
Sumoylation in Physiology, Pathology and Therapy


Abstract
:1. Introduction
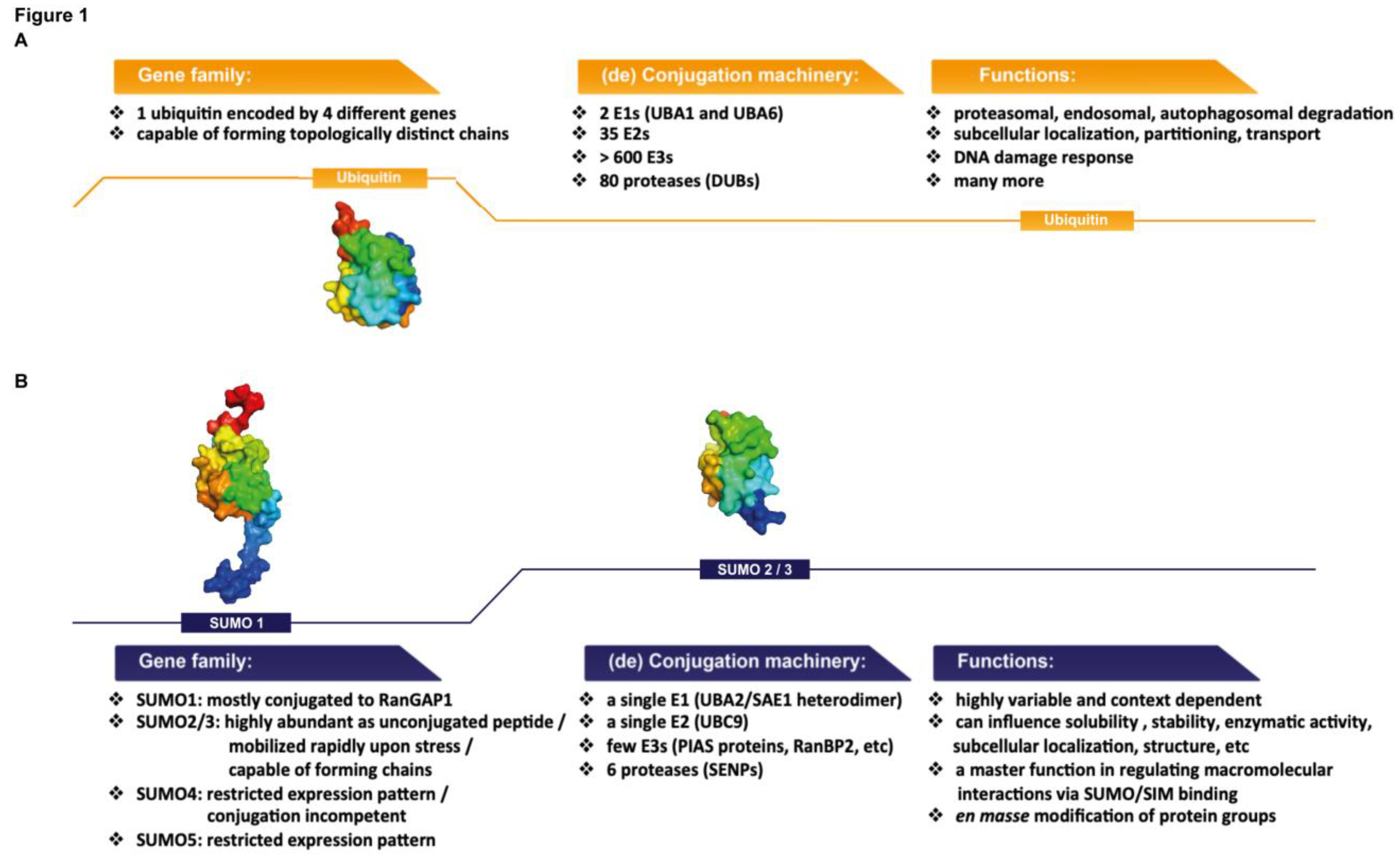
1.1. Sumoylation: Basic Mechanisms and Major Players
1.2. SUMO Proteins: Similarities with and Differences from Ubiquitin
1.3. SUMO Proteases: The Dynamic Nature of SUMO Modification
1.4. Protein Group Sumoylation Concept
1.5. SUMO Chain Formation and the Role of SUMO-Interaction Motifs
2. SUMO in Stress
3. SUMO in Development and Physiology
4. SUMO in Cancer and Therapy
5. SUMO in Neurodegenerative Disorders
6. Concluding Remarks

{kind=link}
| Disease or Physiologıcal Process. | Protein(s). Involved | Role of Sumoylation | References |
|---|---|---|---|
| Acute promyelocytic leukemia (APL) | PML/RARA | Arsenic-enhanced PML/RARA sumoylation promotes degradation and underlies the therapeutic effect of this drug in APL | [52,53,150,152] |
| cute promyelocytic leukemia (APL) | PML/RARA | K160, a major sumoylation site on PML/RARA is essential for cellular transformation in APL | [159] |
| Prostate cancer | Androgen receptor (AR) | Modification by SUMO1 attenuates AR’s transcriptional activity | [177,178,179] |
| Prostate cancer | Androgen receptor (AR) | IRC117539-induced AR sumoylation induces AR degradation and causes loss-of-viability in prostate cancer cell lines | [184] |
| Adult T-cell lymphoma (ATL) | Tax | The arsenic/interferon combination regimen induces Tax sumoylation, followed by degradation, causing loss-of-viability in transformed ATL cells | [163] |
| Various cancers | P53 and regulators | Collective sumoylation in situ in PML NBs may serve to accomplish full P53 activation | [57,63,64,65,66,67] |
| Breast and ovarian cancer | BRCA1 | Sumoylation upregulates BRCA1’s E3 ligase activity | [187] |
| Metastasis, angiogenesis, various cancers | VHL and HIF1α | During hypoxia, sumoylation inactivates VHL, causing HIF1α stabilization | [70] |
| B cell lymphoma | MYC | Sumoylation enhances MYC stability; in MYC-driven B cell lymphomas, MYC further upregulates sumoylation; inhibition of sumoylation diminishes the survival of MYC-driven B cell lymphomas | [171,172] |
| Colon and various cancers | β-catenin | Sumoylation prevents β-catenin ubiquitylation and degradation | [175] |
| Cancer stem cells, various cancers | SUMO E1 and E2 enzymes, various SUMO E3 ligases (i.e. PIAS proteins) | Upregulated expression levels possibly maintain cancer stem cells | [117,188,189,190,191,192,193,194] |
| Neural stem cells | UBC9, global sumoylation, interferons | Enhanced sumoylation may promote neural stem cell maintenance and neuronal recovery in neurodegeneration | [87,222,223] |
| Spinocerebellar ataxia | Ataxin 1 | Sumoylation promotes degradation, reducing aggregate burden | [62] |
| Parkinson’s disease | α-synuclein | Sumoylation enhances protein solubility and opposes toxicity | [219,220] |
| Huntington’s disease | Huntingtin | Sumoylation by PIAS-1 increases aggregation tendency and toxicity | [216] |
| Huntington’s disease, autophagy | Huntingtin | Inhibition of sumoylation enhances autophagy, promotes Huntingtin clearance | [229] |
| Alzheimer’s disease | Tau | Sumoylation prevents ubiquitylation and degradation, thereby promoting aggregation | [211,218] |
| Various neurodegenerative diseases | SUMO/SIM interactions | May promote liquid-liquid phase separation, cytotoxic protein aggregation | [40,205,206] |
Funding
Conflicts of Interest
References
- Cappadocia, L.; Lima, C.D. Ubiquitin-like Protein Conjugation: Structures, Chemistry, and Mechanism. Chem. Rev. 2018, 118, 889–918. [Google Scholar] [CrossRef]
- Hochstrasser, M. Origin and function of ubiquitin-like proteins. Nature 2009, 458, 422–429. [Google Scholar] [CrossRef] [Green Version]
- Celen, A.B.; Sahin, U. Sumoylation on its 25th anniversary: Mechanisms, pathology, and emerging concepts. FEBS J. 2020, 287, 3110–3140. [Google Scholar] [CrossRef]
- Meluh, P.B.; Koshland, D. Evidence that the MIF2 gene of Saccharomyces cerevisiae encodes a centromere protein with homology to the mammalian centromere protein CENP-C. Mol. Biol. Cell 1995, 6, 793–807. [Google Scholar] [CrossRef]
- Shen, Z.; Pardington-Purtymun, P.E.; Comeaux, J.C.; Moyzis, R.K.; Chen, D.J. UBL1: A human ubiquitin-like protein associating with human RAD51/RAD52 proteins. Genomics 1996, 36, 271–279. [Google Scholar] [CrossRef] [Green Version]
- Okura, T.; Gong, L.; Kamitani, T.; Wada, T.; Okura, I.; Wei, C.F.; Chang, H.M.; Yeh, E.T. Protection against Fas/APO-1- and tumor necrosis factor-mediated cell death by a novel protein. Sentrin. J. Immunol. 1996, 157, 4277–4281. [Google Scholar]
- Boddy, M.N.; Howe, K.; Etkin, L.D.; Solomon, E.; Freemont, P.S. PIC 1: A novel ubiquitin-like protein which interacts with the PML component of a multiprotein complex that is disrupted in acute promyelocytic leukaemia. Oncogene 1996, 13, 971–982. [Google Scholar]
- Matunis, M.J.; Coutavas, E.; Blobel, G. A novel ubiquitin-like modification modulates the partitioning of the Ran-GTPase-activating protein RanGAP1 between the cytosol and the nuclear pore complex. J. Cell Biol. 1996, 135, 1457–1470. [Google Scholar] [CrossRef]
- Mahajan, R.; Delphin, C.; Guan, T.; Gerace, L.; Melchior, F. A small ubiquitin-related polypeptide involved in targeting RanGAP1 to nuclear pore complex protein RanBP2. Cell 1997, 88, 97–107. [Google Scholar] [CrossRef] [Green Version]
- Bayer, P.; Arndt, A.; Metzger, S.; Mahajan, R.; Melchior, F.; Jaenicke, R.; Becker, J. Structure determination of the small ubiquitin-related modifier SUMO-1. J. Mol. Biol. 1998, 280, 275–286. [Google Scholar] [CrossRef]
- Vijay-Kumar, S.; Bugg, C.E.; Cook, W.J. Structure of ubiquitin refined at 1.8 A resolution. J. Mol. Biol. 1987, 194, 531–544. [Google Scholar] [CrossRef]
- Vijay-Kumar, S.; Bugg, C.E.; Wilkinson, K.D.; Vierstra, R.D.; Hatfield, P.M.; Cook, W.J. Comparison of the three-dimensional structures of human. yeast, and oat ubiquitin. J. Biol. Chem. 1987, 262, 6396–6399. [Google Scholar] [CrossRef]
- Hay, R.T. Protein modification by SUMO. Trends Biochem. Sci. 2001, 26, 332–333. [Google Scholar] [CrossRef]
- Owerbach, D.; McKay, E.M.; Yeh, E.T.; Gabbay, K.H.; Bohren, K.M. A proline-90 residue unique to SUMO-4 prevents maturation and sumoylation. Biochem. Biophys. Res. Commun. 2005, 337, 517–520. [Google Scholar] [CrossRef]
- Liang, Y.C.; Lee, C.C.; Yao, Y.L.; Lai, C.C.; Schmitz, M.L.; Yang, W.M. SUMO5: A Novel Poly-SUMO Isoform, Regulates PML Nuclear Bodies. Sci. Rep. 2016, 6, 26509. [Google Scholar] [CrossRef]
- Gareau, J.R.; Lima, C.D. The SUMO pathway: Emerging mechanisms that shape specificity. conjugation and recognition. Nat. Rev. Mol. Cell Biol 2010, 11, 861–871. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez, M.S.; Dargemont, C.; Hay, R.T. SUMO-1 conjugation in vivo requires both a consensus modification motif and nuclear targeting. J. Biol. Chem. 2001, 276, 12654–12659. [Google Scholar] [CrossRef] [Green Version]
- Sampson, D.A.; Wang, M.; Matunis, M.J. The small ubiquitin-like modifier-1 (SUMO-1) consensus sequence mediates Ubc9 binding and is essential for SUMO-1 modification. J. Biol. Chem. 2001, 276, 21664–21669. [Google Scholar] [CrossRef] [Green Version]
- Tatham, M.H.; Chen, Y.; Hay, R.T. Role of two residues proximal to the active site of Ubc9 in substrate recognition by the Ubc9.SUMO-1 thiolester complex. Biochemistry 2003, 42, 3168–3179. [Google Scholar] [CrossRef]
- Tatham, M.H.; Kim, S.; Yu, B.; Jaffray, E.; Song, J.; Zheng, J.; Rodriguez, M.S.; Hay, R.T.; Chen, Y. Role of an N-terminal site of Ubc9 in SUMO-1. -2, and -3 binding and conjugation. Biochemistry 2003, 42, 9959–9969. [Google Scholar] [CrossRef]
- Lin, D.; Tatham, M.H.; Yu, B.; Kim, S.; Hay, R.T.; Chen, Y. Identification of a substrate recognition site on Ubc9. J. Biol. Chem. 2002, 277, 21740–21748. [Google Scholar] [CrossRef] [Green Version]
- Giraud, M.F.; Desterro, J.M.; Naismith, J.H. Structure of ubiquitin-conjugating enzyme 9 displays significant differences with other ubiquitin-conjugating enzymes which may reflect its specificity for sumo rather than ubiquitin. Acta Cryst. D Biol. Cryst. 1998, 54, 891–898. [Google Scholar] [CrossRef] [Green Version]
- Tong, H.; Hateboer, G.; Perrakis, A.; Bernards, R.; Sixma, T.K. Crystal structure of murine/human Ubc9 provides insight into the variability of the ubiquitin-conjugating system. J. Biol. Chem. 1997, 272, 21381–21387. [Google Scholar] [CrossRef] [Green Version]
- Eifler, K.; Vertegaal, A.C. Mapping the SUMOylated landscape. FEBS J. 2015, 282, 3669–3680. [Google Scholar] [CrossRef]
- Yang, X.J.; Gregoire, S. A recurrent phospho-sumoyl switch in transcriptional repression and beyond. Mol. Cell 2006, 23, 779–786. [Google Scholar] [CrossRef]
- Hietakangas, V.; Anckar, J.; Blomster, H.A.; Fujimoto, M.; Palvimo, J.J.; Nakai, A.; Sistonen, L. PDSM: A motif for phosphorylation-dependent SUMO modification. Proc. Natl. Acad. Sci. USA 2006, 103, 45–50. [Google Scholar] [CrossRef] [Green Version]
- Desterro, J.M.; Thomson, J.; Hay, R.T. Ubch9 conjugates SUMO but not ubiquitin. FEBS Lett. 1997, 417, 297–300. [Google Scholar] [CrossRef]
- Johnson, E.S.; Schwienhorst, I.; Dohmen, R.J.; Blobel, G. The ubiquitin-like protein Smt3p is activated for conjugation to other proteins by an Aos1p/Uba2p heterodimer. EMBO J. 1997, 16, 5509–5519. [Google Scholar] [CrossRef] [Green Version]
- Desterro, J.M.; Rodriguez, M.S.; Kemp, G.D.; Hay, R.T. Identification of the enzyme required for activation of the small ubiquitin-like protein SUMO-1. J. Biol. Chem. 1999, 274, 10618–10624. [Google Scholar] [CrossRef] [Green Version]
- Kunz, K.; Piller, T.; Muller, S. SUMO-specific proteases and isopeptidases of the SENP family at a glance. J. Cell Sci. 2018, 131, jcs211904. [Google Scholar] [CrossRef] [Green Version]
- Yeh, E.T. SUMOylation and De-SUMOylation: Wrestling with life’s processes. J. Biol Chem 2009, 284, 8223–8227. [Google Scholar] [CrossRef] [Green Version]
- Hendriks, I.A.; Lyon, D.; Su, D.; Skotte, N.H.; Daniel, J.A.; Jensen, L.J.; Nielsen, M.L. Site-specific characterization of endogenous SUMOylation across species and organs. Nat. Commun. 2018, 9, 2456. [Google Scholar] [CrossRef] [Green Version]
- Geiss-Friedlander, R.; Melchior, F. Concepts in sumoylation: A decade on. Nat. Rev. Mol. Cell Biol. 2007, 8, 947–956. [Google Scholar] [CrossRef]
- Wilkinson, K.A.; Henley, J.M. Mechanisms. Regulation and consequences of protein SUMOylation. Biochem J. 2010, 428, 133–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hay, R.T. SUMO: A history of modification. Mol. Cell 2005, 18, 1–12. [Google Scholar] [CrossRef]
- Psakhye, I.; Jentsch, S. Identification of Substrates of Protein-Group SUMOylation. Methods Mol. Biol. 2016, 1475, 219–231. [Google Scholar]
- Jentsch, S.; Psakhye, I. Control of nuclear activities by substrate-selective and protein-group SUMOylation. Annu. Rev. Genet. 2013, 47, 167–186. [Google Scholar] [CrossRef] [PubMed]
- Psakhye, I.; Jentsch, S. Protein group modification and synergy in the SUMO pathway as exemplified in DNA repair. Cell 2012, 151, 807–820. [Google Scholar] [CrossRef] [Green Version]
- Cremona, C.A.; Sarangi, P.; Yang, Y.; Hang, L.E.; Rahman, S.; Zhao, X. Extensive DNA damage-induced sumoylation contributes to replication and repair and acts in addition to the mec1 checkpoint. Mol. Cell 2012, 45, 422–432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keiten-Schmitz, J.; Roder, L.; Hornstein, E.; Muller-McNicoll, M.; Muller, S. SUMO: Glue or Solvent for Phase-Separated Ribonucleoprotein Complexes and Molecular Condensates? Front. Mol. Biosci. 2021, 8, 673038. [Google Scholar] [CrossRef]
- Banani, S.F.; Rice, A.M.; Peeples, W.B.; Lin, Y.; Jain, S.; Parker, R.; Rosen, M.K. Compositional Control of Phase-Separated Cellular Bodies. Cell 2016, 166, 651–663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tatham, M.H.; Jaffray, E.; Vaughan, O.A.; Desterro, J.M.; Botting, C.H.; Naismith, J.H.; Hay, R.T. Polymeric chains of SUMO-2 and SUMO-3 are conjugated to protein substrates by SAE1/SAE2 and Ubc9. J. Biol. Chem. 2001, 276, 35368–35374. [Google Scholar] [CrossRef] [Green Version]
- Hendriks, I.A.; D’Souza, R.C.; Yang, B.; Verlaan-de Vries, M.; Mann, M.; Vertegaal, A.C. Uncovering global SUMOylation signaling networks in a site-specific manner. Nat. Struct. Mol. Biol. 2014, 21, 927–936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hendriks, I.A.; Vertegaal, A.C. A comprehensive compilation of SUMO proteomics. Nat. Rev. Mol. Cell Biol. 2016, 17, 581–595. [Google Scholar] [CrossRef] [PubMed]
- Matic, I.; Schimmel, J.; Hendriks, I.A.; van Santen, M.A.; van de Rijke, F.; van Dam, H.; Gnad, F.; Mann, M.; Vertegaal, A.C. Site-specific identification of SUMO-2 targets in cells reveals an inverted SUMOylation motif and a hydrophobic cluster SUMOylation motif. Mol. Cell 2010, 39, 641–652. [Google Scholar] [CrossRef] [PubMed]
- Praefcke, G.J.; Hofmann, K.; Dohmen, R.J. SUMO playing tag with ubiquitin. Trends Biochem. Sci. 2012, 37, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, J.; Shi, Y.; Valin, A.; Xuan, Y.; Gill, G. Direct binding of CoREST1 to SUMO-2/3 contributes to gene-specific repression by the LSD1/CoREST1/HDAC complex. Mol. Cell 2009, 34, 145–154. [Google Scholar] [CrossRef] [Green Version]
- Meulmeester, E.; Kunze, M.; Hsiao, H.H.; Urlaub, H.; Melchior, F. Mechanism and consequences for paralog-specific sumoylation of ubiquitin-specific protease 25. Mol. Cell 2008, 30, 610–619. [Google Scholar] [CrossRef] [Green Version]
- Miteva, M.; Keusekotten, K.; Hofmann, K.; Praefcke, G.J.; Dohmen, R.J. Sumoylation as a signal for polyubiquitylation and proteasomal degradation. Subcell Biochem. 2010, 54, 195–214. [Google Scholar]
- Heideker, J.; Perry, J.J.; Boddy, M.N. Genome stability roles of SUMO-targeted ubiquitin ligases. DNA Repair 2009, 8, 517–524. [Google Scholar] [CrossRef] [Green Version]
- Yin, Y.; Seifert, A.; Chua, J.S.; Maure, J.F.; Golebiowski, F.; Hay, R.T. SUMO-targeted ubiquitin E3 ligase RNF4 is required for the response of human cells to DNA damage. Genes Dev. 2012, 26, 1196–1208. [Google Scholar] [CrossRef] [Green Version]
- Tatham, M.H.; Geoffroy, M.C.; Shen, L.; Plechanovova, A.; Hattersley, N.; Jaffray, E.G.; Palvimo, J.J.; Hay, R.T. RNF4 is a poly-SUMO-specific E3 ubiquitin ligase required for arsenic-induced PML degradation. Nat. Cell Biol. 2008, 10, 538–546. [Google Scholar] [CrossRef]
- Lallemand-Breitenbach, V.; Jeanne, M.; Benhenda, S.; Nasr, R.; Lei, M.; Peres, L.; Zhou, J.; Zhu, J.; Raught, B.; de The, H. Arsenic degrades PML or PML-RARalpha through a SUMO-triggered RNF4/ubiquitin-mediated pathway. Nat. Cell Biol. 2008, 10, 547–555. [Google Scholar] [CrossRef]
- Kumar, R.; Gonzalez-Prieto, R.; Xiao, Z.; Verlaan-de Vries, M.; Vertegaal, A.C.O. The STUbL RNF4 regulates protein group SUMOylation by targeting the SUMO conjugation machinery. Nat. Commun. 2017, 8, 1809. [Google Scholar] [CrossRef] [Green Version]
- Fulda, S.; Gorman, A.M.; Hori, O.; Samali, A. Cellular stress responses: Cell survival and cell death. Int. J. Cell Biol. 2010, 2010, 214074. [Google Scholar] [CrossRef] [Green Version]
- Ryu, H.Y.; Ahn, S.H.; Hochstrasser, M. SUMO and cellular adaptive mechanisms. Exp. Mol. Med. 2020, 52, 931–939. [Google Scholar] [CrossRef]
- Sahin, U.; de The, H.; Lallemand-Breitenbach, V. PML nuclear bodies: Assembly and oxidative stress-sensitive sumoylation. Nucleus 2014, 5, 499–507. [Google Scholar] [CrossRef] [Green Version]
- Sahin, U.; Lallemand-Breitenbach, V.; de The, H. PML nuclear bodies: Regulation. function and therapeutic perspectives. J. Pathol 2014, 234, 289–291. [Google Scholar] [CrossRef]
- Sahin, U.; Ferhi, O.; Jeanne, M.; Benhenda, S.; Berthier, C.; Jollivet, F.; Niwa-Kawakita, M.; Faklaris, O.; Setterblad, N.; de The, H.; et al. Oxidative stress-induced assembly of PML nuclear bodies controls sumoylation of partner proteins. J. Cell Biol. 2014, 204, 931–945. [Google Scholar] [CrossRef] [Green Version]
- Niwa-Kawakita, M.; Wu, H.C.; The, H.; Lallemand-Breitenbach, V. PML nuclear bodies. membrane-less domains acting as ROS sensors? Semin. Cell Dev. Biol. 2018, 80, 29–34. [Google Scholar] [CrossRef]
- Wang, P.; Benhenda, S.; Wu, H.; Lallemand-Breitenbach, V.; Zhen, T.; Jollivet, F.; Peres, L.; Li, Y.; Chen, S.J.; Chen, Z.; et al. RING tetramerization is required for nuclear body biogenesis and PML sumoylation. Nat. Commun. 2018, 9, 1277. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Giasson, B.I.; Glavis-Bloom, A.; Brewer, M.D.; Shorter, J.; Gitler, A.D.; Yang, X. A cellular system that degrades misfolded proteins and protects against neurodegeneration. Mol. Cell 2014, 55, 15–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ivanschitz, L.; Takahashi, Y.; Jollivet, F.; Ayrault, O.; Le Bras, M.; de The, H. PML IV/ARF interaction enhances p53 SUMO-1 conjugation: Activation, and senescence. Proc. Natl. Acad. Sci. USA 2015, 112, 14278–14283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niwa-Kawakita, M.; Ferhi, O.; Soilihi, H.; Le Bras, M.; Lallemand-Breitenbach, V.; de The, H. PML is a ROS sensor activating p53 upon oxidative stress. J. Exp. Med. 2017, 214, 3197–3206. [Google Scholar] [CrossRef] [Green Version]
- Pearson, M.; Carbone, R.; Sebastiani, C.; Cioce, M.; Fagioli, M.; Saito, S.; Higashimoto, Y.; Appella, E.; Minucci, S.; Pandolfi, P.P.; et al. PML regulates p53 acetylation and premature senescence induced by oncogenic Ras. Nature 2000, 406, 207–210. [Google Scholar] [CrossRef]
- Rodriguez, M.S.; Desterro, J.M.; Lain, S.; Midgley, C.A.; Lane, D.P.; Hay, R.T. SUMO-1 modification activates the transcriptional response of p53. EMBO J. 1999, 18, 6455–6461. [Google Scholar] [CrossRef] [Green Version]
- Gostissa, M.; Hengstermann, A.; Fogal, V.; Sandy, P.; Schwarz, S.E.; Scheffner, M.; Del Sal, G. Activation of p53 by conjugation to the ubiquitin-like protein SUMO-1. EMBO J. 1999, 18, 6462–6471. [Google Scholar] [CrossRef] [Green Version]
- Muller, S.; Berger, M.; Lehembre, F.; Seeler, J.S.; Haupt, Y.; Dejean, A. c-Jun and p53 activity is modulated by SUMO-1 modification. J. Biol. Chem. 2000, 275, 13321–13329. [Google Scholar] [CrossRef] [Green Version]
- Masoud, G.N.; Li, W. HIF-1alpha pathway: Role. Regulation and intervention for cancer therapy. Acta Pharm. Sin. B 2015, 5, 378–389. [Google Scholar] [CrossRef] [Green Version]
- Cai, Q.; Verma, S.C.; Kumar, P.; Ma, M.; Robertson, E.S. Hypoxia inactivates the VHL tumor suppressor through PIASy-mediated SUMO modification. PLoS ONE 2010, 5, e9720. [Google Scholar] [CrossRef] [Green Version]
- Desterro, J.M.; Rodriguez, M.S.; Hay, R.T. SUMO-1 modification of IkappaBalpha inhibits NF-kappaB activation. Mol. Cell 1998, 2, 233–239. [Google Scholar] [CrossRef]
- Saitoh, H.; Hinchey, J. Functional heterogeneity of small ubiquitin-related protein modifiers SUMO-1 versus SUMO-2/3. J. Biol. Chem. 2000, 275, 6252–6258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lapenta, V.; Chiurazzi, P.; van der Spek, P.; Pizzuti, A.; Hanaoka, F.; Brahe, C. SMT3A: A human homologue of the S 1997, cerevisiae SMT3 gene, maps to chromosome 21qter and defines a novel gene family. Genomics 1997, 40, 362–366. [Google Scholar] [CrossRef] [PubMed]
- Kamitani, T.; Kito, K.; Nguyen, H.P.; Fukuda-Kamitani, T.; Yeh, E.T. Characterization of a second member of the sentrin family of ubiquitin-like proteins. J. Biol. Chem. 1998, 273, 11349–11353. [Google Scholar] [CrossRef] [Green Version]
- Golebiowski, F.; Matic, I.; Tatham, M.H.; Cole, C.; Yin, Y.; Nakamura, A.; Cox, J.; Barton, G.J.; Mann, M.; Hay, R.T. System-wide changes to SUMO modifications in response to heat shock. Sci. Signal. 2009, 2, ra24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tammsalu, T.; Matic, I.; Jaffray, E.G.; Ibrahim, A.F.M.; Tatham, M.H.; Hay, R.T. Proteome-wide identification of SUMO2 modification sites. Sci. Signal. 2014, 7, rs2. [Google Scholar] [CrossRef] [Green Version]
- Hendriks, I.A.; D’Souza, R.C.; Chang, J.G.; Mann, M.; Vertegaal, A.C. System-wide identification of wild-type SUMO-2 conjugation sites. Nat. Commun 2015, 6, 7289. [Google Scholar] [CrossRef]
- Tatham, M.H.; Matic, I.; Mann, M.; Hay, R.T. Comparative proteomic analysis identifies a role for SUMO in protein quality control. Sci. Signal. 2011, 4, rs4. [Google Scholar] [CrossRef] [Green Version]
- Uzunova, K.; Gottsche, K.; Miteva, M.; Weisshaar, S.R.; Glanemann, C.; Schnellhardt, M.; Niessen, M.; Scheel, H.; Hofmann, K.; Johnson, E.S.; et al. Ubiquitin-dependent proteolytic control of SUMO conjugates. J. Biol. Chem. 2007, 282, 34167–34175. [Google Scholar] [CrossRef] [Green Version]
- Seeler, J.S.; Dejean, A. Nuclear and unclear functions of SUMO. Nat. Rev. Mol. Cell Biol. 2003, 4, 690–699. [Google Scholar] [CrossRef]
- Liu, X.; Wang, Q.; Chen, W.; Wang, C. Dynamic regulation of innate immunity by ubiquitin and ubiquitin-like proteins. Cytokine Growth Factor Rev. 2013, 24, 559–570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ribet, D.; Cossart, P. Ubiquitin. SUMO, and NEDD8: Key Targets of Bacterial Pathogens. Trends Cell Biol. 2018, 28, 926–940. [Google Scholar] [CrossRef] [PubMed]
- Ribet, D.; Cossart, P. SUMOylation and bacterial pathogens. Virulence 2010, 1, 532–534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adorisio, S.; Fierabracci, A.; Muscari, I.; Liberati, A.M.; Ayroldi, E.; Migliorati, G.; Thuy, T.T.; Riccardi, C.; Delfino, D.V. SUMO proteins: Guardians of immune system. J. Autoimmun. 2017, 84, 21–28. [Google Scholar] [CrossRef]
- Hannoun, Z.; Maarifi, G.; Chelbi-Alix, M.K. The implication of SUMO in intrinsic and innate immunity. Cytokine Growth Factor Rev. 2016, 29, 3–16. [Google Scholar] [CrossRef] [PubMed]
- Ribet, D.; Hamon, M.; Gouin, E.; Nahori, M.A.; Impens, F.; Neyret-Kahn, H.; Gevaert, K.; Vandekerckhove, J.; Dejean, A.; Cossart, P. Listeria monocytogenes impairs SUMOylation for efficient infection. Nature 2010, 464, 1192–1195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sahin, U.; Ferhi, O.; Carnec, X.; Zamborlini, A.; Peres, L.; Jollivet, F.; Vitaliano-Prunier, A.; de The, H.; Lallemand-Breitenbach, V. Interferon controls SUMO availability via the Lin28 and let-7 axis to impede virus replication. Nat. Commun. 2014, 5, 4187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boutell, C.; Cuchet-Lourenco, D.; Vanni, E.; Orr, A.; Glass, M.; McFarlane, S.; Everett, R.D. A viral ubiquitin ligase has substrate preferential SUMO targeted ubiquitin ligase activity that counteracts intrinsic antiviral defence. PLoS Pathog. 2011, 7, e1002245. [Google Scholar] [CrossRef] [PubMed]
- Everett, R.D.; Boutell, C.; Hale, B.G. Interplay between viruses and host sumoylation pathways. Nat. Rev. Microbiol. 2013, 11, 400–411. [Google Scholar] [CrossRef]
- Chelbi-Alix, M.K.; de The, H. Herpes virus induced proteasome-dependent degradation of the nuclear bodies-associated PML and Sp100 proteins. Oncogene 1999, 18, 935–941. [Google Scholar] [CrossRef] [Green Version]
- Sloan, E.; Tatham, M.H.; Groslambert, M.; Glass, M.; Orr, A.; Hay, R.T.; Everett, R.D. Analysis of the SUMO2 Proteome during HSV-1 Infection. PLoS Pathog. 2015, 11, e1005059. [Google Scholar] [CrossRef] [Green Version]
- Boutell, C.; Sadis, S.; Everett, R.D. Herpes simplex virus type 1 immediate-early protein ICP0 and is isolated RING finger domain act as ubiquitin E3 ligases in vitro. J. Virol. 2002, 76, 841–850. [Google Scholar] [CrossRef] [Green Version]
- Morrell, R.; Sadanandom, A. Dealing with Stress: A Review of Plant SUMO Proteases. Front. Plant. Sci. 2019, 10, 1122. [Google Scholar] [CrossRef] [PubMed]
- Mazur, M.J.; van den Burg, H.A. Global SUMO Proteome Responses Guide Gene Regulation. mRNA Biogenesis, and Plant Stress Responses, Front. Plant. Sci. 2012, 3, 215.
- Augustine, R.C.; Vierstra, R.D. SUMOylation: Re-wiring the plant nucleus during stress and development. Curr. Opin. Plant. Biol. 2018, 45, 143–154. [Google Scholar] [CrossRef] [PubMed]
- Elrouby, N.; Coupland, G. Proteome-wide screens for small ubiquitin-like modifier (SUMO) substrates identify Arabidopsis proteins implicated in diverse biological processes. Proc. Natl. Acad. Sci. USA 2010, 107, 17415–17420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurepa, J.; Walker, J.M.; Smalle, J.; Gosink, M.M.; Davis, S.J.; Durham, T.L.; Sung, D.Y.; Vierstra, R.D. The small ubiquitin-like modifier (SUMO) protein modification system in Arabidopsis. Accumulation of SUMO1 and -2 conjugates is increased by stress. J. Biol. Chem. 2003, 278, 6862–6872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, M.J.; Vierstra, R.D. Mass spectrometric identification of SUMO substrates provides insights into heat stress-induced SUMOylation in plants. Plant. Signal. Behav. 2011, 6, 130–133. [Google Scholar] [CrossRef]
- Miller, M.J.; Scalf, M.; Rytz, T.C.; Hubler, S.L.; Smith, L.M.; Vierstra, R.D. Quantitative proteomics reveals factors regulating RNA biology as dynamic targets of stress-induced SUMOylation in Arabidopsis. Mol. Cell Proteom. 2013, 12, 449–463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shiio, Y.; Eisenman, R.N. Histone sumoylation is associated with transcriptional repression. Proc. Natl. Acad. Sci. USA 2003, 100, 13225–13230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maison, C.; Bailly, D.; Quivy, J.P.; Almouzni, G. The methyltransferase Suv39h1 links the SUMO pathway to HP1alpha marking at pericentric heterochromatin. Nat. Commun. 2016, 7, 12224. [Google Scholar] [CrossRef]
- Maison, C.; Bailly, D.; Roche, D.; Montes de Oca, R.; Probst, A.V.; Vassias, I.; Dingli, F.; Lombard, B.; Loew, D.; Quivy, J.P.; et al. SUMOylation promotes de novo targeting of HP1alpha to pericentric heterochromatin. Nat. Genet. 2011, 43, 220–227. [Google Scholar] [CrossRef] [Green Version]
- Jang, M.S.; Ryu, S.W.; Kim, E. Modification of Daxx by small ubiquitin-related modifier-1. Biochem. Biophys. Res. Commun. 2002, 295, 495–500. [Google Scholar] [CrossRef]
- Lin, D.Y.; Huang, Y.S.; Jeng, J.C.; Kuo, H.Y.; Chang, C.C.; Chao, T.T.; Ho, C.C.; Chen, Y.C.; Lin, T.P.; Fang, H.I.; et al. Role of SUMO-interacting motif in Daxx SUMO modification. subnuclear localization, and repression of sumoylated transcription factors. Mol. Cell 2006, 24, 341–354. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, K.; Shinkai, Y. SETDB1-Mediated Silencing of Retroelements. Viruses 2020, 12, 596. [Google Scholar] [CrossRef] [PubMed]
- Schultz, D.C.; Ayyanathan, K.; Negorev, D.; Maul, G.G.; Rauscher, F.J., 3rd. SETDB1: A novel KAP-1-associated histone H3. lysine 9-specific methyltransferase that contributes to HP1-mediated silencing of euchromatic genes by KRAB zinc-finger proteins. Genes Dev. 2002, 16, 919–932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sripathy, S.P.; Stevens, J.; Schultz, D.C. The KAP1 corepressor functions to coordinate the assembly of de novo HP1-demarcated microenvironments of heterochromatin required for KRAB zinc finger protein-mediated transcriptional repression. Mol. Cell Biol. 2006, 26, 8623–8638. [Google Scholar] [CrossRef] [Green Version]
- Rowe, H.M.; Jakobsson, J.; Mesnard, D.; Rougemont, J.; Reynard, S.; Aktas, T.; Maillard, P.V.; Layard-Liesching, H.; Verp, S.; Marquis, J.; et al. KAP1 controls endogenous retroviruses in embryonic stem cells. Nature 2010, 463, 237–240. [Google Scholar] [CrossRef]
- Li, X.; Lee, Y.K.; Jeng, J.C.; Yen, Y.; Schultz, D.C.; Shih, H.M.; Ann, D.K. Role for KAP1 serine 824 phosphorylation and sumoylation/desumoylation switch in regulating KAP1-mediated transcriptional repression. J. Biol. Chem. 2007, 282, 36177–36189. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.K.; Thomas, S.N.; Yang, A.J.; Ann, D.K. Doxorubicin down-regulates Kruppel-associated box domain-associated protein 1 sumoylation that relieves its transcription repression on p21WAF1/CIP1 in breast cancer MCF-7 cells. J. Biol. Chem. 2007, 282, 1595–1606. [Google Scholar] [CrossRef] [Green Version]
- Yang, B.X.; El Farran, C.A.; Guo, H.C.; Yu, T.; Fang, H.T.; Wang, H.F.; Schlesinger, S.; Seah, Y.F.; Goh, G.Y.; Neo, S.P.; et al. Systematic identification of factors for provirus silencing in embryonic stem cells. Cell 2015, 163, 230–245. [Google Scholar] [CrossRef] [Green Version]
- Mascle, X.H.; Germain-Desprez, D.; Huynh, P.; Estephan, P.; Aubry, M. Sumoylation of the transcriptional intermediary factor 1beta (TIF1beta). the Co-repressor of the KRAB Multifinger proteins, is required for its transcriptional activity and is modulated by the KRAB domain. J. Biol. Chem. 2007, 282, 10190–10202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kagey, M.H.; Melhuish, T.A.; Wotton, D. The polycomb protein Pc2 is a SUMO E3. Cell 2003, 113, 127–137. [Google Scholar] [CrossRef] [Green Version]
- Neyret-Kahn, H.; Benhamed, M.; Ye, T.; Le Gras, S.; Cossec, J.C.; Lapaquette, P.; Bischof, O.; Ouspenskaia, M.; Dasso, M.; Seeler, J.; et al. Sumoylation at chromatin governs coordinated repression of a transcriptional program essential for cell growth and proliferation. Genome Res. 2013, 23, 1563–1579. [Google Scholar] [CrossRef] [Green Version]
- Cossec, J.C.; Theurillat, I.; Chica, C.; Bua Aguin, S.; Gaume, X.; Andrieux, A.; Iturbide, A.; Jouvion, G.; Li, H.; Bossis, G.; et al. SUMO Safeguards Somatic and Pluripotent Cell Identities by Enforcing Distinct Chromatin States. Cell Stem. Cell 2018, 23, 742–757.e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tahmasebi, S.; Ghorbani, M.; Savage, P.; Yan, K.; Gocevski, G.; Xiao, L.; You, L.; Yang, X.J. Sumoylation of Kruppel-like factor 4 inhibits pluripotency induction but promotes adipocyte differentiation. J. Biol. Chem. 2013, 288, 12791–12804. [Google Scholar] [CrossRef] [Green Version]
- Du, L.; Li, Y.J.; Fakih, M.; Wiatrek, R.L.; Duldulao, M.; Chen, Z.; Chu, P.; Garcia-Aguilar, J.; Chen, Y. Role of SUMO activating enzyme in cancer stem cell maintenance and self-renewal. Nat. Commun. 2016, 7, 12326. [Google Scholar] [CrossRef] [Green Version]
- Juarez-Vicente, F.; Luna-Pelaez, N.; Garcia-Dominguez, M. The Sumo protease Senp7 is required for proper neuronal differentiation. Biochim. Biophys. Acta 2016, 1863, 1490–1498. [Google Scholar] [CrossRef]
- Cignarelli, A.; Melchiorre, M.; Peschechera, A.; Conserva, A.; Renna, L.A.; Miccoli, S.; Natalicchio, A.; Perrini, S.; Laviola, L.; Giorgino, F. Role of UBC9 in the regulation of the adipogenic program in 3T3-L1 adipocytes. Endocrinology 2010, 151, 5255–5266. [Google Scholar] [CrossRef] [Green Version]
- Liu, F.; Li, L.; Li, Y.; Ma, X.; Bian, X.; Liu, X.; Wang, G.; Zhang, D. Overexpression of SENP1 reduces the stemness capacity of osteosarcoma stem cells and increases their sensitivity to HSVtk/GCV. Int. J. Oncol. 2018, 53, 2010–2020. [Google Scholar] [CrossRef]
- Gonzalez-Prieto, R.; Cuijpers, S.A.; Kumar, R.; Hendriks, I.A.; Vertegaal, A.C. c-Myc is targeted to the proteasome for degradation in a SUMOylation-dependent manner, regulated by PIAS1, SENP7 and RNF4. Cell Cycle 2015, 14, 1859–1872. [Google Scholar] [CrossRef] [Green Version]
- Sabo, A.; Doni, M.; Amati, B. SUMOylation of Myc-family proteins. PLoS ONE 2014, 9, e91072. [Google Scholar] [CrossRef]
- Kawai-Kowase, K.; Ohshima, T.; Matsui, H.; Tanaka, T.; Shimizu, T.; Iso, T.; Arai, M.; Owens, G.K.; Kurabayashi, M. PIAS1 mediates TGFbeta-induced SM alpha-actin gene expression through inhibition of KLF4 function-expression by protein sumoylation. Arter. Thromb. Vasc. Biol. 2009, 29, 99–106. [Google Scholar] [CrossRef] [Green Version]
- Tsuruzoe, S.; Ishihara, K.; Uchimura, Y.; Watanabe, S.; Sekita, Y.; Aoto, T.; Saitoh, H.; Yuasa, Y.; Niwa, H.; Kawasuji, M.; et al. Inhibition of DNA binding of Sox2 by the SUMO conjugation. Biochem. Biophys. Res. Commun. 2006, 351, 920–926. [Google Scholar] [CrossRef]
- Zhang, Z.; Liao, B.; Xu, M.; Jin, Y. Post-translational modification of POU domain transcription factor Oct-4 by SUMO-1. FASEB J. 2007, 21, 3042–3051. [Google Scholar] [CrossRef]
- Tahmasebi, S.; Ghorbani, M.; Savage, P.; Gocevski, G.; Yang, X.J. The SUMO conjugating enzyme Ubc9 is required for inducing and maintaining stem cell pluripotency. Stem. Cells 2014, 32, 1012–1020. [Google Scholar] [CrossRef] [PubMed]
- Eckersley-Maslin, M.; Alda-Catalinas, C.; Blotenburg, M.; Kreibich, E.; Krueger, C.; Reik, W. Dppa2 and Dppa4 directly regulate the Dux-driven zygotic transcriptional program. Genes Dev. 2019, 33, 194–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Iaco, A.; Coudray, A.; Duc, J.; Trono, D. DPPA2 and DPPA4 are necessary to establish a 2C-like state in mouse embryonic stem cells. EMBO Rep. 2019, 20, 20. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, C.; Wang, Z.; Ramazanov, B.; Tang, Y.; Mehta, S.; Dambrot, C.; Lee, Y.W.; Tessema, K.; Kumar, I.; Astudillo, M.; et al. Dppa2/4 Facilitate Epigenetic Remodeling during Reprogramming to Pluripotency. Cell Stem. Cell 2018, 23, 396–411.e8. [Google Scholar] [CrossRef] [Green Version]
- Yan, Y.L.; Zhang, C.; Hao, J.; Wang, X.L.; Ming, J.; Mi, L.; Na, J.; Hu, X.; Wang, Y. DPPA2/4 and SUMO E3 ligase PIAS4 opposingly regulate zygotic transcriptional program. PLoS Biol. 2019, 17, e3000324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tessier, S.; Ferhi, O.; Geoffroy, M.C.; Gonzalez-Prieto, R.; Canat, A.; Quentin, S.; Pla, M.; Niwa-Kawakita, M.; Bercier, P.; Rerolle, D.; et al. Unbiased in vivo exploration of nuclear bodies-enhanced sumoylation reveals that PML orchestrates embryonic stem cell fate. BioRxiv 2021. [Google Scholar]
- Nacerddine, K.; Lehembre, F.; Bhaumik, M.; Artus, J.; Cohen-Tannoudji, M.; Babinet, C.; Pandolfi, P.P.; Dejean, A. The SUMO pathway is essential for nuclear integrity and chromosome segregation in mice. Dev. Cell 2005, 9, 769–779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Epps, J.L.; Tanda, S. The Drosophila semushi mutation blocks nuclear import of bicoid during embryogenesis. Curr. Biol. 1998, 8, 1277–1280. [Google Scholar] [CrossRef] [Green Version]
- Huang, L.; Ohsako, S.; Tanda, S. The lesswright mutation activates Rel-related proteins, leading to overproduction of larval hemocytes in Drosophila melanogaster. Dev. Biol. 2005, 280, 407–420. [Google Scholar] [CrossRef] [Green Version]
- Nie, M.; Xie, Y.; Loo, J.A.; Courey, A.J. Genetic and proteomic evidence for roles of Drosophila SUMO in cell cycle control, Ras signaling, and early pattern formation. PLoS ONE 2009, 4, e5905. [Google Scholar] [CrossRef] [PubMed]
- Nowak, M.; Hammerschmidt, M. Ubc9 regulates mitosis and cell survival during zebrafish development. Mol. Biol. Cell 2006, 17, 5324–5336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, D.; Crowe, E.; Stevens, T.A.; Candido, E.P. Functional and phylogenetic analysis of the ubiquitylation system in Caenorhabditis elegans: Ubiquitin-conjugating enzymes. ubiquitin-activating enzymes, and ubiquitin-like proteins. Genome Biol. 2002, 3, RESEARCH0002. [Google Scholar]
- Wang, L.; Wansleeben, C.; Zhao, S.; Miao, P.; Paschen, W.; Yang, W. SUMO2 is essential while SUMO3 is dispensable for mouse embryonic development. EMBO Rep. 2014, 15, 878–885. [Google Scholar] [CrossRef] [Green Version]
- Evdokimov, E.; Sharma, P.; Lockett, S.J.; Lualdi, M.; Kuehn, M.R. Loss of SUMO1 in mice affects RanGAP1 localization and formation of PML nuclear bodies, but is not lethal as it can be compensated by SUMO2 or SUMO3. J. Cell Sci. 2008, 121, 4106–4113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, F.P.; Mikkonen, L.; Toppari, J.; Palvimo, J.J.; Thesleff, I.; Janne, O.A. Sumo-1 function is dispensable in normal mouse development. Mol. Cell Biol. 2008, 28, 5381–5390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, J.; Kang, X.; Zhang, S.; Yeh, E.T. SUMO-specific protease 1 is essential for stabilization of HIF1alpha during hypoxia. Cell 2007, 131, 584–595. [Google Scholar] [CrossRef] [Green Version]
- Yamaguchi, T.; Sharma, P.; Athanasiou, M.; Kumar, A.; Yamada, S.; Kuehn, M.R. Mutation of SENP1/SuPr-2 reveals an essential role for desumoylation in mouse development. Mol. Cell Biol. 2005, 25, 5171–5182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, L.; Ji, W.; Zhang, H.; Renda, M.J.; He, Y.; Lin, S.; Cheng, E.C.; Chen, H.; Krause, D.S.; Min, W. SENP1-mediated GATA1 deSUMOylation is critical for definitive erythropoiesis. J. Exp. Med. 2010, 207, 1183–1195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rytinki, M.M.; Kaikkonen, S.; Pehkonen, P.; Jaaskelainen, T.; Palvimo, J.J. PIAS proteins: Pleiotropic interactors associated with SUMO. Cell Mol. Life Sci. 2009, 66, 3029–3041. [Google Scholar] [CrossRef] [PubMed]
- Rabellino, A.; Andreani, C.; Scaglioni, P.P. The Role of PIAS SUMO E3-Ligases in Cancer. Cancer Res. 2017, 77, 1542–1547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tahk, S.; Liu, B.; Chernishof, V.; Wong, K.A.; Wu, H.; Shuai, K. Control of specificity and magnitude of NF-kappa B and STAT1-mediated gene activation through PIASy and PIAS1 cooperation. Proc. Natl. Acad. Sci. USA 2007, 104, 11643–11648. [Google Scholar] [CrossRef] [Green Version]
- Constanzo, J.D.; Deng, M.; Rindhe, S.; Tang, K.J.; Zhang, C.C.; Scaglioni, P.P. Pias1 is essential for erythroid and vascular development in the mouse embryo. Dev. Biol. 2016, 415, 98–110. [Google Scholar] [CrossRef]
- Thornton, J.E.; Gregory, R.I. How does Lin28 let-7 control development and disease? Trends Cell Biol. 2012, 22, 474–482. [Google Scholar] [CrossRef] [Green Version]
- de The, H.; Chen, Z. Acute promyelocytic leukaemia: Novel insights into the mechanisms of cure. Nat. Rev. Cancer 2010, 10, 775–783. [Google Scholar] [CrossRef] [PubMed]
- de The, H.; Le Bras, M.; Lallemand-Breitenbach, V. The cell biology of disease: Acute promyelocytic leukemia: Arsenic, and PML bodies. J. Cell Biol. 2012, 198, 11–21. [Google Scholar] [CrossRef] [Green Version]
- de The, H. Differentiation therapy revisited. Nat. Rev. Cancer 2018, 18, 117–127. [Google Scholar] [CrossRef]
- Lallemand-Breitenbach, V.; Zhu, J.; Puvion, F.; Koken, M.; Honore, N.; Doubeikovsky, A.; Duprez, E.; Pandolfi, P.P.; Puvion, E.; Freemont, P.; et al. Role of promyelocytic leukemia (PML) sumolation in nuclear body formation, 11S proteasome recruitment, and As2O3-induced PML or PML/retinoic acid receptor alpha degradation. J. Exp. Med. 2001, 193, 1361–1371. [Google Scholar] [CrossRef] [PubMed]
- Shen, Z.X.; Shi, Z.Z.; Fang, J.; Gu, B.W.; Li, J.M.; Zhu, Y.M.; Shi, J.Y.; Zheng, P.Z.; Yan, H.; Liu, Y.F.; et al. All-trans retinoic acid/As2O3 combination yields a high quality remission and survival in newly diagnosed acute promyelocytic leukemia. Proc. Natl. Acad. Sci. USA 2004, 101, 5328–5335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de The, H.; Pandolfi, P.P.; Chen, Z. Acute Promyelocytic Leukemia: A Paradigm for Oncoprotein-Targeted Cure. Cancer Cell 2017, 32, 552–560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ablain, J.; de The, H. Retinoic acid signaling in cancer: The parable of acute promyelocytic leukemia. Int. J. Cancer 2014, 135, 2262–2272. [Google Scholar] [CrossRef] [PubMed]
- Jeanne, M.; Lallemand-Breitenbach, V.; Ferhi, O.; Koken, M.; Le Bras, M.; Duffort, S.; Peres, L.; Berthier, C.; Soilihi, H.; Raught, B.; et al. PML/RARA oxidation and arsenic binding initiate the antileukemia response of As2O3. Cancer Cell 2010, 18, 88–98. [Google Scholar] [CrossRef] [PubMed]
- Ablain, J.; Rice, K.; Soilihi, H.; de Reynies, A.; Minucci, S.; de The, H. Activation of a promyelocytic leukemia-tumor protein 53 axis underlies acute promyelocytic leukemia cure. Nat. Med. 2014, 20, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Lo-Coco, F.; Avvisati, G.; Vignetti, M.; Thiede, C.; Orlando, S.M.; Iacobelli, S.; Ferrara, F.; Fazi, P.; Cicconi, L.; Di Bona, E.; et al. Retinoic acid and arsenic trioxide for acute promyelocytic leukemia. N. Engl. J. Med. 2013, 369, 111–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, J.; Zhou, J.; Peres, L.; Riaucoux, F.; Honore, N.; Kogan, S.; de The, H. A sumoylation site in PML/RARA is essential for leukemic transformation. Cancer Cell 2005, 7, 143–153. [Google Scholar] [CrossRef] [Green Version]
- Bazarbachi, A.; Suarez, F.; Fields, P.; Hermine, O. How I treat adult T-cell leukemia/lymphoma. Blood 2011, 118, 1736–1745. [Google Scholar] [CrossRef]
- Ablain, J.; Nasr, R.; Bazarbachi, A.; de The, H. The drug-induced degradation of oncoproteins: An unexpected Achilles’ heel of cancer cells? Cancer Discov. 2011, 1, 117–127. [Google Scholar] [CrossRef] [Green Version]
- Boulanger, M.; Paolillo, R.; Piechaczyk, M.; Bossis, G. The SUMO Pathway in Hematomalignancies and Their Response to Therapies. Int. J. Mol. Sci. 2019, 20, 3895. [Google Scholar] [CrossRef] [Green Version]
- Dassouki, Z.; Sahin, U.; El Hajj, H.; Jollivet, F.; Kfoury, Y.; Lallemand-Breitenbach, V.; Hermine, O.; de The, H.; Bazarbachi, A. ATL response to arsenic/interferon therapy is triggered by SUMO/PML/RNF4-dependent Tax degradation. Blood 2015, 125, 474–482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bossis, G.; Sarry, J.E.; Kifagi, C.; Ristic, M.; Saland, E.; Vergez, F.; Salem, T.; Boutzen, H.; Baik, H.; Brockly, F.; et al. The ROS/SUMO axis contributes to the response of acute myeloid leukemia cells to chemotherapeutic drugs. Cell Rep. 2014, 7, 1815–1823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baik, H.; Boulanger, M.; Hosseini, M.; Kowalczyk, J.; Zaghdoudi, S.; Salem, T.; Sarry, J.E.; Hicheri, Y.; Cartron, G.; Piechaczyk, M.; et al. Targeting the SUMO Pathway Primes All-trans Retinoic Acid-Induced Differentiation of Nonpromyelocytic Acute Myeloid Leukemias. Cancer Res. 2018, 78, 2601–2613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seeler, J.S.; Dejean, A. SUMO and the robustness of cancer. Nat. Rev. Cancer 2017, 17, 184–197. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, M.L.; Xiang, D.; Shigdar, S.; Macdonald, J.; Li, Y.; Wang, T.; Pu, C.; Wang, Z.; Qiao, L.; Duan, W. Cancer stem cells: A contentious hypothesis now moving forward. Cancer Lett. 2014, 344, 180–187. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Ng, S.B.; Chng, W.J. LIN28/LIN28B: An emerging oncogenic driver in cancer stem cells. Int. J. Biochem. Cell Biol. 2013, 45, 973–978. [Google Scholar] [CrossRef] [PubMed]
- Balzeau, J.; Menezes, M.R.; Cao, S.; Hagan, J.P. The LIN28/let-7 Pathway in Cancer. Front. Genet. 2017, 8, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bogachek, M.V.; Park, J.M.; De Andrade, J.P.; Lorenzen, A.W.; Kulak, M.V.; White, J.R.; Gu, V.W.; Wu, V.T.; Weigel, R.J. Inhibiting the SUMO Pathway Represses the Cancer Stem Cell Population in Breast and Colorectal Carcinomas. Stem. Cell Rep. 2016, 7, 1140–1151. [Google Scholar] [CrossRef]
- Rabellino, A.; Melegari, M.; Tompkins, V.S.; Chen, W.; Van Ness, B.G.; Teruya-Feldstein, J.; Conacci-Sorrell, M.; Janz, S.; Scaglioni, P.P. PIAS1 Promotes Lymphomagenesis through MYC Upregulation. Cell Rep. 2016, 15, 2266–2278. [Google Scholar] [CrossRef] [Green Version]
- Hoellein, A.; Fallahi, M.; Schoeffmann, S.; Steidle, S.; Schaub, F.X.; Rudelius, M.; Laitinen, I.; Nilsson, L.; Goga, A.; Peschel, C.; et al. Myc-induced SUMOylation is a therapeutic vulnerability for B-cell lymphoma. Blood 2014, 124, 2081–2090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacDonald, B.T.; Tamai, K.; He, X. Wnt/beta-catenin signaling: Components. mechanisms, and diseases. Dev. Cell 2019, 17, 9–26. [Google Scholar]
- Aberle, H.; Bauer, A.; Stappert, J.; Kispert, A.; Kemler, R. beta-catenin is a target for the ubiquitin-proteasome pathway. EMBO J. 1997, 16, 3797–3804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, H.J.; Zhou, L.L.; Fu, W.J.; Zhang, C.Y.; Jiang, H.; Du, J.; Hou, J. beta-catenin SUMOylation is involved in the dysregulated proliferation of myeloma cells. Am. J. Cancer Res. 2015, 5, 309–320. [Google Scholar]
- Li, J.; Xu, Y.; Long, X.D.; Wang, W.; Jiao, H.K.; Mei, Z.; Yin, Q.Q.; Ma, L.N.; Zhou, A.W.; Wang, L.S.; et al. Cbx4 governs HIF-1alpha to potentiate angiogenesis of hepatocellular carcinoma by its SUMO E3 ligase activity. Cancer Cell 2014, 25, 118–131. [Google Scholar] [CrossRef] [Green Version]
- Poukka, H.; Karvonen, U.; Janne, O.A.; Palvimo, J.J. Covalent modification of the androgen receptor by small ubiquitin-like modifier 1 (SUMO-1). Proc. Natl. Acad. Sci. USA 2000, 97, 14145–14150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van der Steen, T.; Tindall, D.J.; Huang, H. Posttranslational modification of the androgen receptor in prostate cancer. Int. J. Mol. Sci. 2013, 14, 14833–14859. [Google Scholar] [CrossRef] [Green Version]
- Sutinen, P.; Malinen, M.; Heikkinen, S.; Palvimo, J.J. SUMOylation modulates the transcriptional activity of androgen receptor in a target gene and pathway selective manner. Nucleic Acids Res. 2014, 42, 8310–8319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carracedo, A.; Pandolfi, P.P. The PTEN-PI3K pathway: Of feedbacks and cross-talks. Oncogene 2008, 27, 5527–5541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salmena, L.; Carracedo, A.; Pandolfi, P.P. Tenets of PTEN tumor suppression. Cell 2008, 133, 403–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engelman, J.A.; Luo, J.; Cantley, L.C. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat. Rev. Genet. 2006, 7, 606–619. [Google Scholar] [CrossRef]
- Huang, J.; Yan, J.; Zhang, J.; Zhu, S.; Wang, Y.; Shi, T.; Zhu, C.; Chen, C.; Liu, X.; Cheng, J.; et al. SUMO1 modification of PTEN regulates tumorigenesis by controlling its association with the plasma membrane. Nat. Commun. 2012, 3, 911. [Google Scholar] [CrossRef] [PubMed]
- Auvin, S.; Ozturk, H.; Abaci, Y.T.; Mautino, G.; Meyer-Losic, F.; Jollivet, F.; Bashir, T.; de The, H.; Sahin, U. A molecule inducing androgen receptor degradation and selectively targeting prostate cancer cells. Life Sci. Alliance 2019, 2, e201800213. [Google Scholar] [CrossRef] [PubMed]
- Witus, S.R.; Stewart, M.D.; Klevit, R.E. The BRCA1/BARD1 ubiquitin ligase and its substrates. Biochem. J. 2021, 478, 3467–3483. [Google Scholar] [CrossRef] [PubMed]
- Densham, R.M.; Morris, J.R. The BRCA1 Ubiquitin ligase function sets a new trend for remodelling in DNA repair. Nucleus 2017, 8, 116–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, J.R.; Boutell, C.; Keppler, M.; Densham, R.; Weekes, D.; Alamshah, A.; Butler, L.; Galanty, Y.; Pangon, L.; Kiuchi, T.; et al. The SUMO modification pathway is involved in the BRCA1 response to genotoxic stress. Nature 2009, 462, 886–890. [Google Scholar] [CrossRef] [PubMed]
- Mo, Y.Y.; Yu, Y.; Theodosiou, E.; Ee, P.L.; Beck, W.T. A role for Ubc9 in tumorigenesis. Oncogene 2005, 24, 2677–2683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, W.; Wang, L.; Roehn, G.; Pearlstein, R.D.; Ali-Osman, F.; Pan, H.; Goldbrunner, R.; Krantz, M.; Harms, C.; Paschen, W. Small ubiquitin-like modifier 1-3 conjugation [corrected] is activated in human astrocytic brain tumors and is required for glioblastoma cell survival. Cancer Sci. 2013, 104, 70–77. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.F.; Gong, C.; Luo, M.; Yao, H.R.; Zeng, Y.J.; Su, F.X. Ubc9 expression predicts chemoresistance in breast cancer. Chin. J. Cancer 2011, 30, 638–644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moschos, S.J.; Smith, A.P.; Mandic, M.; Athanassiou, C.; Watson-Hurst, K.; Jukic, D.M.; Edington, H.D.; Kirkwood, J.M.; Becker, D. SAGE and antibody array analysis of melanoma-infiltrated lymph nodes: Identification of Ubc9 as an important molecule in advanced-stage melanomas. Oncogene 2007, 26, 4216–4225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shao, D.F.; Wang, X.H.; Li, Z.Y.; Xing, X.F.; Cheng, X.J.; Guo, T.; Du, H.; Hu, Y.; Dong, B.; Ding, N.; et al. High-level SAE2 promotes malignant phenotype and predicts outcome in gastric cancer. Am. J. Cancer Res. 2015, 5, 140–154. [Google Scholar] [PubMed]
- Zhang, H.; Kuai, X.; Ji, Z.; Li, Z.; Shi, R. Over-expression of small ubiquitin-related modifier-1 and sumoylated p53 in colon cancer. Cell Biochem. Biophys. 2013, 67, 1081–1087. [Google Scholar] [CrossRef] [PubMed]
- Kukkula, A.; Ojala, V.K.; Mendez, L.M.; Sistonen, L.; Elenius, K.; Sundvall, M. Therapeutic Potential of Targeting the SUMO Pathway in Cancer. Cancers 2021, 13, 4402. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, I.; Ito, A.; Hirai, G.; Nishimura, S.; Kawasaki, H.; Saitoh, H.; Kimura, K.; Sodeoka, M.; Yoshida, M. Ginkgolic acid inhibits protein SUMOylation by blocking formation of the E1-SUMO intermediate. Chem. Biol. 2009, 16, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Baek, S.H.; Ko, J.H.; Lee, J.H.; Kim, C.; Lee, H.; Nam, D.; Lee, J.; Lee, S.G.; Yang, W.M.; Um, J.Y.; et al. Ginkgolic Acid Inhibits Invasion and Migration and TGF-beta-Induced EMT of Lung Cancer Cells Through PI3K/Akt/mTOR Inactivation. J. Cell Physiol. 2017, 232, 346–354. [Google Scholar] [CrossRef] [PubMed]
- Qiao, L.; Zheng, J.; Jin, X.; Wei, G.; Wang, G.; Sun, X.; Li, X. Ginkgolic acid inhibits the invasiveness of colon cancer cells through AMPK activation. Oncol. Lett. 2017, 14, 5831–5838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Meng, X.; Zhang, D.; Xu, X.; Li, S.; Li, Y. Ginkgolic acid suppresses the invasion of HepG2 cells via downregulation of HGF/cMet signaling. Oncol. Rep. 2019, 41, 369–376. [Google Scholar] [PubMed]
- He, X.; Riceberg, J.; Soucy, T.; Koenig, E.; Minissale, J.; Gallery, M.; Bernard, H.; Yang, X.; Liao, H.; Rabino, C.; et al. Probing the roles of SUMOylation in cancer cell biology by using a selective SAE inhibitor. Nat. Chem. Biol. 2017, 13, 1164–1171. [Google Scholar] [CrossRef] [PubMed]
- Kessler, J.D.; Kahle, K.T.; Sun, T.; Meerbrey, K.L.; Schlabach, M.R.; Schmitt, E.M.; Skinner, S.O.; Xu, Q.; Li, M.Z.; Hartman, Z.C.; et al. A SUMOylation-dependent transcriptional subprogram is required for Myc-driven tumorigenesis. Science 2012, 335, 348–353. [Google Scholar] [CrossRef] [Green Version]
- Langston, S.P.; Grossman, S.; England, D.; Afroze, R.; Bence, N.; Bowman, D.; Bump, N.; Chau, R.; Chuang, B.C.; Claiborne, C.; et al. Discovery of TAK-981: A First-in-Class Inhibitor of SUMO-Activating Enzyme for the Treatment of Cancer. J. Med. Chem. 2021, 64, 2501–2520. [Google Scholar] [CrossRef] [PubMed]
- Du, L.; Liu, W.; Rosen, S.T. Targeting SUMOylation in cancer. Curr. Opin. Oncol. 2021, 33, 520–525. [Google Scholar] [CrossRef]
- Anderson, D.B.; Zanella, C.A.; Henley, J.M.; Cimarosti, H. Sumoylation: Implications for Neurodegenerative Diseases. Adv. Exp. Med. Biol. 2017, 963, 261–281. [Google Scholar] [PubMed]
- Krumova, P.; Weishaupt, J.H. Sumoylation in neurodegenerative diseases. Cell Mol. Life Sci. 2013, 70, 2123–2138. [Google Scholar] [CrossRef] [PubMed]
- Taylor, J.P.; Hardy, J.; Fischbeck, K.H. Toxic proteins in neurodegenerative disease. Science 2002, 296, 1991–1995. [Google Scholar] [CrossRef] [PubMed]
- Elbaum-Garfinkle, S. Matter over mind: Liquid phase separation and neurodegeneration. J. Biol. Chem. 2019, 294, 7160–7168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zbinden, A.; Perez-Berlanga, M.; De Rossi, P.; Polymenidou, M. Phase Separation and Neurodegenerative Diseases: A Disturbance in the Force. Dev. Cell 2020, 55, 45–68. [Google Scholar] [CrossRef] [PubMed]
- Dorval, V.; Fraser, P.E. SUMO on the road to neurodegeneration. Biochim. Biophys. Acta 2007, 1773, 694–706. [Google Scholar] [CrossRef] [Green Version]
- Fei, E.; Jia, N.; Yan, M.; Ying, Z.; Sun, Q.; Wang, H.; Zhang, T.; Ma, X.; Ding, H.; Yao, X.; et al. SUMO-1 modification increases human SOD1 stability and aggregation. Biochem. Biophys. Res. Commun. 2006, 347, 406–412. [Google Scholar] [CrossRef]
- Seyfried, N.T.; Gozal, Y.M.; Dammer, E.B.; Xia, Q.; Duong, D.M.; Cheng, D.; Lah, J.J.; Levey, A.I.; Peng, J. Multiplex SILAC analysis of a cellular TDP-43 proteinopathy model reveals protein inclusions associated with SUMOylation and diverse polyubiquitin chains. Mol. Cell Proteom. 2010, 9, 705–718. [Google Scholar] [CrossRef] [Green Version]
- Dorval, V.; Fraser, P.E. Small ubiquitin-like modifier (SUMO) modification of natively unfolded proteins tau and alpha-synuclein. J. Biol. Chem. 2006, 281, 9919–9924. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.Q.; Sarge, K.D. Sumoylation of amyloid precursor protein negatively regulates Abeta aggregate levels. Biochem. Biophys. Res. Commun. 2008, 374, 673–678. [Google Scholar] [CrossRef] [Green Version]
- Oh, Y.; Kim, Y.M.; Mouradian, M.M.; Chung, K.C. Human Polycomb protein 2 promotes alpha-synuclein aggregate formation through covalent SUMOylation. Brain Res. 2011, 1381, 78–89. [Google Scholar] [CrossRef]
- Riley, B.E.; Zoghbi, H.Y.; Orr, H.T. SUMOylation of the polyglutamine repeat protein: Ataxin-1, is dependent on a functional nuclear localization signal. J. Biol. Chem. 2005, 280, 21942–21948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steffan, J.S.; Agrawal, N.; Pallos, J.; Rockabrand, E.; Trotman, L.C.; Slepko, N.; Illes, K.; Lukacsovich, T.; Zhu, Y.Z.; Cattaneo, E.; et al. SUMO modification of Huntingtin and Huntington’s disease pathology. Science 2004, 304, 100–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Rourke, J.G.; Gareau, J.R.; Ochaba, J.; Song, W.; Rasko, T.; Reverter, D.; Lee, J.; Monteys, A.M.; Pallos, J.; Mee, L.; et al. SUMO-2 and PIAS1 modulate insoluble mutant huntingtin protein accumulation. Cell Rep. 2013, 4, 362–375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Escobedo, A.; Topal, B.; Kunze, M.B.A.; Aranda, J.; Chiesa, G.; Mungianu, D.; Bernardo-Seisdedos, G.; Eftekharzadeh, B.; Gairi, M.; Pierattelli, R.; et al. Side chain to main chain hydrogen bonds stabilize a polyglutamine helix in a transcription factor. Nat. Commun. 2019, 10, 2034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, H.B.; Xia, Y.Y.; Shu, X.J.; Liu, Z.C.; Feng, Y.; Liu, X.H.; Yu, G.; Yin, G.; Xiong, Y.S.; Zeng, K.; et al. SUMOylation at K340 inhibits tau degradation through deregulating its phosphorylation and ubiquitination. Proc. Natl. Acad. Sci. USA 2014, 111, 16586–16591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stefanis, L. Alpha-Synuclein in Parkinson’s disease. Cold Spring Harb. Perspect Med. 2012, 2, a009399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krumova, P.; Meulmeester, E.; Garrido, M.; Tirard, M.; Hsiao, H.H.; Bossis, G.; Urlaub, H.; Zweckstetter, M.; Kugler, S.; Melchior, F.; et al. Sumoylation inhibits alpha-synuclein aggregation and toxicity. J. Cell Biol. 2011, 194, 49–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morales, A.V.; Mira, H. Adult Neural Stem Cells: Born to Last. Front. Cell Dev. Biol. 2019, 7, 96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, X.; Moore, D.L. Neural stem cells: Developmental mechanisms and disease modeling. Cell Tissue Res. 2018, 371, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vishwakarma, S.K.; Bardia, A.; Tiwari, S.K.; Paspala, S.A.; Khan, A.A. Current concept in neural regeneration research: NSCs isolation. characterization and transplantation in various neurodegenerative diseases and stroke: A review. J. Adv. Res. 2014, 5, 277–294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chort, A.; Alves, S.; Marinello, M.; Dufresnois, B.; Dornbierer, J.G.; Tesson, C.; Latouche, M.; Baker, D.P.; Barkats, M.; El Hachimi, K.H.; et al. Interferon beta induces clearance of mutant ataxin 7 and improves locomotion in SCA7 knock-in mice. Brain 2013, 136, 1732–1745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakamoto, K.M.; Kim, K.B.; Kumagai, A.; Mercurio, F.; Crews, C.M.; Deshaies, R.J. Protacs: Chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proc. Natl. Acad. Sci. USA 2001, 98, 8554–8559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, L.; Liu, W.; Aldana-Masangkay, G.; Pozhitkov, A.; Pichiorri, F.; Chen, Y.; Rosen, S.T. SUMOylation inhibition enhances dexamethasone sensitivity in multiple myeloma. J. Exp. Clin. Cancer Res. 2022, 41, 8. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Zhang, Y.; Deng, T.; Gu, J.; Liu, J.; Yang, H.; Xu, Y.; Yan, Y.; Yang, F.; Zhang, H.; et al. PDPK1 regulates autophagosome biogenesis by binding to PIK3C3. Autophagy 2021, 17, 2166–2183. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Guo, C.; Lao, Y.; Yang, J.; Chen, F.; Zhao, Y.; Yang, Y.; Yang, J.; Yi, J. A fine-tuning mechanism underlying self-control for autophagy: DeSUMOylation of BECN1 by SENP3. Autophagy 2020, 16, 975–990. [Google Scholar] [CrossRef] [PubMed]
- Ramirez-Jarquin, U.N.; Sharma, M.; Zhou, W.; Shahani, N.; Subramaniam, S. Deletion of SUMO1 attenuates behavioral and anatomical deficits by regulating autophagic activities in Huntington disease. Proc. Natl. Acad. Sci. USA 2022, 119, 119. [Google Scholar] [CrossRef]
- Princz, A.; Pelisch, F.; Tavernarakis, N. SUMO promotes longevity and maintains mitochondrial homeostasis during ageing in Caenorhabditis elegans. Sci. Rep. 2020, 10, 15513. [Google Scholar] [CrossRef] [PubMed]
- Benson, M.; Iniguez-Lluhi, J.A.; Martens, J. Sumo Modification of Ion Channels. Adv. Exp. Med. Biol. 2017, 963, 127–141. [Google Scholar]
- Kho, C.; Lee, A.; Jeong, D.; Oh, J.G.; Gorski, P.A.; Fish, K.; Sanchez, R.; DeVita, R.J.; Christensen, G.; Dahl, R.; et al. Small-molecule activation of SERCA2a SUMOylation for the treatment of heart failure. Nat. Commun. 2015, 6, 7229. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sahin, U.; de Thé, H.; Lallemand-Breitenbach, V. Sumoylation in Physiology, Pathology and Therapy. Cells 2022, 11, 814. https://doi.org/10.3390/cells11050814
Sahin U, de Thé H, Lallemand-Breitenbach V. Sumoylation in Physiology, Pathology and Therapy. Cells. 2022; 11(5):814. https://doi.org/10.3390/cells11050814
Chicago/Turabian StyleSahin, Umut, Hugues de Thé, and Valérie Lallemand-Breitenbach. 2022. "Sumoylation in Physiology, Pathology and Therapy" Cells 11, no. 5: 814. https://doi.org/10.3390/cells11050814
APA StyleSahin, U., de Thé, H., & Lallemand-Breitenbach, V. (2022). Sumoylation in Physiology, Pathology and Therapy. Cells, 11(5), 814. https://doi.org/10.3390/cells11050814