green Version]
- The Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014, 511, 543–550. [Google Scholar] [CrossRef]
- Singh, A.; Ham, J.; Po, J.; Niles, N.; Roberts, T.; Lee, C. The Genomic Landscape of Thyroid Cancer Tumourigenesis and Implications for Immunotherapy. Cells 2021, 10, 1082. [Google Scholar] [CrossRef]
- Mermel, C.H.; Schumacher, S.E.; Hill, B.; Meyerson, M.L.; Beroukhim, R.; Getz, G. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol. 2011, 12, R41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, Z.; Eils, R.; Schlesner, M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 2016, 32, 2847–2849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dolezal, J.M.; Wang, H.; Kulkarni, S.; Jackson, L.; Lu, J.; Ranganathan, S.; Goetzman, E.S.; Bharathi, S.S.; Beezhold, K.; Byersdorfer, C.A.; et al. Sequential adaptive changes in a c-Myc-driven model of hepatocellular carcinoma. J. Biol. Chem. 2017, 292, 10068–10086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leder, A.; Pattengale, P.K.; Kuo, A.; Stewart, T.A.; Leder, P. Consequences of widespread deregulation of the c-myc gene in transgenic mice: Multiple neoplasms and normal development. Cell 1986, 45, 485–495. [Google Scholar] [CrossRef]
- Shachaf, C.M.; Kopelman, A.M.; Arvanitis, C.; Karlsson, A.; Beer, S.; Mandl, S.; Bachmann, M.; Borowsky, A.D.; Ruebner, B.; Cardiff, R.D.; et al. MYC inactivation uncovers pluripotent differentiation and tumour dormancy in hepatocellular cancer. Nature 2004, 431, 1112–1117. [Google Scholar] [CrossRef]
- Tao, J.; Calvisi, D.F.; Ranganathan, S.; Cigliano, A.; Zhou, L.; Singh, S.; Jiang, L.; Fan, B.; Terracciano, L.; Armeanu–Ebinger, S.; et al. Activation of β-Catenin and Yap1 in Human Hepatoblastoma and Induction of Hepatocarcinogenesis in Mice. Gastroenterology 2014, 147, 690–701. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Meyfeldt, J.; Wang, H.; Kulkarni, S.; Lu, J.; Mandel, J.A.; Marburger, B.; Liu, Y.; Gorka, J.E.; Ranganathan, S.; et al. β-Catenin mutations as determinants of hepatoblastoma phenotypes in mice. J. Biol. Chem. 2019, 294, 17524–17542. [Google Scholar] [CrossRef]
- Soucek, L.; Evan, G.I. The ups and downs of Myc biology. Curr. Opin. Genet. Dev. 2010, 20, 91–95. [Google Scholar] [CrossRef] [Green Version]
- Brito, J.P.; Asi, N.; Bancos, I.; Gionfriddo, M.R.; Zeballos-Palacios, C.L.; Leppin, A.L.; Undavalli, C.; Wang, Z.; Domecq, J.P.; Prustsky, G.; et al. Testing for germline mutations in sporadic pheochromocytoma/paraganglioma: A systematic review. Clin. Endocrinol. 2015, 82, 338–345. [Google Scholar] [CrossRef]
- Burnichon, N.; Cascón, A.; Schiavi, F.; Morales, N.P.; Comino-Mendez, I.; Abermil, N.; Inglada, L.; de Cubas, A.A.; Amar, L.; Barontini, M.; et al. MAX Mutations Cause Hereditary and Sporadic Pheochromocytoma and Paraganglioma. Clin. Cancer Res. 2012, 18, 2828–2837. [Google Scholar] [CrossRef] [Green Version]
- Comino-Mendez, I.; Gracia-Aznárez, F.J.; Schiavi, F.; Landa, I.; Leandro-Garcia, L.; Letón, R.; Honrado, E.; Ramos-Medina, R.; Caronia, D.; Pita, G.; et al. Exome sequencing identifies MAX mutations as a cause of hereditary pheochromocytoma. Nat. Genet. 2011, 43, 663–667. [Google Scholar] [CrossRef] [PubMed]
- Galan, S.R.; Kann, P.H. Genetics and molecular pathogenesis of pheochromocytoma and paraganglioma. Clin. Endocrinol. 2013, 78, 165–175. [Google Scholar] [CrossRef] [PubMed]
- Korpershoek, E.; Koffy, D.; Eussen, B.H.; Oudijk, L.; Papathomas, T.G.; Van Nederveen, F.H.; Belt, E.J.T.; Franssen, G.J.H.; Restuccia, D.F.J.; Krol, N.M.G.; et al. Complex MAX Rearrangement in a Family With Malignant Pheochromocytoma, Renal Oncocytoma, and Erythrocytosis. J. Clin. Endocrinol. Metab. 2016, 101, 453–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roszko, K.L.; Blouch, E.; Blake, M.; Powers, J.; Tischler, A.; Hodin, R.; Sadow, P.; Lawson, E.A. Case Report of a Prolactinoma in a Patient With a Novel MAX Mutation and Bilateral Pheochromocytomas. J. Endocr. Soc. 2017, 1, 1401–1407. [Google Scholar] [CrossRef] [Green Version]
- Welander, J.; Andreasson, A.; Juhlin, C.C.; Wiseman, R.W.; Bäckdahl, M.; Höög, A.; Larsson, C.; Gimm, O.; Söderkvist, P. Rare Germline Mutations Identified by Targeted Next-Generation Sequencing of Susceptibility Genes in Pheochromocytoma and Paraganglioma. J. Clin. Endocrinol. Metab. 2014, 99, E1352–E1360. [Google Scholar] [CrossRef]
- Boxer, L.M.; Dang, C.V. Translocations involving c-myc and c-myc function. Oncogene 2001, 20, 5595–5610. [Google Scholar] [CrossRef] [Green Version]
- Jovanović, K.K.; Roche-Lestienne, C.; Ghobrial, I.M.; Facon, T.; Quesnel, B.; Manier, S. Targeting MYC in multiple myeloma. Leukemia 2018, 32, 1295–1306. [Google Scholar] [CrossRef]
- Liu, Y.; Barta, S.K. Diffuse large B-cell lymphoma: 2019 update on diagnosis, risk stratification, and treatment. Am. J. Hematol. 2019, 94, 604–616. [Google Scholar] [CrossRef] [Green Version]
- Schmitz, R.; Ceribelli, M.; Pittaluga, S.; Wright, G.; Staudt, L.M. Oncogenic Mechanisms in Burkitt Lymphoma. Cold Spring Harb. Perspect. Med. 2014, 4, a014282. [Google Scholar] [CrossRef] [Green Version]
- Herranz, D.; Ambesi-Impiombato, A.; Palomero, T.; Schnell, S.A.; Belver, L.; Wendorff, A.A.; Xu, L.; Castillo-Martin, M.; Llobet-Navás, D.; Cordon-Cardo, C.; et al. A NOTCH1-driven MYC enhancer promotes T cell development, transformation and acute lymphoblastic leukemia. Nat. Med. 2014, 20, 1130–1137. [Google Scholar] [CrossRef] [Green Version]
- Kaur, M.; Cole, M.D. MYC Acts via the PTEN Tumor Suppressor to Elicit Autoregulation and Genome-Wide Gene Repression by Activation of the Ezh2 Methyltransferase. Cancer Res. 2013, 73, 695–705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilkins, J.A.; Sansom, O.J. C-Myc Is a Critical Mediator of the Phenotypes of Apc Loss in the Intestine: Figure 1. Cancer Res. 2008, 68, 4963–4966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gebhardt, A.; Frye, M.; Herold, S.; Benitah, S.A.; Braun, K.; Samans, B.; Watt, F.; Elsasser, H.-P.; Eilers, M. Myc regulates keratinocyte adhesion and differentiation via complex formation with Miz1. J. Cell Biol. 2006, 172, 139–149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seoane, J.; Pouponnot, C.; Staller, P.; Schader, M.; Eilers, M.; Massague, J. TGFβ influences Myc, Miz-1 and Smad to control the CDK inhibitor p15INK4b. Nat. Cell Biol. 2001, 3, 400–408. [Google Scholar] [CrossRef]
- Si, J.; Yu, X.; Zhang, Y.; DeWille, J.W. Myc interacts with Max and Miz1 to repress C/EBPδ promoter activity and gene expression. Mol. Cancer 2010, 9, 92. [Google Scholar] [CrossRef] [Green Version]
- Staller, P.; Peukert, K.; Kiermaier, A.; Seoane, J.; Lukas, J.; Karsunky, H.; Möröy, T.; Bartek, J.; Massague, J.; Hänel, F.; et al. Repression of p15INK4b expression by Myc through association with Miz-1. Nat. Cell Biol. 2001, 3, 392–399. [Google Scholar] [CrossRef]
- van Riggelen, J.; Müller, J.; Otto, T.; Beuger, V.; Yetil, A.; Choi, P.S.; Kosan, C.; Möröy, T.; Felsher, D.W.; Eilers, M. The interaction between Myc and Miz1 is required to antagonize TGFβ-dependent autocrine signaling during lymphoma formation and maintenance. Genes Dev. 2010, 24, 1281–1294. [Google Scholar] [CrossRef] [Green Version]
- Gartel, A.L.; Shchors, K. Mechanisms of c-myc-mediated transcriptional repression of growth arrest genes. Exp. Cell Res. 2003, 283, 17–21. [Google Scholar] [CrossRef]
- Gartel, A.L.; Ye, X.; Goufman, E.; Shianov, P.; Hay, N.; Najmabadi, F.; Tyner, A. Myc represses the p21(WAF1/CIP1) promoter and interacts with Sp1/Sp3. Proc. Natl. Acad. Sci. USA 2001, 98, 4510–4515. [Google Scholar] [CrossRef] [Green Version]
- Adhikary, S.; Marinoni, F.; Hock, A.; Hulleman, E.; Popov, N.; Beier, R.; Bernard, S.; Quarto, M.; Capra, M.; Goettig, S.; et al. The Ubiquitin Ligase HectH9 Regulates Transcriptional Activation by Myc and Is Essential for Tumor Cell Proliferation. Cell 2005, 123, 409–421. [Google Scholar] [CrossRef] [Green Version]
- Faiola, F.; Liu, X.; Lo, S.; Pan, S.; Zhang, K.; Lymar, E.; Farina, A.; Martinez, E. Dual Regulation of c-Myc by p300 via Acetylation-Dependent Control of Myc Protein Turnover and Coactivation of Myc-Induced Transcription. Mol. Cell. Biol. 2005, 25, 10220–10234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Z.; Traugh, J.A.; Bishop, J.M. Negative Control of the Myc Protein by the Stress-Responsive Kinase Pak2. Mol. Cell. Biol. 2004, 24, 1582–1594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uribesalgo, I.; Benitah, S.A.; Di Croce, L. From oncogene to tumor suppressor: The dual role of Myc in leukemia. Cell Cycle 2012, 11, 1757–1764. [Google Scholar] [CrossRef] [Green Version]
- Uribesalgo, I.; Buschbeck, M.; Gutiérrez, A.; Teichmann, S.; Demajo, S.; Kuebler, B.; Nomdedeu, J.; Martín-Caballero, J.; Roma, G.; Benitah, S.A.; et al. E-box-independent regulation of transcription and differentiation by MYC. Nat. Cell Biol. 2011, 13, 1443–1449. [Google Scholar] [CrossRef]
- Hörlein, A.J.; Näär, A.M.; Heinzel, T.; Torchia, J.; Gloss, B.; Kurokawa, R.; Ryan, A.; Kamei, Y.; Soderstrom, M.; Glass, C.K.; et al. Ligand-independent repression by the thyroid hormone receptor mediated by a nuclear receptor co-repressor. Nature 1995, 377, 397–404. [Google Scholar] [CrossRef] [PubMed]
- Nisimoto, Y.; Ogawa, H. Interaction between p21-activated protein kinase and Rac during differentiation of HL-60 human promyelocytic leukemia cell induced by all-trans-retinoic acid. Eur. J. Biochem. 2002, 269, 2622–2629. [Google Scholar] [CrossRef] [PubMed]
- Benitah, S.A.; Frye, M.; Glogauer, M.; Watt, F.M. Stem Cell Depletion through Epidermal Deletion of Rac1. Science 2005, 309, 933–935. [Google Scholar] [CrossRef]
- Huang, S.; Spector, D.L. Nascent pre-mRNA transcripts are associated with nuclear regions enriched in splicing factors. Genes Dev. 1991, 5, 2288–2302. [Google Scholar] [CrossRef] [Green Version]
- Watt, F.M.; Frye, M.; Benitah, S.A. MYC in mammalian epidermis: How can an oncogene stimulate differentiation? Nat. Rev. Cancer 2008, 8, 234–242. [Google Scholar] [CrossRef] [Green Version]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [Green Version]
- Chakraborty, A.A.; Scuoppo, C.; Dey, S.; Thomas, L.R.; Lorey, S.L.; Lowe, S.W.; Tansey, W.P. A common functional consequence of tumor-derived mutations within c-MYC. Oncogene 2015, 34, 2406–2409. [Google Scholar] [CrossRef] [Green Version]
- Ayer, D.; Kretzner, L.; Eisenman, R.N. Mad: A heterodimeric partner for Max that antagonizes Myc transcriptional activity. Cell 1993, 72, 211–222. [Google Scholar] [CrossRef]
- Amati, B.; Alevizopoulos, K.; Vlach, J. Myc and the cell cycle. Front. Biosci. 1998, 3, d250–d268. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, S.; Prochownik, E.V. Small-molecule inhibitors of the Myc oncoprotein. Biochim. Biophys. Acta 2015, 1849, 525–543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, H.; Bower, K.E.; Beuscher, A.E.; Zhou, B.; Bobkov, A.A.; Olson, A.J.; Vogt, P.K. Stabilizers of the Max Homodimer Identified in Virtual Ligand Screening Inhibit Myc Function. Mol. Pharmacol. 2009, 76, 491–502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, K.-Y.; Wang, H.; Teriete, P.; Yap, J.L.; Chen, L.; Lanning, M.E.; Hu, A.; Lambert, L.J.; Holien, T.; Sundan, A.; et al. Perturbation of the c-Myc–Max Protein–Protein Interaction via Synthetic α-Helix Mimetics. J. Med. Chem. 2015, 58, 3002–3024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prochownik, E.V.; Vogt, P.K. Therapeutic Targeting of Myc. Genes Cancer 2010, 1, 650–659. [Google Scholar] [CrossRef] [Green Version]
- Whitfield, J.; Beaulieu, M.-E.; Soucek, L. Strategies to Inhibit Myc and Their Clinical Applicability. Front. Cell Dev. Biol. 2017, 5, 10. [Google Scholar] [CrossRef] [Green Version]
- Mukherjee, B.; Morgenbesser, S.D.; DePinho, R.A. Myc family oncoproteins function through a common pathway to transform normal cells in culture: Cross-interference by Max and trans-acting dominant mutants. Genes Dev. 1992, 6, 1480–1492. [Google Scholar] [CrossRef] [Green Version]
- Prendergast, G.C.; Hopewell, R.; Gorham, B.J.; Ziff, E.B. Biphasic effect of Max on Myc cotransformation activity and dependence on amino- and carboxy-terminal Max functions. Genes Dev. 1992, 6, 2429–2439. [Google Scholar] [CrossRef]
- Hopewell, R.; Ziff, E. The nerve growth factor-responsive PC12 cell line does not express the Myc dimerization partner Max. Mol. Cell. Biol. 1995, 15, 3470–3478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jafri, M.; Maher, E.R. Genetics in Endocrinology: The genetics of phaeochromocytoma: Using clinical features to guide genetic testing. Eur. J. Endocrinol. 2012, 166, 151–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flynn, A.; Benn, D.; Clifton-Bligh, R.; Robinson, B.; Trainer, A.H.; James, P.; Hogg, A.; Waldeck, K.; George, J.; Li, J.; et al. The genomic landscape of phaeochromocytoma. J. Pathol. 2015, 236, 78–89. [Google Scholar] [CrossRef]
- Maniam, P.; Zhou, K.; Lonergan, M.; Berg, J.N.; Goudie, D.R.; Newey, P.J. Pathogenicity and Penetrance of Germline SDHA Variants in Pheochromocytoma and Paraganglioma (PPGL). J. Endocr. Soc. 2018, 2, 806–816. [Google Scholar] [CrossRef] [PubMed]
- Daly, A.F.; Castermans, E.; Oudijk, L.; Guitelman, M.A.; Beckers, P.; Potorac, I.; Neggers, S.J.C.M.M.; Sacre, N.; van der Lely, A.-J.; Bours, V.; et al. Pheochromocytomas and pituitary adenomas in three patients with MAX exon deletions. Endocr.-Relat. Cancer 2018, 25, L37–L42. [Google Scholar] [CrossRef] [Green Version]
- Pantaleo, M.A.; Urbini, M.; Indio, V.; Ravegnini, G.; Nannini, M.; De Luca, M.; Tarantino, G.; Angelini, S.; Gronchi, A.; Vincenzi, B.; et al. Genome-Wide Analysis Identifies MEN1 and MAX Mutations and a Neuroendocrine-Like Molecular Heterogeneity in Quadruple WT GIST. Mol. Cancer Res. 2017, 15, 553–562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romero, O.A.; Diz, M.T.; Pros, E.; Savola, S.; Gomez, A.; Moran, S.; Sáez, C.; Iwakawa, R.; Villanueva, A.; Montuenga, L.; et al. MAX Inactivation in Small Cell Lung Cancer Disrupts MYC–SWI/SNF Programs and Is Synthetic Lethal with BRG1. Cancer Discov. 2013, 4, 292–303. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Hashimoto, H.; Zhang, X.; Barwick, B.; Lonial, S.; Boise, L.; Vertino, P.M.; Cheng, X. MAX is an epigenetic sensor of 5-carboxylcytosine and is altered in multiple myeloma. Nucleic Acids Res. 2017, 45, 2396–2407. [Google Scholar] [CrossRef] [Green Version]
- Prendergast, G.C.; Lawe, D.; Ziff, E. Association of Myn, the murine homolog of Max, with c-Myc stimulates methylation-sensitive DNA binding and ras cotransformation. Cell 1991, 65, 395–407. [Google Scholar] [CrossRef]
- Mathsyaraja, H.; Freie, B.; Cheng, P.-F.; Babaeva, E.; Catchpole, J.T.; Janssens, D.; Henikoff, S.; Eisenman, R.N. Max deletion destabilizes MYC protein and abrogates Eµ-Myc lymphomagenesis. Genes Dev. 2019, 33, 1252–1264. [Google Scholar] [CrossRef] [Green Version]
- Thomas, L.R.; Wang, Q.; Grieb, B.; Phan, J.; Foshage, A.M.; Sun, Q.; Olejniczak, E.T.; Clark, T.; Dey, S.; Lorey, S.; et al. Interaction with WDR5 Promotes Target Gene Recognition and Tumorigenesis by MYC. Mol. Cell 2015, 58, 440–452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, L.R.; Adams, C.M.; Wang, J.; Weissmiller, A.M.; Creighton, J.; Lorey, S.L.; Liu, Q.; Fesik, S.W.; Eischen, C.M.; Tansey, W.P. Interaction of the oncoprotein transcription factor MYC with its chromatin cofactor WDR5 is essential for tumor maintenance. Proc. Natl. Acad. Sci. USA 2019, 116, 25260–25268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dammert, M.A.; Brägelmann, J.; Olsen, R.R.; Böhm, S.; Monhasery, N.; Whitney, C.P.; Chalishazar, M.D.; Tumbrink, H.L.; Guthrie, M.R.; Klein, S.; et al. MYC paralog-dependent apoptotic priming orchestrates a spectrum of vulnerabilities in small cell lung cancer. Nat. Commun. 2019, 10, 3485. [Google Scholar] [CrossRef] [PubMed]
- McFadden, D.G.; Papagiannakopoulos, T.; Taylor-Weiner, A.; Stewart, C.; Carter, S.L.; Cibulskis, K.; Bhutkar, A.; McKenna, A.; Dooley, A.; Vernon, A.; et al. Genetic and Clonal Dissection of Murine Small Cell Lung Carcinoma Progression by Genome Sequencing. Cell 2014, 156, 1298–1311. [Google Scholar] [CrossRef] [Green Version]
- Mollaoglu, G.; Guthrie, M.R.; Böhm, S.; Brägelmann, J.; Can, I.; Ballieu, P.M.; Marx, A.; George, J.; Heinen, C.; Chalishazar, M.D.; et al. MYC Drives Progression of Small Cell Lung Cancer to a Variant Neuroendocrine Subtype with Vulnerability to Aurora Kinase Inhibition. Cancer Cell 2017, 31, 270–285. [Google Scholar] [CrossRef] [Green Version]
- Augert, A.; Mathsyaraja, H.; Ibrahim, A.H.; Freie, B.; Geuenich, M.J.; Cheng, P.-F.; Alibeckoff, S.P.; Wu, N.; Hiatt, J.B.; Basom, R.; et al. MAX Functions as a Tumor Suppressor and Rewires Metabolism in Small Cell Lung Cancer. Cancer Cell 2020, 38, 97–114.e7. [Google Scholar] [CrossRef]
- Meuwissen, R.; Linn, S.C.; Linnoila, R.; Zevenhoven, J.; Mooi, W.J.; Berns, A. Induction of small cell lung cancer by somatic inactivation of both Trp53 and Rb1 in a conditional mouse model. Cancer Cell 2003, 4, 181–189. [Google Scholar] [CrossRef] [Green Version]
- Chin, L.; Schreiber-Agus, N.; Pellicer, I.; Chen, K.; Lee, H.W.; Dudast, M.; Cordon-Cardo, C.; DePinho, R. Contrasting roles for Myc and Mad proteins in cellular growth and differentiation. Proc. Natl. Acad. Sci. USA 1995, 92, 8488–8492. [Google Scholar] [CrossRef] [Green Version]
- Västrik, I.; Kaipainen, A.; Penttilä, T.L.; Lymboussakis, A.; Alitalo, R.; Parvinen, M.; Alitalo, K. Expression of the mad gene during cell differentiation in vivo and its inhibition of cell growth in vitro. J. Cell Biol. 1995, 128, 1197–1208. [Google Scholar] [CrossRef]
- O’Hagan, R.C.; Schreiber-Agus, N.; Chen, K.; David, G.; Engelman, J.A.; Schwab, R.; Alland, L.; Thomson, C.; Ronning, D.R.; Sacchettini, J.C.; et al. Gene-target recognition among members of the Myc superfamily and implications for oncogenesis. Nat. Genet. 2000, 24, 113–119. [Google Scholar] [CrossRef]
- Han, S.; Park, K.; Kim, H.Y.; Lee, M.S.; Kim, H.J.; Kim, Y.D. Expression of Mad1 protein inhibits proliferation of cancer cells and inversely correlated with Myc protein expression in primary gastric cancer. Oncol. Rep. 1999, 6, 569–643. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Park, K.; Kim, H.-Y.; Lee, M.-S.; Kim, Y.-D.; Yuh, Y.J.; Kim, S.R.; Suh, H.S. Clinical implication of altered expression of Mad1 protein in human breast carcinoma. Cancer 2000, 88, 1623–1632. [Google Scholar] [CrossRef]
- Zou, L.; Zhang, P.; Luo, C.; Tu, Z. Mad1 suppresses bladder cancer cell proliferation by inhibiting human telomerase reverse transcriptase transcription and telomerase activity. Urology 2006, 67, 1335–1340. [Google Scholar] [CrossRef] [PubMed]
- Chou, C.-K.; Lee, D.-F.; Sun, H.-L.; Li, L.-Y.; Lin, C.-Y.; Huang, W.-C.; Hsu, J.-M.; Kuo, H.-P.; Yamaguchi, H.; Wang, Y.-N.; et al. The suppression of MAD1 by AKT-mediated phosphorylation activates MAD1 target genes transcription. Mol. Carcinog. 2009, 48, 1048–1058. [Google Scholar] [CrossRef] [Green Version]
- Guo, X.-L.; Pan, L.; Zhang, X.-J.; Suo, X.-H.; Niu, Z.-Y.; Zhang, J.-Y.; Wang, F.; Dong, Z.-R.; Da, W.; Ohno, R. Expression and mutation analysis of genes that encode the Myc antagonists Mad1, Mxi1 and Rox in acute leukaemia. Leuk. Lymphoma 2007, 48, 1200–1207. [Google Scholar] [CrossRef]
- Brunet, A.; Bonni, A.; Zigmond, M.J.; Lin, M.Z.; Juo, P.; Hu, L.S.; Anderson, M.J.; Arden, K.C.; Blenis, J.; Greenberg, M.E. Akt Promotes Cell Survival by Phosphorylating and Inhibiting a Forkhead Transcription Factor. Cell 1999, 96, 857–868. [Google Scholar] [CrossRef] [Green Version]
- Poomakkoth, N.; Issa, A.; Abdulrahman, N.; Abdelaziz, S.G.; Mraiche, F. p90 ribosomal S6 kinase: A potential therapeutic target in lung cancer. J. Transl. Med. 2016, 14, 14. [Google Scholar] [CrossRef] [Green Version]
- Sahin, F.; Kannangai, R.; Adegbola, O.; Wang, J.; Su, G.; Torbenson, M. mTOR and P70 S6 Kinase Expression in Primary Liver Neoplasms. Clin. Cancer Res. 2004, 10, 8421–8425. [Google Scholar] [CrossRef] [Green Version]
- Sulzmaier, F.J.; Ramos, J.W. RSK Isoforms in Cancer Cell Invasion and Metastasis. Cancer Res. 2013, 73, 6099–6105. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.; Blenis, J.; Yuan, J. Activation of PI3K/Akt and MAPK pathways regulates Myc-mediated transcription by phosphorylating and promoting the degradation of Mad1. Proc. Natl. Acad. Sci. USA 2008, 105, 6584–6589. [Google Scholar] [CrossRef] [Green Version]
- Yin, X.-Y.; Landay, M.F.; Han, W.; Levitan, E.S.; Watkins, S.; Levenson, R.M.; Farkas, D.L.; Prochownik, E.V. Dynamic in vivo interactions among Myc network members. Oncogene 2001, 20, 4650–4664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xing, Y.; Johnson, C.V.; Dobner, P.R.; Lawrence, J.B. Higher Level Organization of Individual Gene Transcription and RNA Splicing. Science 1993, 259, 1326–1330. [Google Scholar] [CrossRef] [PubMed]
- Xing, Y.; Johnson, C.V.; Moen, P.T.; McNeil, J.A.; Lawrence, J. Nonrandom gene organization: Structural arrangements of specific pre-mRNA transcription and splicing with SC-35 domains. J. Cell Biol. 1995, 131, 1635–1647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, K.P.; Jr., P.T.M.; Wydner, K.L.; Coleman, J.R.; Lawrence, J.B. Processing of Endogenous Pre-mRNAs in Association with SC-35 Domains Is Gene Specific. J. Cell Biol. 1999, 144, 617–629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomez-Roman, N.; Felton-Edkins, Z.A.; Kenneth, N.; Goodfellow, S.J.; Athineos, D.; Zhang, J.; Ramsbottom, B.A.; Innes, F.; Kantidakis, T.; Kerr, E.R.; et al. Activation by c-Myc of transcription by RNA polymerases I, II and III. Biochem. Soc. Symp. 2006, 73, 141–154. [Google Scholar] [CrossRef] [Green Version]
- Grandori, C.; Gomez-Roman, N.; Felton-Edkins, Z.A.; Ngouenet, C.; Galloway, D.A.; Eisenman, R.N.; White, R.J. c-Myc binds to human ribosomal DNA and stimulates transcription of rRNA genes by RNA polymerase I. Nat. Cell Biol. 2005, 7, 311–318. [Google Scholar] [CrossRef]
- Lafita-Navarro, M.D.C.; Blanco, R.; Mata-Garrido, J.; Liano-Pons, J.; Tapia, O.; García-Gutiérrez, L.; García-Alegría, E.; Berciano, M.T.; Lafarga, M.; León, J. MXD1 localizes in the nucleolus, binds UBF and impairs rRNA synthesis. Oncotarget 2016, 7, 69536–69548. [Google Scholar] [CrossRef] [Green Version]
- McStay, B.; Grummt, I. The Epigenetics of rRNA Genes: From Molecular to Chromosome Biology. Annu. Rev. Cell Dev. Biol. 2008, 24, 131–157. [Google Scholar] [CrossRef] [Green Version]
- Koberna, K.; Malínský, J.; Pliss, A.; Masata, M.; Vecerová, J.; Fialová, M.; Bednár, J.; Raška, I. Ribosomal genes in focus: New transcripts label the dense fibrillar components and form clusters indicative of "Christmas trees" in situ. J. Cell Biol. 2002, 157, 743–748. [Google Scholar] [CrossRef] [Green Version]
- Boisvert, F.-M.; Van Koningsbruggen, S.; Navascués, J.; Lamond, A. The multifunctional nucleolus. Nat. Rev. Mol. Cell Biol. 2007, 8, 574–585. [Google Scholar] [CrossRef]
- Moss, T.; Langlois, F.; Gagnon-Kugler, T.; Stefanovsky, V. A housekeeper with power of attorney: The rRNA genes in ribosome biogenesis. Cell. Mol. Life Sci. 2007, 64, 29–49. [Google Scholar] [CrossRef] [PubMed]
- Perry, R.P.; Kelley, D.E. Inhibition of RNA synthesis by actinomycin D: Characteristic dose-response of different RNA species. J. Cell. Physiol. 1970, 76, 127–139. [Google Scholar] [CrossRef] [PubMed]
- Shiue, C.-N.; Nematollahi-Mahani, A.; Wright, A.P. Myc-induced anchorage of the rDNA IGS region to nucleolar matrix modulates growth-stimulated changes in higher-order rDNA architecture. Nucleic Acids Res. 2014, 42, 5505–5517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castellano, L.; Giamas, G.; Jacob, J.; Coombes, R.C.; Lucchesi, W.; Thiruchelvam, P.; Barton, G.; Jiao, L.R.; Wait, R.; Waxman, J.; et al. The estrogen receptor-α-induced microRNA signature regulates itself and its transcriptional response. Proc. Natl. Acad. Sci. USA 2009, 106, 15732–15737. [Google Scholar] [CrossRef] [Green Version]
- Hayashita, Y.; Osada, H.; Tatematsu, Y.; Yamada, H.; Yanagisawa, K.; Tomida, S.; Yatabe, Y.; Kawahara, K.; Sekido, Y.; Takahashi, T. A Polycistronic MicroRNA Cluster, miR-17-92, Is Overexpressed in Human Lung Cancers and Enhances Cell Proliferation. Cancer Res. 2005, 65, 9628–9632. [Google Scholar] [CrossRef] [Green Version]
- Hu, W.; Jin, P.; Ding, C.; Liu, W. miR-19a/b modulates lung cancer cells metastasis through suppression of MXD1 expression. Oncol. Lett. 2016, 12, 1901–1905. [Google Scholar] [CrossRef] [Green Version]
- Lanza, G.; Ferracin, M.; Gafà, R.; Veronese, A.; Spizzo, R.; Pichiorri, F.; Liu, C.-G.; Calin, G.A.; Croce, C.M.; Negrini, M. mRNA/microRNA gene expression profile in microsatellite unstable colorectal cancer. Mol. Cancer 2007, 6, 54. [Google Scholar] [CrossRef] [Green Version]
- Mestdagh, P.; Fredlund, E.; Pattyn, F.; Schulte, J.H.; Muth, D.; Vermeulen, J.; Kumps, C.; Schlierf, S.; De Preter, K.; Van Roy, N.; et al. MYCN/c-MYC-induced microRNAs repress coding gene networks associated with poor outcome in MYCN/c-MYC-activated tumors. Oncogene 2010, 29, 1394–1404. [Google Scholar] [CrossRef] [Green Version]
- O’Donnell, K.A.; Wentzel, E.A.; Zeller, K.I.; Dang, C.; Mendell, J.T. c-Myc-regulated microRNAs modulate E2F1 expression. Nature 2005, 435, 839–843. [Google Scholar] [CrossRef]
- Plum, P.S.; Warnecke-Eberz, U.; Drebber, U.; Chon, S.-H.; Alakus, H.; Hölscher, A.H.; Quaas, A.; Bruns, C.J.; Gockel, I.; Lorenz, D.; et al. Upregulation of miR-17-92 cluster is associated with progression and lymph node metastasis in oesophageal adenocarcinoma. Sci. Rep. 2019, 9, 12113. [Google Scholar] [CrossRef] [Green Version]
- Rinaldi, A.; Poretti, G.; Kwee, I.; Zucca, E.; Catapano, C.V.; Tibiletti, M.G.; Bertoni, F. Concomitant MYC and microRNA cluster miR-17-92 ( C13orf25 ) amplification in human mantle cell lymphoma. Leuk. Lymphoma 2007, 48, 410–412. [Google Scholar] [CrossRef] [PubMed]
- Sandhu, S.K.; Fassan, M.; Volinia, S.; Lovat, F.; Balatti, V.; Pekarsky, Y.; Croce, C.M. B-cell malignancies in microRNA E -miR-17 92 transgenic mice. Proc. Natl. Acad. Sci. USA 2013, 110, 18208–18213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Lu, J.; Mandel, J.A.; Zhang, W.; Schwalbe, M.; Gorka, J.; Liu, Y.; Marburger, B.; Wang, J.; Ranganathan, S.; et al. Patient-Derived Mutant Forms of NFE2L2/NRF2 Drive Aggressive Murine Hepatoblastomas. Cell. Mol. Gastroenterol. Hepatol. 2021, 12, 199–228. [Google Scholar] [CrossRef] [PubMed]
- Yan, S.; Jia, C.; Quan, L.; Zhao, L.; Tian, Y.; Liu, A. Significance of the microRNA-17-92 gene cluster expressed in B-cell non-Hodgkin’s lymphoma. Mol. Med. Rep. 2019, 20, 2459–2467. [Google Scholar] [CrossRef]
- Wu, Q.; Yang, Z.; An, Y.; Hu, H.; Yin, J.; Zhang, P.; Nie, Y.; Wu, K.; Shi, Y.; Fan, D. MiR-19a/b modulate the metastasis of gastric cancer cells by targeting the tumour suppressor MXD1. Cell Death Dis. 2014, 5, e1144. [Google Scholar] [CrossRef]
- Ji, M.; Rao, E.; Ramachandrareddy, H.; Shen, Y.; Jiang, C.; Chen, J.; Hu, Y.; Rizzino, A.; Chan, W.C.; Fu, K.; et al. The miR-17-92 MicroRNA Cluster Is Regulated by Multiple Mechanisms in B-Cell Malignancies. Am. J. Pathol. 2011, 179, 1645–1656. [Google Scholar] [CrossRef]
- Zervos, A.S.; Gyuris, J.; Brent, R. Mxi1, a protein that specifically interacts with Max to bind Myc-Max recognition sites. Cell 1993, 72, 223–232. [Google Scholar] [CrossRef]
- Albarosa, R.; DiDonato, S.; Finocchiaro, G. Redefinition of the coding sequence of the MXI1 gene and identification of a polymorphic repeat in the 3? Non-coding region that allows the detection of loss of heterozygosity of chromosome 10q25 in glioblastomas. Hum. Genet. 1995, 95, 709–711. [Google Scholar] [CrossRef]
- Ariyanayagam-Baksh, S.M.; Baksh, F.K.; Swalsky, P.A.; Finkelstein, S.D. Loss of Heterozygosity in the MXI1 Gene Is a Frequent Occurrence in Melanoma. Mod. Pathol. 2003, 16, 992–995. [Google Scholar] [CrossRef] [Green Version]
- Edelhoff, S.; Sweetser, D.; Disteche, C.M. Mapping of the NEP receptor tyrosine kinase gene to human chromosome 6p21.3 and mouse chromosome 17C. Genomics 1995, 25, 309–311. [Google Scholar] [CrossRef]
- Gray, I.C.; Stewart, L.M.; Phillips, S.M.; Hamilton, J.A.; Gray, N.E.; Watson, G.J.; Spurr, N.K.; Snary, D. Mutation and expression analysis of the putative prostate tumour-suppressor gene PTEN. Br. J. Cancer 1998, 78, 1296–1300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.K.; Ro, J.Y.; Kemp, B.L.; Lee, J.S.; Kwon, T.J.; Hong, W.K.; Mao, L. Identification of two distinct tumor-suppressor loci on the long arm of chromosome 10 in small cell lung cancer. Oncogene 1998, 17, 1749–1753. [Google Scholar] [CrossRef] [PubMed]
- Lacombe, L.; Orlow, I.; Reuter, V.E.; Fair, W.R.; Dalbagni, G.; Zhang, Z.-F.; Cordon-Cardo, C. Microsatellite instability and deletion analysis of chromosome 10 in human prostate cancer. Int. J. Cancer 1996, 69, 110–113. [Google Scholar] [CrossRef]
- Lázcoz, P.; Muñoz, J.; Nistal, M.; Pestaña, Á.; Encío, I.; Castresana, J.S. Loss of heterozygosity and microsatellite instability on chromosome arm 10q in neuroblastoma. Cancer Genet. Cytogenet. 2007, 174, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Matsuyama, H.; Pan, Y.; Yoshihiro, S.; Kudren, D.; Naito, K.; Bergerheim, U.S.; Ekman, P. Clinical significance of chromosome 8p, 10q, and 16q deletions in prostate cancer. Prostate 2003, 54, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Morita, R.; Saito, S.; Ishikawa, J.; Ogawa, O.; Yoshida, O.; Yamakawa, K.; Nakamura, Y. Common regions of deletion on chromosomes 5q, 6q, and 10q in renal cell carcinoma. Cancer Res. 1991, 51, 5817–5820. [Google Scholar] [PubMed]
- Scott, D.K.; Straughton, D.; Bailey, S.; Ellison, D.W.; Clifford, S.C. Identification and Analysis of Tumor Suppressor Loci at Chromosome 10q23.3-10q25.3 in Medulloblastoma. Cell Cycle 2006, 5, 2381–2389. [Google Scholar] [CrossRef] [Green Version]
- Prochownik, E.V.; Grove, L.E.; Deubler, D.; Zhu, X.L.; Stephenson, R.A.; Rohr, L.R.; Yin, X.; Brothman, A.R. Commonly occurring loss and mutation of theMXI1 gene in prostate cancer. Genes Chromosom. Cancer 1998, 22, 295–304. [Google Scholar] [CrossRef]
- Shapiro, D.N.; Valentine, V.; Eagle, L.; Yin, X.; Morris, S.W.; Prochownik, E.V. Assignment of the Human MAD and MXI1 Genes to Chromosomes 2p12-p13 and 10q24-q25. Genomics 1994, 23, 282–285. [Google Scholar] [CrossRef]
- Simon, M.; Kokkino, A.J.; Warnick, R.E.; Tew, J.M., Jr.; von Deimling, A.; Menon, A.G. Role of genomic instability in meningioma progression. Genes Chromosomes Cancer 1996, 16, 265–269. [Google Scholar] [CrossRef]
- Wechsler, D.S.; Shelly, C.A.; Dang, C.V. Genomic Organization of HumanMXI1, a Putative Tumor Suppressor Gene. Genomics 1996, 32, 466–470. [Google Scholar] [CrossRef] [PubMed]
- Eagle, L.R.; Yin, X.; Brothman, A.R.; Williams, B.J.; Atkin, N.B.; Prochownik, E.V. Mutation of the MXI1 gene in prostate cancer. Nat. Genet. 1995, 9, 249–255. [Google Scholar] [CrossRef] [PubMed]
- Kawamata, N.; Park, D.; Wilczynski, S.; Yokota, J.; Koeffler, H.P. Point Mutations of the Mxil Gene are Rare in Prostate Cancers. Prostate 1996, 29, 191–193. [Google Scholar] [CrossRef]
- Li, X.-J.; Wang, D.-Y.; Zhu, Y.; Guo, R.-J.; Wang, X.-D.; Lubomir, K.; Mukai, K.; Sasaki, H.; Yoshida, H.; Oka, T.; et al. Mxi1Mutations in Human Neurofibrosarcomas. Jpn. J. Cancer Res. 1999, 90, 740–746. [Google Scholar] [CrossRef] [PubMed]
- A Kuczyk, M.; Serth, J.; Bokemeyer, C.; Schwede, J.; Herrmann, R.; Machtens, S.; Grünewald, V.; Höfner, K.; Jonas, U. The MXI1 tumor suppressor gene is not mutated in primary prostate cancer. Oncol. Rep. 1998, 5, 213–219. [Google Scholar] [CrossRef]
- Long, Q.; Xu, J.; Osunkoya, A.O.; Sannigrahi, S.; Johnson, B.A.; Zhou, W.; Gillespie, T.; Park, J.Y.; Nam, R.K.; Sugar, L.; et al. Global Transcriptome Analysis of Formalin-Fixed Prostate Cancer Specimens Identifies Biomarkers of Disease Recurrence. Cancer Res. 2014, 74, 3228–3237. [Google Scholar] [CrossRef] [Green Version]
- Rao, U.N.; Bakker, A.; Swalsky, P.A.; Finkelstein, S.D. Max interacting protein 1: Loss of heterozygosity is frequent in desmoplastic melanoma. Mod. Pathol. 1999, 12, 344–350. [Google Scholar]
- Fults, D.; Pedone, C.A.; Thompson, G.E.; Uchiyama, C.M.; Gumpper, K.L.; Iliev, D.; Vinson, V.L.; Tavtigian, S.V.; Perry, W.L., 3rd. Microsatellite deletion mapping on chromosome 10q and mutation analysis of MMAC1, FAS, and MXI1 in human glioblastoma multiforme. Int. J. Oncol. 1998, 12, 905–910. [Google Scholar] [CrossRef]
- Wang, N.-Y.; Xiang, Y.-Y.; Li, X.-J.; Hashimoto, M.; Tanaka, M.; Sugimura, H. Mxi1 is a potential cellular target of carcinogens and frequently mutated in experimental rat tumors and tumor cell lines. Pathol. Int. 2000, 50, 373–383. [Google Scholar] [CrossRef]
- Johnson, A.F.; Nguyen, H.T.; Veitia, R.A. Causes and effects of haploinsufficiency. Biol. Rev. 2019, 94, 1774–1785. [Google Scholar] [CrossRef]
- Sedic, M.; Kuperwasser, C. BRCA1-hapoinsufficiency: Unraveling the molecular and cellular basis for tissue-specific cancer. Cell Cycle 2016, 15, 621–627. [Google Scholar] [CrossRef] [PubMed]
- Schreiber-Agus, N.; Meng, Y.; Hoang, T.; Hou, H., Jr.; Chen, K.; Greenberg, R.; Cordon-Cardo, C.; Lee, H.W.; DePinho, R.A. Role of Mxi1 in ageing organ systems and the regulation of normal and neoplastic growth. Nature 1998, 393, 483–487. [Google Scholar] [CrossRef] [PubMed]
- Kurbegovic, A.; Trudel, M. The master regulators Myc and p53 cellular signaling and functions in polycystic kidney disease. Cell. Signal. 2020, 71, 109594. [Google Scholar] [CrossRef] [PubMed]
- Yoo, K.H.; Sung, Y.H.; Yang, M.H.; Jeon, J.O.; Yook, Y.J.; Woo, Y.M.; Lee, H.-W.; Park, J.H. Inactivation of Mxi1 induces Il-8 secretion activation in polycystic kidney. Biochem. Biophys. Res. Commun. 2007, 356, 85–90. [Google Scholar] [CrossRef]
- Grantham, J.J.; Ye, M.; Davidow, C.; Holub, B.; Sharma, M. Evidence for a potent lipid secretagogue in the cyst fluids of patients with autosomal dominant polycystic kidney disease. J. Am. Soc. Nephrol. 1995, 6, 1242–1249. [Google Scholar] [CrossRef]
- Nichols, M.T.; Gidey, E.; Matzakos, T.; Dahl, R.; Stiegmann, G.; Shah, R.J.; Grantham, J.J.; Fitz, J.G.; Brian, R. Secretion of cytokines and growth factors into autosomal dominant polycystic kidney disease liver cyst fluid. Hepatology 2004, 40, 836–846. [Google Scholar] [CrossRef]
- Lubarsky, B.; Krasnow, M.A. Tube Morphogenesis: Making and Shaping Biological Tubes. Cell 2003, 112, 19–28. [Google Scholar] [CrossRef] [Green Version]
- Song, S.A.; Yoo, K.H.; Ko, J.Y.; Kim, B.H.; Yook, Y.J.; Park, J.H. Over-expression of Mxi1 represses renal epithelial tubulogenesis through the reduction of matrix metalloproteinase 9. Biochem. Biophys. Res. Commun. 2012, 419, 459–465. [Google Scholar] [CrossRef]
- Liu, Z.; Greco, A.J.; Hellman, N.E.; Spector, J.; Robinson, J.; Tang, O.T.; Lipschutz, J.H. Intracellular signaling via ERK/MAPK completes the pathway for tubulogenic fibronectin in MDCK cells. Biochem. Biophys. Res. Commun. 2007, 353, 793–798. [Google Scholar] [CrossRef] [Green Version]
- Ye, P.; Habib, S.L.; Ricono, J.M.; Kim, N.-H.; Choudhury, G.G.; Barnes, J.L.; Abboud, H.E.; Arar, M.Y. Fibronectin induces ureteric bud cells branching and cellular cord and tubule formation. Kidney Int. 2004, 66, 1356–1364. [Google Scholar] [CrossRef] [Green Version]
- Hydbring, P.; Larsson, L.-G. Cdk2: A key regulator of the senescence control function of Myc. Aging 2010, 2, 244–250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hydbring, P.; Castell, A.; Larsson, L.-G. MYC Modulation around the CDK2/p27/SKP2 Axis. Genes 2017, 8, 174. [Google Scholar] [CrossRef]
- Lee, T.C.; Ziff, E.B. Mxi1 Is a Repressor of the c-myc Promoter and Reverses Activation by USF. J. Biol. Chem. 1999, 274, 595–606. [Google Scholar] [CrossRef] [Green Version]
- Taj, M.M.; Tawil, R.J.; Engstrom, L.D.; Zeng, Z.; Hwang, C.; Sanda, M.G.; Wechsler, D.S. Mxi1, a Myc antagonist, suppresses proliferation of DU145 human prostate cells. Prostate 2001, 47, 194–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manni, I.; Tunici, P.; Cirenei, N.; Albarosa, R.; Colombo, B.M.; Roz, L.; Sacchi, A.; Piaggio, G.; Finocchiaro, G. Mxi1 inhibits the proliferation of U87 glioma cells through down-regulation of cyclin B1 gene expression. Br. J. Cancer 2002, 86, 477–484. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.; Grove, L.; Datta, N.S.; Long, M.W.; Prochownik, E.V. C-myc overexpression and p53 loss cooperate to promote genomic instability. Oncogene 1999, 18, 1177–1184. [Google Scholar] [CrossRef] [Green Version]
- Prochownik, E.V. c-Myc: Linking transformation and genomic instability. Curr. Mol. Med. 2008, 8, 446–458. [Google Scholar] [CrossRef]
- Ganem, N.; Storchova, Z.; Pellman, D.; Ganem, N.; Storchova, Z.; Pellman, D. Tetraploidy, aneuploidy and cancer. Curr. Opin. Genet. Dev. 2007, 17, 157–162. [Google Scholar] [CrossRef]
- Hayashi, M.; Karlseder, J. DNA damage associated with mitosis and cytokinesis failure. Oncogene 2013, 32, 4593–4601. [Google Scholar] [CrossRef] [Green Version]
- Margolis, R.L. Tetraploidy and tumor development. Cancer Cell 2005, 8, 353–354. [Google Scholar] [CrossRef] [Green Version]
- Potapova, T.A.; Zhu, J.; Li, R. Aneuploidy and chromosomal instability: A vicious cycle driving cellular evolution and cancer genome chaos. Cancer Metastasis Rev. 2013, 32, 377–389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juergens, K.; Rust, B.; Pieler, T.; Henningfeld, K.A. Isolation and comparative expression analysis of the Myc-regulatory proteins Mad1, Mad3, and Mnt duringXenopus development. Dev. Dyn. 2005, 233, 1554–1559. [Google Scholar] [CrossRef] [PubMed]
- Kenney, A.M.; Cole, M.D.; Rowitch, D.H. Nmycupregulation by sonic hedgehog signaling promotes proliferation in developing cerebellar granule neuron precursors. Development 2003, 130, 15–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, P.; Du, F.; Yuelling, L.W.; Lin, T.; Muradimova, R.E.; Tricarico, R.; Wang, J.; Enikolopov, G.; Bellacosa, A.; Wechsler-Reya, R.J.; et al. A population of Nestin-expressing progenitors in the cerebellum exhibits increased tumorigenicity. Nat. Neurosci. 2013, 16, 1737–1744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliver, T.; Grasfeder, L.L.; Carroll, A.L.; Kaiser, C.; Gillingham, C.L.; Lin, S.M.; Wickramasinghe, R.; Scott, M.P.; Wechsler-Reya, R.J. Transcriptional profiling of the Sonic hedgehog response: A critical role for N-myc in proliferation of neuronal precursors. Proc. Natl. Acad. Sci. USA 2003, 100, 7331–7336. [Google Scholar] [CrossRef] [Green Version]
- Yun, J.-S.; Rust, J.M.; Ishimaru, T.; Díaz, E. A Novel Role of the Mad Family Member Mad3 in Cerebellar Granule Neuron Precursor Proliferation. Mol. Cell. Biol. 2007, 27, 8178–8189. [Google Scholar] [CrossRef] [Green Version]
- Fox, E.J.; Wright, S.C. S-phase-specific expression of the Mad3 gene in proliferating and differentiating cells. Biochem. J. 2001, 359, 361–367. [Google Scholar] [CrossRef]
- Fox, E.J.; Wright, S.C. The transcriptional repressor gene Mad3 is a novel target for regulation by E2F1. Biochem. J. 2003, 370, 307–313. [Google Scholar] [CrossRef] [Green Version]
- Gore, Y.; Lantner, F.; Hart, G.; Shachar, I. Mad3 Negatively Regulates B Cell Differentiation in the Spleen by Inducing Id2 Expression. Mol. Biol. Cell 2010, 21, 1864–1871. [Google Scholar] [CrossRef] [Green Version]
- Barisone, G.A.; Satake, N.; Lewis, C.; Duong, C.; Chen, C.; Lam, K.S.; Nolta, J.; Dίaz, E. Loss of MXD3 induces apoptosis of Reh human precursor B acute lymphoblastic leukemia cells. Blood Cells Mol. Dis. 2015, 54, 329–335. [Google Scholar] [CrossRef] [Green Version]
- Barisone, G.A.; Yun, J.-S.; Díaz, E. From cerebellar proliferation to tumorigenesis: New insights into the role of Mad3. Cell Cycle 2008, 7, 423–427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ngo, T.; Barisone, G.A.; Lam, K.S.; Dίaz, E. MXD3 regulation of DAOY cell proliferation dictated by time course of activation. BMC Cell Biol. 2014, 15, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ngo, T.; Corrales, A.; Bourne, T.; Elmojahid, S.; Lam, K.S.; Díaz, E. Alternative Splicing of MXD3 and Its Regulation of MXD3 Levels in Glioblastoma. Front. Mol. Biosci. 2019, 6, 5. [Google Scholar] [CrossRef] [PubMed]
- Belle, I.; Zhuang, Y. E Proteins in Lymphocyte Development and Lymphoid Diseases. Curr. Top. Dev. Biol. 2014, 110, 153–187. [Google Scholar] [CrossRef] [PubMed]
- Ogiwara, H.; Sasaki, M.; Mitachi, T.; Oike, T.; Higuchi, S.; Tominaga, Y.; Kohno, T. Targeting p300 Addiction in CBP-Deficient Cancers Causes Synthetic Lethality by Apoptotic Cell Death due to Abrogation of MYC Expression. Cancer Discov. 2016, 6, 430–445. [Google Scholar] [CrossRef] [Green Version]
- De Thé, H. Differentiation therapy revisited. Nat. Rev. Cancer 2018, 18, 117–127. [Google Scholar] [CrossRef]
- Pirozzi, C.J.; Yan, H. The implications of IDH mutations for cancer development and therapy. Nat. Rev. Clin. Oncol. 2021, 18, 645–661. [Google Scholar] [CrossRef]
- Kime, L.; Wright, S.C. Mad4 is regulated by a transcriptional repressor complex that contains Miz-1 and c-Myc. Biochem. J. 2003, 370, 291–298. [Google Scholar] [CrossRef] [Green Version]
- Coppola, J.A.; Cole, M.D. Constitutive c-myc oncogene expression blocks mouse erythroleukaemia cell differentiation but not commitment. Nature 1986, 320, 760–763. [Google Scholar] [CrossRef]
- Prochownik, E.V.; Kukowska, J. Deregulated expression of c-myc by murine erythroleukaemia cells prevents differentiation. Nature 1986, 322, 848–850. [Google Scholar] [CrossRef]
- Boros, K.; Lacaud, G.; Kouskoff, V. The transcription factor Mxd4 controls the proliferation of the first blood precursors at the onset of hematopoietic development in vitro. Exp. Hematol. 2011, 39, 1090–1100. [Google Scholar] [CrossRef] [PubMed]
- Mateyak, M.; Obaya, A.J.; Sedivy, J.M. c-Myc Regulates Cyclin D-Cdk4 and -Cdk6 Activity but Affects Cell Cycle Progression at Multiple Independent Points. Mol. Cell. Biol. 1999, 19, 4672–4683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, W.; Shen, J.; Wu, M.; Arsura, M.; Fitzgerald, M.; Suldan, Z.; Kim, D.W.; Hofmann, C.S.; Pianetti, S.; Romieu-Mourez, R.; et al. Repression of transcription of the p27Kip1 cyclin-dependent kinase inhibitor gene by c-Myc. Oncogene 2001, 20, 1688–1702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marcotte, R.; Chen, J.M.; Huard, S.; Wang, E. c-Myc creates an activation loop by transcriptionally repressing its own functional inhibitor, hMad4, in young fibroblasts, a loop lost in replicatively senescent fibroblasts. J. Cell. Biochem. 2005, 96, 1071–1085. [Google Scholar] [CrossRef]
- Meroni, G.; Reymond, A.; Alcalay, M.; Borsani, G.; Tanigami, A.; Tonlorenzi, R.; Nigro, C.L.; Messali, S.; Zollo, M.; Ledbetter, D.H.; et al. Rox, a novel bHLHZip protein expressed in quiescent cells that heterodimerizes with Max, binds a non-canonical E box and acts as a transcriptional repressor. EMBO J. 1997, 16, 2892–2906. [Google Scholar] [CrossRef]
- Wahlström, T.; Henriksson, M. Mnt Takes Control as Key Regulator of the Myc/Max/Mxd Network. In Advances in Cancer Research; Elsevier: Amsterdam, The Netherlands, 2007. [Google Scholar] [CrossRef]
- Lafita-Navarro, M.C.; Liano-Pons, J.; Quintanilla, A.; Varela, I.; Blanco, R.; Ourique, F.; Bretones, G.; Aresti, J.; Molina, E.; Carroll, P.; et al. The MNT transcription factor autoregulates its expression and supports proliferation in MYC-associated factor X (MAX)-deficient cells. J. Biol. Chem. 2020, 295, 2001–2017. [Google Scholar] [CrossRef]
- Nilsson, J.A.; Maclean, K.H.; Keller, U.B.; Pendeville, H.; Baudino, T.A.; Cleveland, J.L. Mnt Loss Triggers Myc Transcription Targets, Proliferation, Apoptosis, and Transformation. Mol. Cell. Biol. 2004, 24, 1560–1569. [Google Scholar] [CrossRef] [Green Version]
- Evan, G.I.; Littlewood, T.D. The role of c-myc in cell growth. Curr. Opin. Genet. Dev. 1993, 3, 44–49. [Google Scholar] [CrossRef]
- Pelengaris, S.; Khan, M.; Evan, G. c-MYC: More than just a matter of life and death. Nat. Rev. Cancer 2002, 2, 764–776. [Google Scholar] [CrossRef]
- Zhuang, D.; Mannava, S.; Grachtchouk, V.; Tang, W.-H.; Patil, S.; Wawrzyniak, J.A.; Berman, A.E.; Giordano, T.; Prochownik, E.V.; Soengas, M.; et al. C-MYC overexpression is required for continuous suppression of oncogene-induced senescence in melanoma cells. Oncogene 2008, 27, 6623–6634. [Google Scholar] [CrossRef] [Green Version]
- Dezfouli, S.; Bakke, A.; Huang, J.; Wynshaw-Boris, A.; Hurlin, P.J. Inflammatory Disease and Lymphomagenesis Caused by Deletion of the Myc Antagonist Mnt in T Cells. Mol. Cell. Biol. 2006, 26, 2080–2092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Felsher, D.W.; Bishop, J. Reversible Tumorigenesis by MYC in Hematopoietic Lineages. Mol. Cell 1999, 4, 199–207. [Google Scholar] [CrossRef]
- Link, J.; Ota, S.; Zhou, Z.-Q.; Daniel, C.J.; Sears, R.C.; Hurlin, P.J. A critical role for Mnt in Myc-driven T-cell proliferation and oncogenesis. Proc. Natl. Acad. Sci. USA 2012, 109, 19685–19690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campbell, K.J.; Vandenberg, C.J.; Anstee, N.; Hurlin, P.J.; Cory, S. Mnt modulates Myc-driven lymphomagenesis. Cell Death Differ. 2017, 24, 2117–2126. [Google Scholar] [CrossRef] [Green Version]
- Smith, D.P.; Bath, M.L.; Harris, A.W.; Cory, S. T-cell lymphomas mask slower developing B-lymphoid and myeloid tumours in transgenic mice with broad haemopoietic expression of MYC. Oncogene 2005, 24, 3544–3553. [Google Scholar] [CrossRef] [Green Version]
- Toyooka, K.; Bowen, T.J.; Hirotsune, S.; Li, Z.; Jain, S.; Ota, S.; Lozach, L.E.; Bassett, I.G.; Rosenfeld, M.G.; Glass, C.K.; et al. Mnt-Deficient Mammary Glands Exhibit Impaired Involution and Tumors with Characteristics of Myc Overexpression. Cancer Res. 2006, 66, 5565–5573. [Google Scholar] [CrossRef] [Green Version]
- Hutchinson, J.N.; Muller, W.J. Transgenic mouse models of human breast cancer. Oncogene 2000, 19, 6130–6137. [Google Scholar] [CrossRef] [Green Version]
- Cornelis, R.S.; Vanvliet, M.; Vos, C.B.J.; Cletonjansen, A.M.; Vandevijver, M.J.; Peterse, J.L.; Khan, P.M.; Borresen, A.L.; Cornelisse, C.J.; Devilee, P. Evidence for a Gene on 17p13.3, Distal to Tp53, as a Target for Allele Loss in Breast-Tumors without P53 Mutations. Cancer Res. 1994, 54, 4200–4206. [Google Scholar] [CrossRef]
- Hoff, C.; Seranski, P.; Mollenhauer, J.; Korn, B.; Detzel, T.; Reinhardt, R.; Ramser, J.; Poustka, A. Physical and Transcriptional Mapping of the 17p13.3 Region That Is Frequently Deleted in Human Cancer. Genomics 2000, 70, 26–33. [Google Scholar] [CrossRef]
- Nigro, C.L.; Venesio, T.; Reymond, A.; Meroni, G.; Alberici, P.; Cainarca, S.; Enrico, F.; Stack, M.; Ledbetter, D.H.; Liscia, D.S.; et al. The Human ROX Gene: Genomic Structure and Mutation Analysis in Human Breast Tumors. Genomics 1998, 49, 275–282. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Sun, H.; Dai, H.; Walsh, R.M.; Imakura, M.; Schelter, J.; Burchard, J.; Dai, X.; Chang, A.N.; Diaz, R.L.; et al. MicroRNA miR-210 modulates cellular response to hypoxia through the MYC antagonist MNT. Cell Cycle 2009, 8, 2756–2768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karenko, L.; Hahtola, S.; Päivinen, S.; Karhu, R.; Syrjä, S.; Kähkönen, M.; Nedoszytko, B.; Kytölä, S.; Zhou, Y.; Blazevic, V.; et al. Primary Cutaneous T-Cell Lymphomas Show a Deletion or Translocation AffectingNAV3, the HumanUNC-53Homologue. Cancer Res. 2005, 65, 8101–8110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kari, L.; Loboda, A.; Nebozhyn, M.; Rook, A.H.; Vonderheid, E.C.; Nichols, C.; Virok, D.; Chang, C.; Horng, W.-H.; Johnston, J.; et al. Classification and Prediction of Survival in Patients with the Leukemic Phase of Cutaneous T Cell Lymphoma. J. Exp. Med. 2003, 197, 1477–1488. [Google Scholar] [CrossRef] [PubMed]
- Mao, X.; Orchard, G.; Lillington, D.M.; Russell-Jones, R.; Young, B.D.; Whittaker, S.J. Amplification and overexpression of JUNB is associated with primary cutaneous T-cell lymphomas. Blood 2003, 101, 1513–1519. [Google Scholar] [CrossRef] [Green Version]
- Van Doorn, R.; Dijkman, R.; Vermeer, M.; Out-Luiting, J.J.; Van Der Raaij-Helmer, E.M.H.; Willemze, R.; Tensen, C. Aberrant Expression of the Tyrosine Kinase Receptor EphA4 and the Transcription Factor Twist in Sézary Syndrome Identified by Gene Expression Analysis. Cancer Res. 2004, 64, 5578–5586. [Google Scholar] [CrossRef] [Green Version]
- Vermeer, M.; Van Doorn, R.; Dijkman, R.; Mao, X.; Whittaker, S.; Vader, P.C.V.V.; Gerritsen, M.-J.P.; Geerts, M.-L.; Gellrich, S.; Söderberg, O.; et al. Novel and Highly Recurrent Chromosomal Alterations in Sezary Syndrome. Cancer Res. 2008, 68, 2689–2698. [Google Scholar] [CrossRef] [Green Version]
- Boonk, S.E.; Zoutman, W.H.; Marie-Cardine, A.; van der Fits, L.; Out-Luiting, J.J.; Mitchell, T.; Tosi, I.; Morris, S.L.; Moriarty, B.; Booken, N.; et al. Evaluation of Immunophenotypic and Molecular Biomarkers for Sézary Syndrome Using Standard Operating Procedures: A Multicenter Study of 59 Patients. J. Investig. Dermatol. 2016, 136, 1364–1372. [Google Scholar] [CrossRef] [Green Version]
- Edelmann, J.; Holzmann, K.; Miller, F.; Winkler, D.; Bühler, A.; Zenz, T.; Bullinger, L.; Kühn, M.W.M.; Gerhardinger, A.; Bloehdorn, J.; et al. High-resolution genomic profiling of chronic lymphocytic leukemia reveals new recurrent genomic alterations. Blood 2012, 120, 4783–4794. [Google Scholar] [CrossRef] [Green Version]
- Aref, S.; Fouda, M.; El-Dosoky, E.; Menessy, A.; Mabed, M.; Saleeb, M.; Zalata, K. c-Myc oncogene and Cdc25A cell activating phosphatase expression in non-Hodgkin’s lymphoma. Hematology 2003, 8, 183–190. [Google Scholar] [CrossRef]
- Huh, Y.O.; Lin, K.I.-C.; Vega, F.; Schlette, E.; Yin, C.C.; Keating, M.J.; Luthra, R.; Medeiros, L.J.; Abruzzo, L.V. MYC translocation in chronic lymphocytic leukaemia is associated with increased prolymphocytes and a poor prognosis. Br. J. Haematol. 2008, 142, 36–44. [Google Scholar] [CrossRef]
- Nagy, B.; Lundán, T.; Larramendy, M.L.; Aalto, Y.; Zhu, Y.; Niini, T.; Edgren, H.; Ferrer, A.; Vilpo, J.; Elonen, E.; et al. Abnormal expression of apoptosis-related genes in haematological malignancies: Overexpression of MYC is poor prognostic sign in mantle cell lymphoma. Br. J. Haematol. 2003, 120, 434–441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; Kater, A.P.; Widhopf, G.F.; Chuang, H.-Y.; Enzler, T.; James, D.F.; Poustovoitov, M.; Tseng, P.-H.; Janz, S.; Hoh, C.; et al. B-cell activating factor and v-Myc myelocytomatosis viral oncogene homolog (c-Myc) influence progression of chronic lymphocytic leukemia. Proc. Natl. Acad. Sci. USA 2010, 107, 18956–18960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lavin, D.P.; Abassi, L.; Inayatullah, M.; Tiwari, V.K. Mnt Represses Epithelial Identity To Promote Epithelial-to-Mesenchymal Transition. Mol. Cell. Biol. 2021, 41, e0018321. [Google Scholar] [CrossRef] [PubMed]
- Christiansen, J.J.; Rajasekaran, A.K. Reassessing Epithelial to Mesenchymal Transition as a Prerequisite for Carcinoma Invasion and Metastasis. Cancer Res. 2006, 66, 8319–8326. [Google Scholar] [CrossRef] [Green Version]
- Jang, M.H.; Kim, H.J.; Kim, E.J.; Chung, Y.R.; Park, S.Y. Expression of epithelial-mesenchymal transition–related markers in triple-negative breast cancer: ZEB1 as a potential biomarker for poor clinical outcome. Hum. Pathol. 2015, 46, 1267–1274. [Google Scholar] [CrossRef]
- Thompson, E.W.; Newgreen, D.F. Carcinoma Invasion and Metastasis: A Role for Epithelial-Mesenchymal Transition? Cancer Res. 2005, 65, 5991–5995. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Li, H.; Ren, G. Epithelial-mesenchymal transition and drug resistance in breast cancer (Review). Int. J. Oncol. 2015, 47, 840–848. [Google Scholar] [CrossRef] [Green Version]
- Gooding, A.J.; Schiemann, W.P. Epithelial–Mesenchymal Transition Programs and Cancer Stem Cell Phenotypes: Mediators of Breast Cancer Therapy Resistance. Mol. Cancer Res. 2020, 18, 1257–1270. [Google Scholar] [CrossRef]
- Raja, R.; Pandey, A.; Kumar, P. Epithelial to mesenchymal plasticity role in cancer progression. Front. Biosci. 2020, 25, 838–873. [Google Scholar] [CrossRef]
- Nguyen, H.V.; Vandenberg, C.J.; Ng, A.P.; Robati, M.R.; Anstee, N.S.; Rimes, J.; Hawkins, E.D.; Cory, S. Development and survival of MYC-driven lymphomas require the MYC antagonist MNT to curb MYC-induced apoptosis. Blood 2020, 135, 1019–1031. [Google Scholar] [CrossRef]
- Vasilevsky, N.A.; Ruby, C.E.; Hurlin, P.J.; Weinberg, A.D. OX40 engagement stabilizes Mxd4 and Mnt protein levels in antigen-stimulated T cells leading to an increase in cell survival. Eur. J. Immunol. 2011, 41, 1024–1034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuzyk, A.; Mai, S. c-MYC-Induced Genomic Instability. Cold Spring Harb. Perspect. Med. 2014, 4, a014373. [Google Scholar] [CrossRef] [PubMed]
- Basu, A.; Haldar, S. The relationship between BcI2, Bax and p53: Consequences for cell cycle progression and cell death. Mol. Hum. Reprod. 1998, 4, 1099–1109. [Google Scholar] [CrossRef] [PubMed]
- Sebé-Pedrós, A.; Ruiz-Trillo, I. Evolution and Classification of the T-Box Transcription Factor Family. Curr. Top. Dev. Biol. 2017, 122, 1–26. [Google Scholar] [CrossRef]
- Blum, M.; Chang, H.-Y.; Chuguransky, S.; Grego, T.; Kandasaamy, S.; Mitchell, A.; Nuka, G.; Paysan-Lafosse, T.; Qureshi, M.; Raj, S.; et al. The InterPro protein families and domains database: 20 years on. Nucleic Acids Res. 2021, 49, D344–D354. [Google Scholar] [CrossRef]
- Kispert, A.; Herrmann, B. The Brachyury gene encodes a novel DNA binding protein. EMBO J. 1993, 12, 3211–3220. [Google Scholar] [CrossRef]
- Müller, C.W.; Herrmann, B.G. Crystallographic structure of the T domain–DNA complex of the Brachyury transcription factor. Nature 1997, 389, 884–888. [Google Scholar] [CrossRef]
- Papaioannou, V.E. The T-box gene family: Emerging roles in development, stem cells and cancer. Development 2014, 141, 3819–3833. [Google Scholar] [CrossRef] [Green Version]
- Wilson, V.; Conlon, F.L. The T-box family. Genome Biol. 2002, 3, REVIEWS3008. [Google Scholar] [CrossRef]
- Burn, S.F.; Washkowitz, A.J.; Gavrilov, S.; Papaioannou, V.E. Postimplantation Mga expression and embryonic lethality of two gene-trap alleles. Patterns 2018, 27, 31–35. [Google Scholar] [CrossRef]
- Washkowitz, A.J.; Schall, C.; Zhang, K.; Wurst, W.; Floss, T.; Mager, J.; Papaioannou, V.E. Mga is essential for the survival of pluripotent cells during peri-implantation development. Development 2015, 142, 31–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Paoli, L.; Cerri, M.; Monti, S.; Rasi, S.; Spina, V.; Bruscaggin, A.; Greco, M.; Ciardullo, C.; Famà, R.; Cresta, S.; et al. MGA, a suppressor of MYC, is recurrently inactivated in high risk chronic lymphocytic leukemia. Leuk. Lymphoma 2013, 54, 1087–1090. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Hu, B.; Wang, F.; Yan, Y.; Kim, E.; Vitale, C.; Patel, K.P.; Strati, P.; Gumbs, C.; Little, L.; et al. Clinical implications of cancer gene mutations in patients with chronic lymphocytic leukemia treated with lenalidomide. Blood 2018, 131, 1820–1832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, L.; Gu, Z.-H.; Yan, Z.-X.; Zhao, X.; Xie, Y.-Y.; Zhang, Z.-G.; Pan, C.-M.; Hu, Y.; Cai, C.-P.; Dong, Y.; et al. Exome sequencing identifies somatic mutations of DDX3X in natural killer/T-cell lymphoma. Nat. Genet. 2015, 47, 1061–1066. [Google Scholar] [CrossRef]
- Montes-Mojarro, I.A.; Chen, B.-J.; Ramirez-Ibarguen, A.F.; Quezada-Fiallos, C.M.; Pérez-Báez, W.B.; Dueñas, D.; Casavilca-Zambrano, S.; Ortiz-Mayor, M.; Rojas-Bilbao, E.; Rivello, H.G.; et al. Mutational profile and EBV strains of extranodal NK/T-cell lymphoma, nasal type in Latin America. Mod. Pathol. 2020, 33, 781–791. [Google Scholar] [CrossRef]
- Amin, N.A.; Seymour, E.; Saiya-Cork, K.; Parkin, B.; Shedden, K.; Malek, S.N. A Quantitative Analysis of Subclonal and Clonal Gene Mutations before and after Therapy in Chronic Lymphocytic Leukemia. Clin. Cancer Res. 2016, 22, 4525–4535. [Google Scholar] [CrossRef] [Green Version]
- Campbell, J.D.; Alexandrov, A.; Kim, J.; Wala, J.; Berger, A.H.; Pedamallu, C.S.; Shukla, S.A.; Guo, G.; Brooks, A.N.; Murray, B.A.; et al. Distinct patterns of somatic genome alterations in lung adenocarcinomas and squamous cell carcinomas. Nat. Genet. 2016, 48, 607–616. [Google Scholar] [CrossRef] [Green Version]
- Cooke, S.L.; Shlien, A.; Marshall, J.; Pipinikas, C.P.; Martincorena, I.; Tubio, J.; Li, Y.; Menzies, A.; Mudie, L.; Ramakrishna, M.; et al. Processed pseudogenes acquired somatically during cancer development. Nat. Commun. 2014, 5, 3644. [Google Scholar] [CrossRef]
- Jo, Y.S.; Kim, M.S.; Yoo, N.J.; Lee, S.H. Somatic mutation of a candidate tumour suppressor MGA gene and its mutational heterogeneity in colorectal cancers. Pathology 2016, 48, 525–527. [Google Scholar] [CrossRef]
- Madan, V.; Han, L.; Hattori, N.; Teoh, W.W.; Mayakonda, A.; Sun, Q.-Y.; Ding, L.-W.; Nordin, H.B.M.; Lim, S.L.; Shyamsunder, P.; et al. ASXL2 regulates hematopoiesis in mice and its deficiency promotes myeloid expansion. Haematologica 2018, 103, 1980–1990. [Google Scholar] [CrossRef] [Green Version]
- Park, C.; Ha, S.Y.; Kim, S.T.; Kim, H.C.; Heo, J.S.; Park, Y.S.; Lauwers, G.; Lee, J.; Kim, K.-M. Identification of the BRAF V600E mutation in gastroenteropancreatic neuroendocrine tumors. Oncotarget 2016, 7, 4024–4035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stratmann, S.; Yones, S.A.; Mayrhofer, M.; Norgren, N.; Skaftason, A.; Sun, J.; Smolinska, K.; Komorowski, J.; Herlin, M.K.; Sundström, C.; et al. Genomic characterization of relapsed acute myeloid leukemia reveals novel putative therapeutic targets. Blood Adv. 2021, 5, 900–912. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.-Y.; Ding, L.-W.; Tan, K.-T.; Chien, W.; Mayakonda, A.; Lin, D.-C.; Loh, X.-Y.; Xiao, J.-F.; Meggendorfer, M.; Alpermann, T.; et al. Ordering of mutations in acute myeloid leukemia with partial tandem duplication of MLL (MLL-PTD). Leukemia 2017, 31, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Veeramachaneni, R.; Walker, T.; Revil, T.; De Weck, A.; Badescu, D.; O’Sullivan, J.; Higgins, C.; Elliott, L.; Liloglou, T.; Risk, J.M.; et al. Analysis of head and neck carcinoma progression reveals novel and relevant stage-specific changes associated with immortalisation and malignancy. Sci. Rep. 2019, 9, 11992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Ma, Y.; Li, Y.; Shen, X.; Yu, Y.; Pan, Y.; Zhang, Y.; Zheng, D.; Zhao, Y.; Ye, T.; et al. Comparative analysis of co-occurring mutations of specific tumor suppressor genes in lung adenocarcinoma between Asian and Caucasian populations. J. Cancer Res. Clin. Oncol. 2019, 145, 747–757. [Google Scholar] [CrossRef]
- Ohanian, M.; Rozovski, U.; Kanagal-Shamanna, R.; Abruzzo, L.V.; Loghavi, S.; Kadia, T.; Futreal, A.; Bhalla, K.; Zuo, Z.; Huh, Y.O.; et al. MYC protein expression is an important prognostic factor in acute myeloid leukemia. Leuk. Lymphoma 2019, 60, 37–48. [Google Scholar] [CrossRef]
- Basit, F.; Andersson, M.; Hultquist, A. The Myc/Max/Mxd Network Is a Target of Mutated Flt3 Signaling in Hematopoietic Stem Cells in Flt3-ITD-Induced Myeloproliferative Disease. Stem Cells Int. 2018, 2018, 3286949. [Google Scholar] [CrossRef]
- Lee, M.J.; Koff, J.L.; Switchenko, J.M.; Jhaney, C.I.; Ba, R.A.H.; Patel, S.; Dave, S.S.; Flowers, C.R. Genome-defined African ancestry is associated with distinct mutations and worse survival in patients with diffuse large B-cell lymphoma. Cancer 2020, 126, 3493–3503. [Google Scholar] [CrossRef]
- Reddy, A.; Zhang, J.; Davis, N.S.; Moffitt, A.; Love, C.L.; Waldrop, A.; Leppä, S.; Pasanen, A.; Meriranta, L.; Karjalainen-Lindsberg, M.-L.; et al. Genetic and Functional Drivers of Diffuse Large B Cell Lymphoma. Cell 2017, 171, 481–494.e15. [Google Scholar] [CrossRef] [Green Version]
- Morkel, M.; Wenkel, J.; Bannister, A.; Kouzarides, T.; Hagemeier, C. An E2F-like repressor of transcription. Nature 1997, 390, 567–568. [Google Scholar] [CrossRef]
- Scelfo, A.; Fernández-Pérez, D.; Tamburri, S.; Zanotti, M.; Lavarone, E.; Soldi, M.; Bonaldi, T.; Ferrari, K.; Pasini, D. Functional Landscape of PCGF Proteins Reveals Both RING1A/B-Dependent-and RING1A/B-Independent-Specific Activities. Mol. Cell 2019, 74, 1037–1052.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stielow, B.; Finkernagel, F.; Stiewe, T.; Nist, A.; Suske, G. MGA, L3MBTL2 and E2F6 determine genomic binding of the non-canonical Polycomb repressive complex PRC1.6. PLoS Genet. 2018, 14, e1007193. [Google Scholar] [CrossRef] [PubMed]
- Trojer, P.; Cao, A.R.; Gao, Z.; Li, Y.; Zhang, J.; Xu, X.; Li, G.; Losson, R.; Erdjument-Bromage, H.; Tempst, P.; et al. L3MBTL2 Protein Acts in Concert with PcG Protein-Mediated Monoubiquitination of H2A to Establish a Repressive Chromatin Structure. Mol. Cell 2011, 42, 438–450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Wijnen, A.J.; Bagheri, L.; Badreldin, A.A.; Larson, A.N.; Dudakovic, A.; Thaler, R.; Paradise, C.R.; Wu, Z. Biological functions of chromobox (CBX) proteins in stem cell self-renewal, lineage-commitment, cancer and development. Bone 2021, 143, 115659. [Google Scholar] [CrossRef] [PubMed]
- Donehower, L.A.; Harvey, M.; Slagle, B.L.; McArthur, M.J.; Montgomery, C.A., Jr.; Butel, J.; Bradley, A. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature 1992, 356, 215–221. [Google Scholar] [CrossRef]
- Jackson, E.L.; Willis, N.; Mercer, K.; Bronson, R.T.; Crowley, D.; Montoya, R.; Jacks, T.; Tuveson, D.A. Analysis of lung tumor initiation and progression using conditional expression of oncogenic K-ras. Genes Dev. 2001, 15, 3243–3248. [Google Scholar] [CrossRef] [Green Version]
- Sánchez-Rivera, F.J.; Papagiannakopoulos, T.; Romero, R.; Tammela, T.; Bauer, M.R.; Bhutkar, A.; Joshi, N.; Subbaraj, L.; Bronson, R.T.; Xue, W.; et al. Rapid modelling of cooperating genetic events in cancer through somatic genome editing. Nature 2014, 516, 428–431. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, A.; Hirasaki, M.; Hishida, T.; Wu, J.; Okamura, D.; Ueda, A.; Nishimoto, M.; Nakachi, Y.; Mizuno, Y.; Okazaki, Y.; et al. Loss of MAX results in meiotic entry in mouse embryonic and germline stem cells. Nat. Commun. 2016, 7, 11056. [Google Scholar] [CrossRef] [Green Version]
- Gao, Z.; Zhang, J.; Bonasio, R.; Strino, F.; Sawai, A.; Parisi, F.; Kluger, Y.; Reinberg, D. PCGF Homologs, CBX Proteins, and RYBP Define Functionally Distinct PRC1 Family Complexes. Mol. Cell 2012, 45, 344–356. [Google Scholar] [CrossRef] [Green Version]
- Kawaguchi, T.; Takenoshita, M.; Kabashima, T.; Uyeda, K. Glucose and cAMP regulate the L-type pyruvate kinase gene by phosphorylation/dephosphorylation of the carbohydrate response element binding protein. Proc. Natl. Acad. Sci. USA 2001, 98, 13710–13715. [Google Scholar] [CrossRef] [Green Version]
- O’Callaghan, B.L.; Koo, S.-H.; Wu, Y.; Freake, H.C.; Towle, H.C. Glucose Regulation of the Acetyl-CoA Carboxylase Promoter PI in Rat Hepatocytes. J. Biol. Chem. 2001, 276, 16033–16039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishii, S.; Iizuka, K.; Miller, B.C.; Uyeda, K. Carbohydrate response element binding protein directly promotes lipogenic enzyme gene transcription. Proc. Natl. Acad. Sci. USA 2004, 101, 15597–15602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rufo, C.; Teran-Garcia, M.; Nakamura, M.T.; Koo, S.-H.; Towle, H.C.; Clarke, S.D. Involvement of a Unique Carbohydrate-responsive Factor in the Glucose Regulation of Rat Liver Fatty-acid Synthase Gene Transcription. J. Biol. Chem. 2001, 276, 21969–21975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shih, H.; Towle, H. Definition of the carbohydrate response element of the rat S14 gene. Evidence for a common factor required for carbohydrate regulation of hepatic genes. J. Biol. Chem. 1992, 267, 13222–13228. [Google Scholar] [CrossRef]
- Li, M.V.; Chen, W.; Poungvarin, N.; Imamura, M.; Chan, L. Glucose-Mediated Transactivation of Carbohydrate Response Element-Binding Protein Requires Cooperative Actions from Mondo Conserved Regions and EssentialTrans-Acting Factor 14-3-3. Mol. Endocrinol. 2008, 22, 1658–1672. [Google Scholar] [CrossRef] [Green Version]
- Merla, G.; Howald, C.; Antonarakis, S.; Reymond, A. The subcellular localization of the ChoRE-binding protein, encoded by the Williams–Beuren syndrome critical region gene 14, is regulated by 14-3-3. Hum. Mol. Genet. 2004, 13, 1505–1514. [Google Scholar] [CrossRef]
- Sakiyama, H.; Wynn, R.M.; Lee, W.-R.; Fukasawa, M.; Mizuguchi, H.; Gardner, K.H.; Repa, J.J.; Uyeda, K. Regulation of Nuclear Import/Export of Carbohydrate Response Element-binding Protein (ChREBP): Interaction of an alpha-helix of ChREBP with the 14-3-3 proteins and regulation by phosphorylation. J. Biol. Chem. 2008, 283, 24899–24908. [Google Scholar] [CrossRef] [Green Version]
- Falcicchio, M.; Ward, J.A.; Macip, S.; Doveston, R.G. Regulation of p53 by the 14-3-3 protein interaction network: New opportunities for drug discovery in cancer. Cell Death Discov. 2020, 6, 126. [Google Scholar] [CrossRef]
- Pennington, K.L.; Chan, T.Y.; Torres, M.; Andersen, J.L. The dynamic and stress-adaptive signaling hub of 14-3-3: Emerging mechanisms of regulation and context-dependent protein–protein interactions. Oncogene 2018, 37, 5587–5604. [Google Scholar] [CrossRef] [Green Version]
- Grompe, M.; Lindstedt, S.; Al-Dhalimy, M.; Kennaway, N.G.; Papaconstantinou, J.; Torres-Ramos, C.A.; Ou, C.-N.; Finegold, M.J. Pharmacological correction of neonatal lethal hepatic dysfunction in a murine model of hereditary tyrosinaemia type I. Nat. Genet. 1995, 10, 453–460. [Google Scholar] [CrossRef]
- Lindqvist, L.M.; Tandoc, K.; Topisirovic, I.; Furic, L. Cross-talk between protein synthesis, energy metabolism and autophagy in cancer. Curr. Opin. Genet. Dev. 2017, 48, 104–111. [Google Scholar] [CrossRef]
- Metukuri, M.R.; Zhang, P.; Stewart, A.F.; Vasavada, R.C.; Garcia-Ocaña, A.; Scott, D.K.; Basantani, M.K.; Chin, C.; Stamateris, R.E.; Alonso, L.C.; et al. ChREBP Mediates Glucose-Stimulated Pancreatic β-Cell Proliferation. Diabetes 2012, 61, 2004–2015. [Google Scholar] [CrossRef] [Green Version]
- Ran, H.; Lu, Y.; Zhang, Q.; Hu, Q.; Zhao, J.; Wang, K.; Tong, X.; Su, Q. MondoA Is Required for Normal Myogenesis and Regulation of the Skeletal Muscle Glycogen Content in Mice. Diabetes Metab. J. 2021, 45, 439–451. [Google Scholar] [CrossRef]
- Eilers, A.L.; Sundwall, E.; Lin, M.; Sullivan, A.A.; Ayer, D.E. A Novel Heterodimerization Domain, CRM1, and 14-3-3 Control Subcellular Localization of the MondoA-Mlx Heterocomplex. Mol. Cell. Biol. 2002, 22, 8514–8526. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.; Holmgren, A. The thioredoxin antioxidant system. Free Radic. Biol. Med. 2014, 66, 75–87. [Google Scholar] [CrossRef]
- Chen, Y.; Ning, J.; Cao, W.; Wang, S.; Du, T.; Jiang, J.; Feng, X.; Zhang, B. Research Progress of TXNIP as a Tumor Suppressor Gene Participating in the Metabolic Reprogramming and Oxidative Stress of Cancer Cells in Various Cancers. Front. Oncol. 2020, 10, 568574. [Google Scholar] [CrossRef]
- Jeon, J.-H.; Lee, K.-N.; Hwang, C.Y.; Kwon, K.-S.; You, K.-H.; Choi, I.; Bradshaw, T.D.; Matthews, C.S.; Cookson, J.; Chew, E.-H.; et al. Tumor Suppressor VDUP1 Increases p27kip1 Stability by Inhibiting JAB1. Cancer Res. 2005, 65, 4485–4489. [Google Scholar] [CrossRef] [Green Version]
- Arakaki, A.K.S.; Pan, W.-A.; Wedegaertner, H.; Roca-Mercado, I.; Chinn, L.; Gujral, T.S.; Trejo, J. α-Arrestin ARRDC3 tumor suppressor function is linked to GPCR-induced TAZ activation and breast cancer metastasis. J. Cell Sci. 2021, 134, jcs254888. [Google Scholar] [CrossRef]
- Xiao, J.; Shi, Q.; Li, W.; Mu, X.; Peng, J.; Li, M.; Chen, M.; Huang, H.; Wang, C.; Gao, K.; et al. ARRDC1 and ARRDC3 act as tumor suppressors in renal cell carcinoma by facilitating YAP1 degradation. Am. J. Cancer Res. 2018, 8, 132–143. [Google Scholar]
- Zheng, D.; Wu, W.; Dong, N.; Jiang, X.; Xu, J.; Zhan, X.; Zhang, Z.; Hu, Z. Mxd1 mediates hypoxia-induced cisplatin resistance in osteosarcoma cells by repression of the PTEN tumor suppressor gene. Mol. Carcinog. 2017, 56, 2234–2244. [Google Scholar] [CrossRef]
- Zheng, Y.; Lin, Z.-Y.; Xie, J.-J.; Jiang, F.-N.; Chen, C.-J.; Li, J.-X.; Zhou, X.; Zhong, W.-D.; Zheng, Z.-Y.L.Y. ARRDC3 Inhibits the Progression of Human Prostate Cancer Through ARRDC3-ITGβ4 Pathway. Curr. Mol. Med. 2017, 17, 221–229. [Google Scholar] [CrossRef]
- Rhein, P.; Scheid, S.; Ratei, R.; Hagemeier, C.; Seeger, K.; Kirschner-Schwabe, R.; Moericke, A.; Schrappe, M.; Spang, R.; Ludwig, W.-D.; et al. Gene expression shift towards normal B cells, decreased proliferative capacity and distinct surface receptors characterize leukemic blasts persisting during induction therapy in childhood acute lymphoblastic leukemia. Leukemia 2007, 21, 897–905. [Google Scholar] [CrossRef]
- Parmenter, T.J.; Kleinschmidt, M.; Kinross, K.M.; Bond, S.T.; Li, J.; Kaadige, M.R.; Rao, A.; Sheppard, K.; Hugo, W.; Pupo, G.M.; et al. Response of BRAF-Mutant Melanoma to BRAF Inhibition Is Mediated by a Network of Transcriptional Regulators of Glycolysis. Cancer Discov. 2014, 4, 423–433. [Google Scholar] [CrossRef] [Green Version]
- Bollag, G.; Hirth, P.; Tsai, J.; Zhang, J.; Ibrahim, P.N.; Cho, H.; Spevak, W.; Zhang, C.; Zhang, Y.; Habets, G.; et al. Clinical efficacy of a RAF inhibitor needs broad target blockade in BRAF-mutant melanoma. Nature 2010, 467, 596–599. [Google Scholar] [CrossRef]
- Carlino, M.S.; Saunders, C.A.; Haydu, L.E.; Menzies, A.M.; Curtis, C.M.; Lebowitz, P.F.; Kefford, R.F.; Long, G.V. 18F-labelled fluorodeoxyglucose–positron emission tomography (FDG–PET) heterogeneity of response is prognostic in dabrafenib treated BRAF mutant metastatic melanoma. Eur. J. Cancer 2013, 49, 395–402. [Google Scholar] [CrossRef]
- McArthur, G.A.; Puzanov, I.; Amaravadi, R.; Ribas, A.; Chapman, P.; Kim, K.B.; Sosman, J.A.; Lee, R.J.; Nolop, K.; Flaherty, K.T.; et al. Marked, Homogeneous, and Early [18F]Fluorodeoxyglucose–Positron Emission Tomography Responses to Vemurafenib in BRAF-Mutant Advanced Melanoma. J. Clin. Oncol. 2012, 30, 1628–1634. [Google Scholar] [CrossRef] [Green Version]
- Zheng, J. Energy metabolism of cancer: Glycolysis versus oxidative phosphorylation (Review). Oncol. Lett. 2012, 4, 1151–1157. [Google Scholar] [CrossRef] [Green Version]
- Goetzman, E.S.; Prochownik, E.V. The Role for Myc in Coordinating Glycolysis, Oxidative Phosphorylation, Glutaminolysis, and Fatty Acid Metabolism in Normal and Neoplastic Tissues. Front. Endocrinol. 2018, 9, 129. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [Green Version]
- Altman, B.J.; Stine, Z.E.; Dang, C.V. Erratum: From Krebs to clinic: Glutamine metabolism to cancer therapy. Nat. Rev. Cancer 2016, 16, 773. [Google Scholar] [CrossRef] [Green Version]
- Graves, J.A.; Wang, Y.; Sims-Lucas, S.; Cherok, E.; Rothermund, K.; Branca, M.F.; Elster, J.; Beer-Stolz, D.; Van Houten, B.; Vockley, J.; et al. Mitochondrial Structure, Function and Dynamics Are Temporally Controlled by c-Myc. PLoS ONE 2012, 7, e37699. [Google Scholar] [CrossRef] [Green Version]
- Hsieh, A.L.; Walton, Z.E.; Altman, B.; Stine, Z.E.; Dang, C.V. MYC and metabolism on the path to cancer. Semin. Cell Dev. Biol. 2015, 43, 11–21. [Google Scholar] [CrossRef] [Green Version]
- Shim, H.; Dolde, C.; Lewis, B.C.; Wu, C.-S.; Dang, G.; Jungmann, R.A.; Dalla-Favera, R.; Dang, C.V. c-Myc transactivation of LDH-A: Implications for tumor metabolism and growth. Proc. Natl. Acad. Sci. USA 1997, 94, 6658–6663. [Google Scholar] [CrossRef] [Green Version]
- Meroni, G.; Cairo, S.; Merla, G.; Messali, S.; Brent, R.; Ballabio, A.; Reymond, A. Mlx, a new Max-like bHLHZip family member: The center stage of a novel transcription factors regulatory pathway? Oncogene 2000, 19, 3266–3277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lytle, N.K.; Barber, A.G.; Reya, T. Stem cell fate in cancer growth, progression and therapy resistance. Nat. Rev. Cancer 2018, 18, 669–680. [Google Scholar] [CrossRef]
- Nedachi, T.; Kadotani, A.; Ariga, M.; Katagiri, H.; Kanzaki, M. Ambient glucose levels qualify the potency of insulin myogenic actions by regulating SIRT1 and FoxO3a in C2C12 myocytes. Am. J. Physiol. Endocrinol. Metab. 2008, 294, E668–E678. [Google Scholar] [CrossRef] [Green Version]
- Hunt, L.C.; Xu, B.; Finkelstein, D.; Fan, Y.; Carroll, P.A.; Cheng, P.-F.; Eisenman, R.N.; Demontis, F. The glucose-sensing transcription factor MLX promotes myogenesis via myokine signaling. Genes Dev. 2015, 29, 2475–2489. [Google Scholar] [CrossRef] [Green Version]
- Rai, M.; Demontis, F. Systemic Nutrient and Stress Signaling via Myokines and Myometabolites. Annu. Rev. Physiol. 2016, 78, 85–107. [Google Scholar] [CrossRef] [PubMed]
- Falcone, G.; Tato, F.; Alemà, S. Distinctive effects of the viral oncogenes myc, erb, fps, and src on the differentiation program of quail myogenic cells. Proc. Natl. Acad. Sci. USA 1985, 82, 426–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donato, M.; Jahromi, A.H.; Andrade, A.I.; Kim, R.; Chaudhery, S.I.; Sangster, G. Hepatic Adenomatosis: A Rare but Important Liver Disease With Severe Clinical Implications. Int. Surg. 2015, 100, 903–907. [Google Scholar] [CrossRef] [Green Version]
- Karim, S.; Adams, D.H.; Lalor, P.F. Hepatic expression and cellular distribution of the glucose transporter family. World J. Gastroenterol. 2012, 18, 6771–6781. [Google Scholar] [CrossRef] [PubMed]
- Torbenson, M. Hepatic Adenomas: Classification, Controversies, and Consensus. Surg. Pathol. Clin. 2018, 11, 351–366. [Google Scholar] [CrossRef] [PubMed]
- Brunt, E.M.; Wong, V.W.S.; Nobili, V.; Day, C.P.; Sookoian, S.; Maher, J.J.; Bugianesi, E.; Sirlin, C.B.; Neuschwander-Tetri, B.A.; Rinella, M.E. Nonalcoholic fatty liver disease. Nat. Rev. Dis. Prim. 2015, 1, 15080. [Google Scholar] [CrossRef]
- Marengo, A.; Rosso, C.; Bugianesi, E. Liver Cancer: Connections with Obesity, Fatty Liver, and Cirrhosis. Annu. Rev. Med. 2016, 67, 103–117. [Google Scholar] [CrossRef]
- Edmunds, L.R.; Otero, P.A.; Sharma, L.; D’Souza, S.; Dolezal, J.M.; David, S.; Lu, J.; Lamm, L.; Basantani, M.; Zhang, P.; et al. Abnormal lipid processing but normal long-term repopulation potential of myc−/− hepatocytes. Oncotarget 2016, 7, 30379–30395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Croce, C.M. Oncogenes and Cancer. N. Engl. J. Med. 2008, 358, 502–511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mandel, J.; Avula, R.; Prochownik, E.V. Sequential analysis of transcript expression patterns improves survival prediction in multiple cancers. BMC Cancer 2020, 20, 297. [Google Scholar] [CrossRef]
- Mandel, J.; Wang, H.; Normolle, D.P.; Chen, W.; Yan, Q.; Lucas, P.; Benos, P.V.; Prochownik, E.V. Expression patterns of small numbers of transcripts from functionally-related pathways predict survival in multiple cancers. BMC Cancer 2019, 19, 686. [Google Scholar] [CrossRef] [Green Version]
- Dolezal, J.M.; Dash, A.P.; Prochownik, E.V. Diagnostic and prognostic implications of ribosomal protein transcript expression patterns in human cancers. BMC Cancer 2018, 18, 275. [Google Scholar] [CrossRef] [Green Version]
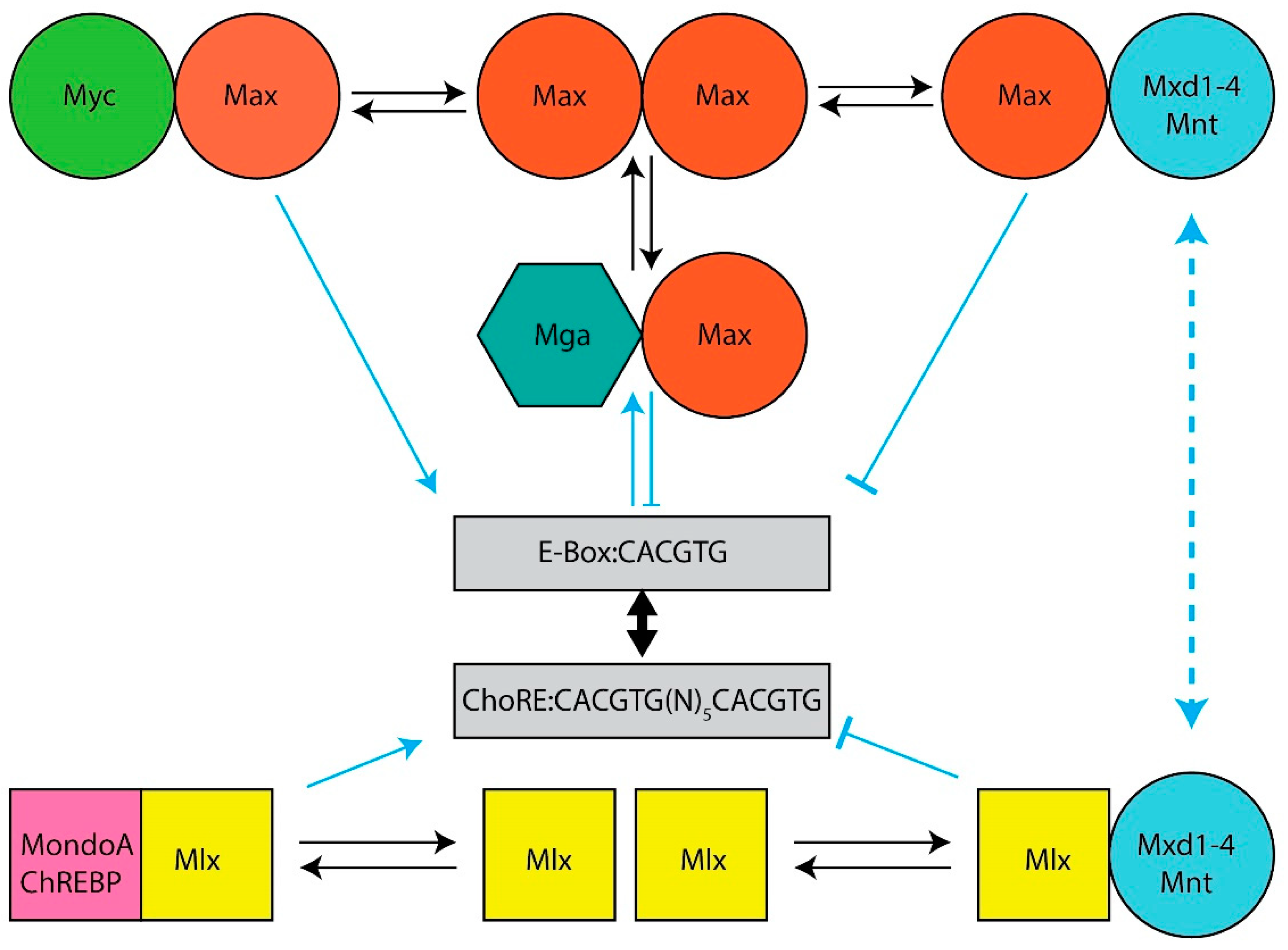

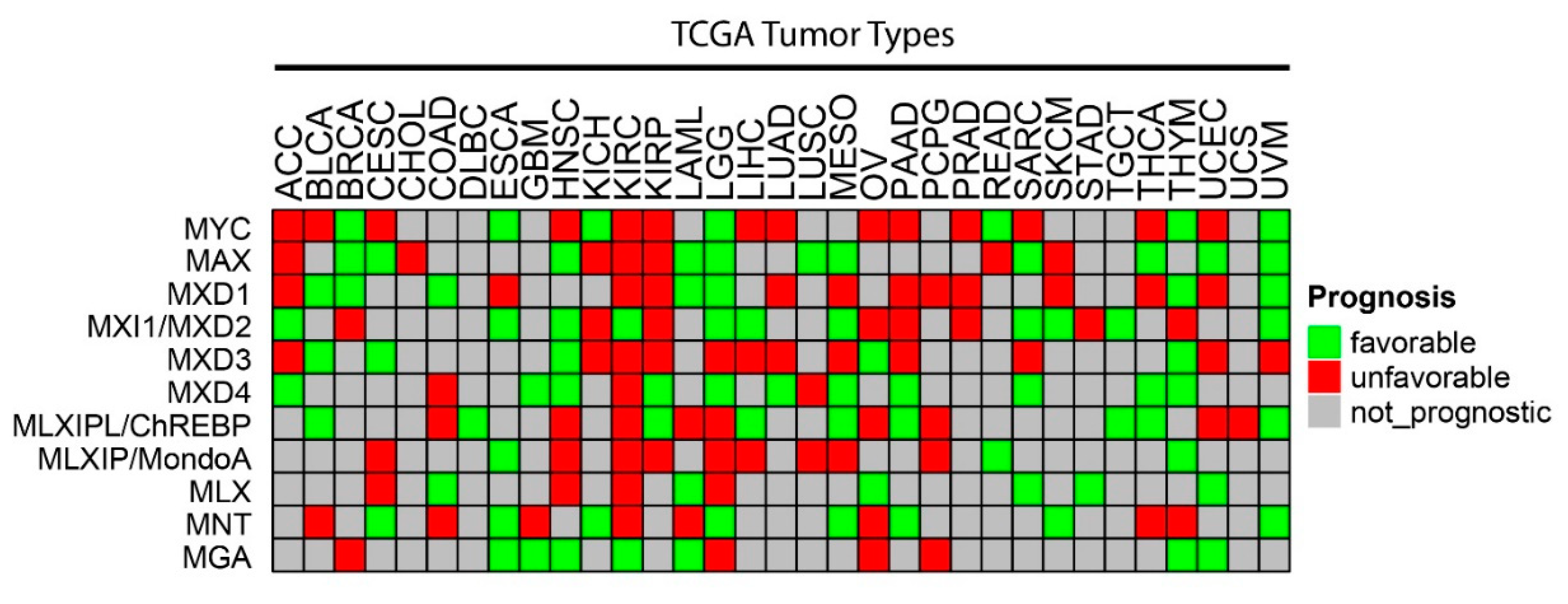
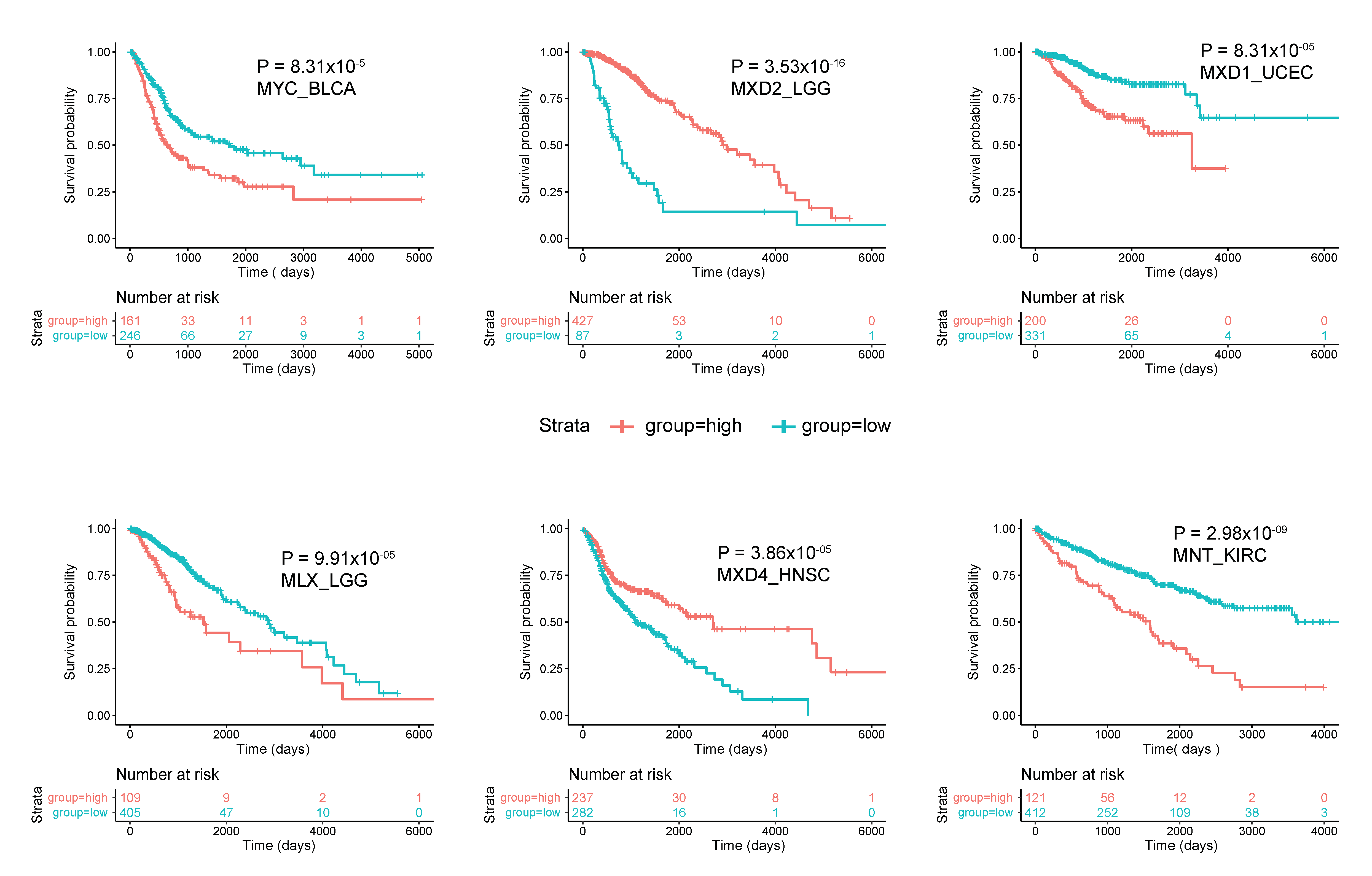

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Prochownik, E.V.; Wang, H. Normal and Neoplastic Growth Suppression by the Extended Myc Network. Cells 2022, 11, 747. https://doi.org/10.3390/cells11040747
Prochownik EV, Wang H. Normal and Neoplastic Growth Suppression by the Extended Myc Network. Cells. 2022; 11(4):747. https://doi.org/10.3390/cells11040747
Chicago/Turabian StyleProchownik, Edward V., and Huabo Wang. 2022. "Normal and Neoplastic Growth Suppression by the Extended Myc Network" Cells 11, no. 4: 747. https://doi.org/10.3390/cells11040747