
Deciphering the Retinal Epigenome during Development, Disease and Reprogramming: Advancements, Challenges and Perspectives


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Temporal Patterning in the Retina
2.1. Molecular Staging and Taxonomic Classification of the Developing Retina
2.2. Temporal Patterning from an Epigenomic Perspective
2.3. A Fine Balance between Reprogramming Capacity and Epigenetic Memory
3. The Epigenome: A Pandora’s Box
3.1. A Truly Unbiased Screening of Open Chromatin States: From Clustering of De Novo Oligonucleotides to the Identification of TFs Binding Patterns in Retinal Cell Fate Specification
3.2. Lhx2 Is Required for the Regionalization of the Optic Vesicle through Cell-Autonomous Regulation of Gene Expression and for the Neuro-Retinal Suppression of Alternative Diencephalic Cell Fates
3.3. Lhx2 Is Expressed in Neuro-Epithelial, Bipotent Progenitors That Give Rise to Segregated, Neuroretina-like and RPE-like Domains in hiPSCs-Derived Optic Cups and Organoids
3.4. Lhx2 and Its Neurogenic Potential
3.5. Lhx2 as a Transcriptional Determinant of Cell Fate Identity
3.6. Chromatin Regulators in Retinal Development
3.7. Leveraging Next-Generation Sequencing towards a Deeper Understanding of Gene Regulatory Networks and Hierarchies

3.8. Footprinting Analysis and Competition for Nucleosome Occupancy by Predicted Pioneer Factors Identify Steric Dependencies and Transcriptional Hierarchies in Cell Fate Specification
4. Tackling the Epigenetic Contribution to Ocular Diseases for Restoration of the Visual Function: From Corrective Therapies to Autologous Cell Replacement through Comparative Genomics
4.1. Target Identification and Repurposing of Epigenetically Relevant Pharmacological Compounds via Drug Delivery Nanosystems
4.2. Expanding the Therapeutic Portfolio from Gene Editing to Genome Editing towards Personalized Medicine
4.3. Agnostic Approaches to Restore the Visual Function, from Prosthetics to Autologous Cell Replacement
4.4. Chasing the Secret to Youth: Is the Evolutionary Conservation of the Regulatory Elements in the Genome Truly Key to Unlock the Regenerative Potential of the Retina?
5. Current Challenges
5.1. Calling for Empirical Diversity and Better Demographic Representation across TFs Position Weight Matrices: The Biases That Underrate the Underdogs
5.2. Zero-Inflation, Pseudo-Replication, and Pseudo-Bulk Aggregation in Single-Cell RNA-Seq: Is Single Cell ATAC-Seq Exempt from Such Biases?
5.3. The 4D Genome: Complementing Single-Cell Sequencing with Spatiotemporal Information by Super-Resolution Imaging
6. Conclusions and Future Perspectives
Harnessing the Full Potential of Next-Generation Sequencing: Resolution vs. Informativity
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Personal Disclosure
References
- Fuhrmann, S. Eye morphogenesis and patterning of the optic vesicle. Curr. Top. Dev. Biol. 2010, 93, 61–84. [Google Scholar] [PubMed] [Green Version]
- Giger, F.A.; Houart, C. The Birth of the Eye Vesicle: When Fate Decision Equals Morphogenesis. Front. Neurosci. 2018, 12, 87. [Google Scholar] [CrossRef] [PubMed]
- Miesfeld, J.B.; Brown, N.L. Eye organogenesis: A hierarchical view of ocular development. Curr. Top. Dev. Biol. 2019, 132, 351–393. [Google Scholar] [PubMed]
- Meunier, D.; Lambiotte, R.; Bullmore, E. Modular and Hierarchically Modular Organization of Brain Networks. Front. Neurosci. 2010, 4, 200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guy, J.; Staiger, J.F. The Functioning of a Cortex without Layers. Front. Neuroanat. 2017, 11, 54. [Google Scholar] [CrossRef] [Green Version]
- Hoon, M.; Okawa, H.; Della Santina, L.; Wong, R.O. Functional architecture of the retina: Development and disease. Prog. Retin. Eye Res. 2014, 42, 44–84. [Google Scholar] [CrossRef] [Green Version]
- Amini, R.; Rocha-Martins, M.; Norden, C. Neuronal Migration and Lamination in the Vertebrate Retina. Front. Neurosci. 2017, 11, 742. [Google Scholar] [CrossRef]
- Donovan, S.L.; Dyer, M.A. Regulation of proliferation during central nervous system development. Semin. Cell Dev. Biol. 2005, 16, 407–421. [Google Scholar] [CrossRef]
- Kohwi, M.; Doe, C.Q. Temporal fate specification and neural progenitor competence during development. Nat. Rev. Neurosci. 2013, 14, 823–838. [Google Scholar] [CrossRef]
- Yoles, E.; Hauben, E.; Palgi, O.; Agranov, E.; Gothilf, A.; Cohen, A.; Kuchroo, V.; Cohen, I.R.; Weiner, H.; Schwartz, M. Protective autoimmunity is a physiological response to CNS trauma. J. Neurosci. 2001, 21, 3740–3748. [Google Scholar] [CrossRef]
- Benowitz, L.; Yin, Y. Rewiring the injured CNS: Lessons from the optic nerve. Exp. Neurol. 2008, 209, 389–398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gadani, S.P.; Walsh, J.T.; Lukens, J.R.; Kipnis, J. Dealing with Danger in the CNS: The Response of the Immune System to Injury. Neuron 2015, 87, 47–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simon, D.W.; McGeachy, M.J.; Bayir, H.; Clark, R.S.; Loane, D.J.; Kochanek, P.M. The far-reaching scope of neuroinflammation after traumatic brain injury. Nat. Rev. Neurol. 2017, 13, 171–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, M.; Luo, C.; Zhao, J.; Devarajan, G.; Xu, H. Immune regulation in the aging retina. Prog. Retin. Eye Res. 2019, 69, 159–172. [Google Scholar] [CrossRef] [Green Version]
- Stepp, M.A.; Menko, A.S. Immune responses to injury and their links to eye disease. Transl. Res. 2021, 236, 52–71. [Google Scholar] [CrossRef]
- London, A.; Benhar, I.; Schwartz, M. The retina as a window to the brain—From eye research to CNS disorders. Nat. Rev. Neurol. 2013, 9, 44–53. [Google Scholar] [CrossRef]
- Nowacka, B.; Lubiński, W.; Honczarenko, K.; Potemkowski, A.; Safranow, K. Ophthalmological features of Parkinson disease. Med. Sci. Monit. 2014, 20, 2243–2249. [Google Scholar]
- Hirose, A.; Hirose, A.; Katagiri, S.; Hayashi, T.; Matsuura, T.; Nagai, N.; Fujinami, K.; Iwata, T.; Tsunoda, K. Progress of macular atrophy during 30 months’ follow-up in a patient with spinocerebellar ataxia type1 (SCA1). Doc. Ophthalmol. 2021, 142, 87–98. [Google Scholar] [CrossRef]
- Liao, C.; Xu, J.; Chen, Y.; Ip, N.Y. Retinal Dysfunction in Alzheimer’s Disease and Implications for Biomarkers. Biomolecules 2021, 11, 8. [Google Scholar] [CrossRef]
- Dhawan, P.S.; Leong, D.; Tapsell, L.; Starling, A.J.; Galetta, S.L.; Balcer, L.J.; Overall, T.L.; Adler, J.S.; Halker-Singh, R.B.; Vargas, B.B.; et al. King-Devick Test identifies real-time concussion and asymptomatic concussion in youth athletes. Neurol. Clin. Pract. 2017, 7, 464–473. [Google Scholar] [CrossRef]
- Fisher, J.B.; Jacobs, D.A.; Markowitz, C.E.; Galetta, S.L.; Volpe, N.J.; Nano-Schiavi, M.L.; Baier, M.L.; Frohman, E.M.; Winslow, H.; Frohman, T.C.; et al. Relation of visual function to retinal nerve fiber layer thickness in multiple sclerosis. Ophthalmology 2006, 113, 324–332. [Google Scholar] [CrossRef]
- Talman, L.S.; Bisker, E.R.; Sackel, D.J.; Long, D.A., Jr.; Galetta, K.M.; Ratchford, J.N.; Lile, D.J.; Farrell, S.K.; Loguidice, M.J.; Remington, G.; et al. Longitudinal study of vision and retinal nerve fiber layer thickness in multiple sclerosis. Ann. Neurol. 2010, 67, 749–760. [Google Scholar] [PubMed] [Green Version]
- Petzold, A.; de Boer, J.F.; Schippling, S.; Vermersch, P.; Kardon, R.; Green, A.; Calabresi, P.A.; Polman, C. Optical coherence tomography in multiple sclerosis: A systematic review and meta-analysis. Lancet Neurol. 2010, 9, 921–932. [Google Scholar] [CrossRef] [Green Version]
- Mailankody, P.; Battu, R.; Khanna, A.; Lenka, A.; Yadav, R.; Pal, P.K. Optical coherence tomography as a tool to evaluate retinal changes in Parkinson’s disease. Parkinsonism Relat. Disord. 2015, 21, 1164–1169. [Google Scholar] [CrossRef] [PubMed]
- Ahn, J.; Lee, J.Y.; Kim, T.W.; Yoon, E.J.; Oh, S.; Kim, Y.K.; Kim, J.M.; Woo, S.J.; Kim, K.W.; Jeon, B. Retinal thinning associates with nigral dopaminergic loss in de novo Parkinson disease. Neurology 2018, 91, e1003–e1012. [Google Scholar] [CrossRef]
- Abd Hamid, M.R.; Wan Hitam, W.H.; Abd Halim, S. Retinal Nerve Fiber Layer and Macular Thickness in Parkinson’s Disease Patients. Cureus 2021, 13, e16224. [Google Scholar] [CrossRef] [PubMed]
- Balcer, L.J.; Raynowska, J.; Nolan, R.; Galetta, S.L.; Kapoor, R.; Benedict, R.; Phillips, G.; LaRocca, N.; Hudson, L.; Rudick, R.; et al. Validity of low-contrast letter acuity as a visual performance outcome measure for multiple sclerosis. Mult. Scler. J. 2017, 23, 734–747. [Google Scholar] [CrossRef]
- Seay, M.; Akhand, O.; Galetta, M.S.; Cobbs, L.; Hasanaj, L.; Amorapanth, P.; Rizzo, J.R.; Nolan, R.; Serrano, L.; Rucker, J.C.; et al. Mobile Universal Lexicon Evaluation System (MULES) in MS: Evaluation of a new visual test of rapid picture naming. J. Neurol. Sci. 2018, 394, 1–5. [Google Scholar] [CrossRef]
- Conway, J.; Ilardi, M.; Gonzalez, C.; Dahan, N.; Fallon, S.; Moehringer, N.; Hasanaj, L.; Joseph, B.; Serrano, L.; Rizzo, J.R.; et al. Rapid picture naming in Parkinson’s disease using the Mobile Universal Lexicon Evaluation System (MULES). J. Neurol. Sci. 2020, 410, 116680. [Google Scholar] [CrossRef]
- O’Bryhim, B.E.; Apte, R.S.; Kung, N.; Coble, D.; Van Stavern, G.P. Association of Preclinical Alzheimer Disease with Optical Coherence Tomographic Angiography Findings. JAMA Ophthalmol. 2018, 136, 1242–1248. [Google Scholar] [CrossRef]
- Kleerekooper, I.; Houston, S.; Dubis, A.M.; Trip, S.A.; Petzold, A. Optical Coherence Tomography Angiography (OCTA) in Multiple Sclerosis and Neuromyelitis Optica Spectrum Disorder. Front. Neurol. 2020, 11, 604049. [Google Scholar] [CrossRef] [PubMed]
- Almonte, M.T.; Capellan, P.; Yap, T.E.; Cordeiro, M.F. Retinal correlates of psychiatric disorders. Ther. Adv. Chronic Dis. 2020, 11, 2040622320905215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silverstein, S.M.; Fradkin, S.I.; Demmin, D.L. Schizophrenia and the retina: Towards a 2020 perspective. Schizophr. Res. 2020, 219, 84–94. [Google Scholar] [CrossRef] [PubMed]
- Arsenault, E.; Lavigne, A.A.; Mansouri, S.; Gagne, A.M.; Francis, K.; Bittar, T.P.; Quessy, F.; Abdallah, K.; Barbeau, A.; Hebert, M.; et al. Sex-Specific Retinal Anomalies Induced by Chronic Social Defeat Stress in Mice. Front. Behav. Neurosci. 2021, 15, 714810. [Google Scholar] [CrossRef] [PubMed]
- Turner, D.L.; Cepko, C.L. A common progenitor for neurons and glia persists in rat retina late in development. Nature 1987, 328, 131–136. [Google Scholar] [CrossRef] [PubMed]
- Ohsawa, R.; Kageyama, R. Regulation of retinal cell fate specification by multiple TFs. Brain Res. 2008, 1192, 90–98. [Google Scholar] [CrossRef]
- Bassett, E.A.; Wallace, V.A. Cell fate determination in the vertebrate retina. Trends Neurosci. 2012, 35, 565–573. [Google Scholar] [CrossRef]
- Mo, A.; Mukamel, E.A.; Davis, F.P.; Luo, C.; Henry, G.L.; Picard, S.; Urich, M.A.; Nery, J.R.; Sejnowski, T.J.; Lister, R.; et al. Epigenomic Signatures of Neuronal Diversity in the Mammalian Brain. Neuron 2015, 86, 1369–1384. [Google Scholar] [CrossRef] [Green Version]
- Young, R.W. Cell differentiation in the retina of the mouse. Anat. Rec. 1985, 212, 199–205. [Google Scholar] [CrossRef]
- Livesey, F.J.; Cepko, C.L. Vertebrate neural cell-fate determination: Lessons from the retina. Nat. Rev. Neurosci. 2001, 2, 109–118. [Google Scholar] [CrossRef]
- Rapaport, D.H.; Wong, L.L.; Wood, E.D.; Yasumura, D.; LaVail, M.M. Timing and topography of cell genesis in the rat retina. J. Comp. Neurol. 2004, 474, 304–324. [Google Scholar] [CrossRef] [PubMed]
- Dyer, M.A.; Martins, R.; da Silva Filho, M.; Muniz, J.A.; Silveira, L.C.; Cepko, C.L.; Finlay, B.L. Developmental sources of conservation and variation in the evolution of the primate eye. Proc. Natl. Acad. Sci. USA 2009, 106, 8963–8968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, J.; Zhang, G.; Almeida, A.D.; Cayouette, M.; Simons, B.D.; Harris, W.A. How variable clones build an invariant retina. Neuron 2012, 75, 786–798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cepko, C. Intrinsically different retinal progenitor cells produce specific types of progeny. Nat. Rev. Neurosci. 2014, 15, 615–627. [Google Scholar] [CrossRef]
- Rossi, A.M.; Fernandes, V.M.; Desplan, C. Timing temporal transitions during brain development. Curr. Opin. Neurobiol. 2017, 42, 84–92. [Google Scholar] [CrossRef] [Green Version]
- Cremer, T.; Kurz, A.; Zirbel, R.; Dietzel, S.; Rinke, B.; Schrock, E.; Speicher, M.R.; Mathieu, U.; Jauch, A.; Emmerich, P.; et al. Role of chromosome territories in the functional compartmentalization of the cell nucleus. Role of chromosome territories in the functional compartmentalization of the cell nucleus. Cold Spring Harb. Symp. Quant. Biol. 1993, 58, 777–792. [Google Scholar] [CrossRef] [Green Version]
- Croft, J.A.; Bridger, J.M.; Boyle, S.; Perry, P.; Teague, P.; Bickmore, W.A. Differences in the localization and morphology of chromosomes in the human nucleus. J. Cell Biol. 1999, 145, 1119–1131. [Google Scholar] [CrossRef] [Green Version]
- Tanabe, H.; Muller, S.; Neusser, M.; von Hase, J.; Calcagno, E.; Cremer, M.; Solovei, I.; Cremer, C.; Cremer, T. Evolutionary conservation of chromosome territory arrangements in cell nuclei from higher primates. Proc. Natl. Acad. Sci. USA 2002, 99, 4424–4429. [Google Scholar] [CrossRef] [Green Version]
- Misteli, T. Beyond the sequence: Cellular organization of genome function. Cell 2007, 128, 787–800. [Google Scholar] [CrossRef] [Green Version]
- Lieberman-Aiden, E.; van Berkum, N.L.; Williams, L.; Imakaev, M.; Ragoczy, T.; Telling, A.; Amit, I.; Lajoie, B.R.; Sabo, P.J.; Dorschner, M.O.; et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 2009, 326, 289–293. [Google Scholar] [CrossRef] [Green Version]
- Smallwood, A.; Ren, B. Genome organization and long-range regulation of gene expression by enhancers. Curr. Opin. Cell Biol. 2013, 25, 387–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rao, S.S.; Huntley, M.H.; Durand, N.C.; Stamenova, E.K.; Bochkov, I.D.; Robinson, J.T.; Sanborn, A.L.; Machol, I.; Omer, A.D.; Lander, E.S.; et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 2014, 159, 1665–1680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bianco, S.; Lupianez, D.G.; Chiariello, A.M.; Annunziatella, C.; Kraft, K.; Schopflin, R.; Wittler, L.; Andrey, G.; Vingron, M.; Pombo, A.; et al. Polymer physics predicts the effects of structural variants on chromatin architecture. Nat. Genet. 2018, 50, 662–667. [Google Scholar] [CrossRef] [PubMed]
- Jerković, I.; Cavalli, G. Understanding 3D genome organization by multidisciplinary methods. Nat. Rev. Mol. Cell Biol. 2021, 22, 511–528. [Google Scholar] [CrossRef] [PubMed]
- Lupianez, D.G.; Kraft, K.; Heinrich, V.; Krawitz, P.; Brancati, F.; Klopocki, E.; Horn, D.; Kayserili, H.; Opitz, J.M.; Laxova, R.; et al. Disruptions of topological chromatin domains cause pathogenic rewiring of gene-enhancer interactions. Cell 2015, 161, 1012–1025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dixon, J.R.; Selvaraj, S.; Yue, F.; Kim, A.; Li, Y.; Shen, Y.; Hu, M.; Liu, J.S.; Ren, B. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 2012, 485, 376–380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hnisz, D.; Abraham, B.J.; Lee, T.I.; Lau, A.; Saint-Andre, V.; Sigova, A.A.; Hoke, H.A.; Young, R.A. Super-enhancers in the control of cell identity and disease. Cell 2013, 155, 934–947. [Google Scholar] [CrossRef] [Green Version]
- Filippova, D.; Patro, R.; Duggal, G.; Kingsford, C. Identification of alternative topological domains in chromatin. Algorithms Mol. Biol. 2014, 9, 14. [Google Scholar] [CrossRef] [Green Version]
- Won, H.; de la Torre-Ubieta, L.; Stein, J.L.; Parikshak, N.N.; Huang, J.; Opland, C.K.; Gandal, M.J.; Sutton, G.J.; Hormozdiari, F.; Lu, D.; et al. Chromosome conformation elucidates regulatory relationships in developing human brain. Nature 2016, 538, 523–527. [Google Scholar] [CrossRef] [Green Version]
- Freire-Pritchett, P.; Schoenfelder, S.; Varnai, C.; Wingett, S.W.; Cairns, J.; Collier, A.J.; Garcia-Vilchez, R.; Furlan-Magaril, M.; Osborne, C.S.; Fraser, P.; et al. Global reorganisation of cis-regulatory units upon lineage commitment of human embryonic stem cells. eLife 2017, 6, e21926. [Google Scholar] [CrossRef]
- Dekker, J.; Belmont, A.S.; Guttman, M.; Leshyk, V.O.; Lis, J.T.; Lomvardas, S.; Mirny, L.A.; O’Shea, C.C.; Park, P.J.; Ren, B.; et al. The 4D nucleome project. Nature 2017, 549, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Macosko, E.Z.; Basu, A.; Satija, R.; Nemesh, J.; Shekhar, K.; Goldman, M.; Tirosh, I.; Bialas, A.R.; Kamitaki, N.; Martersteck, E.M.; et al. Highly Parallel Genome-wide Expression Profiling of Individual Cells Using Nanoliter Droplets. Cell 2015, 161, 1202–1214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoshino, A.; Ratnapriya, R.; Brooks, M.J.; Chaitankar, V.; Wilken, M.S.; Zhang, C.; Starostik, M.R.; Gieser, L.; La Torre, A.; Nishio, M.; et al. Molecular Anatomy of the Developing Human Retina. Dev. Cell 2017, 43, 763–779.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rheaume, B.A.; Jereen, A.; Bolisetty, M.; Sajid, M.S.; Yang, Y.; Renna, K.; Sun, L.; Robson, P.; Trakhtenberg, E.F. Single cell transcriptome profiling of retinal ganglion cells identifies cellular subtypes. Nat. Commun. 2018, 9, 2759. [Google Scholar] [CrossRef]
- Peng, Y.R.; Shekhar, K.; Yan, W.; Herrmann, D.; Sappington, A.; Bryman, G.S.; van Zyl, T.; Do, M.T.H.; Regev, A.; Sanes, J.R. Molecular Classification and Comparative Taxonomics of Foveal and Peripheral Cells in Primate Retina. Cell 2019, 176, 1222–1237.e22. [Google Scholar] [CrossRef] [Green Version]
- Laboissonniere, L.A.; Goetz, J.J.; Martin, G.M.; Bi, R.; Lund, T.J.S.; Ellson, L.; Lynch, M.R.; Mooney, B.; Wickham, H.; Liu, P.; et al. Molecular signatures of retinal ganglion cells revealed through single cell profiling. Sci. Rep. 2019, 9, 15778. [Google Scholar] [CrossRef] [Green Version]
- Tran, N.M.; Shekhar, K.; Whitney, I.E.; Jacobi, A.; Benhar, I.; Hong, G.; Yan, W.; Adiconis, X.; Arnold, M.E.; Lee, J.M.; et al. Single-Cell Profiles of Retinal Ganglion Cells Differing in Resilience to Injury Reveal Neuroprotective Genes. Neuron 2019, 104, 1039–1055.e12. [Google Scholar] [CrossRef]
- Kolsch, Y.; Hahn, J.; Sappington, A.; Stemmer, M.; Fernandes, A.M.; Helmbrecht, T.O.; Lele, S.; Butrus, S.; Laurell, E.; Arnold-Ammer, I.; et al. Molecular classification of zebrafish retinal ganglion cells links genes to cell types to behavior. Neuron 2021, 109, 645–662.e9. [Google Scholar] [CrossRef]
- Xie, H.; Zhang, W.; Zhang, M.; Akhtar, T.; Li, Y.; Yi, W.; Sun, X.; Zuo, Z.; Wei, M.; Fang, X.; et al. Chromatin accessibility analysis reveals regulatory dynamics of developing human retina and hiPSC-derived retinal organoids. Sci. Adv. 2020, 6, eaay5247. [Google Scholar] [CrossRef] [Green Version]
- Cherry, T.J.; Yang, M.G.; Harmin, D.A.; Tao, P.; Timms, A.E.; Bauwens, M.; Allikmets, R.; Jones, E.M.; Chen, R.; De Baere, E.; et al. Mapping the cis-regulatory architecture of the human retina reveals noncoding genetic variation in disease. Proc. Natl. Acad. Sci. USA 2020, 117, 9001–9012. [Google Scholar] [CrossRef]
- Sridhar, A.; Hoshino, A.; Finkbeiner, C.R.; Chitsazan, A.; Dai, L.; Haugan, A.K.; Eschenbacher, K.M.; Jackson, D.L.; Trapnell, C.; Bermingham-McDonogh, O.; et al. Single-Cell Transcriptomic Comparison of Human Fetal Retina, hPSC-Derived Retinal Organoids, and Long-Term Retinal Cultures. Cell Rep. 2020, 30, 1644–1659.e4. [Google Scholar] [CrossRef] [PubMed]
- Cowan, C.S.; Renner, M.; De Gennaro, M.; Gross-Scherf, B.; Goldblum, D.; Hou, Y.; Munz, M.; Rodrigues, T.M.; Krol, J.; Szikra, T.; et al. Cell Types of the Human Retina and Its Organoids at Single-Cell Resolution. Cell 2020, 182, 1623–1640.e34. [Google Scholar] [CrossRef] [PubMed]
- Armand, E.J.; Li, J.; Xie, F.; Luo, C.; Mukamel, E.A. Single-Cell Sequencing of Brain Cell Transcriptomes and Epigenomes. Neuron 2021, 109, 11–26. [Google Scholar] [CrossRef] [PubMed]
- Ozel, M.N.; Simon, F.; Jafari, S.; Holguera, I.; Chen, Y.C.; Benhra, N.; El-Danaf, R.N.; Kapuralin, K.; Malin, J.A.; Konstantinides, N.; et al. Neuronal diversity and convergence in a visual system developmental atlas. Nature 2021, 589, 88–95. [Google Scholar] [CrossRef] [PubMed]
- Zibetti, C.; Liu, S.; Wan, J.; Qian, J.; Blackshaw, S. Epigenomic profiling of retinal progenitors reveals LHX2 is required for developmental regulation of open chromatin. Commun. Biol. 2019, 2, 142. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Jia, C.; Jia, C.; Kazmierkiewicz, K.L.; Bowman, A.S.; Tian, L.; Liu, Y.; Gupta, N.A.; Gudiseva, H.V.; Yee, S.S.; et al. Comprehensive analysis of gene expression in human retina and supporting tissues. Hum. Mol. Genet. 2014, 23, 4001–4014. [Google Scholar] [CrossRef] [Green Version]
- Crawford, G.E.; Holt, I.E.; Whittle, J.; Webb, B.D.; Tai, D.; Davis, S.; Margulies, E.H.; Chen, Y.; Bernat, J.A.; Ginsburg, D.; et al. Genome-wide mapping of DNase hypersensitive sites using massively parallel signature sequencing (MPSS). Genome Res. 2006, 16, 123–131. [Google Scholar] [CrossRef] [Green Version]
- Buenrostro, J.D.; Giresi, P.G.; Zaba, L.C.; Chang, H.Y.; Greenleaf, W.J. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods 2013, 10, 1213–1218. [Google Scholar] [CrossRef]
- Ziller, M.J.; Edri, R.; Yaffe, Y.; Donaghey, J.; Pop, R.; Mallard, W.; Issner, R.; Gifford, C.A.; Goren, A.; Xing, J.; et al. Dissecting neural differentiation regulatory networks through epigenetic footprinting. Nature 2015, 518, 355–359. [Google Scholar] [CrossRef] [Green Version]
- Dekker, J.; Rippe, K.; Dekker, M.; Kleckner, N. Capturing chromosome conformation. Science 2002, 295, 1306–1311. [Google Scholar] [CrossRef] [Green Version]
- Dostie, J.; Richmond, T.A.; Arnaout, R.A.; Selzer, R.R.; Lee, W.L.; Honan, T.A.; Rubio, E.D.; Krumm, A.; Lamb, J.; Nusbaum, C.; et al. Chromosome Conformation Capture Carbon Copy (5C): A massively parallel solution for mapping interactions between genomic elements. Genome Res. 2006, 16, 1299–1309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yardimci, G.G.; Noble, W.S. Software tools for visualizing Hi-C data. Genome Biol. 2017, 18, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aldiri, I.; Xu, B.; Wang, L.; Chen, X.; Hiler, D.; Griffiths, L.; Valentine, M.; Shirinifard, A.; Thiagarajan, S.; Sablauer, A.; et al. The Dynamic Epigenetic Landscape of the Retina During Development, Reprogramming, and Tumorigenesis. Neuron 2017, 94, 550–568.e10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ernst, J.; Kellis, M. ChromHMM: Automating chromatin-state discovery and characterization. Nat. Methods 2012, 9, 215–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Creyghton, M.P.; Cheng, A.W.; Welstead, G.G.; Kooistra, T.; Carey, B.W.; Steine, E.J.; Hanna, J.; Lodato, M.A.; Frampton, G.M.; Sharp, P.A.; et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc. Natl. Acad. Sci. USA 2010, 107, 21931–21936. [Google Scholar] [CrossRef] [Green Version]
- Visel, A.; Blow, M.J.; Li, Z.; Zhang, T.; Akiyama, J.A.; Holt, A.; Plajzer-Frick, I.; Shoukry, M.; Wright, C.; Chen, F.; et al. ChIP-seq accurately predicts tissue-specific activity of enhancers. Nature 2009, 457, 854–858. [Google Scholar] [CrossRef] [Green Version]
- Brzezinski, J.A.; Kim, E.J.; Johnson, J.E.; Reh, T.A. Ascl1 expression defines a subpopulation of lineage-restricted progenitors in the mammalian retina. Development 2011, 138, 3519–3531. [Google Scholar] [CrossRef] [Green Version]
- Baden, T.; Euler, T.; Berens, P. Understanding the retinal basis of vision across species. Nat. Rev. Neurosci. 2020, 21, 5–20. [Google Scholar] [CrossRef]
- Todd, L.; Reh, T.A. Comparative Biology of Vertebrate Retinal Regeneration: Restoration of Vision through Cellular Reprogramming. Cold Spring Harb. Perspect. Biol. 2021, 14, a040816. [Google Scholar] [CrossRef]
- Land, M.F. The physics and biology of animal reflectors. Prog. Biophys. Mol. Biol. 1972, 24, 75–106. [Google Scholar] [CrossRef]
- Kreysing, M.; Pusch, R.; Haverkate, D.; Landsberger, M.; Engelmann, J.; Ruiter, J.; Mora-Ferrer, C.; Ulbricht, E.; Grosche, J.; Franze, K.; et al. Photonic crystal light collectors in fish retina improve vision in turbid water. Science 2012, 336, 1700–1703. [Google Scholar] [CrossRef] [PubMed]
- Blaszczak, Z.; Kreysing, M.; Guck, J. Direct observation of light focusing by single photoreceptor cell nuclei. Opt. Express 2014, 22, 11043–11060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subramanian, K.; Weigert, M.; Borsch, O.; Petzold, H.; Garcia-Ulloa, A.; Myers, E.W.; Ader, M.; Solovei, I.; Kreysing, M. Rod nuclear architecture determines contrast transmission of the retina and behavioral sensitivity in mice. eLife 2019, 8, e49542. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, K.; Petzold, H.; Seelbinder, B.; Hersemann, L.; Nusslein, I.; Kreysing, M. Optical plasticity of mammalian cells. J. Biophotonics 2021, 14, e202000457. [Google Scholar] [CrossRef] [PubMed]
- Solovei, I.; Kreysing, M.; Lanctot, C.; Kosem, S.; Peichl, L.; Cremer, T.; Guck, J.; Joffe, B. Nuclear architecture of rod photoreceptor cells adapts to vision in mammalian evolution. Cell 2009, 137, 356–368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kreysing, M.; Boyde, L.; Guck, J.; Chalut, K.J. Physical insight into light scattering by photoreceptor cell nuclei. Opt. Lett. 2010, 35, 2639–2641. [Google Scholar] [CrossRef]
- Wang, L.; Hiler, D.; Xu, B.; AlDiri, I.; Chen, X.; Zhou, X.; Griffiths, L.; Valentine, M.; Shirinifard, A.; Sablauer, A.; et al. Retinal Cell Type DNA Methylation and Histone Modifications Predict Reprogramming Efficiency and Retinogenesis in 3D Organoid Cultures. Cell Rep. 2018, 22, 2601–2614. [Google Scholar] [CrossRef] [Green Version]
- Norrie, J.L.; Lupo, M.S.; Xu, B.; Al Diri, I.; Valentine, M.; Putnam, D.; Griffiths, L.; Zhang, J.; Johnson, D.; Easton, J.; et al. Nucleome Dynamics during Retinal Development. Neuron 2019, 104, 512–528.e11. [Google Scholar] [CrossRef]
- Chen, X.; Pappo, A.; Dyer, M.A. Pediatric solid tumor genomics and developmental pliancy. Oncogene 2015, 34, 5207–5215. [Google Scholar] [CrossRef] [Green Version]
- Denny, S.K.; Yang, D.; Chuang, C.H.; Brady, J.J.; Lim, J.S.; Gruner, B.M.; Chiou, S.H.; Schep, A.N.; Baral, J.; Hamard, C.; et al. Nfib Promotes Metastasis through a Widespread Increase in Chromatin Accessibility. Cell 2016, 166, 328–342. [Google Scholar] [CrossRef] [Green Version]
- Grabowska, M.M.; Kelly, S.M.; Reese, A.L.; Cates, J.M.; Case, T.C.; Zhang, J.; DeGraff, D.J.; Strand, D.W.; Miller, N.L.; Clark, P.E.; et al. Nfib Regulates Transcriptional Networks That Control the Development of Prostatic Hyperplasia. Endocrinology 2016, 157, 1094–1109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aldiri, I.; Valentine, M.; Xu, B.; Putnam, D.; Griffiths, L.; Lupo, M.; Norrie, J.; Zhang, J.; Johnson, D.; Easton, J.; et al. The Nucleome of Developing Murine Rod Photoreceptors. bioRxiv 2018. [Google Scholar] [CrossRef] [Green Version]
- Stolt, C.C.; Lommes, P.; Sock, E.; Chaboissier, M.C.; Schedl, A.; Wegner, M. The Sox9 TF determines glial fate choice in the developing spinal cord. Genes Dev. 2003, 17, 1677–1689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elliott, J.; Jolicoeur, C.; Ramamurthy, V.; Cayouette, M. Ikaros confers early temporal competence to mouse retinal progenitor cells. Neuron 2008, 60, 26–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alsio, J.M.; Tarchini, B.; Cayouette, M.; Livesey, F.J. Ikaros promotes early-born neuronal fates in the cerebral cortex. Proc. Natl. Acad. Sci. USA 2013, 110, E716–E725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- La Torre, A.; Georgi, S.; Reh, T.A. Conserved microRNA pathway regulates developmental timing of retinal neurogenesis. Proc. Natl. Acad. Sci. USA 2013, 110, E2362–E2370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saurat, N.; Andersson, T.; Vasistha, N.A.; Molnar, Z.; Livesey, F.J. Dicer is required for neural stem cell multipotency and lineage progression during cerebral cortex development. Neural Dev. 2013, 8, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.; Sengel, C.; Emerson, M.M.; Cepko, C.L. A gene regulatory network controls the binary fate decision of rod and bipolar cells in the vertebrate retina. Dev. Cell 2014, 30, 513–527. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Cepko, C.L. Photoreceptor Fate Determination in the Vertebrate Retina. Investig. Opthalmol. Vis. Sci. 2016, 57, ORSFe1–ORSFe6. [Google Scholar] [CrossRef]
- Stenkamp, D.L. Development of the Vertebrate Eye and Retina. Prog. Mol. Biol. Transl. Sci. 2015, 134, 397–414. [Google Scholar]
- Zhang, X.M.; Yang, X.J. Regulation of retinal ganglion cell production by Sonic hedgehog. Development 2001, 128, 943–957. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, M.; Choi, J.; Park, S.; Sockanathan, S. Gde2 regulates cortical neuronal identity by controlling the timing of cortical progenitor differentiation. Development 2012, 139, 3870–3879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vetter, M.L.; Brown, N.L. The role of basic helix-loop-helix genes in vertebrate retinogenesis. Semin. Cell Dev. Biol. 2001, 12, 491–498. [Google Scholar] [CrossRef] [PubMed]
- Kageyama, R.; Ohtsuka, T.; Hatakeyama, J.; Ohsawa, R. Roles of bHLH genes in neural stem cell differentiation. Exp. Cell Res. 2005, 306, 343–348. [Google Scholar] [CrossRef] [PubMed]
- Masland, R.H. The neuronal organization of the retina. Neuron 2012, 76, 266–280. [Google Scholar] [CrossRef] [Green Version]
- Boije, H.; MacDonald, R.B.; Harris, W.A. Reconciling competence and transcriptional hierarchies with stochasticity in retinal lineages. Curr. Opin. Neurobiol. 2014, 27, 68–74. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Taylor, R.J.; La Torre, A.; Wilken, M.S.; Cox, K.E.; Reh, T.A.; Vetter, M.L. Ezh2 maintains retinal progenitor proliferation, transcriptional integrity, and the timing of late differentiation. Dev. Biol. 2015, 403, 128–138. [Google Scholar] [CrossRef] [Green Version]
- Mellough, C.B.; Bauer, R.; Collin, J.; Dorgau, B.; Zerti, D.; Dolan, D.W.P.; Jones, C.M.; Izuogu, O.G.; Yu, M.; Hallam, D.; et al. An integrated transcriptional analysis of the developing human retina. Development 2019, 146, dev169474. [Google Scholar] [CrossRef] [Green Version]
- VandenBosch, L.S.; Wohl, S.G.; Wilken, M.S.; Hooper, M.; Finkbeiner, C.; Cox, K.; Chipman, L.; Reh, T.A. Developmental changes in the accessible chromatin, transcriptome and Ascl1-binding correlate with the loss in Muller Glial regenerative potential. Sci. Rep. 2020, 10, 13615. [Google Scholar] [CrossRef]
- Corbo, J.C.; Lawrence, K.A.; Karlstetter, M.; Myers, C.A.; Abdelaziz, M.; Dirkes, W.; Weigelt, K.; Seifert, M.; Benes, V.; Fritsche, L.G.; et al. CRX ChIP-seq reveals the cis-regulatory architecture of mouse photoreceptors. Genome Res. 2010, 20, 1512–1525. [Google Scholar] [CrossRef] [Green Version]
- Ruzycki, P.A.; Zhang, X.; Chen, S. CRX directs photoreceptor differentiation by accelerating chromatin remodeling at specific target sites. Epigenet. Chromatin 2018, 11, 42. [Google Scholar] [CrossRef] [PubMed]
- Samuel, A.; Housset, M.; Fant, B.; Lamonerie, T. Otx2 ChIP-seq reveals unique and redundant functions in the mature mouse retina. PLoS ONE 2014, 9, e89110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swaroop, A.; Kim, D.; Forrest, D. Transcriptional regulation of photoreceptor development and homeostasis in the mammalian retina. Nat. Rev. Neurosci. 2010, 11, 563–576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lyu, P.; Hoang, T.; Santiago, C.P.; Thomas, E.D.; Timms, A.E.; Appel, H.; Gimmen, M.; Le, N.; Jiang, L.; Kim, D.W.; et al. Gene regulatory networks controlling temporal patterning, neurogenesis, and cell-fate specification in mammalian retina. Cell Rep. 2021, 37, 109994. [Google Scholar] [CrossRef]
- Andzelm, M.M.; Cherry, T.J.; Harmin, D.A.; Boeke, A.C.; Lee, C.; Hemberg, M.; Pawlyk, B.; Malik, A.N.; Flavell, S.W.; Sandberg, M.A.; et al. MEF2D drives photoreceptor development through a genome-wide competition for tissue-specific enhancers. Neuron 2015, 86, 247–263. [Google Scholar] [CrossRef] [Green Version]
- Konstantinides, N.; Kapuralin, K.; Fadil, C.; Barboza, L.; Satija, R.; Desplan, C. Phenotypic Convergence: Distinct TFs Regulate Common Terminal Features. Cell 2018, 174, 622–635.e13. [Google Scholar] [CrossRef] [Green Version]
- Clark, B.S.; Stein-O’Brien, G.L.; Shiau, F.; Cannon, G.H.; Davis-Marcisak, E.; Sherman, T.; Santiago, C.P.; Hoang, T.V.; Rajaii, F.; James-Esposito, R.E.; et al. Single-Cell RNA-Seq Analysis of Retinal Development Identifies NFI Factors as Regulating Mitotic Exit and Late-Born Cell Specification. Neuron 2019, 102, 1111–1126.e5. [Google Scholar] [CrossRef]
- Lu, Y.; Shiau, F.; Yi, W.; Lu, S.; Wu, Q.; Pearson, J.D.; Kallman, A.; Zhong, S.; Hoang, T.; Zuo, Z.; et al. Single-Cell Analysis of Human Retina Identifies Evolutionarily Conserved and Species-Specific Mechanisms Controlling Development. Dev. Cell 2020, 53, 473–491.e9. [Google Scholar] [CrossRef]
- Brzezinski, J.A.; Park, K.U.; Reh, T.A. Blimp1 (Prdm1) prevents re-specification of photoreceptors into retinal bipolar cells by restricting competence. Dev. Biol. 2013, 384, 194–204. [Google Scholar] [CrossRef] [Green Version]
- Hou, P.S.; Chuang, C.Y.; Kao, C.F.; Chou, S.J.; Stone, L.; Ho, H.N.; Chien, C.L.; Kuo, H.C. LHX2 regulates the neural differentiation of human embryonic stem cells via transcriptional modulation of PAX6 and CER1. Nucleic Acids Res. 2013, 41, 7753–7770. [Google Scholar] [CrossRef] [Green Version]
- Gordon, P.J.; Yun, S.; Clark, A.M.; Monuki, E.S.; Murtaugh, L.C.; Levine, E.M. Lhx2 balances progenitor maintenance with neurogenic output and promotes competence state progression in the developing retina. J. Neurosci. 2013, 33, 12197–12207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Melo, J.; Zibetti, C.; Clark, B.S.; Hwang, W.; Miranda-Angulo, A.L.; Qian, J.; Blackshaw, S. Lhx2 Is an Essential Factor for Retinal Gliogenesis and Notch Signaling. J. Neurosci. 2016, 36, 2391–2405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Melo, J.; Clark, B.S.; Blackshaw, S. Multiple intrinsic factors act in concert with Lhx2 to direct retinal gliogenesis. Sci. Rep. 2016, 6, 32757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roy, A.; de Melo, J.; Chaturvedi, D.; Thein, T.; Cabrera-Socorro, A.; Houart, C.; Meyer, G.; Blackshaw, S.; Tole, S. LHX2 is necessary for the maintenance of optic identity and for the progression of optic morphogenesis. J. Neurosci. 2013, 33, 6877–6884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mangale, V.S.; Hirokawa, K.E.; Satyaki, P.R.; Gokulchandran, N.; Chikbire, S.; Subramanian, L.; Shetty, A.S.; Martynoga, B.; Paul, J.; Mai, M.V.; et al. Lhx2 selector activity specifies cortical identity and suppresses hippocampal organizer fate. Science 2008, 319, 304–309. [Google Scholar] [CrossRef] [Green Version]
- Chou, S.J.; Perez-Garcia, C.G.; Kroll, T.T.; O’Leary, D.D. Lhx2 specifies regional fate in Emx1 lineage of telencephalic progenitors generating cerebral cortex. Nat. Neurosci. 2009, 12, 1381–1389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subramanian, L.; Sarkar, A.; Shetty, A.S.; Muralidharan, B.; Padmanabhan, H.; Piper, M.; Monuki, E.S.; Bach, I.; Gronostajski, R.M.; Richards, L.J.; et al. TF Lhx2 is necessary and sufficient to suppress astro-gliogenesis and promote neurogenesis in the developing hippocampus. Proc. Natl. Acad. Sci. USA 2011, 108, E265–E274. [Google Scholar] [CrossRef] [Green Version]
- Chou, S.J.; O’Leary, D.D. Role for Lhx2 in corticogenesis through regulation of progenitor differentiation. Mol. Cell. Neurosci. 2013, 56, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Chinn, G.A.; Hirokawa, K.E.; Chuang, T.M.; Urbina, C.; Patel, F.; Fong, J.; Funatsu, N.; Monuki, E.S. Agenesis of the Corpus Callosum Due to Defective Glial Wedge Formation in Lhx2 Mutant Mice. Cereb. Cortex 2015, 25, 2707–2718. [Google Scholar] [CrossRef] [Green Version]
- Porter, F.D.; Drago, J.; Xu, Y.; Cheema, S.S.; Wassif, C.; Huang, S.P.; Lee, E.; Grinberg, A.; Massalas, J.S.; Bodine, D.; et al. Lhx2, a LIM homeobox gene, is required for eye, forebrain, and definitive erythrocyte development. Development 1997, 124, 2935–2944. [Google Scholar] [CrossRef]
- Tetreault, N.; Champagne, M.P.; Bernier, G. The LIM homeobox TF Lhx2 is required to specify the retina field and synergistically cooperates with Pax6 for Six6 transactivation. Dev. Biol. 2009, 327, 541–550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monuki, E.S.; Porter, F.D.; Walsh, C.A. Patterning of the dorsal telencephalon and cerebral cortex by a roof plate-Lhx2 pathway. Neuron 2001, 32, 591–604. [Google Scholar] [CrossRef] [Green Version]
- Zhong, X.; Gutierrez, C.; Xue, T.; Hampton, C.; Vergara, M.N.; Cao, L.H.; Peters, A.; Park, T.S.; Zambidis, E.T.; Meyer, J.S.; et al. Generation of three-dimensional retinal tissue with functional photoreceptors from human iPSCs. Nat. Commun. 2014, 5, 4047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phillips, M.J.; Perez, E.T.; Martin, J.M.; Reshel, S.T.; Wallace, K.A.; Capowski, E.E.; Singh, R.; Wright, L.S.; Clark, E.M.; Barney, P.M.; et al. Modeling human retinal development with patient-specific induced pluripotent stem cells reveals multiple roles for visual system homeobox 2. Stem Cells 2014, 32, 1480–1492. [Google Scholar] [CrossRef] [Green Version]
- Capowski, E.E.; Simonett, J.M.; Clark, E.M.; Wright, L.S.; Howden, S.E.; Wallace, K.A.; Petelinsek, A.M.; Pinilla, I.; Phillips, M.J.; Meyer, J.S.; et al. Loss of MITF expression during human embryonic stem cell differentiation disrupts retinal pigment epithelium development and optic vesicle cell proliferation. Hum. Mol. Genet. 2014, 23, 6332–6344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raviv, S.; Bharti, K.; Rencus-Lazar, S.; Cohen-Tayar, Y.; Schyr, R.; Evantal, N.; Meshorer, E.; Zilberberg, A.; Idelson, M.; Reubinoff, B.; et al. PAX6 regulates melanogenesis in the retinal pigmented epithelium through feed-forward regulatory interactions with MITF. PLoS Genet. 2014, 10, e1004360. [Google Scholar] [CrossRef] [Green Version]
- Singh, R.K.; Mallela, R.K.; Cornuet, P.K.; Reifler, A.N.; Chervenak, A.P.; West, M.D.; Wong, K.Y.; Nasonkin, I.O. Characterization of Three-Dimensional Retinal Tissue Derived from Human Embryonic Stem Cells in Adherent Monolayer Cultures. Stem Cells Dev. 2015, 24, 2778–2795. [Google Scholar] [CrossRef]
- O’Hara-Wright, M.; Gonzalez-Cordero, A. Retinal organoids: A window into human retinal development. Development 2020, 147, dev189746. [Google Scholar] [CrossRef]
- Eiraku, M.; Takata, N.; Ishibashi, H.; Kawada, M.; Sakakura, E.; Okuda, S.; Sekiguchi, K.; Adachi, T.; Sasai, Y. Self-organizing optic-cup morphogenesis in three-dimensional culture. Nature 2011, 472, 51–56. [Google Scholar] [CrossRef]
- Bharti, K.; Gasper, M.; Ou, J.; Brucato, M.; Clore-Gronenborn, K.; Pickel, J.; Arnheiter, H. A regulatory loop involving PAX6, MITF, and WNT signaling controls retinal pigment epithelium development. PLoS Genet. 2012, 8, e1002757. [Google Scholar] [CrossRef] [Green Version]
- Baumer, N.; Marquardt, T.; Stoykova, A.; Spieler, D.; Treichel, D.; Ashery-Padan, R.; Gruss, P. Retinal pigmented epithelium determination requires the redundant activities of Pax2 and Pax6. Development 2003, 130, 2903–2915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Capowski, E.E.; Samimi, K.; Mayerl, S.J.; Phillips, M.J.; Pinilla, I.; Howden, S.E.; Saha, J.; Jansen, A.D.; Edwards, K.L.; Jager, L.D.; et al. Reproducibility and staging of 3D human retinal organoids across multiple pluripotent stem cell lines. Development 2019, 146, dev171686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Melo, J.; Miki, K.; Rattner, A.; Smallwood, P.; Zibetti, C.; Hirokawa, K.; Monuki, E.S.; Campochiaro, P.A.; Blackshaw, S. Injury-independent induction of reactive gliosis in retina by loss of function of the LIM homeodomain TF Lhx2. Proc. Natl. Acad. Sci. USA 2012, 109, 4657–4662. [Google Scholar] [CrossRef] [Green Version]
- Surzenko, N.; Crowl, T.; Bachleda, A.; Langer, L.; Pevny, L. SOX2 maintains the quiescent progenitor cell state of postnatal retinal Muller glia. Development 2013, 140, 1445–1456. [Google Scholar] [CrossRef] [Green Version]
- Muto, A.; Iida, A.; Satoh, S.; Watanabe, S. The group E Sox genes Sox8 and Sox9 are regulated by Notch signaling and are required for Muller glial cell development in mouse retina. Exp. Eye Res. 2009, 89, 549–558. [Google Scholar] [CrossRef] [PubMed]
- de Melo, J.; Clark, B.S.; Venkataraman, A.; Shiau, F.; Zibetti, C.; Blackshaw, S. Ldb1- and Rnf12-dependent regulation of Lhx2 controls the relative balance between neurogenesis and gliogenesis in the retina. Development 2018, 145, dev159970. [Google Scholar] [CrossRef] [Green Version]
- Brightman, D.S.; Grant, R.L.; Ruzycki, P.A.; Suzuki, R.; Hennig, A.K.; Chen, S. MLL1 is essential for retinal neurogenesis and horizontal inner neuron integrity. Sci. Rep. 2018, 8, 11902. [Google Scholar] [CrossRef]
- Aldiri, I.; Moore, K.B.; Hutcheson, D.A.; Zhang, J.; Vetter, M.L. Polycomb repressive complex PRC2 regulates Xenopus retina development downstream of Wnt/beta-catenin signaling. Development 2013, 140, 2867–2878. [Google Scholar] [CrossRef] [Green Version]
- Fujimura, N.; Kuzelova, A.; Ebert, A.; Strnad, H.; Lachova, J.; Machon, O.; Busslinger, M.; Kozmik, Z. Polycomb repression complex 2 is required for the maintenance of retinal progenitor cells and balanced retinal differentiation. Dev. Biol. 2018, 433, 47–60. [Google Scholar] [CrossRef]
- Iida, A.; Iwagawa, T.; Baba, Y.; Satoh, S.; Mochizuki, Y.; Nakauchi, H.; Furukawa, T.; Koseki, H.; Murakami, A.; Watanabe, S. Roles of histone H3K27 trimethylase Ezh2 in retinal proliferation and differentiation. Dev. Neurobiol. 2015, 75, 947–960. [Google Scholar] [CrossRef]
- Iida, A.; Iwagawa, T.; Kuribayashi, H.; Satoh, S.; Mochizuki, Y.; Baba, Y.; Nakauchi, H.; Furukawa, T.; Koseki, H.; Murakami, A.; et al. Histone demethylase Jmjd3 is required for the development of subsets of retinal bipolar cells. Proc. Natl. Acad. Sci. USA 2014, 111, 3751–3756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, L.; Wong, L.J.; Yan, N.; Han, R.C.; Yu, H.; Guo, C.; Batsuuri, K.; Zinzuwadia, A.; Guan, R.; Cho, K.S.; et al. Ezh2 does not mediate retinal ganglion cell homeostasis or their susceptibility to injury. PLoS ONE 2018, 13, e0191853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, N.; Cheng, L.; Cho, K.; Malik, M.T.; Xiao, L.; Guo, C.; Yu, H.; Zhu, R.; Rao, R.C.; Chen, D.F. Postnatal onset of retinal degeneration by loss of embryonic Ezh2 repression of Six1. Sci. Rep. 2016, 6, 33887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mattar, P.; Stevanovic, M.; Nad, I.; Cayouette, M. Casz1 controls higher-order nuclear organization in rod photoreceptors. Proc. Natl. Acad. Sci. USA 2018, 115, E7987–E7996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cruz-Molina, S.; Respuela, P.; Tebartz, C.; Kolovos, P.; Nikolic, M.; Fueyo, R.; van Ijcken, W.F.J.; Grosveld, F.; Frommolt, P.; Bazzi, H.; et al. PRC2 Facilitates the Regulatory Topology Required for Poised Enhancer Function during Pluripotent Stem Cell Differentiation. Cell Stem Cell 2017, 20, 689–705.e9. [Google Scholar] [CrossRef] [Green Version]
- Barutcu, A.R.; Lajoie, B.R.; Fritz, A.J.; McCord, R.P.; Nickerson, J.A.; van Wijnen, A.J.; Lian, J.B.; Stein, J.L.; Dekker, J.; Stein, G.S.; et al. SMARCA4 regulates gene expression and higher-order chromatin structure in proliferating mammary epithelial cells. Genome Res. 2016, 26, 1188–1201. [Google Scholar] [CrossRef] [Green Version]
- Das, A.V.; James, J.; Bhattacharya, S.; Imbalzano, A.N.; Antony, M.L.; Hegde, G.; Zhao, X.; Mallya, K.; Ahmad, F.; Knudsen, E.; et al. SWI/SNF chromatin remodeling ATPase Brm regulates the differentiation of early retinal stem cells/progenitors by influencing Brn3b expression and Notch signaling. J. Biol. Chem. 2007, 282, 35187–35201. [Google Scholar] [CrossRef] [Green Version]
- Aldiri, I.; Ajioka, I.; Xu, B.; Zhang, J.; Chen, X.; Benavente, C.; Finkelstein, D.; Johnson, D.; Akiyama, J.; Pennacchio, L.A.; et al. Brg1 coordinates multiple processes during retinogenesis and is a tumor suppressor in retinoblastoma. Development 2015, 142, 4092–4106. [Google Scholar] [CrossRef] [Green Version]
- Alver, B.H.; Kim, K.H.; Lu, P.; Wang, X.; Manchester, H.E.; Wang, W.; Haswell, J.R.; Park, P.J.; Roberts, C.W. The SWI/SNF chromatin remodelling complex is required for maintenance of lineage specific enhancers. Nat. Commun. 2017, 8, 14648. [Google Scholar] [CrossRef]
- Perez-Cervantes, C.; Smith, L.A.; Nadadur, R.D.; Hughes, A.E.O.; Wang, S.; Corbo, J.C.; Cepko, C.; Lonfat, N.; Moskowitz, I.P. Enhancer transcription identifies cis-regulatory elements for photoreceptor cell types. Development 2020, 147, dev184432. [Google Scholar] [CrossRef]
- Goodson, N.B.; Kaufman, M.A.; Park, K.U.; Brzezinski, J.A. Simultaneous deletion of Prdm1 and Vsx2 enhancers in the retina alters photoreceptor and bipolar cell fate specification, yet differs from deleting both genes. Development 2020, 147, dev190272. [Google Scholar] [CrossRef] [PubMed]
- Emerson, M.M.; Cepko, C.L. Identification of a retina-specific Otx2 enhancer element active in immature developing photoreceptors. Dev. Biol. 2011, 360, 241–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaufman, M.L.; Goodson, N.B.; Park, K.U.; Schwanke, M.; Office, E.; Schneider, S.R.; Abraham, J.; Hensley, A.; Jones, K.L.; Brzezinski, J.A. Initiation of Otx2 expression in the developing mouse retina requires a unique enhancer and either Ascl1 or Neurog2 activity. Development 2021, 148, dev199399. [Google Scholar] [CrossRef]
- Soufi, A.; Garcia, M.F.; Jaroszewicz, A.; Osman, N.; Pellegrini, M.; Zaret, K.S. Pioneer TFs target partial DNA motifs on nucleosomes to initiate reprogramming. Cell 2015, 161, 555–568. [Google Scholar] [CrossRef] [Green Version]
- Schep, A.N.; Buenrostro, J.D.; Denny, S.K.; Schwartz, K.; Sherlock, G.; Greenleaf, W.J. Structured nucleosome fingerprints enable high-resolution mapping of chromatin architecture within regulatory regions. Genome Res. 2015, 25, 1757–1770. [Google Scholar] [CrossRef] [Green Version]
- Chen, K.; Xi, Y.; Pan, X.; Li, Z.; Kaestner, K.; Tyler, J.; Dent, S.; He, X.; Li, W. DANPOS: Dynamic analysis of nucleosome position and occupancy by sequencing. Genome Res. 2013, 23, 341–351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Huang, J.; Chen, T.; Wang, Y.; Xin, S.; Li, J.; Pei, G.; Kang, J. Yamanaka factors critically regulate the developmental signaling network in mouse embryonic stem cells. Cell Res. 2008, 18, 1177–1189. [Google Scholar] [CrossRef]
- Sherwood, R.I.; Hashimoto, T.; O’Donnell, C.W.; Lewis, S.; Barkal, A.A.; van Hoff, J.P.; Karun, V.; Jaakkola, T.; Gifford, D.K. Discovery of directional and nondirectional pioneer TFs by modeling DNase profile magnitude and shape. Nat. Biotechnol. 2014, 32, 171–178. [Google Scholar] [CrossRef] [Green Version]
- Grant, C.E.; Bailey, T.L.; Noble, W.S. FIMO: Scanning for occurrences of a given motif. Bioinformatics 2011, 27, 1017–1018. [Google Scholar] [CrossRef] [Green Version]
- Pique-Regi, R.; Degner, J.F.; Pai, A.A.; Gaffney, D.J.; Gilad, Y.; Pritchard, J.K. Accurate inference of TF binding from DNA sequence and chromatin accessibility data. Genome Res. 2011, 21, 447–455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.; Zibetti, C.; Wan, J.; Wang, G.; Blackshaw, S.; Qian, J. Assessing the model transferability for prediction of TF binding sites based on chromatin accessibility. BMC Bioinform. 2017, 18, 355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.; Thormann, A.; Flicek, P.; Cunningham, F. The Ensembl Variant Effect Predictor. Genome Biol. 2016, 17, 122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iejima, D.; Itabashi, T.; Kawamura, Y.; Noda, T.; Yuasa, S.; Fukuda, K.; Oka, C.; Iwata, T. HTRA1 (high temperature requirement A serine peptidase 1) gene is transcriptionally regulated by insertion/deletion nucleotides located at the 3′ end of the ARMS2 (age-related maculopathy susceptibility 2) gene in patients with age-related macular degeneration. J. Biol. Chem. 2015, 290, 2784–2797. [Google Scholar] [PubMed] [Green Version]
- Stein-O’Brien, G.L.; Clark, B.S.; Sherman, T.; Zibetti, C.; Hu, Q.; Sealfon, R.; Liu, S.; Qian, J.; Colantuoni, C.; Blackshaw, S.; et al. Decomposing Cell Identity for Transfer Learning across Cellular Measurements, Platforms, Tissues, and Species. Cell Syst. 2019, 8, 395–411.e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pevny, L.H.; Lovell-Badge, R. Sox genes find their feet. Curr. Opin. Genet. Dev. 1997, 7, 338–344. [Google Scholar] [CrossRef]
- Favaro, R.; Valotta, M.; Ferri, A.L.; Latorre, E.; Mariani, J.; Giachino, C.; Lancini, C.; Tosetti, V.; Ottolenghi, S.; Taylor, V.; et al. Hippocampal development and neural stem cell maintenance require Sox2-dependent regulation of Shh. Nat. Neurosci. 2009, 12, 1248–1256. [Google Scholar] [CrossRef]
- Ferri, A.L.; Cavallaro, M.; Braida, D.; Di Cristofano, A.; Canta, A.; Vezzani, A.; Ottolenghi, S.; Pandolfi, P.P.; Sala, M.; DeBiasi, S.; et al. Sox2 deficiency causes neurodegeneration and impaired neurogenesis in the adult mouse brain. Development 2004, 131, 3805–3819. [Google Scholar] [CrossRef] [Green Version]
- Wegner, M.; Stolt, C.C. From stem cells to neurons and glia: A Soxist’s view of neural development. Trends Neurosci. 2005, 28, 583–588. [Google Scholar] [CrossRef]
- Wegner, M. SOX after SOX: SOXession regulates neurogenesis. Genes Dev. 2011, 25, 2423–2428. [Google Scholar] [CrossRef] [Green Version]
- Taranova, O.V.; Magness, S.T.; Fagan, B.M.; Wu, Y.; Surzenko, N.; Hutton, S.R.; Pevny, L.H. SOX2 is a dose-dependent regulator of retinal neural progenitor competence. Genes Dev. 2006, 20, 1187–1202. [Google Scholar] [CrossRef] [Green Version]
- Lodato, M.A.; Ng, C.W.; Wamstad, J.A.; Cheng, A.W.; Thai, K.K.; Fraenkel, E.; Jaenisch, R.; Boyer, L.A. SOX2 co-occupies distal enhancer elements with distinct POU factors in ESCs and NPCs to specify cell state. PLoS Genet. 2013, 9, e1003288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Y.P.; Ouchi, Y.; Satoh, S.; Watanabe, S. Sox2 plays a role in the induction of amacrine and Muller glial cells in mouse retinal progenitor cells. Investig. Opthalmol. Vis. Sci. 2009, 50, 68–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wohl, S.G.; Hooper, M.J.; Reh, T.A. MicroRNAs miR-25, let-7 and miR-124 regulate the neurogenic potential of Muller glia in mice. Development 2019, 146, dev179556. [Google Scholar] [CrossRef] [Green Version]
- Poche, R.A.; Furuta, Y.; Chaboissier, M.C.; Schedl, A.; Behringer, R.R. Sox9 is expressed in mouse multipotent retinal progenitor cells and functions in Muller glial cell development. J. Comp. Neurol. 2008, 510, 237–250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masuda, T.; Wahlin, K.; Wan, J.; Hu, J.; Maruotti, J.; Yang, X.; Iacovelli, J.; Wolkow, N.; Kist, R.; Dunaief, J.L.; et al. TF SOX9 plays a key role in the regulation of visual cycle gene expression in the retinal pigment epithelium. J. Biol. Chem. 2014, 289, 12908–12921. [Google Scholar] [CrossRef] [Green Version]
- Cohen-Tayar, Y.; Cohen, H.; Mitiagin, Y.; Abravanel, Z.; Levy, C.; Idelson, M.; Reubinoff, B.; Itzkovitz, S.; Raviv, S.; Kaestner, K.H.; et al. Pax6 regulation of Sox9 in the mouse retinal pigmented epithelium controls its timely differentiation and choroid vasculature development. Development 2018, 145, dev163691. [Google Scholar]
- Menzel-Severing, J.; Zenkel, M.; Polisetti, N.; Sock, E.; Wegner, M.; Kruse, F.E.; Schlotzer-Schrehardt, U. TF profiling identifies Sox9 as regulator of proliferation and differentiation in corneal epithelial stem/progenitor cells. Sci. Rep. 2018, 8, 10268. [Google Scholar] [CrossRef] [PubMed]
- Vong, K.I.; Leung, C.K.; Behringer, R.R.; Kwan, K.M. Sox9 is critical for suppression of neurogenesis but not initiation of gliogenesis in the cerebellum. Mol. Brain 2015, 8, 25. [Google Scholar] [CrossRef] [Green Version]
- Kang, P.; Lee, H.K.; Glasgow, S.M.; Finley, M.; Donti, T.; Gaber, Z.B.; Graham, B.H.; Foster, A.E.; Novitch, B.G.; Gronostajski, R.M.; et al. Sox9 and NFIA coordinate a transcriptional regulatory cascade during the initiation of gliogenesis. Neuron 2012, 74, 79–94. [Google Scholar] [CrossRef] [Green Version]
- Tomita, K.; Nakanishi, S.; Guillemot, F.; Kageyama, R. Mash1 promotes neuronal differentiation in the retina. Genes Cells 1996, 1, 765–774. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, R.; Fausett, B.V.; Goldman, D. Ascl1a regulates Muller glia dedifferentiation and retinal regeneration through a Lin-28-dependent, let-7 microRNA signalling pathway. Nat. Cell Biol. 2010, 12, 1101–1107. [Google Scholar] [CrossRef] [PubMed]
- Jorstad, N.L.; Wilken, M.S.; Grimes, W.N.; Wohl, S.G.; VandenBosch, L.S.; Yoshimatsu, T.; Wong, R.O.; Rieke, F.; Reh, T.A. Stimulation of functional neuronal regeneration from Muller glia in adult mice. Nature 2017, 548, 103–107. [Google Scholar] [CrossRef]
- Hjelm, B.E.; Rosenberg, J.B.; Szelinger, S.; Sue, L.I.; Beach, T.G.; Huentelman, M.J.; Craig, D.W. Induction of pluripotent stem cells from autopsy donor-derived somatic cells. Neurosci. Lett. 2011, 502, 219–224. [Google Scholar] [CrossRef] [Green Version]
- Wernig, M.; Lengner, C.J.; Hanna, J.; Lodato, M.A.; Steine, E.; Foreman, R.; Staerk, J.; Markoulaki, S.; Jaenisch, R. A drug-inducible transgenic system for direct reprogramming of multiple somatic cell types. Nat. Biotechnol. 2008, 26, 916–924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, X.; Wang, X.; Xiong, W.; Chen, J. In vivo reprogramming reactive glia into iPSCs to produce new neurons in the cortex following traumatic brain injury. Sci. Rep. 2016, 6, 22490. [Google Scholar] [CrossRef] [Green Version]
- Todd, L.; Fischer, A.J. Hedgehog signaling stimulates the formation of proliferating Muller glia-derived progenitor cells in the chick retina. Development 2015, 142, 2610–2622. [Google Scholar]
- Todd, L.; Suarez, L.; Quinn, C.; Fischer, A.J. Retinoic Acid-Signaling Regulates the Proliferative and Neurogenic Capacity of Muller Glia-Derived Progenitor Cells in the Avian Retina. Stem Cells 2018, 36, 392–405. [Google Scholar] [CrossRef] [Green Version]
- Zelinka, C.P.; Volkov, L.; Goodman, Z.A.; Todd, L.; Palazzo, I.; Bishop, W.A.; Fischer, A.J. mTor signaling is required for the formation of proliferating Muller glia-derived progenitor cells in the chick retina. Development 2016, 143, 1859–1873. [Google Scholar] [CrossRef] [Green Version]
- Moore, D.L.; Blackmore, M.G.; Hu, Y.; Kaestner, K.H.; Bixby, J.L.; Lemmon, V.P.; Goldberg, J.L. KLF family members regulate intrinsic axon regeneration ability. Science 2009, 326, 298–301. [Google Scholar] [CrossRef] [Green Version]
- Fang, J.; Shaw, P.X.; Wang, Y.; Goldberg, J.L. Kruppel-Like Factor 4 (KLF4) Is Not Required for Retinal Cell Differentiation. eNeuro 2016, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marcos-Mondejar, P.; Peregrin, S.; Li, J.Y.; Carlsson, L.; Tole, S.; Lopez-Bendito, G. The Lhx2 TF controls thalamocortical axonal guidance by specific regulation of robo1 and robo2 receptors. J. Neurosci. 2012, 32, 4372–4385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Sullivan, M.L.; Punal, V.M.; Kerstein, P.C.; Brzezinski, J.A.; Glaser, T.; Wright, K.M.; Kay, J.N. Astrocytes follow ganglion cell axons to establish an angiogenic template during retinal development. Glia 2017, 65, 1697–1716. [Google Scholar] [CrossRef] [PubMed]
- Rocha-Martins, M.; de Toledo, B.C.; Santos-Franca, P.L.; Oliveira-Valenca, V.M.; Vieira-Vieira, C.H.; Matos-Rodrigues, G.E.; Linden, R.; Norden, C.; Martins, R.A.P.; Silveira, M.S. De novo genesis of retinal ganglion cells by targeted expression of Klf4 in vivo. Development 2019, 146, dev176586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, N.L.; Patel, S.; Brzezinski, J.; Glaser, T. Math5 is required for retinal ganglion cell and optic nerve formation. Development 2001, 128, 2497–2508. [Google Scholar] [CrossRef] [PubMed]
- Pan, L.; Deng, M.; Xie, X.; Gan, L. ISL1 and BRN3B co-regulate the differentiation of murine retinal ganglion cells. Development 2008, 135, 1981–1990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brzezinski, J.A.; Prasov, L.; Glaser, T. Math5 defines the ganglion cell competence state in a subpopulation of retinal progenitor cells exiting the cell cycle. Dev. Biol. 2012, 365, 395–413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, F.; Kaczynski, T.J.; Sethuramanujam, S.; Li, R.; Jain, V.; Slaughter, M.; Mu, X. Two TFs, Pou4f2 and Isl1, are sufficient to specify the retinal ganglion cell fate. Proc. Natl. Acad. Sci. USA 2015, 112, E1559–E1568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miesfeld, J.B.; Ghiasvand, N.M.; Marsh-Armstrong, B.; Marsh-Armstrong, N.; Miller, E.B.; Zhang, P.; Manna, S.K.; Zawadzki, R.J.; Brown, N.L.; Glaser, T. The Atoh7 remote enhancer provides transcriptional robustness during retinal ganglion cell development. Proc. Natl. Acad. Sci. USA 2020, 117, 21690–21700. [Google Scholar] [CrossRef]
- Dupacova, N.; Antosova, B.; Paces, J.; Kozmik, Z. Meis homeobox genes control progenitor competence in the retina. Proc. Natl. Acad. Sci. USA 2021, 118, e2013136118. [Google Scholar] [CrossRef]
- Hoang, T.; Wang, J.; Boyd, P.; Wang, F.; Santiago, C.; Jiang, L.; Yoo, S.; Lahne, M.; Todd, L.J.; Jia, M.; et al. Gene regulatory networks controlling vertebrate retinal regeneration. Science 2020, 370. [Google Scholar] [CrossRef] [PubMed]
- Deneen, B.; Ho, R.; Lukaszewicz, A.; Hochstim, C.J.; Gronostajski, R.M.; Anderson, D.J. The TF NFIA controls the onset of gliogenesis in the developing spinal cord. Neuron 2006, 52, 953–968. [Google Scholar] [CrossRef] [Green Version]
- Matuzelski, E.; Bunt, J.; Harkins, D.; Lim, J.W.C.; Gronostajski, R.M.; Richards, L.J.; Harris, L.; Piper, M. Transcriptional regulation of Nfix by NFIB drives astrocytic maturation within the developing spinal cord. Dev. Biol. 2017, 432, 286–297. [Google Scholar] [CrossRef] [PubMed]
- Nagao, M.; Ogata, T.; Sawada, Y.; Gotoh, Y. Zbtb20 promotes astrocytogenesis during neocortical development. Nat. Commun. 2016, 7, 11102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jorstad, N.L.; Wilken, M.S.; Todd, L.; Finkbeiner, C.; Nakamura, P.; Radulovich, N.; Hooper, M.J.; Chitsazan, A.; Wilkerson, B.A.; Rieke, F.; et al. STAT Signaling Modifies Ascl1 Chromatin Binding and Limits Neural Regeneration from Muller Glia in Adult Mouse Retina. Cell Rep. 2020, 30, 2195–2208.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Zibetti, C.; Shang, P.; Sripathi, S.R.; Zhang, P.; Cano, M.; Hoang, T.; Xia, S.; Ji, H.; Merbs, S.L.; et al. ATAC-Seq analysis reveals a widespread decrease of chromatin accessibility in age-related macular degeneration. Nat. Commun. 2018, 9, 1364. [Google Scholar] [CrossRef] [Green Version]
- Bourne, R.R.A.; Flaxman, S.R.; Braithwaite, T.; Cicinelli, M.V.; Das, A.; Jonas, J.B.; Keeffe, J.; Kempen, J.H.; Leasher, J.; Limburg, H.; et al. Magnitude, temporal trends, and projections of the global prevalence of blindness and distance and near vision impairment: A systematic review and meta-analysis. Lancet Glob. Health 2017, 5, e888–e897. [Google Scholar] [CrossRef] [Green Version]
- Jonas, J.B.; Cheung, C.M.G.; Panda-Jonas, S. Updates on the Epidemiology of Age-Related Macular Degeneration. Asia Pac. J. Ophthalmol. 2017, 6, 493–497. [Google Scholar]
- Cachafeiro, M.; Bemelmans, A.P.; Samardzija, M.; Afanasieva, T.; Pournaras, J.A.; Grimm, C.; Kostic, C.; Philippe, S.; Wenzel, A.; Arsenijevic, Y. Hyperactivation of retina by light in mice leads to photoreceptor cell death mediated by VEGF and retinal pigment epithelium permeability. Cell Death Dis. 2013, 4, e781. [Google Scholar] [CrossRef]
- Raoul, W.; Keller, N.; Rodero, M.; Behar-Cohen, F.; Sennlaub, F.; Combadiere, C. Role of the chemokine receptor CX3CR1 in the mobilization of phagocytic retinal microglial cells. J. Neuroimmunol. 2008, 198, 56–61. [Google Scholar] [CrossRef] [Green Version]
- Ma, W.; Zhang, Y.; Gao, C.; Fariss, R.N.; Tam, J.; Wong, W.T. Monocyte infiltration and proliferation reestablish myeloid cell homeostasis in the mouse retina following retinal pigment epithelial cell injury. Sci. Rep. 2017, 7, 8433. [Google Scholar] [CrossRef]
- Thakkinstian, A.; McEvoy, M.; Chakravarthy, U.; Chakrabarti, S.; McKay, G.J.; Ryu, E.; Silvestri, G.; Kaur, I.; Francis, P.; Iwata, T.; et al. The association between complement component 2/complement factor B polymorphisms and age-related macular degeneration: A HuGE review and meta-analysis. Am. J. Epidemiol. 2012, 176, 361–372. [Google Scholar] [CrossRef] [PubMed]
- Szatmari-Toth, M.; Kristof, E.; Vereb, Z.; Akhtar, S.; Facsko, A.; Fesus, L.; Kauppinen, A.; Kaarniranta, K.; Petrovski, G. Clearance of autophagy-associated dying retinal pigment epithelial cells—A possible source for inflammation in age-related macular degeneration. Cell Death Dis. 2016, 7, e2367. [Google Scholar] [CrossRef] [PubMed]
- Porter, L.F.; Saptarshi, N.; Fang, Y.; Rathi, S.; den Hollander, A.I.; de Jong, E.K.; Clark, S.J.; Bishop, P.N.; Olsen, T.W.; Liloglou, T.; et al. Whole-genome methylation profiling of the retinal pigment epithelium of individuals with age-related macular degeneration reveals differential methylation of the SKI, GTF2H4, and TNXB genes. Clin. Epigenet. 2019, 11, 6. [Google Scholar] [CrossRef] [PubMed]
- Menon, M.; Mohammadi, S.; Davila-Velderrain, J.; Goods, B.A.; Cadwell, T.D.; Xing, Y.; Stemmer-Rachamimov, A.; Shalek, A.K.; Love, J.C.; Kellis, M.; et al. Single-cell transcriptomic atlas of the human retina identifies cell types associated with age-related macular degeneration. Nat. Commun. 2019, 10, 4902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sahaboglu, A.; Paquet-Durand, O.; Dietter, J.; Dengler, K.; Bernhard-Kurz, S.; Ekstrom, P.A.; Hitzmann, B.; Ueffing, M.; Paquet-Durand, F. Retinitis pigmentosa: Rapid neurodegeneration is governed by slow cell death mechanisms. Cell Death Dis. 2013, 4, e488. [Google Scholar] [CrossRef] [PubMed]
- Potic, J.; Mbefo, M.; Berger, A.; Nicolas, M.; Wanner, D.; Kostic, C.; Matet, A.; Behar-Cohen, F.; Moulin, A.; Arsenijevic, Y. An In Vitro Model of Human Retinal Detachment Reveals Successive Death Pathway Activations. Front. Neurosci. 2020, 14, 571293. [Google Scholar] [CrossRef] [PubMed]
- Aapola, U.; Liiv, I.; Peterson, P. Imprinting regulator DNMT3L is a transcriptional repressor associated with histone deacetylase activity. Nucleic Acids Res. 2002, 30, 3602–3608. [Google Scholar] [CrossRef]
- Rountree, M.R.; Bachman, K.E.; Baylin, S.B. DNMT1 binds HDAC2 and a new co-repressor, DMAP1, to form a complex at replication foci. Nat. Genet. 2000, 25, 269–277. [Google Scholar] [CrossRef]
- Wahlin, K.J.; Enke, R.A.; Fuller, J.A.; Kalesnykas, G.; Zack, D.J.; Merbs, S.L. Epigenetics and cell death: DNA hypermethylation in programmed retinal cell death. PLoS ONE 2013, 8, e79140. [Google Scholar] [CrossRef] [Green Version]
- Farinelli, P.; Perera, A.; Arango-Gonzalez, B.; Trifunovic, D.; Wagner, M.; Carell, T.; Biel, M.; Zrenner, E.; Michalakis, S.; Paquet-Durand, F.; et al. DNA methylation and differential gene regulation in photoreceptor cell death. Cell Death Dis. 2014, 5, e1558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corso-Diaz, X.; Jaeger, C.; Chaitankar, V.; Swaroop, A. Epigenetic control of gene regulation during development and disease: A view from the retina. Prog. Retin. Eye Res. 2018, 65, 1–27. [Google Scholar] [CrossRef] [PubMed]
- Gemenetzi, M.; Lotery, A.J. The role of epigenetics in age-related macular degeneration. Eye 2014, 28, 1407–1417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pennington, K.L.; DeAngelis, M.M. Epidemiology of age-related macular degeneration (AMD): Associations with cardiovascular disease phenotypes and lipid factors. Eye Vis. 2016, 3, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, L.; Liu, B.; Tuo, J.; Shen, D.; Chen, P.; Li, Z.; Liu, X.; Ni, J.; Dagur, P.; Sen, H.N.; et al. Hypomethylation of the IL17RC promoter associates with age-related macular degeneration. Cell Rep. 2012, 2, 1151–1158. [Google Scholar] [CrossRef] [Green Version]
- Biswas, S.; Thomas, A.A.; Chen, S.; Aref-Eshghi, E.; Feng, B.; Gonder, J.; Sadikovic, B.; Chakrabarti, S. MALAT1: An Epigenetic Regulator of Inflammation in Diabetic Retinopathy. Sci. Rep. 2018, 8, 6526. [Google Scholar] [CrossRef]
- Sancho-Pelluz, J.; Alavi, M.V.; Sahaboglu, A.; Kustermann, S.; Farinelli, P.; Azadi, S.; van Veen, T.; Romero, F.J.; Paquet-Durand, F.; Ekstrom, P. Excessive HDAC activation is critical for neurodegeneration in the rd1 mouse. Cell Death Dis. 2010, 1, e24. [Google Scholar] [CrossRef] [Green Version]
- Zheng, S.; Xiao, L.; Liu, Y.; Wang, Y.; Cheng, L.; Zhang, J.; Yan, N.; Chen, D. DZNep inhibits H3K27me3 deposition and delays retinal degeneration in the rd1 mice. Cell Death Dis. 2018, 9, 310. [Google Scholar] [CrossRef] [Green Version]
- Trifunovic, D.; Sahaboglu, A.; Kaur, J.; Mencl, S.; Zrenner, E.; Ueffing, M.; Arango-Gonzalez, B.; Paquet-Durand, F. Neuroprotective strategies for the treatment of inherited photoreceptor degeneration. Curr. Mol. Med. 2012, 12, 598–612. [Google Scholar] [CrossRef]
- Mitton, K.P.; Guzman, A.E.; Deshpande, M.; Byrd, D.; DeLooff, C.; Mkoyan, K.; Zlojutro, P.; Wallace, A.; Metcalf, B.; Laux, K.; et al. Different effects of valproic acid on photoreceptor loss in Rd1 and Rd10 retinal degeneration mice. Mol. Vis. 2014, 20, 1527–1544. [Google Scholar]
- Trifunovic, D.; Arango-Gonzalez, B.; Comitato, A.; Barth, M.; Del Amo, E.M.; Kulkarni, M.; Sahaboglu, A.; Hauck, S.M.; Urtti, A.; Arsenijevic, Y.; et al. HDAC inhibition in the cpfl1 mouse protects de-generating cone photoreceptors in vivo. Hum. Mol. Genet. 2016, 25, 4462–4472. [Google Scholar] [PubMed] [Green Version]
- Klingeborn, M.; Dismuke, W.M.; Bowes Rickman, C.; Stamer, W.D. Roles of exosomes in the normal and diseased eye. Prog. Retin. Eye Res. 2017, 59, 158–177. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.H.; Ling, D.; Tu, L.; van Wijngaarden, P.; Dusting, G.J.; Liu, G.S. Gene therapy for diabetic retinopathy: Are we ready to make the leap from bench to bedside? Pharmacol. Ther. 2017, 173, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Fernandez, F.M.; Bianchera, A.; Gasco, P.; Nicoli, S.; Pescina, S. Lipid-Based Nanocarriers for Ophthalmic Administration: Towards Experimental Design Implementation. Pharmaceutics 2021, 13, 447. [Google Scholar] [CrossRef]
- Amadio, M.; Pascale, A.; Cupri, S.; Pignatello, R.; Osera, C.; D’Agata, V.; D’Amico, A.G.; Leggio, G.M.; Ruozi, B.; Govoni, S.; et al. Nanosystems based on siRNA silencing HuR expression counteract diabetic retinopathy in rat. Pharmacol. Res. 2016, 111, 713–720. [Google Scholar] [CrossRef]
- Campochiaro, P.A. Potential applications for RNAi to probe pathogenesis and develop new treatments for ocular disorders. Gene Ther. 2006, 13, 559–562. [Google Scholar] [CrossRef] [Green Version]
- Haurigot, V.; Villacampa, P.; Ribera, A.; Bosch, A.; Ramos, D.; Ruberte, J.; Bosch, F. Long-term retinal PEDF overexpression prevents neovascularization in a murine adult model of retinopathy. PLoS ONE 2012, 7, e41511. [Google Scholar] [CrossRef] [Green Version]
- Achberger, K.; Cipriano, M.; Duchs, M.J.; Schon, C.; Michelfelder, S.; Stierstorfer, B.; Lamla, T.; Kauschke, S.G.; Chuchuy, J.; Roosz, J.; et al. Human stem cell-based retina on chip as new translational model for validation of AAV retinal gene therapy vectors. Stem Cell Rep. 2021, 16, 2242–2256. [Google Scholar] [CrossRef]
- Wiley, L.A.; Burnight, E.R.; Kaalberg, E.E.; Jiao, C.; Riker, M.J.; Halder, J.A.; Luse, M.A.; Han, I.C.; Russell, S.R.; Sohn, E.H.; et al. Assessment of Adeno-Associated Virus Serotype Tropism in Human Retinal Explants. Hum. Gene Ther. 2018, 29, 424–436. [Google Scholar] [CrossRef]
- Chung, S.H.; Sin, T.N.; Ngo, T.; Yiu, G. CRISPR Technology for Ocular Angiogenesis. Front. Genome Ed. 2020, 2, 594984. [Google Scholar] [CrossRef]
- Fenner, B.J.; Tan, T.E.; Barathi, A.V.; Tun, S.B.B.; Yeo, S.W.; Tsai, A.S.H.; Lee, S.Y.; Cheung, C.M.G.; Chan, C.M.; Mehta, J.S.; et al. Gene-Based Therapeutics for Inherited Retinal Diseases. Front. Genet. 2021, 12, 794805. [Google Scholar] [CrossRef] [PubMed]
- Russell, S.; Bennett, J.; Wellman, J.A.; Chung, D.C.; Yu, Z.F.; Tillman, A.; Wittes, J.; Pappas, J.; Elci, O.; McCague, S.; et al. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: A randomised, controlled, open-label, phase 3 trial. Lancet 2017, 390, 849–860. [Google Scholar] [CrossRef]
- Latella, M.C.; Di Salvo, M.T.; Cocchiarella, F.; Benati, D.; Grisendi, G.; Comitato, A.; Marigo, V.; Recchia, A. In Vivo Editing of the Human Mutant Rhodopsin Gene by Electroporation of Plasmid-based CRISPR/Cas9 in the Mouse Retina. Mol. Ther. Nucleic Acids 2016, 5, e389. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.; Zode, G.; Kasetti, R.B.; Ran, F.A.; Yan, W.; Sharma, T.P.; Bugge, K.; Searby, C.C.; Fingert, J.H.; Zhang, F.; et al. CRISPR-Cas9-based treatment of myocilin-associated glaucoma. Proc. Natl. Acad. Sci. USA 2017, 114, 11199–11204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cideciyan, A.V.; Sudharsan, R.; Dufour, V.L.; Massengill, M.T.; Iwabe, S.; Swider, M.; Lisi, B.; Sumaroka, A.; Marinho, L.F.; Appelbaum, T.; et al. Mutation-independent rhodopsin gene therapy by knock-down and replacement with a single AAV vector. Proc. Natl. Acad. Sci. USA 2018, 115, E8547–E8556. [Google Scholar] [CrossRef] [Green Version]
- Woo, M. Eyes hint at hidden mental-health conditions. Nature 2019. [Google Scholar] [CrossRef]
- Wu, W.H.; Tsai, Y.T.; Huang, I.W.; Cheng, C.H.; Hsu, C.W.; Cui, X.; Ryu, J.; Quinn, P.M.J.; Caruso, S.M.; Lin, C.S.; et al. CRISPR genome surgery in a novel humanized model for autosomal dominant retinitis pigmentosa. Mol. Ther. 2022. [Google Scholar] [CrossRef]
- Moreno, A.M.; Fu, X.; Zhu, J.; Katrekar, D.; Shih, Y.V.; Marlett, J.; Cabotaje, J.; Tat, J.; Naughton, J.; Lisowski, L.; et al. In Situ Gene Therapy via AAV-CRISPR-Cas9-Mediated Targeted Gene Regulation. Mol. Ther. 2018, 26, 1818–1827. [Google Scholar] [CrossRef] [Green Version]
- Zetsche, B.; Volz, S.E.; Zhang, F. A split-Cas9 architecture for inducible genome editing and transcription modulation. Nat. Biotechnol. 2015, 33, 139–142. [Google Scholar] [CrossRef] [Green Version]
- Keser, V.; Khan, A.; Siddiqui, S.; Lopez, I.; Ren, H.; Qamar, R.; Nadaf, J.; Majewski, J.; Chen, R.; Koenekoop, R.K. The Genetic Causes of Nonsyndromic Congenital Retinal Detachment: A Genetic and Phenotypic Study of Pakistani Families. Investig. Opthalmol. Vis. Sci. 2017, 58, 1028–1036. [Google Scholar] [CrossRef] [Green Version]
- Ghiasvand, N.M.; Rudolph, D.D.; Mashayekhi, M.; Brzezinski, J.A.; Goldman, D.; Glaser, T. Deletion of a remote enhancer near ATOH7 disrupts retinal neurogenesis, causing NCRNA disease. Nat. Neurosci. 2011, 14, 578–586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhatia, S.; Bengani, H.; Fish, M.; Brown, A.; Divizia, M.T.; de Marco, R.; Damante, G.; Grainger, R.; van Heyningen, V.; Kleinjan, D.A. Disruption of autoregulatory feedback by a mutation in a remote, ultraconserved PAX6 enhancer causes aniridia. Am. J. Hum. Genet. 2013, 93, 1126–1134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bremond-Gignac, D.; Bitoun, P.; Reis, L.M.; Copin, H.; Murray, J.C.; Semina, E.V. Identification of dominant FOXE3 and PAX6 mutations in patients with congenital cataract and aniridia. Mol. Vis. 2010, 16, 1705–1711. [Google Scholar] [PubMed]
- Van Schil, K.; Karlstetter, M.; Aslanidis, A.; Dannhausen, K.; Azam, M.; Qamar, R.; Leroy, B.P.; Depasse, F.; Langmann, T.; De Baere, E. Autosomal recessive retinitis pigmentosa with homozygous rhodopsin mutation E150K and non-coding cis-regulatory variants in CRX-binding regions of SAMD7. Sci. Rep. 2016, 6, 21307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Bruijn, S.E.; Fiorentino, A.; Ottaviani, D.; Fanucchi, S.; Melo, U.S.; Corral-Serrano, J.C.; Mulders, T.; Georgiou, M.; Rivolta, C.; Pontikos, N.; et al. Structural Variants Create New Topological-Associated Domains and Ectopic Retinal Enhancer-Gene Contact in Dominant Retinitis Pigmentosa. Am. J. Hum. Genet. 2020, 107, 802–814. [Google Scholar] [CrossRef] [PubMed]
- Zalatan, J.G.; Lee, M.E.; Almeida, R.; Gilbert, L.A.; Whitehead, E.H.; La Russa, M.; Tsai, J.C.; Weissman, J.S.; Dueber, J.E.; Qi, L.S.; et al. Engineering complex synthetic transcriptional programs with CRISPR RNA scaffolds. Cell 2015, 160, 339–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nunez, J.K.; Chen, J.; Pommier, G.C.; Cogan, J.Z.; Replogle, J.M.; Adriaens, C.; Ramadoss, G.N.; Shi, Q.; Hung, K.L.; Samelson, A.J.; et al. Genome-wide programmable transcriptional memory by CRISPR-based epigenome editing. Cell 2021, 184, 2503–2519.e17. [Google Scholar] [CrossRef]
- Zhuo, C.; Zhang, J.; Lee, J.H.; Jiao, J.; Cheng, D.; Liu, L.; Kim, H.W.; Tao, Y.; Li, M. Spatiotemporal control of CRISPR/Cas9 gene editing. Signal Transduct. Target. Ther. 2021, 6, 238. [Google Scholar] [CrossRef]
- Mandegar, M.A.; Huebsch, N.; Frolov, E.B.; Shin, E.; Truong, A.; Olvera, M.P.; Chan, A.H.; Miyaoka, Y.; Holmes, K.; Spencer, C.I.; et al. CRISPR Interference Efficiently Induces Specific and Reversible Gene Silencing in Human iPSCs. Cell Stem Cell 2016, 18, 541–553. [Google Scholar] [CrossRef] [Green Version]
- Konermann, S.; Brigham, M.D.; Trevino, A.E.; Joung, J.; Abudayyeh, O.O.; Barcena, C.; Hsu, P.D.; Habib, N.; Gootenberg, J.S.; Nishimasu, H.; et al. Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature 2015, 517, 583–588. [Google Scholar] [CrossRef] [Green Version]
- Bin Moon, S.; Lee, J.M.; Kang, J.G.; Lee, N.E.; Ha, D.I.; Kim, D.Y.; Kim, S.H.; Yoo, K.; Kim, D.; Ko, J.H.; et al. Highly efficient genome editing by CRISPR-Cpf1 using CRISPR RNA with a uridinylate-rich 3′-overhang. Nat. Commun. 2018, 9, 3651. [Google Scholar] [CrossRef] [PubMed]
- Sahel, J.A.; Boulanger-Scemama, E.; Pagot, C.; Arleo, A.; Galluppi, F.; Martel, J.N.; Esposti, S.D.; Delaux, A.; de Saint Aubert, J.B.; de Montleau, C.; et al. Partial recovery of visual function in a blind patient after optogenetic therapy. Nat. Med. 2021, 27, 1223–1229. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, E. Development of visual Neuroprostheses: Trends and challenges. Bioelectron. Med. 2018, 4, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kashani, A.H.; Lebkowski, J.S.; Rahhal, F.M.; Avery, R.L.; Salehi-Had, H.; Dang, W.; Lin, C.M.; Mitra, D.; Zhu, D.; Thomas, B.B.; et al. A bioengineered retinal pigment epithelial monolayer for advanced, dry age-related macular degeneration. Sci. Transl. Med. 2018, 10, eaao4097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikelle, L.; Al-Ubaidi, M.R.; Naash, M.I. Pluripotent Stem Cells for the Treatment of Retinal Degeneration: Current Strategies and Future Directions. Front. Cell Dev. Biol. 2020, 8, 743. [Google Scholar] [CrossRef]
- Miyake, A.; Araki, M. Retinal stem/progenitor cells in the ciliary marginal zone complete retinal regeneration: A study of retinal regeneration in a novel animal model. Dev. Neurobiol. 2014, 74, 739–756. [Google Scholar] [CrossRef]
- Marcucci, F.; Murcia-Belmonte, V.; Wang, Q.; Coca, Y.; Ferreiro-Galve, S.; Kuwajima, T.; Khalid, S.; Ross, M.E.; Mason, C.; Herrera, E. The Ciliary Margin Zone of the Mammalian Retina Generates Retinal Ganglion Cells. Cell Rep. 2016, 17, 3153–3164. [Google Scholar] [CrossRef]
- Reh, T.A.; Nagy, T. A possible role for the vascular membrane in retinal regeneration in Rana catesbienna tadpoles. Dev. Biol. 1987, 122, 471–482. [Google Scholar] [CrossRef]
- Mitashov, V.I. Mechanisms of retina regeneration in urodeles. Int. J. Dev. Biol. 1996, 40, 833–844. [Google Scholar]
- Del Rio-Tsonis, K.; Tsonis, P.A. Eye regeneration at the molecular age. Dev. Dyn. 2003, 226, 211–224. [Google Scholar] [CrossRef]
- Klein, L.R.; MacLeish, P.R.; Wiesel, T.N. Immunolabelling by a newt retinal pigment epithelium antibody during retinal development and regeneration. J. Comp. Neurol. 1990, 293, 331–339. [Google Scholar] [CrossRef] [PubMed]
- Casco-Robles, M.M.; Nakamura, K.; Casco-Robles, M.M.; Kunahong, A.; Inami, W.; Toyama, F.; Maruo, F.; Chiba, C. Turning the fate of reprogramming cells from retinal disorder to regeneration by Pax6 in newts. Sci. Rep. 2016, 6, 33761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Islam, M.R.; Nakamura, K.; Casco-Robles, M.M.; Kunahong, A.; Inami, W.; Toyama, F.; Maruo, F.; Chiba, C. The newt reprograms mature RPE cells into a unique multipotent state for retinal regeneration. Sci. Rep. 2014, 4, 6043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maier, W.; Wolburg, H. Regeneration of the goldfish retina after exposure to different doses of ouabain. Cell Tissue Res. 1979, 202, 99–118. [Google Scholar] [CrossRef] [PubMed]
- Knight, J.K.; Raymond, P.A. Retinal pigmented epithelium does not transdifferentiate in adult goldfish. J. Neurobiol. 1995, 27, 447–456. [Google Scholar] [CrossRef] [Green Version]
- Coulombre, J.L.; Coulombre, A.J. Regeneration of neural retina from the pigmented epithelium in the chick embryo. Dev. Biol. 1965, 12, 79–92. [Google Scholar] [CrossRef]
- Zhao, S.; Thornquist, S.C.; Barnstable, C.J. In vitro transdifferentiation of embryonic rat retinal pigment epithelium to neural retina. Brain Res. 1995, 677, 300–310. [Google Scholar] [CrossRef]
- Moshiri, A.; Reh, T.A. Persistent progenitors at the retinal margin of ptc+/− mice. J. Neurosci. 2004, 24, 229–237. [Google Scholar] [CrossRef] [Green Version]
- Fischer, A.J.; Reh, T.A. Muller glia are a potential source of neural regeneration in the postnatal chicken retina. Nat. Neurosci. 2001, 4, 247–252. [Google Scholar] [CrossRef]
- Fausett, B.V.; Goldman, D. A role for alpha1 tubulin-expressing Muller glia in regeneration of the injured zebrafish retina. J. Neurosci. 2006, 26, 6303–6313. [Google Scholar] [CrossRef] [Green Version]
- Lahne, M.; Brecker, M.; Jones, S.E.; Hyde, D.R. The Regenerating Adult Zebrafish Retina Recapitulates Developmental Fate Specification Programs. Front. Cell Dev. Biol. 2020, 8, 617923. [Google Scholar] [CrossRef] [PubMed]
- Fuhrmann, S.; Zou, C.; Levine, E.M. Retinal pigment epithelium development, plasticity, and tissue homeostasis. Exp. Eye Res. 2014, 123, 141–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dvoriantchikova, G.; Seemungal, R.J.; Ivanov, D. The epigenetic basis for the impaired ability of adult murine retinal pigment epithelium cells to regenerate retinal tissue. Sci. Rep. 2019, 9, 3860. [Google Scholar] [CrossRef] [PubMed]
- Levine, E.M.; Close, J.; Fero, M.; Ostrovsky, A.; Reh, T.A. p27(Kip1) regulates cell cycle withdrawal of late multipotent progenitor cells in the mammalian retina. Dev. Biol. 2000, 219, 299–314. [Google Scholar] [CrossRef] [Green Version]
- Joly, S.; Pernet, V.; Samardzija, M.; Grimm, C. Pax6-positive Muller glia cells express cell cycle markers but do not proliferate after photoreceptor injury in the mouse retina. Glia 2011, 59, 1033–1046. [Google Scholar] [CrossRef] [PubMed]
- Beveridge, N.J.; Tooney, P.A.; Carroll, A.P.; Tran, N.; Cairns, M.J. Down-regulation of miR-17 family expression in response to retinoic acid induced neuronal differentiation. Cell. Signal. 2009, 21, 1837–1845. [Google Scholar] [CrossRef] [PubMed]
- Foshay, K.M.; Gallicano, G.I. miR-17 family miRNAs are expressed during early mammalian development and regulate stem cell differentiation. Dev. Biol. 2009, 326, 431–443. [Google Scholar] [CrossRef] [Green Version]
- Naka-Kaneda, H.; Nakamura, S.; Igarashi, M.; Aoi, H.; Kanki, H.; Tsuyama, J.; Tsutsumi, S.; Aburatani, H.; Shimazaki, T.; Okano, H. The miR-17/106-p38 axis is a key regulator of the neurogenic-to-gliogenic transition in developing neural stem/progenitor cells. Proc. Natl. Acad. Sci. USA 2014, 111, 1604–1609. [Google Scholar] [CrossRef] [Green Version]
- Trompeter, H.I.; Abbad, H.; Iwaniuk, K.M.; Hafner, M.; Renwick, N.; Tuschl, T.; Schira, J.; Muller, H.W.; Wernet, P. MicroRNAs MiR-17, MiR-20a, and MiR-106b act in concert to modulate E2F activity on cell cycle arrest during neuronal lineage differentiation of USSC. PLoS ONE 2011, 6, e16138. [Google Scholar] [CrossRef]
- Yang, Y.; Sun, B.; Huang, J.; Xu, L.; Pan, J.; Fang, C.; Li, M.; Li, G.; Tao, Y.; Yang, X.; et al. Up-regulation of miR-325-3p suppresses pineal aralkylamine N-acetyltransferase (Aanat) after neonatal hypoxia-ischemia brain injury in rats. Brain Res. 2017, 1668, 28–35. [Google Scholar] [CrossRef]
- Roesch, K.; Jadhav, A.P.; Trimarchi, J.M.; Stadler, M.B.; Roska, B.; Sun, B.B.; Cepko, C.L. The transcriptome of retinal Muller glial cells. J. Comp. Neurol. 2008, 509, 225–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karl, M.O.; Hayes, S.; Nelson, B.R.; Tan, K.; Buckingham, B.; Reh, T.A. Stimulation of neural regeneration in the mouse retina. Proc. Natl. Acad. Sci. USA 2008, 105, 19508–19513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ueki, Y.; Wilken, M.S.; Cox, K.E.; Chipman, L.; Jorstad, N.; Sternhagen, K.; Simic, M.; Ullom, K.; Nakafuku, M.; Reh, T.A. Transgenic expression of the proneural TF Ascl1 in Muller glia stimulates retinal regeneration in young mice. Proc. Natl. Acad. Sci. USA 2015, 112, 13717–13722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilken, M.S.; Reh, T.A. Retinal regeneration in birds and mice. Curr. Opin. Genet. Dev. 2016, 40, 57–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turkalj, B.; Quallich, D.; Bessert, D.A.; Kramer, A.C.; Cook, T.A.; Thummel, R. Development and characterization of a chronic photoreceptor degeneration model in adult zebrafish that does not trigger a regenerative response. Exp. Eye Res. 2021, 209, 108630. [Google Scholar] [CrossRef] [PubMed]
- Singh, H.P.; Wang, S.; Stachelek, K.; Lee, S.; Reid, M.W.; Thornton, M.E.; Craft, C.M.; Grubbs, B.H.; Cobrinik, D. Developmental stage-specific proliferation and retinoblastoma genesis in RB-deficient human but not mouse cone precursors. Proc. Natl. Acad. Sci. USA 2018, 115, E9391–E9400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aydin, B.; Kakumanu, A.; Rossillo, M.; Moreno-Estelles, M.; Garipler, G.; Ringstad, N.; Flames, N.; Mahony, S.; Mazzoni, E.O. Proneural factors Ascl1 and Neurog2 contribute to neuronal subtype identities by establishing distinct chromatin landscapes. Nat. Neurosci. 2019, 22, 897–908. [Google Scholar] [CrossRef] [PubMed]
- Yao, K.; Qiu, S.; Tian, L.; Snider, W.D.; Flannery, J.G.; Schaffer, D.V.; Chen, B. Wnt Regulates Proliferation and Neurogenic Potential of Muller Glial Cells via a Lin28/let-7 miRNA-Dependent Pathway in Adult Mammalian Retinas. Cell Rep. 2016, 17, 165–178. [Google Scholar] [CrossRef] [Green Version]
- Rueda, E.M.; Hall, B.M.; Hill, M.C.; Swinton, P.G.; Tong, X.; Martin, J.F.; Poche, R.A. The Hippo Pathway Blocks Mammalian Retinal Muller Glial Cell Reprogramming. Cell Rep. 2019, 27, 1637–1649.e6. [Google Scholar] [CrossRef] [Green Version]
- Todd, L.; Hooper, M.J.; Haugan, A.K.; Finkbeiner, C.; Jorstad, N.; Radulovich, N.; Wong, C.K.; Donaldson, P.C.; Jenkins, W.; Chen, Q.; et al. Efficient stimulation of retinal regeneration from Muller glia in adult mice using combinations of proneural bHLH TFs. Cell Rep. 2021, 37, 109857. [Google Scholar] [CrossRef]
- Lu, Y.; Brommer, B.; Tian, X.; Krishnan, A.; Meer, M.; Wang, C.; Vera, D.L.; Zeng, Q.; Yu, D.; Bonkowski, M.S.; et al. Reprogramming to recover youthful epigenetic information and restore vision. Nature 2020, 588, 124–129. [Google Scholar] [CrossRef] [PubMed]
- Heinz, S.; Benner, C.; Spann, N.; Bertolino, E.; Lin, Y.C.; Laslo, P.; Cheng, J.X.; Murre, C.; Singh, H.; Glass, C.K. Simple combinations of lineage-determining TFs prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 2010, 38, 576–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berger, M.F.; Bulyk, M.L. Protein binding microarrays (PBMs) for rapid, high-throughput characterization of the sequence specificities of DNA binding proteins. Methods Mol. Biol. 2006, 338, 245–260. [Google Scholar]
- Tuerk, C.; Gold, L. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science 1990, 249, 505–510. [Google Scholar] [CrossRef] [PubMed]
- O’Malley, R.C.; Huang, S.C.; Song, L.; Lewsey, M.G.; Bartlett, A.; Nery, J.R.; Galli, M.; Gallavotti, A.; Ecker, J.R. Cistrome and Epicistrome Features Shape the Regulatory DNA Landscape. Cell 2016, 166, 1598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartlett, A.; O’Malley, R.C.; Huang, S.C.; Galli, M.; Nery, J.R.; Gallavotti, A.; Ecker, J.R. Mapping genome-wide transcription-factor binding sites using DAP-seq. Nat. Protoc. 2017, 12, 1659–1672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baumgart, L.A.; Lee, J.E.; Salamov, A.; Dilworth, D.J.; Na, H.; Mingay, M.; Blow, M.J.; Zhang, Y.; Yoshinaga, Y.; Daum, C.G.; et al. Persistence and plasticity in bacterial gene regulation. Nat. Methods 2021, 18, 1499–1505. [Google Scholar] [CrossRef] [PubMed]
- Orenstein, Y.; Shamir, R. A comparative analysis of TF binding models learned from PBM, HT-SELEX and ChIP data. Nucleic Acids Res. 2014, 42, e63. [Google Scholar] [CrossRef] [Green Version]
- van Helden, J. Regulatory sequence analysis tools. Nucleic Acids Res. 2003, 31, 3593–3596. [Google Scholar] [CrossRef] [Green Version]
- Matys, V.; Kel-Margoulis, O.V.; Fricke, E.; Liebich, I.; Land, S.; Barre-Dirrie, A.; Reuter, I.; Chekmenev, D.; Krull, M.; Hornischer, K.; et al. TRANSFAC and its module TRANSCompel: Transcriptional gene regulation in eukaryotes. Nucleic Acids Res. 2006, 34, D108–D110. [Google Scholar] [CrossRef] [Green Version]
- Mathelier, A.; Fornes, O.; Arenillas, D.J.; Chen, C.Y.; Denay, G.; Lee, J.; Shi, W.; Shyr, C.; Tan, G.; Worsley-Hunt, R.; et al. JASPAR 2016: A major expansion and update of the open-access database of TF binding profiles. Nucleic Acids Res. 2016, 44, D110–D115. [Google Scholar] [CrossRef] [Green Version]
- Kulakovskiy, I.V.; Vorontsov, I.E.; Yevshin, I.S.; Sharipov, R.N.; Fedorova, A.D.; Rumynskiy, E.I.; Medvedeva, Y.A.; Magana-Mora, A.; Bajic, V.B.; Papatsenko, D.A.; et al. HOCOMOCO: Towards a complete collection of TF binding models for human and mouse via large-scale ChIP-Seq analysis. Nucleic Acids Res. 2018, 46, D252–D259. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.; Fornes, O.; Stigliani, A.; Gheorghe, M.; Castro-Mondragon, J.A.; van der Lee, R.; Bessy, A.; Cheneby, J.; Kulkarni, S.R.; Tan, G.; et al. JASPAR 2018: Update of the open-access data-base of transcription factor binding profiles and its web framework. Nucleic Acids Res. 2018, 46, D260–D266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castro-Mondragon, J.A.; Riudavets-Puig, R.; Rauluseviciute, I.; Berhanu Lemma, R.; Turchi, L.; Blanc-Mathieu, R.; Lucas, J.; Boddie, P.; Khan, A.; Manosalva Perez, N.; et al. JASPAR 2022: The 9th release of the open-access database of TF binding profiles. Nucleic Acids Res. 2022, 50, D165–D173. [Google Scholar] [CrossRef] [PubMed]
- Desplan, C.; Theis, J.; O’Farrell, P.H. The Drosophila developmental gene, engrailed, encodes a sequence-specific DNA binding activity. Nature 1985, 318, 630–635. [Google Scholar] [CrossRef] [Green Version]
- Treisman, J.; Gonczy, P.; Vashishtha, M.; Harris, E.; Desplan, C. A single amino acid can determine the DNA binding specificity of homeodomain proteins. Cell 1989, 59, 553–562. [Google Scholar] [CrossRef]
- Vermunt, M.W.; Tan, S.C.; Castelijns, B.; Geeven, G.; Reinink, P.; de Bruijn, E.; Kondova, I.; Persengiev, S.; Netherlands Brain, B.; Bontrop, R.; et al. Epigenomic annotation of gene regulatory alterations during evolution of the primate brain. Nat. Neurosci. 2016, 19, 494–503. [Google Scholar] [CrossRef] [PubMed]
- Fish, A.; Chen, L.; Capra, J.A. Gene Regulatory Enhancers with Evolutionarily Conserved Activity Are More Pleiotropic than Those with Species-Specific Activity. Genome Biol. Evol. 2017, 9, 2615–2625. [Google Scholar] [CrossRef] [Green Version]
- Maurano, M.T.; Humbert, R.; Rynes, E.; Thurman, R.E.; Haugen, E.; Wang, H.; Reynolds, A.P.; Sandstrom, R.; Qu, H.; Brody, J.; et al. Systematic localization of common disease-associated variation in regulatory DNA. Science 2012, 337, 1190–1195. [Google Scholar] [CrossRef] [Green Version]
- Leslie, R.; O’Donnell, C.J.; Johnson, A.D. GRASP: Analysis of genotype-phenotype results from 1390 genome-wide association studies and corresponding open access database. Bioinformatics 2014, 30, i185–i194. [Google Scholar] [CrossRef] [Green Version]
- Soldner, F.; Stelzer, Y.; Shivalila, C.S.; Abraham, B.J.; Latourelle, J.C.; Barrasa, M.I.; Goldmann, J.; Myers, R.H.; Young, R.A.; Jaenisch, R. Parkinson-associated risk variant in distal enhancer of alpha-synuclein modulates target gene expression. Nature 2016, 533, 95–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castelijns, B.; Baak, M.L.; Geeven, G.; Vermunt, M.W.; Wiggers, C.R.M.; Timpanaro, I.S.; Kondova, I.; de Laat, W.; Creyghton, M.P. Recently Evolved Enhancers Emerge with High Interindividual Variability and Less Frequently Associate with Disease. Cell Rep. 2020, 31, 107799. [Google Scholar] [CrossRef] [PubMed]
- Villar, D.; Flicek, P.; Odom, D.T. Evolution of TF binding in metazoans—Mechanisms and functional implications. Nat. Rev. Genet. 2014, 15, 221–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villar, D.; Berthelot, C.; Aldridge, S.; Rayner, T.F.; Lukk, M.; Pignatelli, M.; Park, T.J.; Deaville, R.; Erichsen, J.T.; Jasinska, A.J.; et al. Enhancer evolution across 20 mammalian species. Cell 2015, 160, 554–566. [Google Scholar] [CrossRef] [Green Version]
- Pennacchio, L.A.; Bickmore, W.; Dean, A.; Nobrega, M.A.; Bejerano, G. Enhancers: Five essential questions. Nat. Rev. Genet. 2013, 14, 288–295. [Google Scholar] [CrossRef]
- Osterwalder, M.; Barozzi, I.; Tissieres, V.; Fukuda-Yuzawa, Y.; Mannion, B.J.; Afzal, S.Y.; Lee, E.A.; Zhu, Y.; Plajzer-Frick, I.; Pickle, C.S.; et al. Enhancer redundancy provides phenotypic robustness in mammalian development. Nature 2018, 554, 239–243. [Google Scholar] [CrossRef] [Green Version]
- Carelli, F.N.; Liechti, A.; Halbert, J.; Warnefors, M.; Kaessmann, H. Repurposing of promoters and enhancers during mammalian evolution. Nat. Commun. 2018, 9, 4066. [Google Scholar] [CrossRef] [Green Version]
- Gupta, S.; Stamatoyannopoulos, J.A.; Bailey, T.L.; Noble, W.S. Quantifying similarity between motifs. Genome Biol. 2007, 8, R24. [Google Scholar] [CrossRef] [Green Version]
- Castro-Mondragon, J.A.; Jaeger, S.; Thieffry, D.; Thomas-Chollier, M.; van Helden, J. RSAT matrix-clustering: Dynamic exploration and redundancy reduction of TF binding motif collections. Nucleic Acids Res. 2017, 45, e119. [Google Scholar] [CrossRef]
- La Manno, G.; Soldatov, R.; Zeisel, A.; Braun, E.; Hochgerner, H.; Petukhov, V.; Lidschreiber, K.; Kastriti, M.E.; Lonnerberg, P.; Furlan, A.; et al. RNA velocity of single cells. Nature 2018, 560, 494–498. [Google Scholar] [CrossRef] [Green Version]
- Bergen, V.; Lange, M.; Peidli, S.; Wolf, F.A.; Theis, F.J. Generalizing RNA velocity to transient cell states through dynamical modeling. Nat. Biotechnol. 2020, 38, 1408–1414. [Google Scholar] [CrossRef] [PubMed]
- Tedesco, M.; Giannese, F.; Lazarevic, D.; Giansanti, V.; Rosano, D.; Monzani, S.; Catalano, I.; Grassi, E.; Zanella, E.R.; Botrugno, O.A.; et al. Chromatin Velocity reveals epigenetic dynamics by single-cell profiling of heterochromatin and euchromatin. Nat. Biotechnol. 2021, 40, 235–244. [Google Scholar] [CrossRef] [PubMed]
- Worley, B.; Powers, R. A Sequential Algorithm for Multiblock Orthogonal Projections to Latent Structures. Chemom. Intell. Lab. Syst. 2015, 149, 33–39. [Google Scholar] [CrossRef] [Green Version]
- Zimmerman, K.D.; Espeland, M.A.; Langefeld, C.D. A practical solution to pseudoreplication bias in single-cell studies. Nat. Commun. 2021, 12, 738. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Sheng, Q.; Ping, J.; Ramirez, M.A.; Lau, K.S.; Coffey, R.J.; Shyr, Y. scRNABatchQC: Multi-samples quality control for single cell RNA-seq data. Bioinformatics 2019, 35, 5306–5308. [Google Scholar] [CrossRef] [PubMed]
- Hashimshony, T.; Senderovich, N.; Avital, G.; Klochendler, A.; de Leeuw, Y.; Anavy, L.; Gennert, D.; Li, S.; Livak, K.J.; Rozenblatt-Rosen, O.; et al. CEL-Seq2: Sensitive highly-multiplexed single-cell RNA-Seq. Genome Biol. 2016, 17, 77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finak, G.; McDavid, A.; Yajima, M.; Deng, J.; Gersuk, V.; Shalek, A.K.; Slichter, C.K.; Miller, H.W.; McElrath, M.J.; Prlic, M.; et al. MAST: A flexible statistical framework for assessing transcriptional changes and characterizing heterogeneity in single-cell RNA sequencing data. Genome Biol. 2015, 16, 278. [Google Scholar] [CrossRef] [Green Version]
- Raj, A.; van Oudenaarden, A. Nature, nurture, or chance: Stochastic gene expression and its consequences. Cell 2008, 135, 216–226. [Google Scholar] [CrossRef] [Green Version]
- Van den Berge, K.; Perraudeau, F.; Soneson, C.; Love, M.I.; Risso, D.; Vert, J.P.; Robinson, M.D.; Dudoit, S.; Clement, L. Observation weights unlock bulk RNA-seq tools for zero inflation and single-cell applications. Genome Biol. 2018, 19, 24. [Google Scholar] [CrossRef] [Green Version]
- Pierson, E.; Yau, C. ZIFA: Dimensionality reduction for zero-inflated single-cell gene expression analysis. Genome Biol. 2015, 16, 241. [Google Scholar] [CrossRef] [Green Version]
- Lin, P.; Troup, M.; Ho, J.W. CIDR: Ultrafast and accurate clustering through imputation for single-cell RNA-seq data. Genome Biol. 2017, 18, 59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Svensson, V. Droplet scRNA-seq is not zero-inflated. Nat. Biotechnol. 2020, 38, 147–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lun, A.T.; Bach, K.; Marioni, J.C. Pooling across cells to normalize single-cell RNA sequencing data with many zero counts. Genome Biol. 2016, 17, 75. [Google Scholar] [CrossRef] [PubMed]
- Lun, A.T.L.; Marioni, J.C. Overcoming confounding plate effects in differential expression analyses of single-cell RNA-seq data. Biostatistics 2017, 18, 451–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crowell, H.L.; Soneson, C.; Germain, P.L.; Calini, D.; Collin, L.; Raposo, C.; Malhotra, D.; Robinson, M.D. muscat detects subpopulation-specific state transitions from multi-sample multi-condition single-cell transcriptomics data. Nat. Commun. 2020, 11, 6077. [Google Scholar] [CrossRef] [PubMed]
- Cusanovich, D.A.; Hill, A.J.; Aghamirzaie, D.; Daza, R.M.; Pliner, H.A.; Berletch, J.B.; Filippova, G.N.; Huang, X.; Christiansen, L.; DeWitt, W.S.; et al. A Single-Cell Atlas of In Vivo Mammalian Chromatin Accessibility. Cell 2018, 174, 1309–1324.e18. [Google Scholar] [CrossRef] [Green Version]
- Swanson, E.; Lord, C.; Reading, J.; Heubeck, A.T.; Genge, P.C.; Thomson, Z.; Weiss, M.D.; Li, X.J.; Savage, A.K.; Green, R.R.; et al. Simultaneous trimodal single-cell measurement of transcripts, epitopes, and chromatin accessibility using TEA-seq. eLife 2021, 10, e63632. [Google Scholar] [CrossRef]
- Fang, R.; Preissl, S.; Li, Y.; Hou, X.; Lucero, J.; Wang, X.; Motamedi, A.; Shiau, A.K.; Zhou, X.; Xie, F.; et al. Comprehensive analysis of single cell ATAC-seq data with SnapATAC. Nat. Commun. 2021, 12, 1337. [Google Scholar] [CrossRef]
- Kiselev, V.Y.; Andrews, T.S.; Hemberg, M. Challenges in unsupervised clustering of single-cell RNA-seq data. Nat. Rev. Genet. 2019, 20, 273–282. [Google Scholar] [CrossRef]
- McInnes, L.; Healy, J.; Melville, J. Umap: Uniform manifold approximation and projection for dimension reduction. arXiv 2018, arXiv:1802.03426. Available online: https://arxiv.org/abs/1802.03426 (accessed on 13 January 2021).
- Becht, E.; McInnes, L.; Healy, J.; Dutertre, C.A.; Kwok, I.W.H.; Ng, L.G.; Ginhoux, F.; Newell, E.W. Dimensionality reduction for visualizing single-cell data using UMAP. Nat. Biotechnol. 2018, 37, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Lareau, C.; Andreani, T.; Vinyard, M.E.; Garcia, S.P.; Clement, K.; Andrade-Navarro, M.A.; Buenrostro, J.D.; Pinello, L. Assessment of computational methods for the analysis of single-cell ATAC-seq data. Genome Biol. 2019, 20, 241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiang, R.; Wang, W.; Yang, L.; Wang, S.; Xu, C.; Chen, X. A Comparison for Dimensionality Reduction Methods of Single-Cell RNA-seq Data. Front. Genet. 2021, 12, 646936. [Google Scholar] [CrossRef] [PubMed]
- Linderman, G.C. Dimensionality Reduction of Single-Cell RNA-Seq Data. Methods Mol. Biol. 2021, 2284, 331–342. [Google Scholar]
- Ma, K.Y.; Schonnesen, A.A.; Brock, A.; Van Den Berg, C.; Eckhardt, S.G.; Liu, Z.; Jiang, N. Single-cell RNA sequencing of lung adenocarcinoma reveals heterogeneity of immune response-related genes. JCI Insight 2019, 4, e121387. [Google Scholar] [CrossRef]
- Mendoza-Parra, M.A.; Van Gool, W.; Mohamed Saleem, M.A.; Ceschin, D.G.; Gronemeyer, H. A quality control system for profiles obtained by ChIP sequencing. Nucleic Acids Res. 2013, 41, e196. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Brown, J.B.; Huang, H.; Bickel, P.J. Measuring reproducibility of high-throughput experiments. Ann. Appl. Stat. 2011, 5, 1752–1779. [Google Scholar] [CrossRef]
- Thanawala, S.U.; Rister, J.; Goldberg, G.W.; Zuskov, A.; Olesnicky, E.C.; Flowers, J.M.; Jukam, D.; Purugganan, M.D.; Gavis, E.R.; Desplan, C.; et al. Regional modulation of a stochastically expressed factor determines photoreceptor subtypes in the Drosophila retina. Dev. Cell 2013, 25, 93–105. [Google Scholar] [CrossRef] [Green Version]
- Martin-Gayo, E.; Cole, M.B.; Kolb, K.E.; Ouyang, Z.; Cronin, J.; Kazer, S.W.; Ordovas-Montanes, J.; Lichterfeld, M.; Walker, B.D.; Yosef, N.; et al. A Reproducibility-Based Computational Framework Identifies an Inducible, Enhanced Antiviral State in Dendritic Cells from HIV-1 Elite Controllers. Genome Biol. 2018, 19, 10. [Google Scholar] [CrossRef]
- Karabacak Calviello, A.; Hirsekorn, A.; Wurmus, R.; Yusuf, D.; Ohler, U. Reproducible inference of TF footprints in ATAC-seq and DNase-seq datasets using protocol-specific bias modeling. Genome Biol. 2019, 20, 42. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, Y.; Zhu, X.; Purmann, C.; Haney, M.S.; Ward, T.; Khechaduri, A.; Yao, J.; Weissman, S.M.; Urban, A.E. Local and global chromatin interactions are altered by large genomic deletions associated with human brain development. Nat. Commun. 2018, 9, 5356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spielmann, M.; Lupianez, D.G.; Mundlos, S. Structural variation in the 3D genome. Nat. Rev. Genet. 2018, 19, 453–467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, H.; Xie, W. The role of 3D genome organization in development and cell differentiation. Nat. Rev. Mol. Cell Biol. 2019, 20, 535–550. [Google Scholar] [CrossRef] [PubMed]
- Mateo, L.J.; Murphy, S.E.; Hafner, A.; Cinquini, I.S.; Walker, C.A.; Boettiger, A.N. Visualizing DNA folding and RNA in embryos at single-cell resolution. Nature 2019, 568, 49–54. [Google Scholar] [CrossRef]
- Takei, Y.; Shah, S.; Harvey, S.; Qi, L.S.; Cai, L. Multiplexed Dynamic Imaging of Genomic Loci by Combined CRISPR Imaging and DNA Sequential FISH. Biophys. J. 2017, 112, 1773–1776. [Google Scholar] [CrossRef]
- Boettiger, A.; Murphy, S. Advances in Chromatin Imaging at Kilobase-Scale Resolution. Trends Genet. 2020, 36, 273–287. [Google Scholar] [CrossRef] [Green Version]
- Takei, Y.; Zheng, S.; Yun, J.; Shah, S.; Pierson, N.; White, J.; Schindler, S.; Tischbirek, C.H.; Yuan, G.C.; Cai, L. Single-cell nuclear architecture across cell types in the mouse brain. Science 2021, 374, 586–594. [Google Scholar] [CrossRef]
- Betzig, E.; Patterson, G.H.; Sougrat, R.; Lindwasser, O.W.; Olenych, S.; Bonifacino, J.S.; Davidson, M.W.; Lippincott-Schwartz, J.; Hess, H.F. Imaging intracellular fluorescent proteins at nanometer resolution. Science 2006, 313, 1642–1645. [Google Scholar] [CrossRef] [Green Version]
- Beliveau, B.J.; Boettiger, A.N.; Nir, G.; Bintu, B.; Yin, P.; Zhuang, X.; Wu, C.T. In Situ Super-Resolution Imaging of Genomic DNA with OligoSTORM and OligoDNA-PAINT. Methods Mol. Biol. 2017, 1663, 231–252. [Google Scholar]
- Beliveau, B.J.; Joyce, E.F.; Apostolopoulos, N.; Yilmaz, F.; Fonseka, C.Y.; McCole, R.B.; Chang, Y.; Li, J.B.; Senaratne, T.N.; Williams, B.R.; et al. Versatile design and synthesis platform for visualizing genomes with Oligopaint FISH probes. Proc. Natl. Acad. Sci. USA 2012, 109, 21301–21306. [Google Scholar] [CrossRef] [Green Version]
- Jungmann, R.; Avendano, M.S.; Woehrstein, J.B.; Dai, M.; Shih, W.M.; Yin, P. Multiplexed 3D cellular super-resolution imaging with DNA-PAINT and Exchange-PAINT. Nat. Methods 2014, 11, 313–318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rust, M.J.; Bates, M.; Zhuang, X. Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM). Nat. Methods 2006, 3, 793–796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, B.; Wang, W.; Bates, M.; Zhuang, X. Three-dimensional super-resolution imaging by stochastic optical reconstruction microscopy. Science 2008, 319, 810–813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, M.; Ren, B. The Three-Dimensional Organization of Mammalian Genomes. Annu. Rev. Cell Dev. Biol. 2017, 33, 265–289. [Google Scholar] [CrossRef]
- Cavalli, G.; Heard, E. Advances in epigenetics link genetics to the environment and disease. Nature 2019, 571, 489–499. [Google Scholar] [CrossRef] [Green Version]
- Bintu, B.; Mateo, L.J.; Su, J.H.; Sinnott-Armstrong, N.A.; Parker, M.; Kinrot, S.; Yamaya, K.; Boettiger, A.N.; Zhuang, X. Super-resolution chromatin tracing reveals domains and cooperative interactions in single cells. Science 2018, 362, eaau1783. [Google Scholar] [CrossRef] [Green Version]
- Barth, R.; Fourel, G.; Shaban, H.A. Dynamics as a cause for the nanoscale organization of the genome. Nucleus 2020, 11, 83–98. [Google Scholar] [CrossRef]
- Su, J.H.; Zheng, P.; Kinrot, S.S.; Bintu, B.; Zhuang, X. Genome-Scale Imaging of the 3D Organization and Transcriptional Activity of Chromatin. Cell 2020, 182, 1641–1659.e26. [Google Scholar] [CrossRef]
- Liu, Y.; Yang, M.; Deng, Y.; Su, G.; Enninful, A.; Guo, C.C.; Tebaldi, T.; Zhang, D.; Kim, D.; Bai, Z.; et al. High-Spatial-Resolution Multi-Omics Sequencing via Deterministic Barcoding in Tissue. Cell 2020, 183, 1665–1681.e18. [Google Scholar] [CrossRef]
- Stevens, T.J.; Lando, D.; Basu, S.; Atkinson, L.P.; Cao, Y.; Lee, S.F.; Leeb, M.; Wohlfahrt, K.J.; Boucher, W.; O’Shaughnessy-Kirwan, A.; et al. 3D structures of individual mammalian genomes studied by single-cell Hi-C. Nature 2017, 544, 59–64. [Google Scholar] [CrossRef] [Green Version]
- Nagano, T.; Lubling, Y.; Varnai, C.; Dudley, C.; Leung, W.; Baran, Y.; Mendelson Cohen, N.; Wingett, S.; Fraser, P.; Tanay, A. Cell-cycle dynamics of chromosomal organization at single-cell resolution. Nature 2017, 547, 61–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flyamer, I.M.; Gassler, J.; Imakaev, M.; Brandao, H.B.; Ulianov, S.V.; Abdennur, N.; Razin, S.V.; Mirny, L.A.; Tachibana-Konwalski, K. Single-nucleus Hi-C reveals unique chromatin reorganization at oocyte-to-zygote transition. Nature 2017, 544, 110–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, L.; Xing, D.; Chang, C.H.; Li, H.; Xie, X.S. Three-dimensional genome structures of single diploid human cells. Science 2018, 361, 924–928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ou, H.D.; Phan, S.; Deerinck, T.J.; Thor, A.; Ellisman, M.H.; O’Shea, C.C. ChromEMT: Visualizing 3D chromatin structure and compaction in interphase and mitotic cells. Science 2017, 357. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.V.; Corces, V.G. Seeing Is Believing: ORCA Allows Visualization of Three-Dimensional Genome Organization at Single-Cell Resolution. Biochemistry 2019, 58, 3477–3479. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Su, J.H.; Beliveau, B.J.; Bintu, B.; Moffitt, J.R.; Wu, C.T.; Zhuang, X. Spatial organization of chromatin domains and compartments in single chromosomes. Science 2016, 353, 598–602. [Google Scholar] [CrossRef] [Green Version]
- Nir, G.; Farabella, I.; Perez Estrada, C.; Ebeling, C.G.; Beliveau, B.J.; Sasaki, H.M.; Lee, S.D.; Nguyen, S.C.; McCole, R.B.; Chattoraj, S.; et al. Walking along chromosomes with super-resolution imaging, contact maps, and integrative modeling. PLoS Genet. 2018, 14, e1007872. [Google Scholar] [CrossRef] [Green Version]
- Boettiger, A.N.; Bintu, B.; Moffitt, J.R.; Wang, S.; Beliveau, B.J.; Fudenberg, G.; Imakaev, M.; Mirny, L.A.; Wu, C.T.; Zhuang, X. Super-resolution imaging reveals distinct chromatin folding for different epigenetic states. Nature 2016, 529, 418–422. [Google Scholar] [CrossRef] [Green Version]
- Kundu, S.; Ji, F.; Sunwoo, H.; Jain, G.; Lee, J.T.; Sadreyev, R.I.; Dekker, J.; Kingston, R.E. Polycomb Repressive Complex 1 Generates Discrete Compacted Domains that Change during Differentiation. Mol. Cell 2017, 65, 432–446.e5. [Google Scholar] [CrossRef] [Green Version]
- Tasic, B.; Menon, V.; Nguyen, T.N.; Kim, T.K.; Jarsky, T.; Yao, Z.; Levi, B.; Gray, L.T.; Sorensen, S.A.; Dolbeare, T.; et al. Adult mouse cortical cell taxonomy revealed by single cell transcriptomics. Nat. Neurosci. 2016, 19, 335–346. [Google Scholar] [CrossRef] [Green Version]
- Payne, A.C.; Chiang, Z.D.; Reginato, P.L.; Mangiameli, S.M.; Murray, E.M.; Yao, C.C.; Markoulaki, S.; Earl, A.S.; Labade, A.S.; Jaenisch, R.; et al. In situ genome sequencing resolves DNA sequence and structure in intact biological samples. Science 2021, 371. [Google Scholar] [CrossRef] [PubMed]
- Cao, B.; Coelho, S.; Li, J.; Wang, G.; Pertsinidis, A. Volumetric interferometric lattice light-sheet imaging. Nat. Biotechnol. 2021, 39, 1385–1393. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Eshein, A.; Virk, R.K.A.; Eid, A.; Wu, W.; Frederick, J.; VanDerway, D.; Gladstein, S.; Huang, K.; Shim, A.R.; et al. Nanoscale chromatin imaging and analysis platform bridges 4D chromatin organization with molecular function. Sci. Adv. 2021, 7. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.P.; Tirosh, I.; Trombetta, J.J.; Shalek, A.K.; Gillespie, S.M.; Wakimoto, H.; Cahill, D.P.; Nahed, B.V.; Curry, W.T.; Martuza, R.L.; et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 2014, 344, 1396–1401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Usoskin, D.; Furlan, A.; Islam, S.; Abdo, H.; Lonnerberg, P.; Lou, D.; Hjerling-Leffler, J.; Haeggstrom, J.; Kharchenko, O.; Kharchenko, P.V.; et al. Unbiased classification of sensory neuron types by large-scale single-cell RNA sequencing. Nat. Neurosci. 2015, 18, 145–153. [Google Scholar] [CrossRef]
- Buettner, F.; Natarajan, K.N.; Casale, F.P.; Proserpio, V.; Scialdone, A.; Theis, F.J.; Teichmann, S.A.; Marioni, J.C.; Stegle, O. Computational analysis of cell-to-cell heterogeneity in single-cell RNA-sequencing data reveals hidden subpopulations of cells. Nat. Biotechnol. 2015, 33, 155–160. [Google Scholar] [CrossRef]
- Kolodziejczyk, A.A.; Kim, J.K.; Tsang, J.C.; Ilicic, T.; Henriksson, J.; Natarajan, K.N.; Tuck, A.C.; Gao, X.; Buhler, M.; Liu, P.; et al. Single Cell RNA-Sequencing of Pluripotent States Unlocks Modular Transcriptional Variation. Cell Stem Cell 2015, 17, 471–485. [Google Scholar] [CrossRef] [Green Version]
- Nowakowski, T.J.; Bhaduri, A.; Pollen, A.A.; Alvarado, B.; Mostajo-Radji, M.A.; Di Lullo, E.; Haeussler, M.; Sandoval-Espinosa, C.; Liu, S.J.; Velmeshev, D.; et al. Spatiotemporal gene expression trajectories reveal developmental hierarchies of the human cortex. Science 2017, 358, 1318–1323. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Dong, J.; Yan, L.; Yong, J.; Liu, X.; Hu, Y.; Fan, X.; Wu, X.; Guo, H.; Wang, X.; et al. Single-Cell RNA-Seq Analysis Maps Development of Human Germline Cells and Gonadal Niche Interactions. Cell Stem Cell 2017, 20, 891–892. [Google Scholar] [CrossRef] [Green Version]
- Mayer, C.; Hafemeister, C.; Bandler, R.C.; Machold, R.; Batista Brito, R.; Jaglin, X.; Allaway, K.; Butler, A.; Fishell, G.; Satija, R. Developmental diversification of cortical inhibitory interneurons. Nature 2018, 555, 457–462. [Google Scholar] [CrossRef]
- Zhong, S.; Zhang, S.; Fan, X.; Wu, Q.; Yan, L.; Dong, J.; Zhang, H.; Li, L.; Sun, L.; Pan, N.; et al. A single-cell RNA-seq survey of the developmental landscape of the human prefrontal cortex. Nature 2018, 555, 524–528. [Google Scholar] [CrossRef] [PubMed]
- Polioudakis, D.; de la Torre-Ubieta, L.; Langerman, J.; Elkins, A.G.; Shi, X.; Stein, J.L.; Vuong, C.K.; Nichterwitz, S.; Gevorgian, M.; Opland, C.K.; et al. A Single-Cell Transcriptomic Atlas of Human Neocortical Development during Mid-gestation. Neuron 2019, 103, 785–801.e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Setty, M.; Kiseliovas, V.; Levine, J.; Gayoso, A.; Mazutis, L.; Pe’er, D. Characterization of cell fate probabilities in single-cell data with Palantir. Nat. Biotechnol. 2019, 37, 451–460. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Hocker, J.D.; Miller, M.; Hou, X.; Chiou, J.; Poirion, O.B.; Qiu, Y.; Li, Y.E.; Gaulton, K.J.; Wang, A.; et al. A single-cell atlas of chromatin accessibility in the human genome. Cell 2021, 184, 5985–6001.e19. [Google Scholar] [CrossRef]
- Zibetti, C.; Liu, S.; Wan, J.; Qian, J.; Blackshaw, S. Lhx2 regulates temporal changes in chromatin accessibility and transcription factor binding in retinal progenitor cells. bioRxiv 2017. [Google Scholar] [CrossRef] [Green Version]
- Steiner, F.A.; Talbert, P.B.; Kasinathan, S.; Deal, R.B.; Henikoff, S. Cell-type-specific nuclei purification from whole animals for genome-wide expression and chromatin profiling. Genome Res. 2012, 22, 766–777. [Google Scholar] [CrossRef] [Green Version]
- Schreiber, J.; Bilmes, J.; Noble, W.S. Prioritizing transcriptomic and epigenomic experiments using an optimization strategy that leverages imputed data. Bioinformatics 2021, 37, 439–447. [Google Scholar] [CrossRef]
- Wahls, W.P. The NIH must reduce disparities in funding to maximize its return on investments from taxpayers. eLife 2018, 7, e34965. [Google Scholar] [CrossRef]
- Barnett, A.G.; Clarke, P.; Vaquette, C.; Graves, N. Using democracy to award research funding: An observational study. Res. Integr. Peer Rev. 2017, 2, 16. [Google Scholar] [CrossRef] [Green Version]
- Gasparyan, A.Y.; Yessirkepov, M.; Voronov, A.A.; Gerasimov, A.N.; Kostyukova, E.I.; Kitas, G.D. Preserving the Integrity of Citations and References by All Stakeholders of Science Communication. J. Korean Med. Sci. 2015, 30, 1545–1552. [Google Scholar] [CrossRef] [Green Version]
- Dworkin, J.D.; Linn, K.A.; Teich, E.G.; Zurn, P.; Shinohara, R.T.; Bassett, D.S. The extent and drivers of gender imbalance in neuroscience reference lists. Nat. Neurosci. 2020, 23, 918–926. [Google Scholar] [CrossRef] [PubMed]
- Zurn, P.; Bassett, D.S.; Rust, N.C. The Citation Diversity Statement: A Practice of Transparency, A Way of Life. Trends Cogn. Sci. 2020, 24, 669–672. [Google Scholar] [CrossRef] [PubMed]
- Scanff, A.; Naudet, F.; Cristea, I.A.; Moher, D.; Bishop, D.V.M.; Locher, C. A survey of biomedical journals to detect editorial bias and nepotistic behavior. PLoS Biol. 2021, 19, e3001133. [Google Scholar] [CrossRef]
- Tiokhin, L.; Panchanathan, K.; Lakens, D.; Vazire, S.; Morgan, T.; Zollman, K. Honest signaling in academic publishing. PLoS ONE 2021, 16, e0246675. [Google Scholar] [CrossRef] [PubMed]
- McConnell, S.C.; Westerman, E.L.; Pierre, J.F.; Heckler, E.J.; Schwartz, N.B. United States National Postdoc Survey results and the interaction of gender, career choice and mentor impact. eLife 2018, 7, e40189. [Google Scholar] [CrossRef] [PubMed]
- Davis, S.M.; Singh, H.; Weismann, C.M.; Bankston, A.; Ruiz Villalobos, J.P. Actionable recommendations from trainees to improve science training. eLife 2020, 9, e59806. [Google Scholar]
- National Academies of Sciences, Engineering, and Medicine. The Science of Effective Mentorship in STEMM.; Byars-Winston, A., Dahlberg, M.L., Eds.; The National Academies Press: Washington, DC, USA, 2019; 306p. [Google Scholar]
- Barres, B.A. Stop blocking postdocs’ paths to success. Nature 2017, 548, 517–519. [Google Scholar] [CrossRef] [Green Version]
- Kaptein, M. Developing and testing a measure for the ethical culture of organizations: The corporate ethical virtues model. J. Organ. Behav. 2008, 29, 923–947. [Google Scholar] [CrossRef] [Green Version]
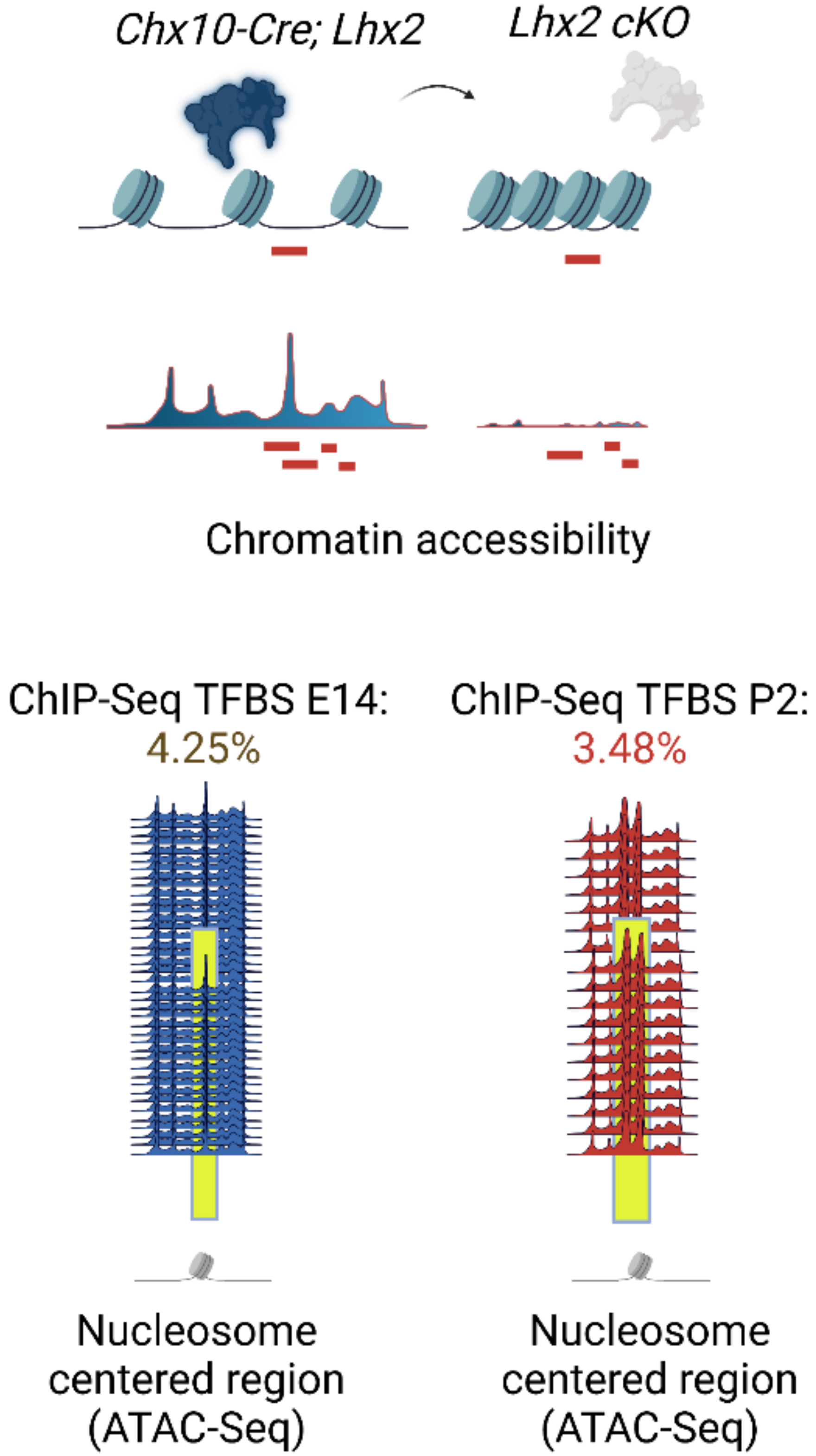
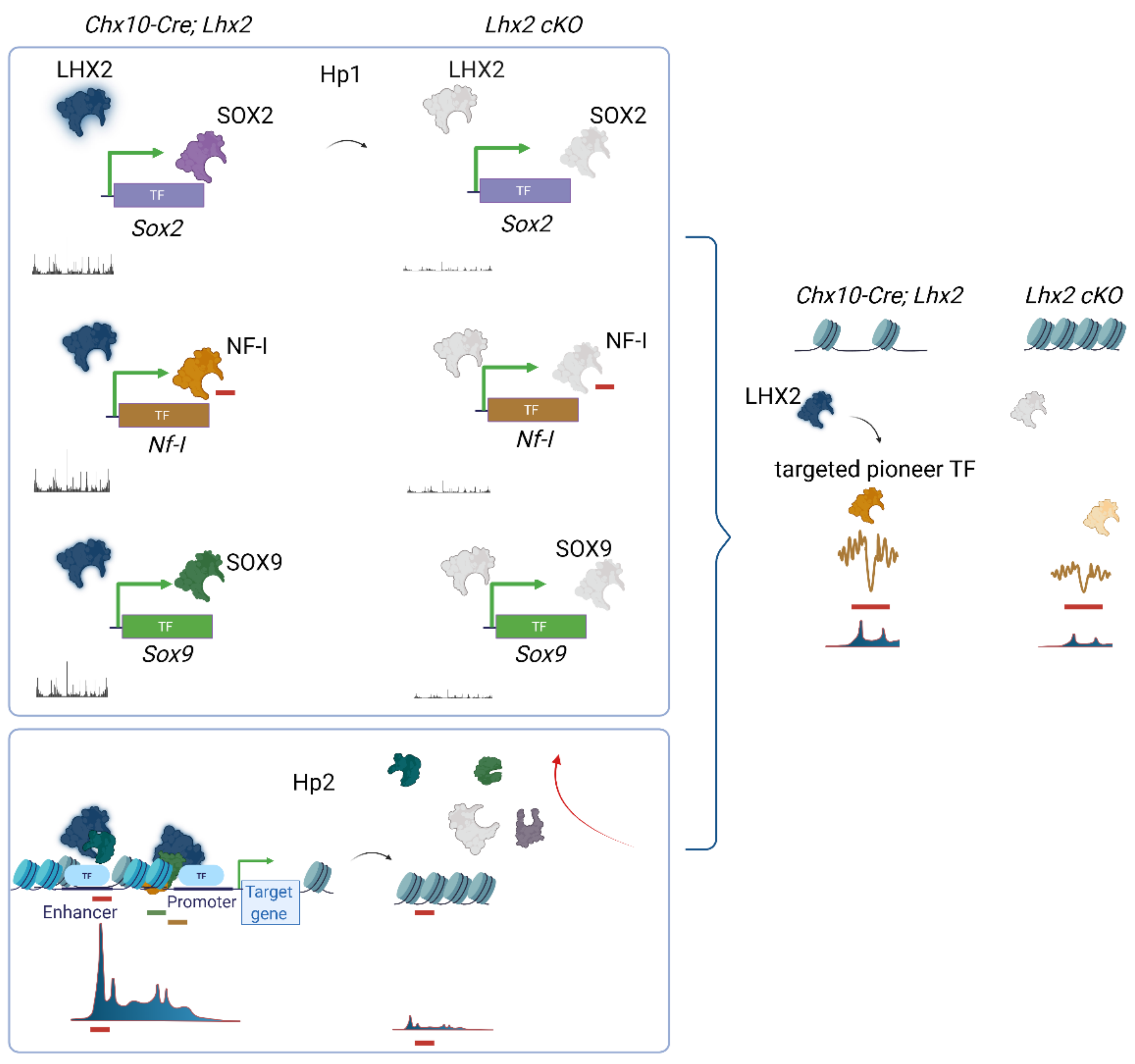
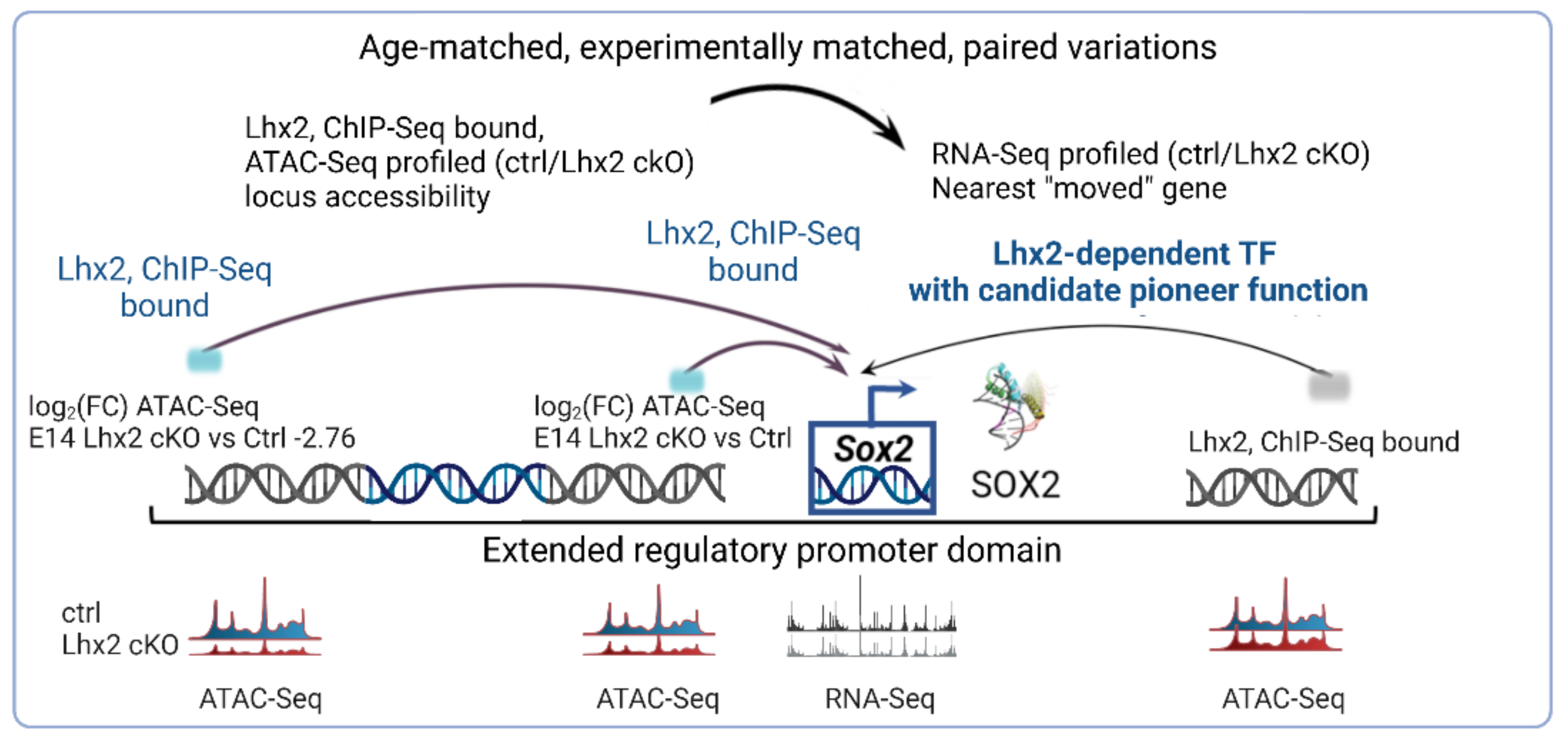
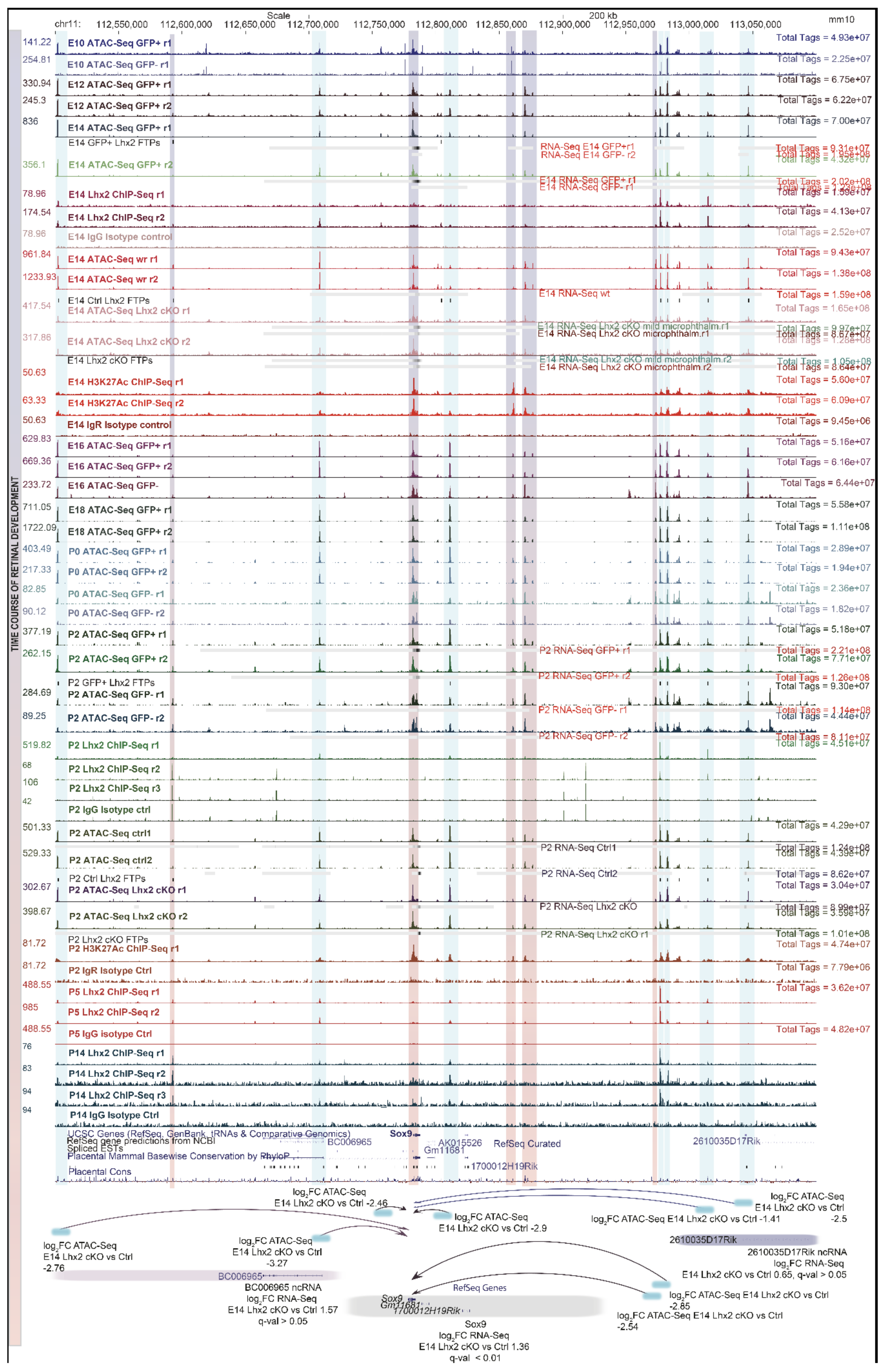
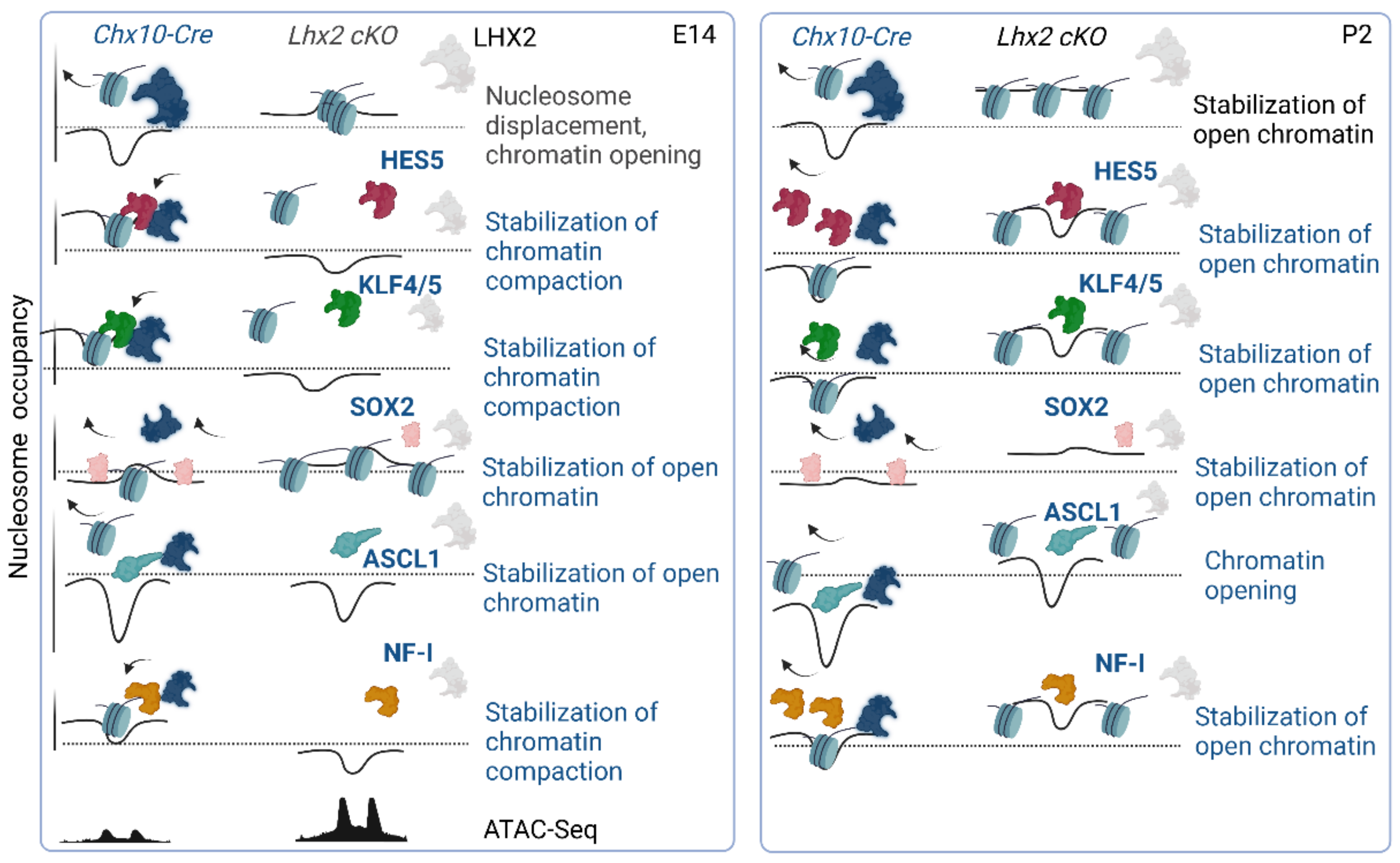

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zibetti, C. Deciphering the Retinal Epigenome during Development, Disease and Reprogramming: Advancements, Challenges and Perspectives. Cells 2022, 11, 806. https://doi.org/10.3390/cells11050806
Zibetti C. Deciphering the Retinal Epigenome during Development, Disease and Reprogramming: Advancements, Challenges and Perspectives. Cells. 2022; 11(5):806. https://doi.org/10.3390/cells11050806
Chicago/Turabian StyleZibetti, Cristina. 2022. "Deciphering the Retinal Epigenome during Development, Disease and Reprogramming: Advancements, Challenges and Perspectives" Cells 11, no. 5: 806. https://doi.org/10.3390/cells11050806