
Waste Clearance in the Brain and Neuroinflammation: A Novel Perspective on Biomarker and Drug Target Discovery in Alzheimer’s Disease

Abstract
:1. Introduction
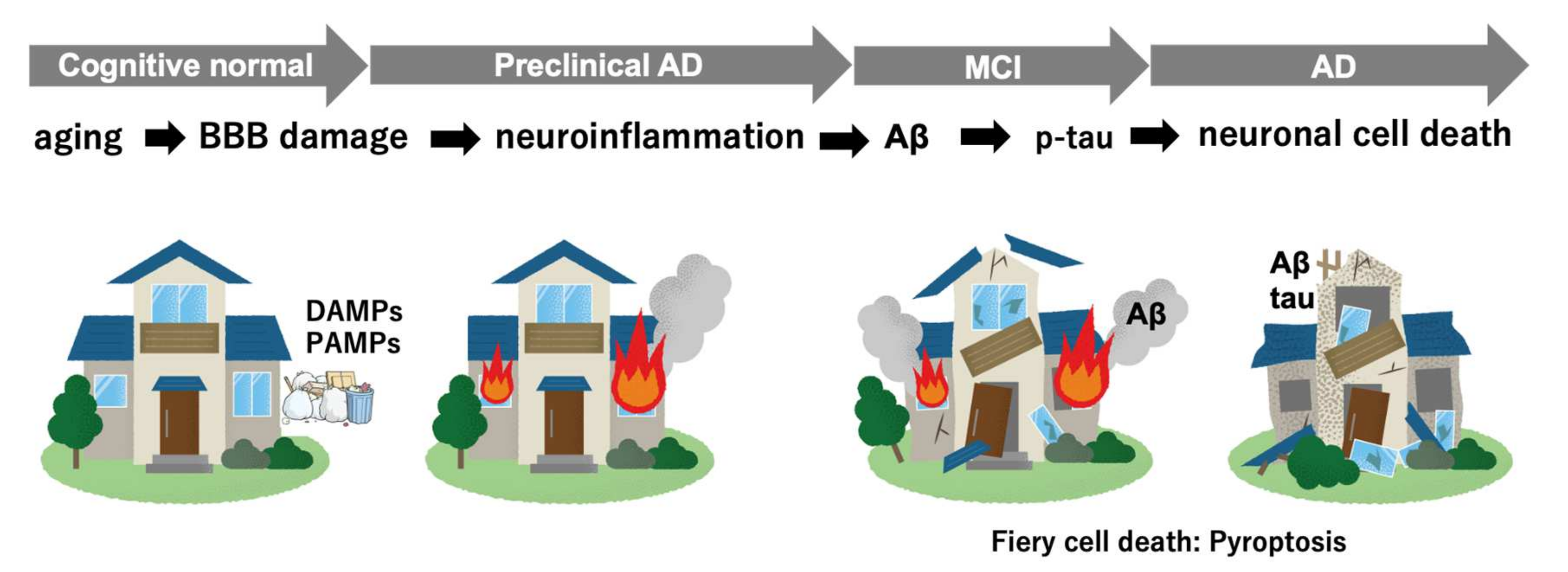
2. Multifactorial Pathobiology in AD
2.1. The Danger Signal Activates Neuroinflammation and Induces Pyroptosis
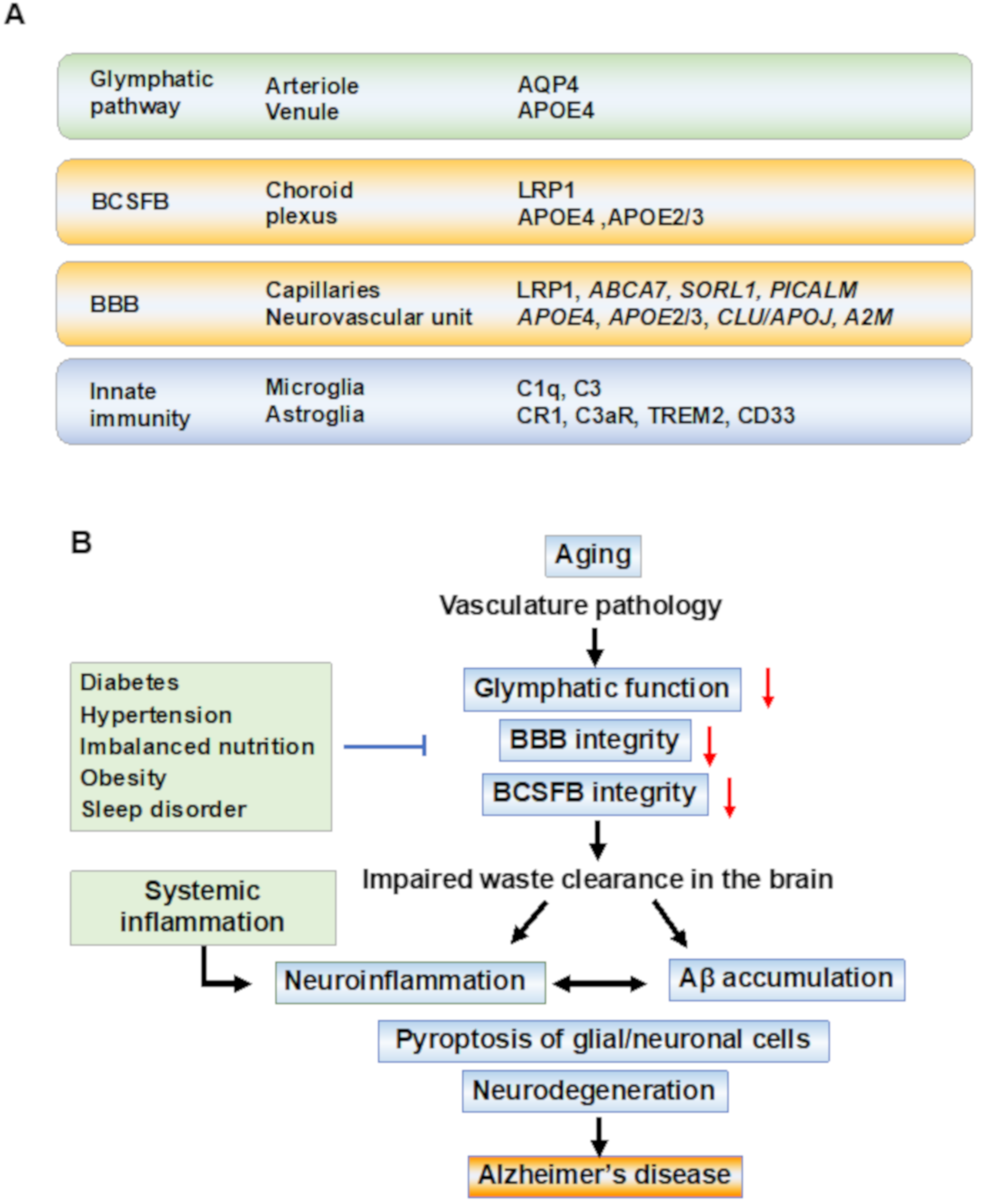
2.2. The Glymphatic System for Clearance of Brain Waste
2.3. BBB and Blood–CSF Barrier in AD Pathobiology
2.4. Microglial Activation for Waste Clearance in the Brain
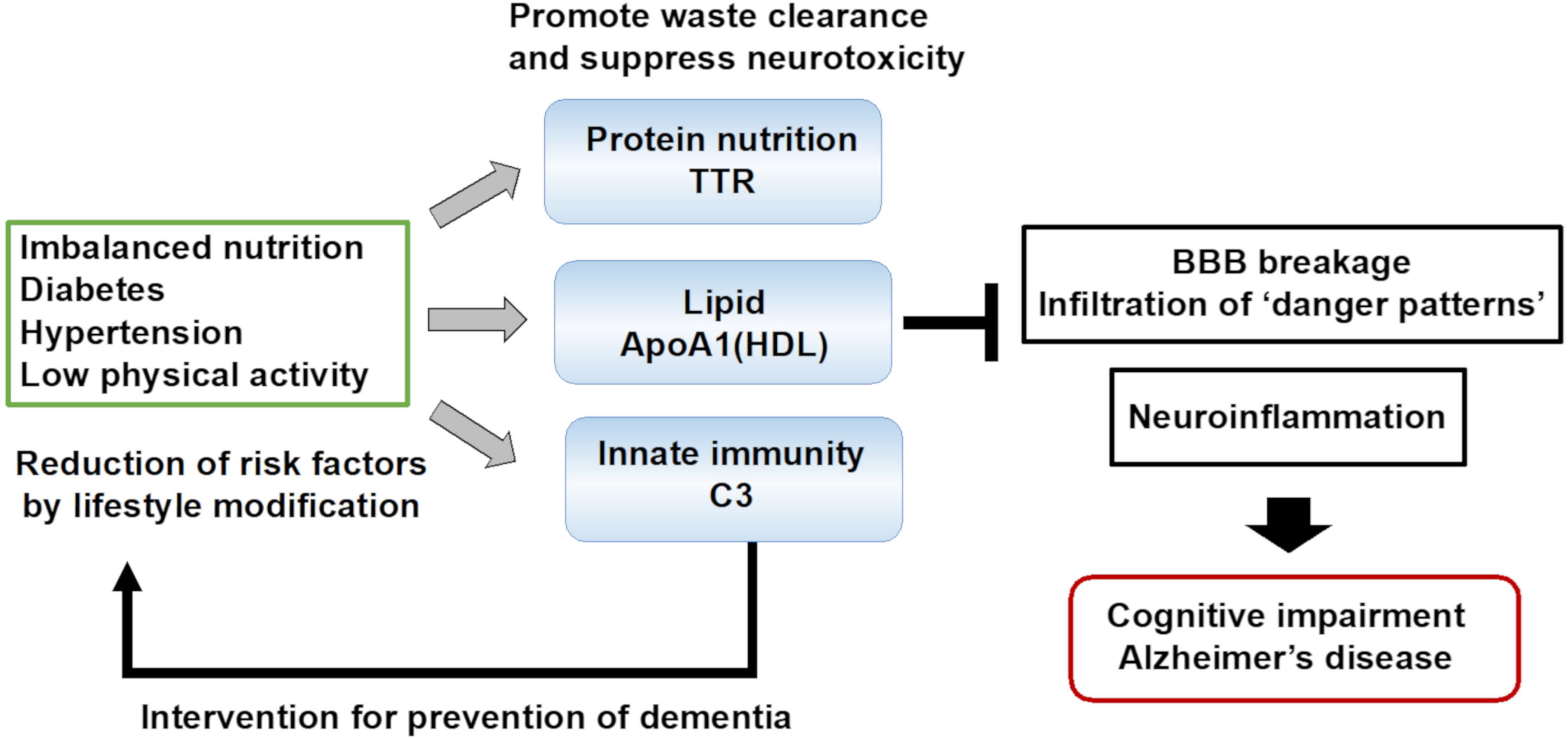
3. Biomarkers for the Waste Clearance Dysfunction in AD
4. Future Perspectives
Funding
Conflicts of Interest
References
- GBD 2016 Dementia Collaborators. Global, Regional, and National Burden of Alzheimer’s Disease and Other Dementias, 1990–2016: A Systematic Analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019, 18, 88–106. [Google Scholar] [CrossRef] [Green Version]
- Gauthier, S.; Rosa-Neto, P.; Morais, J.A.; Webster, C. World Alzheimer Report 2021: Journey through the Diagnosis of Dementia; Alzheimer’s Disease International: London, UK, 2021. [Google Scholar]
- Sweeney, M.D.; Montagne, A.; Sagare, A.P.; Nation, D.A.; Schneider, L.S.; Chui, H.C.; Harrington, M.G.; Pa, J.; Law, M.; Wang, D.J.J.; et al. Vascular Dysfunction-The Disregarded Partner of Alzheimer’s Disease. Alzheimers Dement. 2019, 15, 158–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cummings, J.; Lee, G.; Zhong, K.; Fonseca, J.; Taghva, K. Alzheimer’s Disease Drug Development Pipeline: 2021. Alzheimers Dement. 2021, 7, e12179. [Google Scholar] [CrossRef] [PubMed]
- Alexander, G.C.; Knopman, D.S.; Emerson, S.S.; Ovbiagele, B.; Kryscio, R.J.; Perlmutter, J.S.; Kesselheim, A.S. Revisiting FDA Approval of Aducanumab. N. Engl. J. Med. 2021, 385, 769–771. [Google Scholar] [CrossRef] [PubMed]
- Ross, C.A.; Poirier, M.A. Protein Aggregation and Neurodegenerative Disease. Nat. Med. 2004, 10, S10–S17. [Google Scholar] [CrossRef] [PubMed]
- Arai, T.; Hasegawa, M.; Nonoka, T.; Kametani, F.; Yamashita, M.; Hosokawa, M.; Niizato, K.; Tsuchiya, K.; Kobayashi, Z.; Ikeda, K.; et al. Phosphorylated and Cleaved TDP-43 in ALS, FTLD and Other Neurodegenerative Disorders and in Cellular Models of TDP-43 Proteinopathy. Neuropathology 2010, 30, 170–181. [Google Scholar] [CrossRef] [PubMed]
- Jack, C.R., Jr.; Knopman, D.S.; Jagust, W.J.; Shaw, L.M.; Aisen, P.S.; Weiner, M.W.; Petersen, R.C.; Trojanowski, J.Q. Hypothetical Model of Dynamic Biomarkers of the Alzheimer’s Pathological Cascade. Lancet Neurol. 2010, 9, 119–128. [Google Scholar] [CrossRef] [Green Version]
- Sperling, R.A.; Aisen, P.S.; Beckett, L.A.; Bennett, D.A.; Craft, S.; Fagan, A.M.; Iwatsubo, T.; Jack, C.R., Jr.; Kaye, J.; Montine, T.J.; et al. Toward Defining the Preclinical Stages of Alzheimer’s Disease: Recommendations from the National Institute on Aging-Alzheimer’s Association Workgroups on Diagnostic Guidelines for Alzheimer’s Disease. Alzheimers Dement. 2011, 7, 280–292. [Google Scholar] [CrossRef] [Green Version]
- Mawuenyega, K.G.; Sigurdson, W.; Ovod, V.; Munsell, L.; Kasten, T.; Morris, J.C.; Yarasheski, K.E.; Bateman, R.J. Decreased Clearance of CNS Beta-Amyloid in Alzheimer’s Disease. Science 2010, 330, 1774. [Google Scholar] [CrossRef] [Green Version]
- Livingston, G.; Huntley, J.; Sommerlad, A.; Ames, D.; Ballard, C.; Banerjee, S.; Brayne, C.; Burns, A.; Cohen-Mansfield, J.; Cooper, C.; et al. Dementia Prevention, Intervention, and Care: 2020 Report of the Lancet Commission. Lancet 2020, 396, 413–446. [Google Scholar] [CrossRef]
- Iliff, J.J.; Wang, M.; Liao, Y.; Plogg, B.A.; Peng, W.; Gundersen, G.A.; Benveniste, H.; Vates, G.E.; Deane, R.; Goldman, S.A.; et al. A Paravascular Pathway Facilitates CSF Flow through the Brain Parenchyma and the Clearance of Interstitial Solutes, Including Amyloid β. Sci. Transl. Med. 2012, 4, 147ra111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nedergaard, M.; Goldman, S.A. Glymphatic Failure as a Final Common Pathway to Dementia. Science 2020, 370, 50–56. [Google Scholar] [CrossRef]
- Bergsbaken, T.; Fink, S.L.; Cookson, B.T. Pyroptosis: Host Cell Death and Inflammation. Nat. Rev. Microbiol. 2009, 7, 99–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voet, S.; Srinivasan, S.; Lamkanfi, M.; van Loo, G. Inflammasomes in Neuroinflammatory and Neurodegenerative Diseases. EMBO Mol. Med. 2019, 11, e10248. [Google Scholar] [CrossRef] [PubMed]
- Tan, M.-S.; Tan, L.; Jiang, T.; Zhu, X.-C.; Wang, H.-F.; Jia, C.-D.; Yu, J.-T. Amyloid-β Induces NLRP1-Dependent Neuronal Pyroptosis in Models of Alzheimer’s Disease. Cell Death Dis. 2014, 5, e1382. [Google Scholar] [CrossRef]
- Heneka, M.T.; Kummer, M.P.; Stutz, A.; Delekate, A.; Schwartz, S.; Vieira-Saecker, A.; Griep, A.; Axt, D.; Remus, A.; Tzeng, T.-C.; et al. NLRP3 Is Activated in Alzheimer’s Disease and Contributes to Pathology in APP/PS1 Mice. Nature 2012, 493, 674–678. [Google Scholar] [CrossRef]
- Jha, N.K.; Jha, S.K.; Kar, R.; Nand, P.; Swati, K.; Goswami, V.K. Nuclear Factor-Kappa β as a Therapeutic Target for Alzheimer’s Disease. J. Neurochem. 2019, 150, 113–137. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Zhao, Y.; Zhang, J.; Yang, G. Mechanisms of NLRP3 Inflammasome Activation: Its Role in the Treatment of Alzheimer’s Disease. Neurochem. Res. 2020, 45, 2560–2572. [Google Scholar] [CrossRef]
- Panza, F.; Lozupone, M.; Logroscino, G.; Imbimbo, B.P. A Critical Appraisal of Amyloid-β-Targeting Therapies for Alzheimer Disease. Nat. Rev. Neurol. 2019, 15, 73–88. [Google Scholar] [CrossRef]
- Itzhaki, R.F.; Golde, T.E.; Heneka, M.T.; Readhead, B. Do Infections Have a Role in the Pathogenesis of Alzheimer Disease? Nat. Rev. Neurol. 2020, 16, 193–197. [Google Scholar] [CrossRef]
- Wozniak, M.A.; Mee, A.P.; Itzhaki, R.F. Herpes Simplex Virus Type 1 DNA Is Located within Alzheimer’s Disease Amyloid Plaques. J. Pathol. 2009, 217, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Zhan, X.; Stamova, B.; Jin, L.-W.; DeCarli, C.; Phinney, B.; Sharp, F.R. Gram-Negative Bacterial Molecules Associate with Alzheimer Disease Pathology. Neurology 2016, 87, 2324–2332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lövheim, H.; Gilthorpe, J.; Adolfsson, R.; Nilsson, L.-G.; Elgh, F. Reactivated Herpes Simplex Infection Increases the Risk of Alzheimer’s Disease. Alzheimers Dement. 2015, 11, 593–599. [Google Scholar] [CrossRef] [PubMed]
- Butler, L.; Walker, K.A. The Role of Chronic Infection in Alzheimer’s Disease: Instigators, Co-Conspirators, or Bystanders? Curr. Clin. Microbiol. Rep. 2021, 8, 199–212. [Google Scholar] [CrossRef] [PubMed]
- Kamer, A.R.; Craig, R.G.; Dasanayake, A.P.; Brys, M.; Glodzik-Sobanska, L.; de Leon, M.J. Inflammation and Alzheimer’s Disease: Possible Role of Periodontal Diseases. Alzheimers Dement. 2008, 4, 242–250. [Google Scholar] [CrossRef]
- Kamer, A.R.; Craig, R.G.; Niederman, R.; Fortea, J.; de Leon, M.J. Periodontal Disease as a Possible Cause for Alzheimer’s Disease. Periodontol. 2000 2020, 83, 242–271. [Google Scholar] [CrossRef]
- Dominy, S.S.; Lynch, C.; Ermini, F.; Benedyk, M.; Marczyk, A.; Konradi, A.; Nguyen, M.; Haditsch, U.; Raha, D.; Griffin, C.; et al. Porphyromonas Gingivalis in Alzheimer’s Disease Brains: Evidence for Disease Causation and Treatment with Small-Molecule Inhibitors. Sci. Adv. 2019, 5, eaau3333. [Google Scholar] [CrossRef] [Green Version]
- Bettcher, B.M.; Tansey, M.G.; Dorothée, G.; Heneka, M.T. Peripheral and Central Immune System Crosstalk in Alzheimer Disease—A Research Prospectus. Nat. Rev. Neurol. 2021, 17, 689–701. [Google Scholar] [CrossRef]
- Qin, L.; Wu, X.; Block, M.L.; Liu, Y.; Breese, G.R.; Hong, J.-S.; Knapp, D.J.; Crews, F.T. Systemic LPS Causes Chronic Neuroinflammation and Progressive Neurodegeneration. Glia 2007, 55, 453–462. [Google Scholar] [CrossRef] [Green Version]
- Semmler, A.; Okulla, T.; Sastre, M.; Dumitrescu-Ozimek, L.; Heneka, M.T. Systemic Inflammation Induces Apoptosis with Variable Vulnerability of Different Brain Regions. J. Chem. Neuroanat. 2005, 30, 144–157. [Google Scholar] [CrossRef]
- Cunningham, C.; Wilcockson, D.C.; Campion, S.; Lunnon, K.; Perry, V.H. Central and Systemic Endotoxin Challenges Exacerbate the Local Inflammatory Response and Increase Neuronal Death during Chronic Neurodegeneration. J. Neurosci. 2005, 25, 9275–9284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, D.E.; Pietrzik, C.U.; Baum, L.; Chevallier, N.; Merriam, D.E.; Kounnas, M.Z.; Wagner, S.L.; Troncoso, J.C.; Kawas, C.H.; Katzman, R.; et al. Modulation of Amyloid Beta-Protein Clearance and Alzheimer’s Disease Susceptibility by the LDL Receptor-Related Protein Pathway. J. Clin. Investig. 2000, 106, 1159–1166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiang, Y.; Bu, X.-L.; Liu, Y.-H.; Zhu, C.; Shen, L.-L.; Jiao, S.-S.; Zhu, X.-Y.; Giunta, B.; Tan, J.; Song, W.-H.; et al. Physiological Amyloid-Beta Clearance in the Periphery and Its Therapeutic Potential for Alzheimer’s Disease. Acta Neuropathol. 2015, 130, 487–499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harrison, I.F.; Ismail, O.; Machhada, A.; Colgan, N.; Ohene, Y.; Nahavandi, P.; Ahmed, Z.; Fisher, A.; Meftah, S.; Murray, T.K.; et al. Impaired Glymphatic Function and Clearance of Tau in an Alzheimer’s Disease Model. Brain 2020, 143, 2576–2593. [Google Scholar] [CrossRef] [PubMed]
- Nedergaard, M. Garbage Truck of the Brain. Science 2013, 340, 1529–1530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kress, B.T.; Iliff, J.J.; Xia, M.; Wang, M.; Wei, H.S.; Zeppenfeld, D.; Xie, L.; Kang, H.; Xu, Q.; Liew, J.A.; et al. Impairment of Paravascular Clearance Pathways in the Aging Brain. Ann. Neurol. 2014, 76, 845–861. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Cai, J.; Zhang, W.; Gong, X.; Yan, S.; Zhang, K.; Luo, Z.; Sun, J.; Jiang, Q.; Lou, M. Impairment of the Glymphatic Pathway and Putative Meningeal Lymphatic Vessels in the Aging Human. Ann. Neurol. 2020, 87, 357–369. [Google Scholar] [CrossRef]
- Rasmussen, M.K.; Mestre, H.; Nedergaard, M. The Glymphatic Pathway in Neurological Disorders. Lancet Neurol. 2018, 17, 1016–1024. [Google Scholar] [CrossRef] [Green Version]
- Jessen, N.A.; Munk, A.S.F.; Lundgaard, I.; Nedergaard, M. The Glymphatic System: A Beginner’s Guide. Neurochem. Res. 2015, 40, 2583–2599. [Google Scholar] [CrossRef] [Green Version]
- Nation, D.A.; Sweeney, M.D.; Montagne, A.; Sagare, A.P.; D’Orazio, L.M.; Pachicano, M.; Sepehrband, F.; Nelson, A.R.; Buennagel, D.P.; Harrington, M.G.; et al. Blood-Brain Barrier Breakdown Is an Early Biomarker of Human Cognitive Dysfunction. Nat. Med. 2019, 25, 270–276. [Google Scholar] [CrossRef]
- Montagne, A.; Barnes, S.R.; Sweeney, M.D.; Halliday, M.R.; Sagare, A.P.; Zhao, Z.; Toga, A.W.; Jacobs, R.E.; Liu, C.Y.; Amezcua, L.; et al. Blood-Brain Barrier Breakdown in the Aging Human Hippocampus. Neuron 2015, 85, 296–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montagne, A.; Nation, D.A.; Sagare, A.P.; Barisano, G.; Sweeney, M.D.; Chakhoyan, A.; Pachicano, M.; Joe, E.; Nelson, A.R.; D’Orazio, L.M.; et al. APOE4 Leads to Blood-Brain Barrier Dysfunction Predicting Cognitive Decline. Nature 2020, 581, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; Golenbock, D.T.; Latz, E. Innate Immunity in Alzheimer’s Disease. Nat. Immunol. 2015, 16, 229–236. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Holtzman, D.M. Interplay between Innate Immunity and Alzheimer Disease: APOE and TREM2 in the Spotlight. Nat. Rev. Immunol. 2018, 18, 759–772. [Google Scholar] [CrossRef]
- Sarlus, H.; Heneka, M.T. Microglia in Alzheimer’s Disease. J. Clin. Investig. 2017, 127, 3240–3249. [Google Scholar] [CrossRef]
- Perry, V.H.; Newman, T.A.; Cunningham, C. The Impact of Systemic Infection on the Progression of Neurodegenerative Disease. Nat. Rev. Neurosci. 2003, 4, 103–112. [Google Scholar] [CrossRef]
- Holmes, C.; Cunningham, C.; Zotova, E.; Woolford, J.; Dean, C.; Kerr, S.; Culliford, D.; Perry, V.H. Systemic Inflammation and Disease Progression in Alzheimer Disease. Neurology 2009, 73, 768–774. [Google Scholar] [CrossRef] [Green Version]
- Tejera, D.; Mercan, D.; Sanchez-Caro, J.M. Systemic Inflammation Impairs Microglial Aβ Clearance through NLRP 3 Inflammasome. EMBO J. 2019, 38, e101064. [Google Scholar] [CrossRef]
- Harold, D.; Abraham, R.; Hollingworth, P.; Sims, R.; Gerrish, A.; Hamshere, M.L.; Pahwa, J.S.; Moskvina, V.; Dowzell, K.; Williams, A.; et al. Genome-Wide Association Study Identifies Variants at CLU and PICALM Associated with Alzheimer’s Disease. Nat. Genet. 2009, 41, 1088–1093. [Google Scholar] [CrossRef] [Green Version]
- Hollingworth, P.; Harold, D.; Sims, R.; Gerrish, A.; Lambert, J.-C.; Carrasquillo, M.M.; Abraham, R.; Hamshere, M.L.; Pahwa, J.S.; Moskvina, V.; et al. Common Variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP Are Associated with Alzheimer’s Disease. Nat. Genet. 2011, 43, 429–435. [Google Scholar] [CrossRef] [Green Version]
- Lambert, J.-C.; Heath, S.; Even, G.; Campion, D.; Sleegers, K.; Hiltunen, M.; Combarros, O.; Zelenika, D.; Bullido, M.J.; Tavernier, B.; et al. Genome-Wide Association Study Identifies Variants at CLU and CR1 Associated with Alzheimer’s Disease. Nat. Genet. 2009, 41, 1094–1099. [Google Scholar] [CrossRef]
- Rainey-Smith, S.R.; Mazzucchelli, G.N.; Villemagne, V.L.; Brown, B.M.; Porter, T.; Weinborn, M.; Bucks, R.S.; Milicic, L.; Sohrabi, H.R.; Taddei, K.; et al. Genetic Variation in Aquaporin-4 Moderates the Relationship between Sleep and Brain Aβ-Amyloid Burden. Transl. Psychiatry 2018, 8, 47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandra, A.; Farrell, C.; Wilson, H.; Dervenoulas, G.; De Natale, E.R.; Politis, M. Alzheimer’s Disease Neuroimaging Initiative Aquaporin-4 Polymorphisms Predict Amyloid Burden and Clinical Outcome in the Alzheimer’s Disease Spectrum. Neurobiol. Aging 2021, 97, 1–9. [Google Scholar] [CrossRef]
- Lambert, J.C.; Ibrahim-Verbaas, C.A.; Harold, D.; Naj, A.C.; Sims, R.; Bellenguez, C.; DeStafano, A.L.; Bis, J.C.; Beecham, G.W.; Grenier-Boley, B.; et al. Meta-Analysis of 74,046 Individuals Identifies 11 New Susceptibility Loci for Alzheimer’s Disease. Nat. Genet. 2013, 45, 1452–1458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willer, C.J.; Schmidt, E.M.; Sengupta, S.; Peloso, G.M.; Gustafsson, S.; Kanoni, S.; Ganna, A.; Chen, J.; Buchkovich, M.L.; Mora, S.; et al. Discovery and Refinement of Loci Associated with Lipid Levels. Nat. Genet. 2013, 45, 1274–1283. [Google Scholar] [PubMed] [Green Version]
- Kunkle, B.W.; Grenier-Boley, B.; Sims, R.; Bis, J.C.; Damotte, V.; Naj, A.C.; Boland, A.; Vronskaya, M.; van der Lee, S.J.; Amlie-Wolf, A.; et al. Genetic Meta-Analysis of Diagnosed Alzheimer’s Disease Identifies New Risk Loci and Implicates Aβ, Tau, Immunity and Lipid Processing. Nat. Genet. 2019, 51, 414–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guerreiro, R.; Wojtas, A.; Bras, J.; Carrasquillo, M.; Rogaeva, E.; Majounie, E.; Cruchaga, C.; Sassi, C.; Kauwe, J.S.K.; Younkin, S.; et al. TREM2 Variants in Alzheimer’s Disease. N. Engl. J. Med. 2013, 368, 117–127. [Google Scholar] [CrossRef] [Green Version]
- Jonsson, T.; Stefansson, H.; Steinberg, S.; Jonsdottir, I.; Jonsson, P.V.; Snaedal, J.; Bjornsson, S.; Huttenlocher, J.; Levey, A.I.; Lah, J.J.; et al. Variant of TREM2 Associated with the Risk of Alzheimer’s Disease. N. Engl. J. Med. 2013, 368, 107–116. [Google Scholar] [CrossRef] [Green Version]
- Jäkel, L.; De Kort, A.M.; Klijn, C.J.M.; Schreuder, F.H.B.M.; Verbeek, M.M. Prevalence of Cerebral Amyloid Angiopathy: A Systematic Review and Meta-Analysis. Alzheimers Dement. 2022, 18, 10–28. [Google Scholar] [CrossRef]
- Hampel, H.; Müller-Spahn, F.; Berger, C.; Haberl, A.; Ackenheil, M.; Hock, C. Evidence of Blood-Cerebrospinal Fluid-Barrier Impairment in a Subgroup of Patients with Dementia of the Alzheimer Type and Major Depression: A Possible Indicator for Immunoactivation. Dement. Geriatr. Cogn. Disord. 1995, 6, 348–354. [Google Scholar] [CrossRef]
- Süssmuth, S.D.; Reiber, H.; Tumani, H. Tau Protein in Cerebrospinal Fluid (CSF): A Blood–CSF Barrier Related Evaluation in Patients with Various Neurological Diseases. Neurosci. Lett. 2001, 300, 95–98. [Google Scholar] [CrossRef]
- Pascale, C.L.; Miller, M.C.; Chiu, C.; Boylan, M.; Caralopoulos, I.N.; Gonzalez, L.; Johanson, C.E.; Silverberg, G.D. Amyloid-Beta Transporter Expression at the Blood-CSF Barrier Is Age-Dependent. Fluids Barriers CNS 2011, 8, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanekiyo, T.; Bu, G. The Low-Density Lipoprotein Receptor-Related Protein 1 and Amyloid-β Clearance in Alzheimer’s Disease. Front. Aging Neurosci. 2014, 6, 93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shibata, M.; Yamada, S.; Kumar, S.R.; Calero, M.; Bading, J.; Frangione, B.; Holtzman, D.M.; Miller, C.A.; Strickland, D.K.; Ghiso, J.; et al. Clearance of Alzheimer’s Amyloid-Ss(1-40) Peptide from Brain by LDL Receptor-Related Protein-1 at the Blood-Brain Barrier. J. Clin. Investig. 2000, 106, 1489–1499. [Google Scholar] [CrossRef] [Green Version]
- González-Marrero, I.; Giménez-Llort, L.; Johanson, C.E.; Carmona-Calero, E.M.; Castañeyra-Ruiz, L.; Brito-Armas, J.M.; Castañeyra-Perdomo, A.; Castro-Fuentes, R. Choroid Plexus Dysfunction Impairs Beta-Amyloid Clearance in a Triple Transgenic Mouse Model of Alzheimer’s Disease. Front. Cell Neurosci. 2015, 9, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seppälä, T.T.; Koivisto, A.M.; Hartikainen, P.; Helisalmi, S.; Soininen, H.; Herukka, S.-K. Longitudinal Changes of CSF Biomarkers in Alzheimer’s Disease. J. Alzheimers Dis. 2011, 25, 583–594. [Google Scholar] [CrossRef] [PubMed]
- Maccioni, R.B.; Lavados, M.; Guillón, M.; Mujica, C.; Bosch, R.; Farías, G.; Fuentes, P. Anomalously Phosphorylated Tau and Aβ Fragments in the CSF Correlates with Cognitive Impairment in MCI Subjects. Neurobiol. Aging 2006, 27, 237–244. [Google Scholar] [CrossRef]
- Tahira, A.; Marques, F.; Lisboa, B.; Feltrin, A.; Barbosa, A.; de Oliveira, K.C.; de Bragança Pereira, C.A.; Leite, R.; Grinberg, L.; Suemoto, C.; et al. Are the 50’s, the Transition Decade, in Choroid Plexus Aging? Geroscience 2021, 43, 225–237. [Google Scholar] [CrossRef]
- Kant, S.; Stopa, E.G.; Johanson, C.E.; Baird, A.; Silverberg, G.D. Choroid Plexus Genes for CSF Production and Brain Homeostasis Are Altered in Alzheimer’s Disease. Fluids Barriers CNS 2018, 15, 34. [Google Scholar] [CrossRef] [Green Version]
- Sousa, J.C.; Cardoso, I.; Marques, F.; Saraiva, M.J.; Palha, J.A. Transthyretin and Alzheimer’s Disease: Where in the Brain? Neurobiol. Aging 2007, 28, 713–718. [Google Scholar] [CrossRef]
- Buxbaum, J.N.; Ye, Z.; Reixach, N.; Friske, L.; Levy, C.; Das, P.; Golde, T.; Masliah, E.; Roberts, A.R.; Bartfai, T. Transthyretin Protects Alzheimer’s Mice from the Behavioral and Biochemical Effects of Aβ Toxicity. Proc. Natl. Acad. Sci. USA 2008, 105, 2681–2686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, Q.; Anderson, D.H.; Liang, W.Y.; Chou, J.; Saelices, L. The Inhibition of Cellular Toxicity of Amyloid-β by Dissociated Transthyretin. J. Biol. Chem. 2020, 295, 14015–14024. [Google Scholar] [CrossRef] [PubMed]
- Klunk, W.E.; Engler, H.; Nordberg, A.; Wang, Y.; Blomqvist, G.; Holt, D.P.; Bergström, M.; Savitcheva, I.; Huang, G.-F.; Estrada, S.; et al. Imaging Brain Amyloid in Alzheimer’s Disease with Pittsburgh Compound-B. Ann. Neurol. 2004, 55, 306–319. [Google Scholar] [CrossRef] [PubMed]
- Shoghi-Jadid, K.; Small, G.W.; Agdeppa, E.D.; Kepe, V.; Ercoli, L.M.; Siddarth, P.; Read, S.; Satyamurthy, N.; Petric, A.; Huang, S.-C.; et al. Localization of Neurofibrillary Tangles and Beta-Amyloid Plaques in the Brains of Living Patients with Alzheimer Disease. Am. J. Geriatr. Psychiatry 2002, 10, 24–35. [Google Scholar] [CrossRef] [PubMed]
- Palmert, M.R.; Podlisny, M.B.; Witker, D.S.; Oltersdorf, T.; Younkin, L.H.; Selkoe, D.J.; Younkin, S.G. The Beta-Amyloid Protein Precursor of Alzheimer Disease Has Soluble Derivatives Found in Human Brain and Cerebrospinal Fluid. Proc. Natl. Acad. Sci. USA 1989, 86, 6338–6342. [Google Scholar] [CrossRef] [Green Version]
- Vigo-Pelfrey, C.; Lee, D.; Keim, P.; Lieberburg, I.; Schenk, D.B. Characterization of Beta-Amyloid Peptide from Human Cerebrospinal Fluid. J. Neurochem. 1993, 61, 1965–1968. [Google Scholar] [CrossRef] [PubMed]
- Tamaoka, A.; Fukushima, T.; Sawamura, N.; Ishikawa, K.; Oguni, E.; Komatsuzaki, Y.; Shoji, S. Amyloid Beta Protein in Plasma from Patients with Sporadic Alzheimer’s Disease. J. Neurol. Sci. 1996, 141, 65–68. [Google Scholar] [CrossRef]
- Scheuner, D.; Eckman, C.; Jensen, M.; Song, X.; Citron, M.; Suzuki, N.; Bird, T.D.; Hardy, J.; Hutton, M.; Kukull, W.; et al. Secreted Amyloid Beta-Protein Similar to That in the Senile Plaques of Alzheimer’s Disease Is Increased in Vivo by the Presenilin 1 and 2 and APP Mutations Linked to Familial Alzheimer’s Disease. Nat. Med. 1996, 2, 864–870. [Google Scholar] [CrossRef]
- Mattsson, N.; Zetterberg, H.; Janelidze, S.; Insel, P.S.; Andreasson, U.; Stomrud, E.; Palmqvist, S.; Baker, D.; Tan Hehir, C.A.; Jeromin, A.; et al. Plasma Tau in Alzheimer Disease. Neurology 2016, 87, 1827–1835. [Google Scholar] [CrossRef] [Green Version]
- Thijssen, E.H.; La Joie, R.; Strom, A.; Fonseca, C.; Iaccarino, L.; Wolf, A.; Spina, S.; Allen, I.E.; Cobigo, Y.; Heuer, H.; et al. Plasma Phosphorylated Tau 217 and Phosphorylated Tau 181 as Biomarkers in Alzheimer’s Disease and Frontotemporal Lobar Degeneration: A Retrospective Diagnostic Performance Study. Lancet Neurol. 2021, 20, 739–752. [Google Scholar] [CrossRef]
- Palmqvist, S.; Zetterberg, H.; Mattsson, N.; Johansson, P.; Minthon, L.; Blennow, K.; Olsson, M.; Hansson, O.; Alzheimer’s Disease Neuroimaging Initiative. Swedish BioFINDER Study Group Detailed Comparison of Amyloid PET and CSF Biomarkers for Identifying Early Alzheimer Disease. Neurology 2015, 85, 1240–1249. [Google Scholar] [CrossRef] [PubMed]
- Mattsson, N.; Andreasson, U.; Zetterberg, H.; Blennow, K. Alzheimer’s Disease Neuroimaging Initiative Association of Plasma Neurofilament Light with Neurodegeneration in Patients with Alzheimer Disease. JAMA Neurol. 2017, 74, 557–566. [Google Scholar] [CrossRef] [PubMed]
- Cicognola, C.; Janelidze, S.; Hertze, J.; Zetterberg, H.; Blennow, K.; Mattsson-Carlgren, N.; Hansson, O. Plasma Glial Fibrillary Acidic Protein Detects Alzheimer Pathology and Predicts Future Conversion to Alzheimer Dementia in Patients with Mild Cognitive Impairment. Alzheimers Res. Ther. 2021, 13, 68. [Google Scholar] [CrossRef] [PubMed]
- Benedet, A.L.; Milà-Alomà, M.; Vrillon, A.; Ashton, N.J.; Pascoal, T.A.; Lussier, F.; Karikari, T.K.; Hourregue, C.; Cognat, E.; Dumurgier, J.; et al. Differences Between Plasma and Cerebrospinal Fluid Glial Fibrillary Acidic Protein Levels Across the Alzheimer Disease Continuum. JAMA Neurol. 2021, 78, 1471–1483. [Google Scholar] [CrossRef]
- Beydoun, M.A.; Noren Hooten, N.; Beydoun, H.A.; Maldonado, A.I.; Weiss, J.; Evans, M.K.; Zonderman, A.B. Plasma Neurofilament Light as a Potential Biomarker for Cognitive Decline in a Longitudinal Study of Middle-Aged Urban Adults. Transl. Psychiatry 2021, 11, 436. [Google Scholar] [CrossRef]
- Pereira, J.B.; Janelidze, S.; Smith, R.; Mattsson-Carlgren, N.; Palmqvist, S.; Teunissen, C.E.; Zetterberg, H.; Stomrud, E.; Ashton, N.J.; Blennow, K.; et al. Plasma GFAP Is an Early Marker of Amyloid-β but Not Tau Pathology in Alzheimer’s Disease. Brain 2021, 144, 3505–3516. [Google Scholar] [CrossRef]
- Geekiyanage, H.; Jicha, G.A.; Nelson, P.T.; Chan, C. Blood Serum MiRNA: Non-Invasive Biomarkers for Alzheimer’s Disease. Exp. Neurol. 2012, 235, 491–496. [Google Scholar] [CrossRef] [Green Version]
- Cha, D.J.; Mengel, D.; Mustapic, M.; Liu, W.; Selkoe, D.J.; Kapogiannis, D.; Galasko, D.; Rissman, R.A.; Bennett, D.A.; Walsh, D.M. MiR-212 and MiR-132 Are Downregulated in Neurally Derived Plasma Exosomes of Alzheimer’s Patients. Front. Neurosci. 2019, 13, 1208. [Google Scholar] [CrossRef] [Green Version]
- Badhwar, A.; Haqqani, A.S. Biomarker Potential of Brain-Secreted Extracellular Vesicles in Blood in Alzheimer’s Disease. Alzheimers Dement. 2020, 12, e12001. [Google Scholar] [CrossRef]
- Salta, E.; De Strooper, B. MicroRNA-132: A Key Noncoding RNA Operating in the Cellular Phase of Alzheimer’s Disease. FASEB J. 2017, 31, 424–433. [Google Scholar] [CrossRef]
- Salta, E.; Sierksma, A.; Vanden Eynden, E.; De Strooper, B. MiR-132 Loss De-represses ITPKB and Aggravates Amyloid and TAU Pathology in Alzheimer’s Brain. EMBO Mol. Med. 2016, 8, 1005–1018. [Google Scholar] [CrossRef] [PubMed]
- Uchida, K.; Shan, L.; Suzuki, H.; Tabuse, Y.; Nishimura, Y.; Hirokawa, Y.; Mizukami, K.; Akatsu, H.; Meno, K.; Asada, T. Amyloid-β Sequester Proteins as Blood-Based Biomarkers of Cognitive Decline. Alzheimers Dement. 2015, 1, 270–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.; Suzuki, H.; Ito, H.; Korenaga, T.; Akatsu, H.; Meno, K.; Uchida, K. Serum Levels of Proteins Involved in Amyloid-β Clearance Are Related to Cognitive Decline and Neuroimaging Changes in Mild Cognitive Impairment. Alzheimers Dement. 2019, 11, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Zuin, M.; Cervellati, C.; Trentini, A.; Passaro, A.; Rosta, V.; Zimetti, F.; Zuliani, G. Association between Serum Concentrations of Apolipoprotein A-I (ApoA-I) and Alzheimer’s Disease: Systematic Review and Meta-Analysis. Diagnostics 2021, 11, 984. [Google Scholar] [CrossRef] [PubMed]
- Koch, M.; DeKosky, S.T.; Fitzpatrick, A.L.; Furtado, J.D.; Lopez, O.L.; Kuller, L.H.; Mackey, R.H.; Hughes, T.M.; Mukamal, K.J.; Jensen, M.K. Apolipoproteins and Alzheimer’s Pathophysiology. Alzheimers Dement. 2018, 10, 545–553. [Google Scholar] [CrossRef]
- Fu, H.; Liu, B.; Frost, J.L.; Hong, S.; Jin, M.; Ostaszewski, B.; Shankar, G.M.; Costantino, I.M.; Carroll, M.C.; Mayadas, T.N.; et al. Complement Component C3 and Complement Receptor Type 3 Contribute to the Phagocytosis and Clearance of Fibrillar Aβ by Microglia. Glia 2012, 60, 993–1003. [Google Scholar] [CrossRef] [Green Version]
- Morgan, A.R.; Touchard, S.; Leckey, C.; O’Hagan, C.; Nevado-Holgado, A.J.; Barkhof, F.; Bertram, L.; Blin, O.; Bos, I.; NIMA Consortium; et al. Inflammatory Biomarkers in Alzheimer’s Disease Plasma. Alzheimers Dement. 2019, 15, 776–787. [Google Scholar] [CrossRef]
- Hakobyan, S.; Harding, K.; Aiyaz, M.; Hye, A.; Dobson, R.; Baird, A.; Liu, B.; Harris, C.L.; Lovestone, S.; Morgan, B.P. Complement Biomarkers as Predictors of Disease Progression in Alzheimer’s Disease. J. Alzheimers Dis. 2016, 54, 707–716. [Google Scholar] [CrossRef] [Green Version]
- Biere, A.L.; Ostaszewski, B.; Stimson, E.R.; Hyman, B.T.; Maggio, J.E.; Selkoe, D.J. Amyloid β-Peptide Is Transported on Lipoproteins and Albumin in Human Plasma. J. Biol. Chem. 1996, 271, 32916–32922. [Google Scholar] [CrossRef] [Green Version]
- Velayudhan, L.; Killick, R.; Hye, A.; Kinsey, A.; Güntert, A.; Lynham, S.; Ward, M.; Leung, R.; Lourdusamy, A.; To, A.W.M.; et al. Plasma Transthyretin as a Candidate Marker for Alzheimer’s Disease. J. Alzheimers Dis. 2012, 28, 369–375. [Google Scholar] [CrossRef] [Green Version]
- Serot, J.M.; Christmann, D.; Dubost, T.; Couturier, M. Cerebrospinal Fluid Transthyretin: Aging and Late Onset Alzheimer’s Disease. J. Neurol. Neurosurg. Psychiatry 1997, 63, 506–508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, S.H.; Jung, E.S.; Sohn, J.H.; Hong, H.J.; Hong, H.S.; Kim, J.W.; Na, D.L.; Kim, M.; Kim, H.; Ha, H.J.; et al. Human Serum Transthyretin Levels Correlate Inversely with Alzheimer’s Disease. J. Alzheimers. Dis. 2011, 25, 77–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ribeiro, C.A.; Santana, I.; Oliveira, C.; Baldeiras, I.; Moreira, J.; Saraiva, M.J.; Cardoso, I. Transthyretin Decrease in Plasma of MCI and AD Patients: Investigation of Mechanisms for Disease Modulation. Curr. Alzheimer Res. 2012, 9, 881–889. [Google Scholar] [CrossRef]
- Uchida, Y.; Kan, H.; Sakurai, K.; Arai, N.; Inui, S.; Kobayashi, S.; Kato, D.; Ueki, Y.; Matsukawa, N. Iron Leakage Owing to blood-brain Barrier Disruption in Small Vessel Disease CADASIL. Neurology 2020, 95, e1188–e1198. [Google Scholar] [CrossRef] [PubMed]
- Montagne, A.; Nikolakopoulou, A.M.; Zhao, Z.; Sagare, A.P.; Si, G.; Lazic, D.; Barnes, S.R.; Daianu, M.; Ramanathan, A.; Go, A.; et al. Pericyte Degeneration Causes White Matter Dysfunction in the Mouse Central Nervous System. Nat. Med. 2018, 24, 326–337. [Google Scholar] [CrossRef] [PubMed]
- Taoka, T.; Masutani, Y.; Kawai, H.; Nakane, T. Evaluation of Glymphatic System Activity with the Diffusion MR Technique: Diffusion Tensor Image Analysis along the Perivascular Space (DTI-ALPS) in Alzheimer’s Disease Cases. Jpn. J. Radiol. 2017, 35, 172–178. [Google Scholar] [CrossRef]
- Watts, R.; Steinklein, J.M.; Waldman, L.; Zhou, X.; Filippi, C.G. Measuring Glymphatic Flow in Man Using Quantitative Contrast-Enhanced MRI. Am. J. Neuroradiol. 2019, 40, 648–651. [Google Scholar] [CrossRef] [Green Version]
- Ringstad, G.; Vatnehol, S.A.S.; Eide, P.K. Glymphatic MRI in Idiopathic Normal Pressure Hydrocephalus. Brain 2017, 140, 2691–2705. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Wolk, D.A.; Reddin, J.S.; Korczykowski, M.; Martinez, P.M.; Musiek, E.S.; Newberg, A.B.; Julin, P.; Arnold, S.E.; Greenberg, J.H.; et al. Voxel-Level Comparison of Arterial Spin-Labeled Perfusion MRI and FDG-PET in Alzheimer Disease. Neurology 2011, 77, 1977–1985. [Google Scholar] [CrossRef] [Green Version]
- Dyrba, M.; Ewers, M.; Wegrzyn, M.; Kilimann, I.; Plant, C.; Oswald, A.; Meindl, T.; Pievani, M.; Bokde, A.L.W.; Fellgiebel, A.; et al. Combining DTI and MRI for the Automated Detection of Alzheimer’s Disease Using a Large European Multicenter Dataset. In Proceedings of the Second International Conference on Multimodal Brain Image Analysis (MBIA 2012), Nice, France, 1–5 October 2012; Lecture Notes in Computer Science. Yap, P.T., Liu, T., Shen, D., Westin, C.F., Shen, L., Eds.; Springer: Berlin/Heidelberg, Germany, 2012; Volume 7509, pp. 18–28. [Google Scholar]
- Thijssen, E.H.; La Joie, R.; Wolf, A.; Strom, A.; Wang, P.; Iaccarino, L.; Bourakova, V.; Cobigo, Y.; Heuer, H.; Spina, S.; et al. Diagnostic Value of Plasma Phosphorylated Tau181 in Alzheimer’s Disease and Frontotemporal Lobar Degeneration. Nat. Med. 2020, 26, 387–397. [Google Scholar] [CrossRef]
- Guideline Development Group, World Health Organization. Risk Reduction of Cognitive Decline and Dementia: WHO Guidelines; World Health Organization: Geneva, Switzerland, 2019. [Google Scholar]
- Tarasoff-Conway, J.M.; Carare, R.O.; Osorio, R.S.; Glodzik, L.; Butler, T.; Fieremans, E.; Axel, L.; Rusinek, H.; Nicholson, C.; Zlokovic, B.V.; et al. Clearance Systems in the Brain—Implications for Alzheimer Disease. Nat. Rev. Neurol. 2015, 11, 457–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klostranec, J.M.; Vucevic, D.; Bhatia, K.D.; Kortman, H.G.J.; Krings, T.; Murphy, K.P.; terBrugge, K.G.; Mikulis, D.J. Current Concepts in Intracranial Interstitial Fluid Transport and the Glymphatic System: Part II-Imaging Techniques and Clinical Applications. Radiology 2021, 301, 516–532. [Google Scholar] [CrossRef] [PubMed]
- Silva, C.S.; Eira, J.; Ribeiro, C.A.; Oliveira, Â.; Sousa, M.M.; Cardoso, I.; Liz, M.A. Transthyretin Neuroprotection in Alzheimer’s Disease Is Dependent on Proteolysis. Neurobiol. Aging 2017, 59, 10–14. [Google Scholar] [CrossRef] [PubMed]
- Tsuzuki, K.; Fukatsu, R.; Yamaguchi, H.; Tateno, M.; Imai, K.; Fujii, N.; Yamauchi, T. Transthyretin Binds Amyloid β Peptides, Aβ1--42 and Aβ1--40 to Form Complex in the Autopsied Human Kidney--Possible Role of Transthyretin for Aβ Sequestration. Neurosci. Lett. 2000, 281, 171–174. [Google Scholar] [CrossRef]
- Alemi, M.; Gaiteiro, C.; Ribeiro, C.A.; Santos, L.M.; Gomes, J.R.; Oliveira, S.M.; Couraud, P.-O.; Weksler, B.; Romero, I.; Saraiva, M.J.; et al. Transthyretin Participates in Beta-Amyloid Transport from the Brain to the Liver—Involvement of the Low-Density Lipoprotein Receptor-Related Protein 1? Sci. Rep. 2016, 6, 20164. [Google Scholar] [CrossRef] [Green Version]
- Stanyon, H.F.; Viles, J.H. Human Serum Albumin Can Regulate Amyloid-β Peptide Fiber Growth in the Brain Interstitium: Implications for Alzheimer Disease. J. Biol. Chem. 2012, 287, 28163–28168. [Google Scholar] [CrossRef] [Green Version]
- Boada, M.; López, O.L.; Olazarán, J.; Núñez, L.; Pfeffer, M.; Paricio, M.; Lorites, J.; Piñol-Ripoll, G.; Gámez, J.E.; Anaya, F.; et al. A Randomized, Controlled Clinical Trial of Plasma Exchange with Albumin Replacement for Alzheimer’s Disease: Primary Results of the AMBAR Study. Alzheimers Dement. 2020, 16, 1412–1425. [Google Scholar] [CrossRef]
- Koldamova, R.P.; Lefterov, I.M.; Lefterova, M.I.; Lazo, J.S. Apolipoprotein A-I Directly Interacts with Amyloid Precursor Protein and Inhibits A Beta Aggregation and Toxicity. Biochemistry 2001, 40, 3553–3560. [Google Scholar] [CrossRef]
- Bell, R.D.; Sagare, A.P.; Friedman, A.E.; Bedi, G.S.; Holtzman, D.M.; Deane, R.; Zlokovic, B.V. Transport Pathways for Clearance of Human Alzheimer’s Amyloid β-Peptide and Apolipoproteins E and J in the Mouse Central Nervous System. J. Cereb. Blood Flow Metab. 2007, 27, 909–918. [Google Scholar] [CrossRef] [Green Version]
- Schilling, S.; Tzourio, C.; Soumaré, A.; Kaffashian, S.; Dartigues, J.-F.; Ancelin, M.-L.; Samieri, C.; Dufouil, C.; Debette, S. Differential Associations of Plasma Lipids with Incident Dementia and Dementia Subtypes in the 3C Study: A Longitudinal, Population-Based Prospective Cohort Study. PLoS Med. 2017, 14, e1002265. [Google Scholar] [CrossRef]
- Rasmussen, K.L.; Tybjærg-Hansen, A.; Nordestgaard, B.G.; Frikke-Schmidt, R. Plasma Apolipoprotein E Levels and Risk of Dementia: A Mendelian Randomization Study of 106,562 Individuals. Alzheimers Dement. 2018, 14, 71–80. [Google Scholar] [CrossRef] [PubMed]
- Slot, R.E.R.; Van Harten, A.C.; Kester, M.I.; Jongbloed, W.; Bouwman, F.H.; Teunissen, C.E.; Scheltens, P.; Veerhuis, R.; van der Flier, W.M. Apolipoprotein A1 in Cerebrospinal Fluid and Plasma and Progression to Alzheimer’s Disease in Non-Demented Elderly. J. Alzheimers Dis. 2017, 56, 687–697. [Google Scholar] [CrossRef] [PubMed]
- Crane, A.; Brubaker, W.D.; Johansson, J.U.; Trigunaite, A.; Ceballos, J.; Bradt, B.; Glavis-Bloom, C.; Wallace, T.L.; Tenner, A.J.; Rogers, J. Peripheral Complement Interactions with Amyloid β Peptide in Alzheimer’s Disease: 2. Relationship to Amyloid β Immunotherapy. Alzheimers Dement. 2018, 14, 243–252. [Google Scholar] [CrossRef] [PubMed]
- Brubaker, W.D.; Crane, A.; Johansson, J.U.; Yen, K.; Garfinkel, K.; Mastroeni, D.; Asok, P.; Bradt, B.; Sabbagh, M.; Wallace, T.L.; et al. Peripheral Complement Interactions with Amyloid β Peptide: Erythrocyte Clearance Mechanisms. Alzheimers Dement. 2017, 13, 1397–1409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rasmussen, K.L.; Nordestgaard, B.G.; Frikke-Schmidt, R.; Nielsen, S.F. An Updated Alzheimer Hypothesis: Complement C3 and Risk of Alzheimer’s Disease—A Cohort Study of 95,442 Individuals. Alzheimers Dement. 2018, 14, 1589–1601. [Google Scholar] [CrossRef] [PubMed]
- Winblad, B.; Amouyel, P.; Andrieu, S.; Ballard, C.; Brayne, C.; Brodaty, H.; Cedazo-Minguez, A.; Dubois, B.; Edvardsson, D.; Feldman, H.; et al. Defeating Alzheimer’s Disease and Other Dementias: A Priority for European Science and Society. Lancet Neurol. 2016, 15, 455–532. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Yu, Z.; Su, W.; Isquith, D.A.; Neradilek, M.B.; Lu, N.; Gu, F.; Li, H.; Zhao, X.-Q. Low Levels of ApoA1 Improve Risk Prediction of Type 2 Diabetes Mellitus. J. Clin. Lipidol. 2017, 11, 362–368. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Yu, Z.; Wen, S.; Isquith, D.A.; Neradilek, M.B.; Lu, N.; Gu, F.; Li, H.; Zhao, X.-Q. Development of Type-2 Diabetes Mellitus Is Associated with Low Levels of ApoA1. J. Diabetes Metab. 2016, 7, 1000669. [Google Scholar] [CrossRef] [Green Version]
- Brahimaj, A.; Ligthart, S.; Ikram, M.A.; Hofman, A.; Franco, O.H.; Sijbrands, E.J.G.; Kavousi, M.; Dehghan, A. Serum Levels of Apolipoproteins and Incident Type 2 Diabetes: A Prospective Cohort Study. Diabetes Care 2017, 40, 346–351. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
{kind=link}
| Biomarker | Mechanism | Method/Target | References |
|---|---|---|---|
| Aβ | aggregation neurotoxic | PET CSF plasma EVs | [74,75] [76,77] [78,79] |
| Total tau P-tau | aggregation neurotoxic | PET plasma CSF EVs | [80,81,82] |
| NFL GFAP | neurodegeneration | serum/plasma CSF | [83,84,85,86,87] |
| miR-132, miR-124 miR-132, miR-146 miR-1908, miR-205 | Aβ and tau pathology neuroinflammation cholesterol metabolism | serum EVs | [88,89,90,91,92] |
| ApoE ApoA1 ApoJ | lipid metabolism blood–CSF barrier integrity waste clearance | serum/plasma CSF | [65,93,94,95,96] |
| C3 C5 Clusterin | innate immunity waste clearance | serum/plasma CSF | [93,94,97,98,99] |
| TTR Albumin | blood–CSF barrier waste clearance | serum/plasma CSF | [93,94,100,101,102,103,104] |
| BBB breakage | BBB waste clearance | DCE-MRI | [41,42,43,105,106] |
| CSF–ISF exchange | glymphatic system waste clearance | gadolinium-enhanced glymphatic MRI DTI MRI DTI-ALPS ASL-MRI | [107,108,109,110,111] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Uchida, K. Waste Clearance in the Brain and Neuroinflammation: A Novel Perspective on Biomarker and Drug Target Discovery in Alzheimer’s Disease. Cells 2022, 11, 919. https://doi.org/10.3390/cells11050919
Uchida K. Waste Clearance in the Brain and Neuroinflammation: A Novel Perspective on Biomarker and Drug Target Discovery in Alzheimer’s Disease. Cells. 2022; 11(5):919. https://doi.org/10.3390/cells11050919
Chicago/Turabian StyleUchida, Kazuhiko. 2022. "Waste Clearance in the Brain and Neuroinflammation: A Novel Perspective on Biomarker and Drug Target Discovery in Alzheimer’s Disease" Cells 11, no. 5: 919. https://doi.org/10.3390/cells11050919
APA StyleUchida, K. (2022). Waste Clearance in the Brain and Neuroinflammation: A Novel Perspective on Biomarker and Drug Target Discovery in Alzheimer’s Disease. Cells, 11(5), 919. https://doi.org/10.3390/cells11050919