
The Role of E3 Ligase Pirh2 in Disease

 , and
, and
Abstract
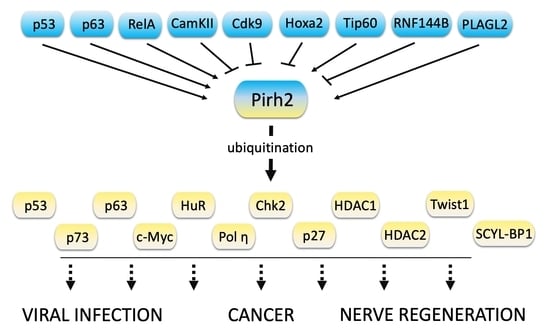
:
1. Introduction
2. The Structure of Pirh2
3. Pirh2 Regulation
4. The Protein Targets of Pirh2
4.1. p53 Family Proteins
4.2. c-Myc Oncogene
4.3. RNA Binding Protein HuR
4.4. DNA Polymerase Eta
4.5. The Chk2 Protein Kinase
4.6. The Cyclin-Dependent Kinase (CDK) Inhibitor p27
4.7. Histone Deacetylases HDAC1 and HDAC2
4.8. Twist1
4.9. SCYL1-BP1
5. The Role of Pirh2 in Different Types of Cancer
5.1. Lung Cancer
5.2. Prostate Cancer
5.3. Oral Cancer
5.4. Hepatocellular Carcinoma
5.5. Head and Neck Cancer
5.6. Glioma
5.7. Breast Cancer
5.8. Multiple Myeloma
6. The Role of Pirh2 in Viral Infections
7. The Role of Pirh2 in Regeneration of Nerve Tissue Damage
8. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Luo, H. Interplay between the virus and the ubiquitin–proteasome system: Molecular mechanism of viral pathogenesis. Curr. Opin. Virol. 2016, 17, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Shen, M.; Schmitt, S.; Buac, D.; Dou, Q.P. Targeting the ubiquitin–proteasome system for cancer therapy. Expert Opin. Ther. Targets 2013, 17, 1091–1108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yadav, D.; Lee, J.Y.; Puranik, N.; Chauhan, P.S.; Chavda, V.; Jin, J.-O.; Lee, P.C. Modulating the Ubiquitin–Proteasome System: A Therapeutic Strategy for Autoimmune Diseases. Cells 2022, 11, 1093. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Q.; Huang, T.; Zhang, L.; Zhou, Y.; Luo, H.; Xu, H.; Wang, X. Dysregulation of ubiquitin-proteasome system in neurodegenerative diseases. Front. Aging Neurosci. 2016, 8, 303. [Google Scholar] [CrossRef] [PubMed]
- Konopleva, M.; Martinelli, G.; Daver, N.; Papayannidis, C.; Wei, A.; Higgins, B.; Ott, M.; Mascarenhas, J.; Andreeff, M. MDM2 inhibition: An important step forward in cancer therapy. Leukemia 2020, 34, 2858–2874. [Google Scholar] [CrossRef] [PubMed]
- Haronikova, L.; Bonczek, O.; Zatloukalova, P.; Kokas-Zavadil, F.; Kucerikova, M.; Coates, P.J.; Fahraeus, R.; Vojtesek, B. Resistance mechanisms to inhibitors of p53-MDM2 interactions in cancer therapy: Can we overcome them? Cell. Mol. Biol. Lett. 2021, 26, 1–33. [Google Scholar] [CrossRef]
- Wang, W.; Qin, J.J.; Rajaei, M.; Li, X.; Yu, X.; Hunt, C.; Zhang, R. Targeting MDM2 for novel molecular therapy: Beyond oncology. Med. Res. Rev. 2020, 40, 856–880. [Google Scholar] [CrossRef] [PubMed]
- Beitel, L.; Elhaji, Y.; Lumbroso, R.; Wing, S.; Panet-Raymond, V.; Gottlieb, B.; Pinsky, L.; Trifiro, M. Cloning and characterization of an androgen receptor N-terminal-interacting protein with ubiquitin-protein ligase activity. J. Mol. Endocrinol. 2002, 29, 41–60. [Google Scholar] [CrossRef]
- Li, W.; Bengtson, M.H.; Ulbrich, A.; Matsuda, A.; Reddy, V.A.; Orth, A.; Chanda, S.K.; Batalov, S.; Joazeiro, C.A. Genome-wide and functional annotation of human E3 ubiquitin ligases identifies MULAN, a mitochondrial E3 that regulates the organelle’s dynamics and signaling. PLoS ONE 2008, 3, e1487. [Google Scholar] [CrossRef]
- Metzger, M.B.; Hristova, V.A.; Weissman, A.M. HECT and RING finger families of E3 ubiquitin ligases at a glance. J. Cell Sci. 2012, 125, 531–537. [Google Scholar] [CrossRef] [Green Version]
- Sheng, Y.; Laister, R.C.; Lemak, A.; Wu, B.; Tai, E.; Duan, S.; Lukin, J.; Sunnerhagen, M.; Srisailam, S.; Karra, M. Molecular basis of Pirh2-mediated p53 ubiquitylation. Nature Struct. Mol. Biol. 2008, 15, 1334. [Google Scholar] [CrossRef] [PubMed]
- Corcoran, C.A.; Montalbano, J.; Sun, H.; He, Q.; Huang, Y.; Sheikh, M.S. Identification and characterization of two novel isoforms of Pirh2 ubiquitin ligase that negatively regulate p53 independent of RING finger domains. J. Biol. Chem. 2009, 284, 21955–21970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, G.; Sun, M.; Zhang, L.; Zhou, J.; Wang, Y.; Huo, K. A novel hPirh2 splicing variant without ubiquitin protein ligase activity interacts with p53 and is down-regulated in hepatocellular carcinoma. FEBS Lett. 2010, 584, 2772–2778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, J.; Huang, Y.; Sheikh, M.S. Identification of Pirh2D, an additional novel isoform of Pirh2 ubiquitin ligase. Mol. Cell. Pharmacol. 2010, 2, 21. [Google Scholar] [PubMed]
- Leng, R.P.; Lin, Y.; Ma, W.; Wu, H.; Lemmers, B.; Chung, S.; Parant, J.M.; Lozano, G.; Hakem, R.; Benchimol, S. Pirh2, a p53-induced ubiquitin-protein ligase, promotes p53 degradation. Cell 2003, 112, 779–791. [Google Scholar] [CrossRef] [Green Version]
- Duan, W.; Gao, L.; Wu, X.; Zhang, Y.; Otterson, G.A.; Villalona-Calero, M.A. Differential response between the p53 ubiquitin–protein ligases Pirh2 and MdM2 following DNA damage in human cancer cells. Exp. Cell Res. 2006, 312, 3370–3378. [Google Scholar] [CrossRef] [PubMed]
- Daks, A.; Petukhov, A.; Shuvalov, O.Y.; Vasil’eva, E.; Melino, G.; Barlev, N.; Fedorova, O. Tumor suppressor p63 regulates Pirh2 ubiquitin ligase expression. Cell Tissue Biol. 2016, 10, 202–205. [Google Scholar] [CrossRef]
- Wang, F.; Zheng, J.; Yang, J.; Luo, T.; Xu, J.; Yang, Y.; Gu, Y.; Zeng, Y. N-α-Acetyltransferase 10 inhibits invasion and metastasis of oral squamous cell carcinoma via regulating Pirh2-p53 signalling pathway. J. Cell. Mol. Med. 2022, 00, 1–14. [Google Scholar] [CrossRef]
- Duan, S.; Yao, Z.; Hou, D.; Wu, Z.; Zhu, W.g.; Wu, M. Phosphorylation of Pirh2 by Calmodulin-dependent kinase II impairs its ability to ubiquitinate p53. EMBO J. 2007, 26, 3062–3074. [Google Scholar] [CrossRef] [Green Version]
- Bagashev, A.; Fan, S.; Mukerjee, R.; Paolo Claudio, P.; Chabrashvili, T.; Leng, R.P.; Benchimol, S.; Sawaya, B.E. Cdk9 phosphorylates Pirh2 protein and prevents degradation of p53 protein. Cell Cycle 2013, 12, 1569–1577. [Google Scholar] [CrossRef] [Green Version]
- Bergiers, I.; Bridoux, L.; Nguyen, N.; Twizere, J.-C.; Rezsöhazy, R. The homeodomain transcription factor Hoxa2 interacts with and promotes the proteasomal degradation of the E3 ubiquitin protein ligase RCHY1. PLoS ONE 2013, 8, e80387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bridoux, L.; Deneyer, N.; Bergiers, I.; Rezsohazy, R. Molecular analysis of the HOXA2-dependent degradation of RCHY1. PLoS ONE 2015, 10, e0141347. [Google Scholar] [CrossRef] [PubMed]
- Logan, I.R.; Sapountzi, V.; Gaughan, L.; Neal, D.E.; Robson, C.N. Control of human PIRH2 protein stability: Involvement of TIP60 and the proteasome. J. Biol. Chem. 2004, 279, 11696–11704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, G.; Ning, J.; Yang, Y.-C. PLAGL2 controls the stability of Pirh2, an E3 ubiquitin ligase for p53. Biochem. Biophys. Res. Commun. 2007, 364, 344–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, G.; Gong, Y.; Wang, Q.; Wang, L.; Zhang, X. miR-100 antagonism triggers apoptosis by inhibiting ubiquitination-mediated p53 degradation. Oncogene 2017, 36, 1023–1037. [Google Scholar] [CrossRef]
- Jung, Y.-S.; Qian, Y.; Chen, X. Pirh2 RING-finger E3 ubiquitin ligase: Its role in tumorigenesis and cancer therapy. FEBS Lett. 2012, 586, 1397–1402. [Google Scholar] [CrossRef] [Green Version]
- Bailey, T.L.; Johnson, J.; Grant, C.E.; Noble, W.S. The MEME suite. Nucleic Acids Res. 2015, 43, 39–49. [Google Scholar] [CrossRef] [Green Version]
- Opazo, P.; Labrecque, S.; Tigaret, C.M.; Frouin, A.; Wiseman, P.W.; De Koninck, P.; Choquet, D. CaMKII triggers the diffusional trapping of surface AMPARs through phosphorylation of stargazin. Neuron 2010, 67, 239–252. [Google Scholar] [CrossRef] [Green Version]
- Sanhueza, M.; Lisman, J. The CaMKII/NMDAR complex as a molecular memory. Mol. Brain 2013, 6, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Bourguignon, L.Y.; Gilad, E.; Brightman, A.; Diedrich, F.; Singleton, P. Hyaluronan-CD44 interaction with leukemia-associated RhoGEF and epidermal growth factor receptor promotes Rho/Ras co-activation, phospholipase Cϵ-Ca2+ signaling, and cytoskeleton modification in head and neck squamous cell carcinoma cells. J. Biol. Chem. 2006, 281, 14026–14040. [Google Scholar] [CrossRef] [Green Version]
- Flentke, G.R.; Garic, A.; Hernandez, M.; Smith, S.M. CaMKII represses transcriptionally active β-catenin to mediate acute ethanol neurodegeneration and can phosphorylate β-catenin. J. Neurochem. 2014, 128, 523–535. [Google Scholar] [PubMed] [Green Version]
- Wheeler, D.G.; Barrett, C.F.; Groth, R.D.; Safa, P.; Tsien, R.W. CaMKII locally encodes L-type channel activity to signal to nuclear CREB in excitation–transcription coupling. J. Cell Biol. 2008, 183, 849–863. [Google Scholar] [CrossRef] [PubMed]
- Daniels, L.; Bell, J.R.; Delbridge, L.; McDonald, F.J.; Lamberts, R.R.; Erickson, J.R. The role of CaMKII in diabetic heart dysfunction. Heart Fail. Rev. 2015, 20, 589–600. [Google Scholar] [CrossRef] [PubMed]
- Pellicena, P.; Schulman, H. CaMKII inhibitors: From research tools to therapeutic agents. Front. Pharmacol. 2014, 5, 21. [Google Scholar] [CrossRef] [Green Version]
- dos Santos Paparidis, N.F.; Durvale, M.C.; Canduri, F. The emerging picture of CDK9/P-TEFb: More than 20 years of advances since PITALRE. Mol. BioSys. 2017, 13, 246–276. [Google Scholar] [CrossRef]
- Price, D.H. P-TEFb, a cyclin-dependent kinase controlling elongation by RNA polymerase II. Mol. Cell. Biol. 2000, 20, 2629–2634. [Google Scholar] [CrossRef] [Green Version]
- Kretz, A.-L.; Schaum, M.; Richter, J.; Kitzig, E.F.; Engler, C.C.; Leithäuser, F.; Henne-Bruns, D.; Knippschild, U.; Lemke, J. CDK9 is a prognostic marker and therapeutic target in pancreatic cancer. Tumor Biol. 2017, 39, 1010428317694304. [Google Scholar] [CrossRef] [Green Version]
- Rahaman, M.H.; Kumarasiri, M.; Mekonnen, L.B.; Yu, M.; Diab, S.; Albrecht, H.; Milne, R.W.; Wang, S. Targeting CDK9: A promising therapeutic opportunity in prostate cancer. Endocr.-Relat. Cancer 2016, 23, T211–T226. [Google Scholar] [CrossRef] [Green Version]
- He, S.; Fang, X.; Xia, X.; Hou, T.; Zhang, T. Targeting CDK9: A novel biomarker in the treatment of endometrial cancer. Oncol. Rep. 2020, 44, 1929–1938. [Google Scholar] [CrossRef]
- Morales, F.; Giordano, A. Overview of CDK9 as a target in cancer research. Cell Cycle 2016, 15, 519–527. [Google Scholar] [CrossRef] [Green Version]
- Boffo, S.; Damato, A.; Alfano, L.; Giordano, A. CDK9 inhibitors in acute myeloid leukemia. J. Exp. Clin. Cancer Res. 2018, 37, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mandal, R.; Becker, S.; Strebhardt, K. Targeting CDK9 for Anti-Cancer Therapeutics. Cancers 2021, 13, 2181. [Google Scholar] [CrossRef] [PubMed]
- Olson, C.M.; Jiang, B.; Erb, M.A.; Liang, Y.; Doctor, Z.M.; Zhang, Z.; Zhang, T.; Kwiatkowski, N.; Boukhali, M.; Green, J.L. Pharmacological perturbation of CDK9 using selective CDK9 inhibition or degradation. Nat. Chem. Biol. 2018, 14, 163–170. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Shen, F.; Wang, H.; Li, A.; Wang, J.; Du, L.; Liu, B.; Zhang, B.; Lian, X.; Pang, B. Abnormally high expression of HOXA2 as an independent factor for poor prognosis in glioma patients. Cell Cycle 2020, 19, 1632–1640. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Bai, Y.; Feng, Z.; Li, W.; Yang, C.; Guo, Y.; Lin, C.; Zhang, Y.; He, Q.; Hu, G. Study of promoter methylation patterns of HOXA2, HOXA5, and HOXA6 and its clinicopathological characteristics in colorectal cancer. Front. Oncol. 2019, 9, 394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.-P.; Peng, C.-C.; Chung, I.-C.; Huang, M.-Y.; Huang, S.-T.; Chen, C.-C.; Chang, K.-P.; Hsu, C.-L.; Chang, Y.-S. Aberrantly hypermethylated Homeobox A2 derepresses metalloproteinase-9 through TBP and promotes invasion in Nasopharyngeal carcinoma. Oncotarget 2013, 4, 2154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deneyer, N.; Bridoux, L.; Bombled, C.; Pringels, T.; Bergiers, I.; dit Ruys, S.P.; Vertommen, D.; Twizere, J.-C.; Rezsohazy, R. HOXA2 activity regulation by cytoplasmic relocation, protein stabilization and post-translational modification. Biochim. Et Biophys. Acta (BBA)-Gene Regul. Mech. 2019, 1862, 194404. [Google Scholar] [CrossRef]
- Bhatlekar, S.; Fields, J.Z.; Boman, B.M. HOX genes and their role in the development of human cancers. J. Mol. Med. 2014, 92, 811–823. [Google Scholar] [CrossRef]
- Tsimokha, A.S.; Kulichkova, V.A.; Karpova, E.V.; Zaykova, J.J.; Aksenov, N.D.; Vasilishina, A.A.; Kropotov, A.V.; Antonov, A.; Barlev, N.A. DNA damage modulates interactions between microRNAs and the 26S proteasome. Oncotarget 2014, 5, 3555. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Jiang, X.; Chen, S.; Fernandes, N.; Price, B.D. A role for the Tip60 histone acetyltransferase in the acetylation and activation of ATM. Proc. Natl. Acad. Sci. USA 2005, 102, 13182–13187. [Google Scholar] [CrossRef] [Green Version]
- Gaughan, L.; Logan, I.R.; Cook, S.; Neal, D.E.; Robson, C.N. Tip60 and histone deacetylase 1 regulate androgen receptor activity through changes to the acetylation status of the receptor. J. Biol. Chem. 2002, 277, 25904–25913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghobashi, A.H.; Kamel, M.A. Tip60: Updates. J. Appl. Genet. 2018, 59, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Ikura, T.; Ogryzko, V.V.; Grigoriev, M.; Groisman, R.; Wang, J.; Horikoshi, M.; Scully, R.; Qin, J.; Nakatani, Y. Involvement of the TIP60 histone acetylase complex in DNA repair and apoptosis. Cell 2000, 102, 463–473. [Google Scholar] [CrossRef] [Green Version]
- Tang, Y.; Luo, J.; Zhang, W.; Gu, W. Tip60-dependent acetylation of p53 modulates the decision between cell-cycle arrest and apoptosis. Mol. Cell 2006, 24, 827–839. [Google Scholar] [CrossRef]
- Halkidou, K.; Gnanapragasam, V.J.; Mehta, P.B.; Logan, I.R.; Brady, M.E.; Cook, S.; Leung, H.Y.; Neal, D.E.; Robson, C.N. Expression of Tip60, an androgen receptor coactivator, and its role in prostate cancer development. Oncogene 2003, 22, 2466–2477. [Google Scholar] [CrossRef] [Green Version]
- Mattera, L.; Escaffit, F.; Pillaire, M.; Selves, J.; Tyteca, S.; Hoffmann, J.; Gourraud, P.; Chevillard-Briet, M.; Cazaux, C.; Trouche, D. The p400/Tip60 ratio is critical for colorectal cancer cell proliferation through DNA damage response pathways. Oncogene 2009, 28, 1506–1517. [Google Scholar] [CrossRef] [Green Version]
- Chen, G.; Cheng, Y.; Tang, Y.; Martinka, M.; Li, G. Role of Tip60 in human melanoma cell migration, metastasis, and patient survival. J. Investig. Dermatol. 2012, 132, 2632–2641. [Google Scholar] [CrossRef] [Green Version]
- Judes, G.; Rifaï, K.; Ngollo, M.; Daures, M.; Bignon, Y.-J.; Penault-Llorca, F.; Bernard-Gallon, D. A bivalent role of TIP60 histone acetyl transferase in human cancer. Epigenomics 2015, 7, 1351–1363. [Google Scholar] [CrossRef]
- Zhou, J.; Liu, H.; Zhang, L.; Liu, X.; Zhang, C.; Wang, Y.; He, Q.; Zhang, Y.; Li, Y.; Chen, Q. DJ-1 promotes colorectal cancer progression through activating PLAGL2/Wnt/BMP4 axis. Cell Death Dis. 2018, 9, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Hu, W.; Zheng, S.; Guo, H.; Dai, B.; Ni, J.; Shi, Y.; Bian, H.; Li, L.; Shen, Y.; Wu, M. PLAGL2-EGFR-HIF-1/2α Signaling Loop Promotes HCC Progression and Erlotinib Insensitivity. Hepatology 2021, 73, 674–691. [Google Scholar] [CrossRef]
- Landrette, S.F.; Kuo, Y.-H.; Hensen, K.; van Waalwijk van Doorn-Khosrovani, S.B.; Perrat, P.N.; Van de Ven, W.J.; Delwel, R.; Castilla, L.H. Plag1 and Plagl2 are oncogenes that induce acute myeloid leukemia in cooperation with Cbfb-MYH11. Blood 2005, 105, 2900–2907. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Zhou, Z.; Han, S.; Chen, J.; Liu, Z.; Zhang, X.; Yuan, W.; Ji, J.; Shu, X. PLAGL2 promotes epithelial–mesenchymal transition and mediates colorectal cancer metastasis via β-catenin-dependent regulation of ZEB1. Br. J. Cancer 2020, 122, 578–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, L.; Zhao, N.; Zhou, Z.; Chen, J.; Han, S.; Zhang, X.; Bao, H.; Yuan, W.; Shu, X. PLAGL2 promotes the proliferation and migration of gastric cancer cells via USP37-mediated deubiquitination of Snail1. Theranostics 2021, 11, 700. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Sun, L.; Liu, R.; Mo, H.; Niu, Y.; Chen, T.; Wang, Y.; Han, S.; Tu, K.; Liu, Q. Long non-coding RNA MAPKAPK5-AS1/PLAGL2/HIF-1α signaling loop promotes hepatocellular carcinoma progression. J. Exp. Clin. Cancer Res. 2021, 40, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Hendriks, I.A.; D’souza, R.C.; Yang, B.; Verlaan-de Vries, M.; Mann, M.; Vertegaal, A.C. Uncovering global SUMOylation signaling networks in a site-specific manner. Nat. Struct. Mol. Biol. 2014, 21, 927–936. [Google Scholar] [CrossRef] [Green Version]
- Akimov, V.; Barrio-Hernandez, I.; Hansen, S.V.; Hallenborg, P.; Pedersen, A.-K.; Bekker-Jensen, D.B.; Puglia, M.; Christensen, S.D.; Vanselow, J.T.; Nielsen, M.M. UbiSite approach for comprehensive mapping of lysine and N-terminal ubiquitination sites. Nat. Struct. Mol. Biol. 2018, 25, 631–640. [Google Scholar] [CrossRef]
- Boeing, S.; Williamson, L.; Encheva, V.; Gori, I.; Saunders, R.E.; Instrell, R.; Aygün, O.; Rodriguez-Martinez, M.; Weems, J.C.; Kelly, G.P. Multiomic analysis of the UV-induced DNA damage response. Cell Rep. 2016, 15, 1597–1610. [Google Scholar] [CrossRef] [Green Version]
- Mertins, P.; Qiao, J.W.; Patel, J.; Udeshi, N.D.; Clauser, K.R.; Mani, D.; Burgess, M.W.; Gillette, M.A.; Jaffe, J.D.; Carr, S.A. Integrated proteomic analysis of post-translational modifications by serial enrichment. Nat. Methods 2013, 10, 634–637. [Google Scholar] [CrossRef]
- Udeshi, N.D.; Svinkina, T.; Mertins, P.; Kuhn, E.; Mani, D.; Qiao, J.W.; Carr, S.A. Refined preparation and use of anti-diglycine remnant (K-ε-GG) antibody enables routine quantification of 10,000 s of ubiquitination sites in single proteomics experiments. Mol. Cell. Proteom. 2013, 12, 825–831. [Google Scholar] [CrossRef] [Green Version]
- Daks, A.; Petukhov, A.; Fedorova, O.; Shuvalov, O.; Kizenko, A.; Tananykina, E.; Vasileva, E.; Semenov, O.; Bottrill, A.; Barlev, N. The RNA-binding protein HuR is a novel target of Pirh2 E3 ubiquitin ligase. Cell Death Dis. 2021, 12, 1–14. [Google Scholar] [CrossRef]
- Bell, H.S.; Ryan, K.M. Targeting the p53 family for cancer therapy:‘big brother’joins the fight. Cell Cycle 2007, 6, 1995–2000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marouco, D.; Garabadgiu, A.V.; Melino, G.; Barlev, N.A. Lysine-specific modifications of p53: A matter of life and death? Oncotarget 2013, 4, 1556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Tavana, O.; Gu, W. p53 modifications: Exquisite decorations of the powerful guardian. J. Mol. Cell Biol. 2019, 11, 564–577. [Google Scholar] [CrossRef] [Green Version]
- Esrig, D.; Spruck, C., 3rd; Nichols, P.W.; Chaiwun, B.; Steven, K.; Groshen, S.; Chen, S.C.; Skinner, D.G.; Jones, P.A.; Cote, R.J. p53 nuclear protein accumulation correlates with mutations in the p53 gene, tumor grade, and stage in bladder cancer. Am. J. Pathol. 1993, 143, 1389. [Google Scholar]
- Daks, A.; Melino, D.; Barlev, N. The role of different E3 ubiquitin ligases in regulation of the P53 tumor suppressor protein. Tsitologiia 2013, 55, 673–687. [Google Scholar] [PubMed]
- Maya, R.; Balass, M.; Kim, S.-T.; Shkedy, D.; Leal, J.-F.M.; Shifman, O.; Moas, M.; Buschmann, T.; Ronai, Z.e.; Shiloh, Y. ATM-dependent phosphorylation of Mdm2 on serine 395: Role in p53 activation by DNA damage. Genes Dev. 2001, 15, 1067–1077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Leary, K.A.; Mendrysa, S.M.; Vaccaro, A.; Perry, M.E. Mdm2 regulates p53 independently of p19ARF in homeostatic tissues. Mol. Cell. Biol. 2004, 24, 186–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.; Gu, W. The multiple levels of regulation by p53 ubiquitination. Cell Death Differ. 2010, 17, 86–92. [Google Scholar] [CrossRef]
- Fedorova, O.; Daks, A.; Petrova, V.; Petukhov, A.; Lezina, L.; Shuvalov, O.; Davidovich, P.; Kriger, D.; Lomert, E.; Tentler, D. Novel isatin-derived molecules activate p53 via interference with Mdm2 to promote apoptosis. Cell Cycle 2018, 17, 1917–1930. [Google Scholar] [CrossRef] [Green Version]
- Tai, E. Characterization of the E3 Ubiquitin Ligase Pirh2. Ph.D Thesis, University of Toronto, Toronto, ON, Canada, 2010. [Google Scholar]
- Canman, C.E.; Lim, D.-S.; Cimprich, K.A.; Taya, Y.; Tamai, K.; Sakaguchi, K.; Appella, E.; Kastan, M.B.; Siliciano, J.D. Activation of the ATM kinase by ionizing radiation and phosphorylation of p53. Science 1998, 281, 1677–1679. [Google Scholar]
- Shieh, S.-Y.; Ikeda, M.; Taya, Y.; Prives, C. DNA damage-induced phosphorylation of p53 alleviates inhibition by MDM2. Cell 1997, 91, 325–334. [Google Scholar] [CrossRef] [Green Version]
- de Oca Luna, R.M.; Wagner, D.S.; Lozano, G. Rescue of early embryonic lethality in mdm2-deficient mice by deletion of p53. Nature 1995, 378, 203–206. [Google Scholar] [CrossRef] [PubMed]
- Hakem, A.; Bohgaki, M.; Lemmers, B.; Tai, E.; Salmena, L.; Matysiak-Zablocki, E.; Jung, Y.-S.; Karaskova, J.; Kaustov, L.; Duan, S. Role of Pirh2 in mediating the regulation of p53 and c-Myc. PLoS Genet. 2011, 7, e1002360. [Google Scholar] [CrossRef] [PubMed]
- Yan, W.; Jung, Y.-S.; Zhang, Y.; Chen, X. Arsenic trioxide reactivates proteasome-dependent degradation of mutant p53 protein in cancer cells in part via enhanced expression of Pirh2 E3 ligase. PLoS ONE 2014, 9, e103497. [Google Scholar] [CrossRef]
- Vijayakumaran, R.; Tan, K.H.; Miranda, P.J.; Haupt, S.; Haupt, Y. Regulation of mutant p53 protein expression. Front. Oncol. 2015, 5, 284. [Google Scholar] [CrossRef] [Green Version]
- Haupt, Y.; Maya, R.; Kazaz, A.; Oren, M. Mdm2 promotes the rapid degradation of p53. Nature 1997, 387, 296–299. [Google Scholar] [CrossRef]
- Kubbutat, M.H.; Jones, S.N.; Vousden, K.H. Regulation of p53 stability by Mdm2. Nature 1997, 387, 299–303. [Google Scholar] [CrossRef]
- Lukashchuk, N.; Vousden, K.H. Ubiquitination and degradation of mutant p53. Mol. Cell. Biol. 2007, 27, 8284–8295. [Google Scholar] [CrossRef] [Green Version]
- McKeon, F.; Melino, G. Fog of war: The emerging p53 family. Cell Cycle 2007, 6, 229–232. [Google Scholar] [CrossRef]
- Mills, A.A.; Zheng, B.; Wang, X.-J.; Vogel, H.; Roop, D.R.; Bradley, A. p63 is a p53 homologue required for limb and epidermal morphogenesis. Nature 1999, 398, 708–713. [Google Scholar] [CrossRef]
- Yang, A.; Walker, N.; Bronson, R.; Kaghad, M.; Oosterwegel, M.; Bonnin, J.; Vagner, C.; Bonnet, H.; Dikkes, P.; Sharpe, A. p73-deficient mice have neurological, pheromonal and inflammatory defects but lack spontaneous tumours. Nature 2000, 404, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Yang, A.; McKeon, F. P63 and P73: P53 mimics, menaces and more. Nat. Rev. Mol. Cell Biol. 2000, 1, 199–207. [Google Scholar] [CrossRef] [PubMed]
- Jung, Y.-S.; Qian, Y.; Yan, W.; Chen, X. Pirh2 E3 ubiquitin ligase modulates keratinocyte differentiation through p63. J. Investig. Dermatol. 2013, 133, 1178–1187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, H.; Abou, Z.R.; Flores, E.R.; Leng, R.P. Pirh2, a ubiquitin E3 ligase, inhibits p73 transcriptional activity by promoting its ubiquitination. Mol. Cancer Res. 2011, 9, 1780–1790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, Y.-S.; Qian, Y.; Chen, X. The p73 tumor suppressor is targeted by Pirh2 RING finger E3 ubiquitin ligase for the proteasome-dependent degradation. J. Biol. Chem. 2011, 286, 35388–35395. [Google Scholar] [CrossRef] [Green Version]
- Abou Zeinab, R.; Wu, H.H.; Abuetabh, Y.; Leng, S.; Sergi, C.; Eisenstat, D.D.; Leng, R.P. Pirh2, an E3 ligase, regulates the AIP4–p73 regulatory pathway by modulating AIP4 expression and ubiquitination. Carcinogenesis 2021, 42, 650–662. [Google Scholar] [CrossRef]
- Jackstadt, R.; Hermeking, H. MicroRNAs as regulators and mediators of c-MYC function. Biochim. Et Biophys. Acta (BBA)-Gene Regul. Mech. 2015, 1849, 544–553. [Google Scholar] [CrossRef]
- McMahon, S.B. MYC and the control of apoptosis. Cold Spring Harb. Perspect. Med. 2014, 4, a014407. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, G.J. Emerging roles of Myc in stem cell biology and novel tumor therapies. J. Exp. Clin. Cancer Res. 2018, 37, 1–20. [Google Scholar]
- Chen, T.; You, Y.; Jiang, H.; Wang, Z.Z. Epithelial–mesenchymal transition (EMT): A biological process in the development, stem cell differentiation, and tumorigenesis. J. Cell. Physiol. 2017, 232, 3261–3272. [Google Scholar] [CrossRef]
- Gordan, J.D.; Thompson, C.B.; Simon, M.C. HIF and c-Myc: Sibling rivals for control of cancer cell metabolism and proliferation. Cancer Cell 2007, 12, 108–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daks, A.; Petukhov, A.; Fedorova, O.; Shuvalov, O.; Merkulov, V.; Vasileva, E.; Antonov, A.; Barlev, N.A. E3 ubiquitin ligase Pirh2 enhances tumorigenic properties of human non-small cell lung carcinoma cells. Genes Cancer 2016, 7, 383. [Google Scholar] [CrossRef] [PubMed]
- Zucal, C.; D’Agostino, V.; Loffredo, R.; Mantelli, B.; Thongon, N.; Lal, P.; Latorre, E.; Provenzani, A. Targeting the multifaceted HuR protein, benefits and caveats. Curr. Drug Targets 2015, 16, 499–515. [Google Scholar] [CrossRef] [PubMed]
- Cherry, J.; Karschner, V.; Jones, H.; Pekala, P.H. HuR, an RNA-binding protein, involved in the control of cellular differentiation. In Vivo 2006, 20, 17–23. [Google Scholar]
- Abdelmohsen, K.; Gorospe, M. Posttranscriptional regulation of cancer traits by HuR. Wiley Interdiscip. Rev. RNA 2010, 1, 214–229. [Google Scholar] [CrossRef]
- Zybura-Broda, K.; Wolder-Gontarek, M.; Ambrozek-Latecka, M.; Choros, A.; Bogusz, A.; Wilemska-Dziaduszycka, J.; Rylski, M. HuR (Elavl1) and HuB (Elavl2) stabilize matrix metalloproteinase-9 mRNA during seizure-induced Mmp-9 expression in neurons. Front. Neurosci. 2018, 12, 224. [Google Scholar] [CrossRef]
- Wang, W.; Caldwell, M.C.; Lin, S.; Furneaux, H.; Gorospe, M. HuR regulates cyclin A and cyclin B1 mRNA stability during cell proliferation. EMBO J. 2000, 19, 2340–2350. [Google Scholar] [CrossRef] [Green Version]
- Galbán, S.; Kuwano, Y.; Pullmann Jr, R.; Martindale, J.L.; Kim, H.H.; Lal, A.; Abdelmohsen, K.; Yang, X.; Dang, Y.; Liu, J.O. RNA-binding proteins HuR and PTB promote the translation of hypoxia-inducible factor 1α. Mol. Cell. Biol. 2008, 28, 93–107. [Google Scholar] [CrossRef] [Green Version]
- Kullmann, M.; Göpfert, U.; Siewe, B.; Hengst, L. ELAV/Hu proteins inhibit p27 translation via an IRES element in the p27 5′ UTR. Genes Dev. 2002, 16, 3087–3099. [Google Scholar] [CrossRef] [Green Version]
- Glorian, V.; Maillot, G.; Polès, S.; Iacovoni, J.; Favre, G.; Vagner, S. HuR-dependent loading of miRNA RISC to the mRNA encoding the Ras-related small GTPase RhoB controls its translation during UV-induced apoptosis. Cell Death Differ. 2011, 18, 1692–1701. [Google Scholar] [CrossRef]
- Kim, H.H.; Kuwano, Y.; Srikantan, S.; Lee, E.K.; Martindale, J.L.; Gorospe, M. HuR recruits let-7/RISC to repress c-Myc expression. Genes Dev. 2009, 23, 1743–1748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, X.; Xu, L. The RNA-binding protein HuR in human cancer: A friend or foe? Adv. Drug Deliv. Rev. 2022, 184, 114179. [Google Scholar] [CrossRef] [PubMed]
- Latorre, E.; Tebaldi, T.; Viero, G.; Spartà, A.M.; Quattrone, A.; Provenzani, A. Downregulation of HuR as a new mechanism of doxorubicin resistance in breast cancer cells. Mol. Cancer 2012, 11, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gallouzi, I.-E.; Brennan, C.M.; Stenberg, M.G.; Swanson, M.S.; Eversole, A.; Maizels, N.; Steitz, J.A. HuR binding to cytoplasmic mRNA is perturbed by heat shock. Proc. Natl. Acad. Sci. USA 2000, 97, 3073–3078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, H.; Kodiha, M.; Lee, S.; Stochaj, U. Identification of novel markers that demarcate the nucleolus during severe stress and chemotherapeutic treatment. PLoS ONE 2013, 8, e80237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biertümpfel, C.; Zhao, Y.; Kondo, Y.; Ramón-Maiques, S.; Gregory, M.; Lee, J.Y.; Masutani, C.; Lehmann, A.R.; Hanaoka, F.; Yang, W. Structure and mechanism of human DNA polymerase η. Nature 2010, 465, 1044–1048. [Google Scholar] [CrossRef] [Green Version]
- Knobel, P.A.; Marti, T.M. Translesion DNA synthesis in the context of cancer research. Cancer Cell Int. 2011, 11, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Saha, P.; Mandal, T.; Talukdar, A.D.; Kumar, D.; Kumar, S.; Tripathi, P.P.; Wang, Q.E.; Srivastava, A.K. DNA polymerase eta: A potential pharmacological target for cancer therapy. J. Cell. Physiol. 2021, 236, 4106–4120. [Google Scholar] [CrossRef]
- Chou, K.-M. DNA polymerase eta and chemotherapeutic agents. Antioxid. Redox Signal. 2011, 14, 2521–2529. [Google Scholar] [CrossRef] [Green Version]
- Goodman, M.F. Error-prone repair DNA polymerases in prokaryotes and eukaryotes. Annu. Rev. Biochem. 2002, 71, 17–50. [Google Scholar] [CrossRef] [Green Version]
- Makridakis, N.M.; Reichardt, J.K. Translesion DNA polymerases and cancer. Front. Genet. 2012, 3, 174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thakur, M.; Wernick, M.; Collins, C.; Limoli, C.L.; Crowley, E.; Cleaver, J.E. DNA polymerase η undergoes alternative splicing, protects against UV sensitivity and apoptosis, and suppresses Mre11-dependent recombination. Genes Chromosomes Cancer 2001, 32, 222–235. [Google Scholar] [CrossRef] [PubMed]
- Masutani, C.; Kusumoto, R.; Yamada, A.; Yuasa, M.; Araki, M.; Nogimori, T.; Yokoi, M.; Eki, T.; Iwai, S.; Hanaoka, F. Xeroderma pigmentosum variant: From a human genetic disorder to a novel DNA polymerase. Cold Spring Harb. Symp. Quant. Biol. 2000, 65, 71–80. [Google Scholar] [CrossRef] [PubMed]
- Loeb, L.A.; Monnat, R.J. DNA polymerases and human disease. Nat. Rev. Genet. 2008, 9, 594–604. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, A.K.; Han, C.; Zhao, R.; Cui, T.; Dai, Y.; Mao, C.; Zhao, W.; Zhang, X.; Yu, J.; Wang, Q.-E. Enhanced expression of DNA polymerase eta contributes to cisplatin resistance of ovarian cancer stem cells. Proc. Natl. Acad. Sci. USA 2015, 112, 4411–4416. [Google Scholar] [CrossRef] [Green Version]
- Jung, Y.-S.; Hakem, A.; Hakem, R.; Chen, X. Pirh2 E3 ubiquitin ligase monoubiquitinates DNA polymerase eta to suppress translesion DNA synthesis. Mol. Cell. Biol. 2011, 31, 3997–4006. [Google Scholar] [CrossRef] [Green Version]
- Jung, Y.-S.; Liu, G.; Chen, X. Pirh2 E3 ubiquitin ligase targets DNA polymerase eta for 20S proteasomal degradation. Mol. Cell. Biol. 2010, 30, 1041–1048. [Google Scholar] [CrossRef] [Green Version]
- Duan, S.; Yao, Z.; Zhu, Y.; Wang, G.; Hou, D.; Wen, L.; Wu, M. The Pirh2–keratin 8/18 interaction modulates the cellular distribution of mitochondria and UV-induced apoptosis. Cell Death Differ. 2009, 16, 826–837. [Google Scholar] [CrossRef] [Green Version]
- Matsuoka, S.; Huang, M.; Elledge, S.J. Linkage of ATM to cell cycle regulation by the Chk2 protein kinase. Science 1998, 282, 1893–1897. [Google Scholar] [CrossRef]
- Chehab, N.H.; Malikzay, A.; Appel, M.; Halazonetis, T.D. Chk2/hCds1 functions as a DNA damage checkpoint in G1 by stabilizing p53. Genes Dev. 2000, 14, 278–288. [Google Scholar] [CrossRef]
- Stevens, C.; Smith, L.; La Thangue, N.B. Chk2 activates E2F-1 in response to DNA damage. Nat. Cell Biol. 2003, 5, 401–409. [Google Scholar] [CrossRef] [PubMed]
- Chou, W.C.; Wang, H.C.; Wong, F.H.; Ding, S.l.; Wu, P.E.; Shieh, S.Y.; Shen, C.Y. Chk2-dependent phosphorylation of XRCC1 in the DNA damage response promotes base excision repair. EMBO J. 2008, 27, 3140–3150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Willers, H.; Feng, Z.; Ghosh, J.C.; Kim, S.; Weaver, D.T.; Chung, J.H.; Powell, S.N.; Xia, F. Chk2 phosphorylation of BRCA1 regulates DNA double-strand break repair. Mol. Cell. Biol. 2004, 24, 708–718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falck, J.; Mailand, N.; Syljuåsen, R.G.; Bartek, J.; Lukas, J. The ATM–Chk2–Cdc25A checkpoint pathway guards against radioresistant DNA synthesis. Nature 2001, 410, 842–847. [Google Scholar] [CrossRef]
- Falck, J.; Petrini, J.H.; Williams, B.R.; Lukas, J.; Bartek, J. The DNA damage-dependent intra–S phase checkpoint is regulated by parallel pathways. Nat. Genet. 2002, 30, 290–294. [Google Scholar] [CrossRef]
- Fernandez-Capetillo, O.; Chen, H.-T.; Celeste, A.; Ward, I.; Romanienko, P.J.; Morales, J.C.; Naka, K.; Xia, Z.; Camerini-Otero, R.D.; Motoyama, N. DNA damage-induced G2–M checkpoint activation by histone H2AX and 53BP1. Nat. Cell Biol. 2002, 4, 993–997. [Google Scholar] [CrossRef]
- Shi, Y.; Felley-Bosco, E.; Marti, T.M.; Orlowski, K.; Pruschy, M.; Stahel, R.A. Starvation-induced activation of ATM/Chk2/p53 signaling sensitizes cancer cells to cisplatin. BMC Cancer 2012, 12, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Zhang, P.; Wang, J.; Gao, W.; Yuan, B.-Z.; Rogers, J.; Reed, E. CHK2 kinase expression is down-regulated due to promoter methylation in non-small cell lung cancer. Mol. Cancer 2004, 3, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Matsuoka, S.; Nakagawa, T.; Masuda, A.; Haruki, N.; Elledge, S.J.; Takahashi, T. Reduced expression and impaired kinase activity of a Chk2 mutant identified in human lung cancer. Cancer Res. 2001, 61, 5362–5365. [Google Scholar]
- Stolz, A.; Ertych, N.; Bastians, H. Tumor suppressor CHK2: Regulator of DNA damage response and mediator of chromosomal stability. Clin. Cancer Res. 2011, 17, 401–405. [Google Scholar] [CrossRef] [Green Version]
- Iacobucci, I.; Di Rorà, A.G.L.; Falzacappa, M.V.V.; Derenzini, E.; Ferrari, A.; Imbrogno, E.; Papayannidis, C.; Venturi, C.; Lonetti, A.; Guadagnuolo, V. Inhibition of Checkpoint Kinase 1 (Chk1) and 2 (Chk2) is a novel therapeutic strategy in B-and T-Acute Lymphoblastic Leukemia (ALL). Cancer Res. 2013, 73, 705. [Google Scholar]
- Wang, W.-J.; Wu, S.-P.; Liu, J.-B.; Shi, Y.-S.; Huang, X.; Zhang, Q.-B.; Yao, K.-T. MYC regulation of CHK1 and CHK2 promotes radioresistance in a stem cell-like population of nasopharyngeal carcinoma cells. Cancer Res. 2013, 73, 1219–1231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ta, H.Q.; Ivey, M.L.; Frierson, H.F.; Conaway, M.R.; Dziegielewski, J.; Larner, J.M.; Gioeli, D. Checkpoint kinase 2 negatively regulates androgen sensitivity and prostate cancer cell growth. Cancer Res. 2015, 75, 5093–5105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, J.H.; Kim, K.-T.; Im, J.; Park, S.; Choi, M.K.; Kim, I.; Nam, K.-Y.; Yoon, J. PHI-101, a potent and novel inhibitor of CHK2 in ovarian and breast cancer cells. Cancer Res. 2021, 81, 1461. [Google Scholar] [CrossRef]
- Smith, H.L.; Southgate, H.; Tweddle, D.A.; Curtin, N.J. DNA damage checkpoint kinases in cancer. Expert Reviews in Molecular Medicine 2020, 22, e2. [Google Scholar] [CrossRef]
- Bohgaki, M.; Hakem, A.; Halaby, M.; Bohgaki, T.; Li, Q.; Bissey, P.; Shloush, J.; Kislinger, T.; Sanchez, O.; Sheng, Y. The E3 ligase PIRH2 polyubiquitylates CHK2 and regulates its turnover. Cell Death Differ. 2013, 20, 812–822. [Google Scholar] [CrossRef] [Green Version]
- Kass, E.M.; Ahn, J.; Tanaka, T.; Freed-Pastor, W.A.; Keezer, S.; Prives, C. Stability of checkpoint kinase 2 is regulated via phosphorylation at serine 456. J. Biol. Chem. 2007, 282, 30311–30321. [Google Scholar] [CrossRef] [Green Version]
- Cardoso, M.C.; Leonhardt, H.; Nadal-Ginard, B. Reversal of terminal differentiation and control of DNA replication: Cyclin A and Cdk2 specifically localize at subnuclear sites of DNA replication. Cell 1993, 74, 979–992. [Google Scholar] [CrossRef] [Green Version]
- Copeland, N.A.; Sercombe, H.E.; Ainscough, J.F.; Coverley, D. Ciz1 cooperates with cyclin-A–CDK2 to activate mammalian DNA replication in vitro. J. Cell Sci. 2010, 123, 1108–1115. [Google Scholar] [CrossRef] [Green Version]
- Grimmler, M.; Wang, Y.; Mund, T.; Cilenšek, Z.; Keidel, E.-M.; Waddell, M.B.; Jäkel, H.; Kullmann, M.; Kriwacki, R.W.; Hengst, L. Cdk-inhibitory activity and stability of p27Kip1 are directly regulated by oncogenic tyrosine kinases. Cell 2007, 128, 269–280. [Google Scholar] [CrossRef] [Green Version]
- Nigg, E.A. Cyclin-dependent protein kinases: Key regulators of the eukaryotic cell cycle. Bioessays 1995, 17, 471–480. [Google Scholar] [CrossRef] [PubMed]
- Abbastabar, M.; Kheyrollah, M.; Azizian, K.; Bagherlou, N.; Tehrani, S.S.; Maniati, M.; Karimian, A. Multiple functions of p27 in cell cycle, apoptosis, epigenetic modification and transcriptional regulation for the control of cell growth: A double-edged sword protein. DNA Repair 2018, 69, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Blain, S.W.; Scher, H.I.; Cordon-Cardo, C.; Koff, A. p27 as a target for cancer therapeutics. Cancer Cell 2003, 3, 111–115. [Google Scholar] [CrossRef] [Green Version]
- Ye, D.; Luo, H.; Lai, Z.; Zou, L.; Zhu, L.; Mao, J.; Jacob, T.; Ye, W.; Wang, L.; Chen, L. ClC-3 chloride channel proteins regulate the cell cycle by up-regulating cyclin D1-CDK4/6 through suppressing p21/p27 expression in nasopharyngeal carcinoma cells. Sci. Rep. 2016, 6, 1–16. [Google Scholar] [CrossRef] [Green Version]
- García-Gutiérrez, L.; Bretones, G.; Molina, E.; Arechaga, I.; Symonds, C.; Acosta, J.C.; Blanco, R.; Fernández, A.; Alonso, L.; Sicinski, P. Myc stimulates cell cycle progression through the activation of Cdk1 and phosphorylation of p27. Sci. Rep. 2019, 9, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Cuadrado, M.; Gutierrez-Martinez, P.; Swat, A.; Nebreda, A.R.; Fernandez-Capetillo, O. p27Kip1 stabilization is essential for the maintenance of cell cycle arrest in response to DNA damage. Cancer Res. 2009, 69, 8726–8732. [Google Scholar] [CrossRef] [Green Version]
- Chen, A.C.H.; Peng, Q.; Fong, S.W.; Lee, K.C.; Yeung, W.S.B.; Lee, Y.L. DNA Damage Response and Cell Cycle Regulation in Pluripotent Stem Cells. Genes 2021, 12, 1548. [Google Scholar] [CrossRef]
- Traub, F.; Mengel, M.; Lück, H.J.; Kreipe, H.H.; von Wasielewski, R. Prognostic impact of Skp2 and p27 in human breast cancer. Breast Cancer Res. Treat. 2006, 99, 185–191. [Google Scholar] [CrossRef]
- Zhuang, Y.; Yin, H.-T.; Yin, X.-L.; Wang, J.; Zhang, D.-P. High p27 expression is associated with a better prognosis in East Asian non-small cell lung cancer patients. Clin. Chim. Acta 2011, 412, 2228–2231. [Google Scholar] [CrossRef]
- Liu, D.F.; Ferguson, K.; Cooper, G.S.; Grady, W.M.; Willis, J. p27 cell-cycle inhibitor is inversely correlated with lymph node metastases in right-sided colon cancer. J. Clin. Lab. Anal. 1999, 13, 291–295. [Google Scholar] [CrossRef]
- Hattori, T.; Isobe, T.; Abe, K.; Kikuchi, H.; Kitagawa, K.; Oda, T.; Uchida, C.; Kitagawa, M. Pirh2 promotes ubiquitin-dependent degradation of the cyclin-dependent kinase inhibitor p27Kip1. Cancer Res. 2007, 67, 10789–10795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelly, R.D.; Cowley, S.M. The physiological roles of histone deacetylase (HDAC) 1 and 2: Complex co-stars with multiple leading parts. Biochem. Soc. Trans. 2013, 41, 741–749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barneda-Zahonero, B.; Parra, M. Histone deacetylases and cancer. Mol. Oncol. 2012, 6, 579–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Seto, E. HDACs and HDAC inhibitors in cancer development and therapy. Cold Spring Harb. Perspect. Med. 2016, 6, a026831. [Google Scholar] [CrossRef] [Green Version]
- Jenke, R.; Reßing, N.; Hansen, F.K.; Aigner, A.; Büch, T. Anticancer therapy with HDAC inhibitors: Mechanism-based combination strategies and future perspectives. Cancers 2021, 13, 634. [Google Scholar] [CrossRef]
- Logan, I.R.; Gaughan, L.; McCracken, S.R.; Sapountzi, V.; Leung, H.Y.; Robson, C.N. Human PIRH2 enhances androgen receptor signaling through inhibition of histone deacetylase 1 and is overexpressed in prostate cancer. Mol. Cell. Biol. 2006, 26, 6502–6510. [Google Scholar] [CrossRef] [Green Version]
- Choi, M.; Choi, Y.M.; An, I.-S.; Bae, S.; Jung, J.H.; An, S. E3 ligase RCHY1 negatively regulates HDAC2. Biochem. Biophys. Res. Commun. 2020, 521, 37–41. [Google Scholar] [CrossRef]
- Zhu, Q.-Q.; Ma, C.; Wang, Q.; Song, Y.; Lv, T. The role of TWIST1 in epithelial-mesenchymal transition and cancers. Tumor Biol. 2016, 37, 185–197. [Google Scholar] [CrossRef]
- Qin, Q.; Xu, Y.; He, T.; Qin, C.; Xu, J. Normal and disease-related biological functions of Twist1 and underlying molecular mechanisms. Cell Res. 2012, 22, 90–106. [Google Scholar] [CrossRef] [Green Version]
- Kress, W.; Schropp, C.; Lieb, G.; Petersen, B.; Büsse-Ratzka, M.; Kunz, J.; Reinhart, E.; Schäfer, W.-D.; Sold, J.; Hoppe, F. Saethre–Chotzen syndrome caused by TWIST 1 gene mutations: Functional differentiation from Muenke coronal synostosis syndrome. Eur. J. Hum. Genet. 2006, 14, 39–48. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Z.; Rahman, M.A.; Chen, Z.G.; Shin, D.M. Multiple biological functions of Twist1 in various cancers. Oncotarget 2017, 8, 20380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang-Hartwich, Y.; Tedja, R.; Roberts, C.M.; Goodner-Bingham, J.; Cardenas, C.; Gurea, M.; Sumi, N.J.; Alvero, A.B.; Glackin, C.A.; Mor, G. p53–Pirh2 complex promotes Twist1 degradation and inhibits EMT. Mol. Cancer Res. 2019, 17, 153–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Li, J.; Wang, C.; Ma, Y.; Huo, K. A new human gene hNTKL-BP1 interacts with hPirh2. Biochem. Biophys. Res. Commun. 2005, 330, 293–297. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chen, X.; Chen, X.; Chen, Q.; Huo, K. Transcriptional profiling and dynamical regulation analysis identify potential kernel target genes of SCYL1-BP1 in HEK293T cells. Mol. Cells 2014, 37, 691. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Zhi, Q.; Ye, Q.; Zhou, C.; Zhang, L.; Yan, W.; Wu, Q.; Zhang, D.; Li, P.; Huo, K. SCYL1-BP1 affects cell cycle arrest in human hepatocellular carcinoma cells via Cyclin F and RRM2. Anti-Cancer Agents Med. Chem. (Former. Curr. Med. Chem.-Anti-Cancer Agents) 2016, 16, 440–446. [Google Scholar] [CrossRef]
- Yan, J.; Di, Y.; Shi, H.; Rao, H.; Huo, K. Overexpression of SCYL1-BP1 stabilizes functional p53 by suppressing MDM2-mediated ubiquitination. FEBS Lett. 2010, 584, 4319–4324. [Google Scholar] [CrossRef] [Green Version]
- Yan, J.; Zhang, D.; Di, Y.; Shi, H.; Rao, H.; Huo, K. A newly identified Pirh2 substrate SCYL1-BP1 can bind to MDM2 and accelerate MDM2 self-ubiquitination. FEBS Lett. 2010, 584, 3275–3278. [Google Scholar] [CrossRef] [Green Version]
- Duan, W.; Gao, L.; Druhan, L.J.; Zhu, W.-G.; Morrison, C.; Otterson, G.A.; Villalona-Calero, M.A. Expression of Pirh2, a newly identified ubiquitin protein ligase, in lung cancer. J. Natl. Cancer Inst. 2004, 96, 1718–1721. [Google Scholar] [CrossRef] [Green Version]
- Su, Y.; Bai, M.; Zhu, L.; Jin, Y.; Zhang, X.; Zhou, Q. Pirh2 shRNA mediated by psiRNA-hH1 vector plasmid effectively inhibits the proliferation of lung carcinoma cells: In vitro and in vivo experiments. Zhonghua Yi Xue Za Zhi 2007, 87, 1199–1203. [Google Scholar]
- Su, Y.; Zhu, L.; Jin, Y.; Zhang, X.; Zhou, Q.; Bai, M. Impact of siRNA targeting pirh2 on proliferation and cell cycle control of the lung adenocarcinoma cell line A549. Front. Med. China 2007, 1, 359–363. [Google Scholar] [CrossRef]
- Fedorova, O.; Gudovich, A.; Daks, A.; Baidyuk, E.; Shuvalov, O.; Petukhov, A.; Parfenyev, S.; Kizenko, A.; Barlev, N.A. Regulation of autophagy flux by E3 ubiquitin ligase Pirh2 in lung cancer. Biochem. Biophys. Res. Commun. 2021, 563, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Wang, F.; Yang, Y.; Xu, J.; Yang, J.; Wang, K.; Liu, Y.; Du, G.; Zeng, Y. Inverse correlation between Naa10p and Pirh2 expression and the combined prognostic value in oral squamous cell carcinoma patients. J. Oral Pathol. Med. 2019, 48, 686–695. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.M.; Yang, L.Y.; Guo, L.; Fan, C.; Wu, F. p53-induced RING-H2 protein, a novel marker for poor survival in hepatocellular carcinoma after hepatic resection. Cancer Interdiscip. Int. J. Am. Cancer Soc. 2009, 115, 4554–4563. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Qian, X.; Cheng, C.; He, S.; Sun, L.; Ke, Q.; Zhang, L.; Pan, X.; He, F.; Wang, Q. Expression of Pirh2, a p27Kip1 ubiquitin ligase, in hepatocellular carcinoma: Correlation with p27Kip1 and cell proliferation. Hum. Pathol. 2011, 42, 507–515. [Google Scholar] [CrossRef]
- Shimada, M.; Kitagawa, K.; Dobashi, Y.; Isobe, T.; Hattori, T.; Uchida, C.; Abe, K.; Kotake, Y.; Oda, T.; Suzuki, H. High expression of Pirh2, an E3 ligase for p27, is associated with low expression of p27 and poor prognosis in head and neck cancers. Cancer Sci. 2009, 100, 866–872. [Google Scholar] [CrossRef] [Green Version]
- Bao, Y.; Wu, X.; Yuan, D.; Shi, W.; Shi, J. High expression of Pirh2 is associated with poor prognosis in glioma. Cell. Mol. Neurobiol. 2017, 37, 1501–1509. [Google Scholar] [CrossRef]
- Xu, X.; Chen, Y.; Wang, X.; Mu, X. Role of Hippo/YAP signaling in irradiation-induced glioma cell apoptosis. Cancer Manag. Res. 2019, 11, 7577. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.; Chen, Y.; Sun, F.; Ni, Q.; Wang, H.; Huang, Y.; Zhang, C.; Liu, K.; Wang, S.; Qiu, J. Downregulated PIRH2 can decrease the proliferation of breast cancer cells. Arch. Med. Res. 2016, 47, 186–195. [Google Scholar] [CrossRef]
- Yang, L.; Chen, J.; Han, X.; Zhang, E.; Huang, X.; Guo, X.; Chen, Q.; Wu, W.; Zheng, G.; He, D. Pirh2 mediates the sensitivity of myeloma cells to bortezomib via canonical NF-κB signaling pathway. Protein Cell 2018, 9, 770–784. [Google Scholar] [CrossRef] [Green Version]
- Reiner, A.S.; Lobaugh, S.M.; Gonen, S.; Diamond, E.L.; Panageas, K.S. A Population-Based Study of Treatment and Survival in Older Glioma Patients. JNCI Cancer Spectr. 2022, 6, pkac010. [Google Scholar] [CrossRef]
- Scott, K.; Hayden, P.J.; Will, A.; Wheatley, K.; Coyne, I. Bortezomib for the treatment of multiple myeloma. Cochrane Database Syst. Rev. 2016, 4, CD010816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.-H.; Li, S.-C.; Huang, L.-H.; Chen, P.-C.; Lin, Y.-Y.; Lin, C.-C.; Kuo, H.-C. Identifying genetic hypomethylation and upregulation of Toll-like receptors in Kawasaki disease. Oncotarget 2017, 8, 11249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoon, C.-H.; Kim, S.-Y.; Byeon, S.E.; Jeong, Y.; Lee, J.; Kim, K.P.; Park, J.; Bae, Y.-S. p53-derived host restriction of HIV-1 replication by protein kinase R-mediated Tat phosphorylation and inactivation. J. Virol. 2015, 89, 4262–4280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gualberto, A.; Baldwin, A.S. p53 and Sp1 Interact and Cooperate in the Tumor Necrosis Factor-induced Transcriptional Activation of the HIV-1 Long Terminal Repeat (∗). J. Biol. Chem. 1995, 270, 19680–19683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bargonetti, J.; Chicas, A.; White, D.; Prives, C. p53 represses Sp1 DNA binding and HIV-LTR directed transcription. Cell. Mol. Biol. (Noisy-Le-Grand Fr.) 1997, 43, 935–949. [Google Scholar]
- Mukerjee, R.; Claudio, P.P.; Chang, J.R.; Del Valle, L.; Sawaya, B.E. Transcriptional regulation of HIV-1 gene expression by p53. Cell Cycle 2010, 9, 4569–4578. [Google Scholar] [CrossRef] [Green Version]
- Shi, B.; Sharifi, H.J.; DiGrigoli, S.; Kinnetz, M.; Mellon, K.; Hu, W.; de Noronha, C. Inhibition of HIV early replication by the p53 and its downstream gene p21. Virol. J. 2018, 15, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Ma-Lauer, Y.; Carbajo-Lozoya, J.; Hein, M.Y.; Müller, M.A.; Deng, W.; Lei, J.; Meyer, B.; Kusov, Y.; Von Brunn, B.; Bairad, D.R. p53 down-regulates SARS coronavirus replication and is targeted by the SARS-unique domain and PLpro via E3 ubiquitin ligase RCHY1. Proc. Natl. Acad. Sci. USA 2016, 113, E5192–E5201. [Google Scholar] [CrossRef]
- Achong, B.; Mansell, P.; Epstein, M.; Clifford, P. An unusual virus in cultures from a human nasopharyngeal carcinoma. J. Natl. Cancer Inst. 1971, 46, 299–307. [Google Scholar]
- Dong, L.; Cheng, Q.; Wang, Z.; Yuan, P.; Li, Z.; Sun, Y.; Han, S.; Yin, J.; Peng, B.; He, X. Human Pirh2 is a novel inhibitor of prototype foamy virus replication. Viruses 2015, 7, 1668–1684. [Google Scholar] [CrossRef] [Green Version]
- Kiupel, M.; Stevenson, G.; Mittal, S.; Clark, E.; Haines, D. Circovirus-like viral associated disease in weaned pigs in Indiana. Vet. Pathol. 1998, 35, 303–307. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhu, Y.; Chen, I.; Lau, J.; He, F.; Lau, A.; Wang, Z.; Karuppannan, A.K.; Kwang, J. The ORF3 protein of porcine circovirus type 2 interacts with porcine ubiquitin E3 ligase Pirh2 and facilitates p53 expression in viral infection. J. Virol. 2007, 81, 9560–9567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karuppannan, A.K.; Liu, S.; Jia, Q.; Selvaraj, M.; Kwang, J. Porcine circovirus type 2 ORF3 protein competes with p53 in binding to Pirh2 and mediates the deregulation of p53 homeostasis. Virology 2010, 398, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugai, A.; Sato, H.; Yoneda, M.; Kai, C. Phosphorylation of measles virus nucleoprotein affects viral growth by changing gene expression and genomic RNA stability. J. Virol. 2013, 87, 11684–11692. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.; Cortay, J.-C.; Logan, I.R.; Sapountzi, V.; Robson, C.N.; Gerlier, D. Inhibition of ubiquitination and stabilization of human ubiquitin E3 ligase PIRH2 by measles virus phosphoprotein. J. Virol. 2005, 79, 11824–11836. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Deng, J.; Chen, H.; Chen, T.; Cao, X.; Hou, H.; Huan, W.; Zhang, G.; Yu, B.; Wang, Y. Dynamic changes of PIRH2 and p27kip1 expression in injured rat sciatic nerve. Neurol. Sci. 2012, 33, 749–757. [Google Scholar] [CrossRef]
- Wu, X.; Shi, W.; Zhao, W.; Shao, B.; Yuan, Q.; Li, C.; Zhang, S.; Sun, B.; Wu, Q.; Chen, J. Changes in Pirh2 and p27kip1 expression following traumatic brain injury in adult rats. J. Mol. Neurosci. 2012, 46, 184–191. [Google Scholar] [CrossRef]
- Scherer, S. Axon-Schwann cell interactions during peripheral nerve degeneration and regeneration. In Glial Cell Development; Jessen, K., Richardson, W., Eds.; Bios Scientific Publishers: Oxford, UK, 1997; pp. 165–196. ISBN 9781872748542. [Google Scholar]
- Chen, J.; Zou, F.; Fu, H.; Mao, H.; Gong, M.; Ni, L.; Xu, X.; Shi, J.; Ke, K.; Cao, M. SCY1-like 1 binding protein 1 (SCYL1-bp1) interacts with p53-induced RING H2 protein (Pirh2) after traumatic brain injury in rats. J. Mol. Histol. 2013, 44, 271–283. [Google Scholar] [CrossRef]
- Liu, Y.; Chen, Y.; Lu, X.; Wang, Y.; Duan, Y.; Cheng, C.; Shen, A. SCYL1BP1 modulates neurite outgrowth and regeneration by regulating the Mdm2/p53 pathway. Mol. Biol. Cell 2012, 23, 4506–4514. [Google Scholar] [CrossRef]
- Davidovich, P.; Aksenova, V.; Petrova, V.; Tentler, D.; Orlova, D.; Smirnov, S.; Gurzhiy, V.; Okorokov, A.; Garabadzhiu, A.; Melino, G. Discovery of novel isatin-based p53 inducers. ACS Med. Chem. Lett. 2015, 6, 856–860. [Google Scholar] [CrossRef] [Green Version]
- Niazi, S.; Purohit, M. Rational design of promiscuous binding modulators of p53 inducing E3 (Ub)-ligases (Mdm2 and Pirh2) as anticancer agents: An in silico approach. MedChemComm 2015, 6, 1959–1968. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Protein | Type of Regulation | Protein Type | Effect | Reference |
|---|---|---|---|---|
| p53 | Transcriptional | Transcription factor | Activates Pirh2 expression in mouse models. The p53-binding site was detected in intron 3 of RCHY1 mouse gene. | [15] |
| p63 | Transcriptional | Transcription factor | TA isoform of p63 activates Pirh2 expression | [17] |
| RelA/p65 subunit of NFkB | Transcriptional | Transcription factor | Activates Pirh2 expression binding to the RCHY1 promoter region | [18] |
| CamKII | Post-translational | Kinase | Phosphorylates Pirh2 at T154 and S155, enhances Pirh2 auto-ubiquitination | [19] |
| Cdk9 | Post-translational | Kinase | Phosphorylates Pirh2 at S211 and T217, enhances Pirh2 auto-ubiquitination | [20] |
| Hoxa2 | Post-translational | Transcription factor | Physically interacts with Pirh2 and 20S proteasome subunits and promotes proteasomal degradation of Pirh2 in a ubiquitin-independent manner | [21] |
| [22] | ||||
| Tip60 | Post-translational | Acetyltransferase | Interacts with and stabilizes Pirh2 | [23] |
| PLAGL2 | Post-translational | Transcription factor | Physically interacts with Pirh2 dimers and this interaction stabilizes Pirh2 protein level | [24] |
| RNF144B | Post-translational | Ubiquitin ligase | Ubiquitinates Pirh2 and targets it for degradation through ubiquitin-proteasome system | [25] |
| Cancer Type | Oncogene or Tumor Suppressor | The Effect of Pirh2 | Reference |
|---|---|---|---|
| Lung cancer | Oncogene | Pirh2 level is upregulated in NSCLC samples, promotes p53 degradation | [16,179] |
| Oncogene | Pirh2 level is upregulated in NSCLC samples, increases cell proliferation | [180,181] | |
| Oncogene | Increases tumorigenic of NSCLC cells, promotes c-Myc expression | [70,103] | |
| Oncogene | Increases autophagy level and doxorubicin resistance of NSCLC cells | [182] | |
| Prostate cancer | Oncogene | Negative prognostic factor, enhances ER-mediated PSA expression and suppresses HDAC1 | [167] |
| Oral cancer | Oncogene | Negative prognostic factor, activates OSCC cell migration | [183] |
| Hepatocellular carcinoma | Oncogene | Negative prognostic factor, Pirh2 level is upregulated in HCC tissues | [184] |
| Oncogene | Negative prognostic factor, downregulates p27 in HCC cells | [185] | |
| Head and neck cancer | Oncogene | Negative prognostic factor, promotes proliferation of HNSCC cells | [186] |
| Glioma | Oncogene | Negative prognostic factor, promotes proliferation and suppresses apoptosis in human glioma cells U87MG | [187] |
| Tumor suppressor | Induces apoptosis level in human glioma cells U251 in response to irradiation | [188] | |
| Breast cancer | Oncogene | Negative prognostic factor, Pirh2 level is upregulated in BC samples, promotes proliferation and stabilizes β-catenin level in BC cells | [189] |
| Multiple myeloma | Tumor suppressor | Pirh2 reduced in bortezomib-resistant MM cells. Pirh2 downregulates NF-κB signaling pathway in MM. Overexpression of Pirh2 overcomes bortezomib resistance. | [190] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Daks, A.; Fedorova, O.; Parfenyev, S.; Nevzorov, I.; Shuvalov, O.; Barlev, N.A. The Role of E3 Ligase Pirh2 in Disease. Cells 2022, 11, 1515. https://doi.org/10.3390/cells11091515
Daks A, Fedorova O, Parfenyev S, Nevzorov I, Shuvalov O, Barlev NA. The Role of E3 Ligase Pirh2 in Disease. Cells. 2022; 11(9):1515. https://doi.org/10.3390/cells11091515
Chicago/Turabian StyleDaks, Alexandra, Olga Fedorova, Sergey Parfenyev, Ivan Nevzorov, Oleg Shuvalov, and Nickolai A. Barlev. 2022. "The Role of E3 Ligase Pirh2 in Disease" Cells 11, no. 9: 1515. https://doi.org/10.3390/cells11091515