Abstract
Primary cilia are non-motile plasma membrane extrusions that display a variety of receptors and mechanosensors. Loss of function results in ciliopathies, which have been strongly linked with congenital heart disease, as well as abnormal development and function of most organ systems. Adults with congenital heart disease have high rates of acquired heart failure, and usually die from a cardiac cause. Here we explore primary cilia’s role in acquired heart disease. Intraflagellar Transport 88 knockout results in reduced primary cilia, and knockout from cardiac endothelium produces myxomatous degeneration similar to mitral valve prolapse seen in adult humans. Induced primary cilia inactivation by other mechanisms also produces excess myocardial hypertrophy and altered scar architecture after ischemic injury, as well as hypertension due to a lack of vascular endothelial nitric oxide synthase activation and the resultant left ventricular dysfunction. Finally, primary cilia have cell-to-cell transmission capacity which, when blocked, leads to progressive left ventricular hypertrophy and heart failure, though this mechanism has not been fully established. Further research is still needed to understand primary cilia’s role in adult cardiac pathology, especially heart failure.
1. Introduction
The incidence of adults with congenital heart disease (CHD) has been progressively increasing for some time, in part driven by significant improvements in the management of these patients as children [1]. A child born with CHD today has a 97% chance of survival to adulthood [2], and, at least since 2010, the number of adults living with CHD has exceeded the number of children [3]. Further improvement will need to come from the ongoing management of these patients as adolescents and adults [2]. Adults with CHD show an increased risk of developing ventricular hypertrophy, heart failure, arrhythmias, and sudden cardiac death later in life than patients born with grossly normal hearts [4,5,6,7]. In fact, a majority of these patients die from cardiac causes [1].
Current strategies for the management of these patients, as well as for risk stratification, are insufficient [8,9]. In order to improve outcomes in these patients, providers and translational scientists need to understand the mechanisms of acquired heart disease in this population. With their strong links to both congenital and acquired heart disease, primary cilia represent an important target for further research and therapeutics.
Primary cilia have been the focus of research since the 1960s, when they were first recognized as distinct from motile cilia and present in most mammalian tissues [10,11]. Diseases related to cilia gene mutations, coined ciliopathies, have since been identified in many organ systems [12,13]. Cilia’s role in the cardiovascular system has been more recently defined, with large studies and reviews describing the occurrence of most, if not all, congenital heart diseases in response to mutations in cilia-related genes [14,15]. Primary cilia have now been recognized to play an important role in acquired heart disease as well, and the etiology of this association remains an active area of research. Here we review the available literature on primary cilia and their role in acquired heart disease, and outline areas where more research is needed.
2. Primary Cilia
2.1. Cilia Structure and Components
Primary cilia are extrusions of the plasma membrane that display a variety of receptors and mechanosensors. The core structure is an axoneme of nine doublet microtubules that extend from a basal body, and they are therefore referred to as “9 + 0” cilia. This distinguishes them from motile “9 + 2” cilia, which have an additional two dynein-associated central microtubules, permitting motion [11].
As primary cilia do not intrinsically have associated ribosomes, they instead rely on the intraflagellar transport (IFT) system to ferry receptors and other proteins into and out of the cilium [12]. This system is capable of bidirectional movement along the length of the flagella, between the outer doublet of microtubules and the flagellar membrane [16,17]. IFT proteins, especially Ift88, are often knockout targets in cilia research, as their inactivation results in the absence of primary cilia in the affected cell [18,19].
At the base of the cilium, near the basal body, an interactome of proteins, coined CPLANE, is responsible for ciliogenesis and intraflagellar transport. (Figure 1) These proteins act at the basal body to recruit IFT-A proteins to the base of the cilium and stabilize and insert complete IFT-A particles into the axoneme. Mutations in these proteins have been associated with a variety of ciliopathies [17]. Numerous other membrane-bound proteins located along the cilia have been associated with ciliopathies as well, including polycystins, known for causing autosomal dominant polycystic kidney disease, and septins, which have been linked with a variety of cancers and neurodegenerative conditions [20,21,22,23,24,25].
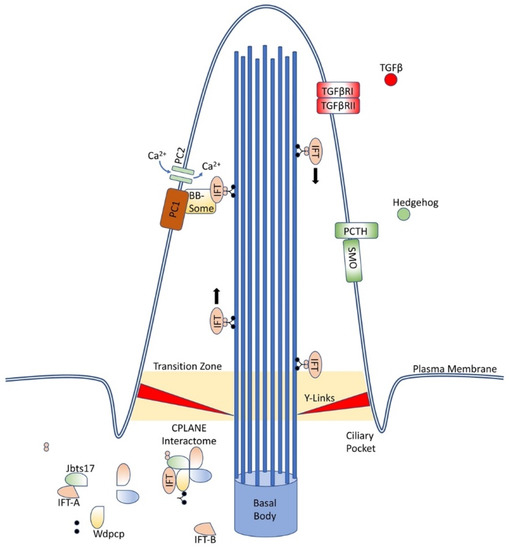
Figure 1.
Primary cilia structure and components. Primary cilia are an extrusion from the cell wall capable of displaying numerous proteins, including those depicted here and many others, and are supported by nine doublet microtubules arising from the basal body. IFT proteins ferry components along the length of the cilia, while the CPLANE interactome remains at the base of the cilia.
2.2. Ciliopathies
For classification purposes, first-order ciliopathies are those diseases which occur due to a mutation in genes required for the proper assembly, maintenance, or function of the cilia or the related centriole; second-order ciliopathies occur due to dysregulation of further upstream factors, such as the nuclear transcription factors Atf3, Tsc22d4, and Cbx5 [26,27]. There are at least 300–1000 first-order, and many more second-order, genes [26,28,29].
Primary cilia play an important role in most mammalian organ systems, so ciliopathies tend to display a variety of multiorgan dysfunction phenotypes. (Table 1) Bardet—Biedl syndrome, for example, is characterized by retinitis pigmentosa, obesity, polydactyly, cognitive impairment, and renal failure [30]. Most ciliopathies show some amount of brain, craniofacial, or endocrine dysfunction, though kidney, reproductive, and heart tissues are also often involved [26].

Table 1.
Known human ciliopathies. A list of known human ciliopathies, though many more are thought to be related to cilia function. Ciliopathies present with significant variation in phenotype depending on the underlying gene mutation and other factors. Due to historical clinical definitions, some syndromes are phenotypes possible from a variety of gene mutations, while others are phenotypic variations of the same genetic defect [42,43].
One of primary cilia’s most important roles, and part of the reason mutations cause such varied phenotypes, is the display of receptors important for cell signaling pathways and the machinery for signal transduction [31]. One of the best studied is Hedgehog (Hh), which is highly dependent on functional primary cilia [19,32,33,34]. The transmembrane protein Smoothened (Smo), which is responsible for Gli protein activation in the Hh pathway, is found at the tip of the cilium [35]. Other pathways, such as Wnt, Notch, and PCP, similarly depend on primary cilia, and ciliopathies can impair their function [36,37].
Autophagy and programmed cell death pathways, which are important for tissue homeostasis and are perturbed in neurodegenerative diseases and cancer, depend on proper ciliary function due to machinery localization to the cilia and interdependent feedback mechanisms [36]. Loss of primary cilia function results in excess cell death from autophagy in mitochondrial stress responses and from mitochondria-dependent apoptosis [38,39]. Finally, extracellular matrix makeup is sensed and regulated through primary cilia [40,41].
2.3. Primary Cilia Locations
Despite their importance for many cellular pathways, primary cilia have not been identified on all cardiac cell types. Primary cilia are displayed on fibroblasts in the heart, Ref. [55] as well as on vascular endothelial cells, though expression on valvular endothelium decreases over time, from abundance in embryologic samples to near absence in adult samples [13,56,57]. Most cardiac interstitial (mesenchymal) cells also display primary cilia [57]. Cardiomyocytes contain primary cilia in embryonic tissue samples and lack them in adult samples, but there is disagreement regarding their presence on neonatal samples, Refs. [55,58] suggesting a possible loss of primary cilia over time.
3. Primary Cilia in Acquired Heart Disease
3.1. Acquired Valvular Heart Disease
The importance of proper cilia function in the embryonic heart has been well established [14,19,59,60]. In a comprehensive analysis of over 87,000 mutagenized mouse fetuses, Li et al. identified 61 genes in which mutations were capable of producing echocardiographically identifiable congenital heart defects, and 35 of these genes encoded either motile or primary cilia proteins. An additional 16 genes were involved in cilia-transduced cell signaling, and 10 regulated vesicular trafficking, which is necessary for proper cilia function [14].
Unlike the congenital defects analyzed by Li et al., mitral valve prolapse (MVP) is not evident on echocardiogram at birth. Instead, it is unusual in infants and children and it is more frequently identified in patients aged 30–80 years of age [61]. This valve pathology is a result of myxomatous degeneration over the lifetime of the patient.
In a genome-wide association study, enrichment for cilia genes was found in patients with MVP, and murine homozygous mutants of the two known familial MVP genes, Dchs1 and Flna, showed decreased primary cilia length on the neonatal mitral valve leaflets [57,62]. Exploring cilia’s mechanistic role in MVP, Toomer et al. showed that the presence of primary cilia on endocardial cells correlated with increased proteoglycan and decreased collagen in the extracellular matrix of valve endocardium. Conditional knockout of intraflagellar transport protein 88 (Ift88) in cardiac endothelial cells in mice resulted in decreased primary cilia counts, increased proteoglycans, and fragmented collagen, i.e., the initiation of myxomatous degeneration [57,63]. While primary cilia abundance on valvular endothelium decreases with age, their effect on the extracellular matrix persists. As adults, these mice show myxomatous mitral valve disease [57].
3.2. Fibrosis
In addition to myxomatous degeneration of the valve, patients with MVP also show progressive left ventricular fibrosis. Cardiac fibrosis is an excessive production and deposition of scar tissue, often a result of conditions such as hypertension or diabetes mellitus, and can lead to increased tissue stiffness, cardiomyocyte atrophy, and arrhythmias [64,65]. The fibrosis observed with MVP is more significant than that seen in patients with primary mitral valve regurgitation from a non-MVP etiology, which may suggest a common cause for both excessive fibrosis and MVP [66].
In cardiac fibroblasts, activation of the transforming growth factor β-1 (TGF-β1) receptor results in production of fibronectin, collagen type I, and collagen type III, which are necessary components of the extracellular matrix in fibrotic tissue [67]. Fibroblasts also undergo transformation to myofibroblasts, which express α-smooth muscle actin (α-SMA) and display contractile ability.
Inactivation of primary cilia by small interference RNA (siRNA) silencing of Polycystin-1 (PC1) in fibroblasts results in a lack of upregulated collagen production in response to TGF-β1. Similarly, siRNA silencing of either PC1 or Ift88 in cardiac fibroblasts results in failure of the fibroblasts to differentiate into myofibroblasts capable of contractile function, which is necessary for standard cardiac remodeling. These mice instead show excess myocardial hypertrophy and altered scar architecture [55].
In addition to native cardiac fibroblast proliferation, endothelial-mesenchymal transition (EndMT) is now recognized as an important source of fibroblasts for perivascular and subendocardial fibrosis [68]. Knockdown of Ift88 in endothelial cells, which results in the absence of primary cilia on these cells, appears to be insufficient to directly induce EndMT in vivo but may prime these cells for EndMT in response to lower stress than would otherwise be required [69,70].
3.3. Vascular Pathology and Cilia
In addition to their role in fibrosis after an ischemic injury, primary cilia also regulate atherosclerosis and, therefore, the risk of ischemic events. Primary cilia serve as mechanosensors in a variety of cell types [71]. In endothelial cells with functional primary cilia, excess shear stress stimulates PC1 interaction with Polycystin-2 (PC2), permitting calcium influx and activating calcium-dependent signaling molecules, including calmodulin and calcium-dependent protein kinase (PKC), that lead to activation of endothelial nitric oxide synthetase (eNOS) and subsequent vasodilation [72,73,74].
Branch points and the lesser curvature of the aorta are at particular risk of atherosclerosis due to relatively low and oscillatory shear stress [75]. These areas also display increased density and stability of primary cilia [76,77]. Initial research suggested that primary cilia may play a role in producing atherosclerosis, as apolipoprotein-E-deficient (Apoe−/−) mice display increased primary cilia as well as increased atherosclerosis at these risk points [63]. However, removing these cilia via knockout of Ift88 results in increased atherosclerosis in Apoe−/− mice in response to a high fat, high cholesterol diet, suggesting this is a protective response mediated by eNOS [78].
PC1 and PC2 gene mutations produce autosomal dominant polycystic kidney disease (ADPKD), which results in hypertension in two-thirds of cases [79]. In addition to the eNOS activation mechanism, primary cilia also protect against hypertension via dopamine receptor 5 (DR5) [74,80]. Stimulation at this receptor results in adenylyl cyclase and PKC activation, leading to vasodilation [81].
3.4. Ventricular Remodeling and Recovery
Cardiomyocyte hypertrophy is an important cell autonomous and non-cell autonomous adaptive response to significant stress, especially hypertension, that is necessary for survival. However prolonged stress and resultant excess hypertrophy and cardiac remodeling can lead to heart failure and sudden cardiac death [82,83,84]. Cardiomyocytes have some ability to sense mechanical forces, including hemodynamic stress, in order to convert stress into intracellular growth signals and induce hypertrophy. However, the molecular identity of the mechanosensor remains elusive. Primary cilia are an attractive candidate as a mechanosensor; however, this has not been demonstrated experimentally.
One possible mechanism appears to be via ciliary extracellular-like vesicles (cELVs) [85]. These vesicles are released from cilia under normal circumstances and at increased rates under fluid shear stress. Blocking ciliary proteins necessary for cELV production using short hairpin RNA (shRNA) prevents cELV production and results in left ventricular hypertrophy, decreasing left ventricular ejection fraction, and, eventually, low blood pressure and cardiovascular collapse [85,86].
3.5. Congenital Heart Disease and Late-Onset Heart Failure
Patients with CHD show a higher risk of heart failure later in life than patients born with grossly normal hearts [4,5,6]. One study showed an overall prevalence of heart failure of 26% in a cohort of patients with surgically corrected CHD [6]. While the highest risk of heart failure is in patients with morphologically right ventricles exposed to systemic pressures, even patients with isolated ventricular septal defect are at higher risk of systolic and diastolic dysfunction 30 or more years after surgical repair [87]. This suggests that either a factor of the surgery can produce ventricular dysfunction decades later, such as the residual scar tissue, or else that a common etiology for both the CHD and ventricular dysfunction exists.
Some familial CHD-producing gene mutations have also been associated with ventricular dysfunction, such as the sarcomeric gene MYH7 [5]. However, we are not aware of any current published research directly linking primary cilia gene mutations with heart failure through a mechanism different than those discussed above. While primary cilia are not displayed on adult cardiomyocytes, many ciliary proteins continue to exist and function at non-cilia locations, and cilia continue to be present in other cell types. Acquired ventricular dysfunction may therefore be mediated by ciliated non-myocytes, or else via cilia-independent functions of cilia proteins in cardiomyocytes. Alternatively, ciliogenesis may be reactivated in de-differentiated cardiomyocytes or cardiomyocyte progenitor cells in response to stress. Another possibility is that primary cilia defects in the developing heart result in permanent differences in the adult myocytes’ response to the stresses discussed above. Additional research is needed to identify the role of primary cilia in heart failure.
4. Concluding Remarks
Primary ciliary gene defects have previously been observed in a variety of syndromes, including ADPKD and Bardet—Biedl, as well as isolated congenital heart diseases. The role of cilia in these congenital conditions has been well defined. However, the role of primary cilia in acquired heart disease has not previously been reviewed.
Here we have reviewed literature exploring the effect of cilia gene knockouts on a variety of acquired cardiac pathologies. Mice with Ift88 knockout in valvular cells show myxomatous degeneration of the mitral valve similar to that observed in adult humans with mitral valve prolapse. Similarly, knockout in endothelial cells increased rates of endothelial to mesenchymal transition and increased fibrotic disease in response to stress. These models also show increased hypertension and atherosclerotic disease. Finally, primary cilia have cell-to-cell transmission capacity which, when blocked, leads to progressive left ventricular hypertrophy and heart failure.
While primary cilia have been linked with conditions that lead to heart failure, such as hypertension or atherosclerotic disease, a mechanistic causal relationship has not yet been fully established. Further research is needed to understand primary cilia’s role in adult cardiac pathology and especially in ventricular dysfunction.
Overall, despite decreased abundance in adult heart tissue, primary cilia continue to play an important role in cardiac homeostasis throughout adult life.
Author Contributions
Conceptualization, J.S.; original draft preparation, Z.E.H.; writing—review and editing Z.E.H. and J.S.; visualization, Z.E.H.; supervision, J.S. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported in part by U.S. Public Health Service Grants HL67724, HL91469, HL102738, HL112330, HL138720, HL144626, HL150881, and AG23039 (J.S.), the American Heart Association Merit Award 20 Merit 35,120,374 (J.S.), and the Fondation Leducq Transatlantic Network of Excellence 15CVD04 (J.S.).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Acknowledgments
The authors thank Daniela Zablocki for critical reading of the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Wu, M.H.; Lu, C.W.; Chen, H.C.; Kao, F.Y.; Huang, S.K. Adult congenital heart disease in a nationwide population 2000–2014: Epidemiological trends, arrhythmia, and standardized mortality ratio. J. Am. Heart Assoc. 2018, 7, e007907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mandalenakis, Z.; Giang, K.W.; Eriksson, P.; Liden, H.; Synnergren, M.; Wåhlander, H.; Fedchenko, M.; Rosengren, A.; Dellborg, M. Survival in children with congenital heart disease: Have we reached a peak at 97%? J. Am. Heart Assoc. 2020, 9, e017704. [Google Scholar] [CrossRef] [PubMed]
- Gilboa, S.M.; Devine, O.J.; Kucik, J.E.; Oster, M.E.; Riehle-Colarusso, T.; Nembhard, W.N.; Xu, P.; Correa, A.; Jenkins, K.; Marelli, A.J. Congenital heart defects in the United States. Circulation 2016, 134, 101–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graham, T.P., Jr.; Bernard, Y.D.; Mellen, B.G.; Celermajer, D.; Baumgartner, H.; Cetta, F.; Connolly, H.M.; Davidson, W.R.; Dellborg, M.; Foster, E.; et al. Long-term outcome in congenitally corrected transposition of the great arteries: A multi-institutional study. J. Am. Coll. Cardiol. 2000, 36, 255–261. [Google Scholar] [CrossRef] [Green Version]
- Hinton, R.B.; Ware, S.M. Heart failure in pediatric patients with congenital heart disease. Circ. Res. 2017, 120, 978–994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Norozi, K.; Wessel, A.; Alpers, V.; Arnhold, J.O.; Geyer, S.; Zoege, M.; Buchhorn, R. Incidence and risk distribution of heart failure in adolescents and adults with congenital heart disease after cardiac surgery. Am. J. Cardiol. 2006, 97, 1238–1243. [Google Scholar] [CrossRef]
- Oliver, J.M.; Gallego, P.; Gonzalez, A.E.; Avila, P.; Alonso, A.; Garcia-Hamilton, D.; Peinado, R.; Dos-Subirà, L.; Pijuan-Domenech, A.; Rueda, J.; et al. Predicting sudden cardiac death in adults with congenital heart disease. Heart 2021, 107, 67–75. [Google Scholar] [CrossRef]
- Budts, W.; Ravekes, W.J.; Danford, D.A.; Kutty, S. Diastolic heart failure in patients with the fontan circulation: A Review. JAMA Cardiol. 2020, 5, 590. [Google Scholar] [CrossRef]
- Inai, K. Biomarkers for heart failure and prognostic prediction in patients with Fontan circulation. Pediatr. Int. 2021. [Google Scholar] [CrossRef]
- Myklebust, R.; Engedal, H.; Saetersdal, T.S.; Ulstein, M. Primary 9+0 cilia in the embryonic and the adult human heart. Anat. Embryol. 1977, 151, 127–139. [Google Scholar] [CrossRef]
- Satir, P.; Pedersen, L.B.; Christensen, S.T. The primary cilium at a glance. J. Cell Sci. 2010, 123, 499–503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fry, A.M.; Leaper, M.J.; Bayliss, R. The primary cilium: Guardian of organ development and homeostasis. Organogenesis 2013, 10, 62–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Yang, S.; Deepak, V.; Chinipardaz, Z.; Yang, S. Identification of cilia in different mouse tissues. Cells 2021, 10, 1623. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Klena, N.T.; Gabriel, G.C.; Liu, X.; Kim, A.J.; Lemke, K.; Chen, Y.; Chatterjee, B.; Devine, W.; Damerla, R.R.; et al. Global genetic analysis in mice unveils central role for cilia in congenital heart disease. Nature 2015, 521, 520–524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gabriel, G.C.; Young, C.B.; Lo, C.W. Role of cilia in the pathogenesis of congenital heart disease. Semin. Cell Dev. Biol. 2020, 110, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Kozminski, K.G.; Johnson, K.A.; Forscher, P.; Rosenbaum, J.L. A motility in the eukaryotic flagellum unrelated to flagellar beating. Proc. Natl. Acad. Sci. USA 1993, 90, 5519–5523. [Google Scholar] [CrossRef] [Green Version]
- Toriyama, M.; Lee, C.; Taylor, S.P.; Duran, I.; Cohn, D.H.; Bruel, A.L.; Tabler, J.M.; Drew, K.; Kelly, M.R.; Kim, S.; et al. The ciliopathy-associated CPLANE proteins direct basal body recruitment of intraflagellar transport machinery. Nat. Genet. 2016, 48, 648–656. [Google Scholar] [CrossRef] [Green Version]
- Luu, V.Z.; Luu, A.Z.; Chowdhury, B.; Elbardisy, O.; Pan, Y.; Al-Omran, M.; Quan, A.; Teoh, H.; Hess, D.A.; Verma, S. Disruption of endothelial cell intraflagellar transport protein 88 exacerbates doxorubicin-induced cardiotoxicity. Life Sci. 2020, 260, 118216. [Google Scholar] [CrossRef]
- Willaredt, M.A.; Gorgas, K.; Gardner, H.A.R.; Tucker, K.L. Multiple essential roles for primary cilia in heart development. Cilia 2012, 1, 23. [Google Scholar] [CrossRef] [Green Version]
- Moore, K.; Moore, R.; Wang, C.; Norris, R.A. Tugging at the heart strings: The septin cytoskeleton in heart development and disease. J. Cardiovasc. Dev. Dis. 2020, 7, 3. [Google Scholar] [CrossRef] [Green Version]
- Douguet, D.; Patel, A.; Honoré, E. Structure and function of polycystins: Insights into polycystic kidney disease. Nat. Rev. Nephrol. 2019, 15, 412–422. [Google Scholar] [CrossRef] [PubMed]
- Kojima, K.; Sakai, I.; Hasegawa, A.; Niiya, H.; Azuma, T.; Matsuo, Y.; Fujii, N.; Tanimoto, M.; Fujita, S. FLJ10849, a septin family gene, fuses MLL in a novel leukemia cell line CNLBC1 derived from chronic neutrophilic leukemia in transformation with t(4;11)(q21;q23). Leukemia 2004, 18, 998–1005. [Google Scholar] [CrossRef] [PubMed]
- Angelis, D.; Spiliotis, E.T. Septin mutations in human cancers. Front. Cell Dev. Biol. 2016, 4, 122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cavini, I.A.; Leonardo, D.A.; Rosa, H.V.D.; Castro, D.K.S.V.; Pereira, H.D.; Valadares, N.F.; Araujo, A.P.U.; Garratt, R.C. The structural biology of septins and their filaments: An update. Front. Cell Dev. Biol. 2021, 9, 9. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, A.; Kinoshita, M.; Akiyama, H.; Tomimoto, H.; Akiguchi, I.; Kumar, S.; Noda, M.; Kimura, J. Identification of septins in neurofibrillary tangles in Alzheimer’s disease. Am. J. Pathol. 1998, 153, 1551–1560. [Google Scholar] [CrossRef]
- Lovera, M.; Lüders, J. The ciliary impact of nonciliary gene mutations. Trends Cell Biol. 2021, 31, 876–887. [Google Scholar] [CrossRef] [PubMed]
- Wheway, G.; Schmidts, M.; Mans, D.A.; Szymanska, K.; Nguyen, T.-M.T.; Racher, H.; Phelps, I.G.; Toedt, G.; Kennedy, J.; Wunderlich, K.A.; et al. An siRNA-based functional genomics screen for the identification of regulators of ciliogenesis and ciliopathy genes. Nat. Cell. Biol. 2015, 17, 1074–1087. [Google Scholar] [CrossRef]
- Van Dam, T.J.; Wheway, G.; Slaats, G.G.; Huynen, M.A.; Giles, R.H. The SYSCILIA gold standard (SCGSv1) of known ciliary components and its applications within a systems biology consortium. Cilia 2013, 2, 7. [Google Scholar] [CrossRef] [Green Version]
- Van Dam, T.J.P.; Kennedy, J.; van der Lee, R.; de Vrieze, E.; Wunderlich, K.A.; Rix, S.; Dougherty, G.W.; Lambacher, N.J.; Li, C.; Jensen, V.L.; et al. CiliaCarta: An integrated and validated compendium of ciliary genes. PLoS ONE 2019, 14, e0216705. [Google Scholar] [CrossRef] [Green Version]
- Delvallée, C.; Nicaise, S.; Antin, M.; Leuvrey, A.-S.; Nourisson, E.; Leitch, C.C.; Kellaris, G.; Stoetzel, C.; Geoffroy, V.; Scheidecker, S.; et al. A BBS1 SVA F retrotransposon insertion is a frequent cause of Bardet-Biedl syndrome. Clin. Genet. 2020, 99, 318–324. [Google Scholar] [CrossRef]
- Malicki, J.J.; Johnson, C.A. The cilium: Cellular antenna and central processing unit. Trends Cell Biol. 2016, 27, 126–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pusapati, G.V.; Kong, J.; Patel, B.B.; Krishnan, A.; Sagner, A.; Kinnebrew, M.; Briscoe, J.; Aravind, L.; Rohatgi, R. CRISPR screens uncover genes that regulate target cell sensitivity to the morphogen sonic hedgehog. Dev. Cell 2018, 44, 113–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breslow, D.K.; Hoogendoorn, S.; Kopp, A.R.; Morgens, D.W.; Vu, B.K.; Kennedy, M.C.; Han, K.; Li, A.; Hess, G.T.; Bassik, M.C.; et al. A CRISPR-based screen for Hedgehog signaling provides insights into ciliary function and ciliopathies. Nat. Genet. 2018, 50, 460–471. [Google Scholar] [CrossRef] [PubMed]
- Larsen, L.J.; Møller, L.B. Crosstalk of hedgehog and mTORC1 pathways. Cells 2020, 9, 2316. [Google Scholar] [CrossRef] [PubMed]
- Bangs, F.; Anderson, K.V. Primary cilia and mammalian hedgehog signaling. Cold Spring Harb. Perspect. Biol. 2017, 9, a028175. [Google Scholar] [CrossRef] [PubMed]
- Morleo, M.; Franco, B. The autophagy-cilia axis: An intricate relationship. Cells 2019, 8, 905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goetz, S.C.; Anderson, K. The primary cilium: A signalling centre during vertebrate development. Nat. Rev. Genet. 2010, 11, 331–344. [Google Scholar] [CrossRef] [PubMed]
- Bae, J.-E.; Kang, G.M.; Min, S.H.; Jo, D.S.; Jung, Y.-K.; Kim, K.; Kim, M.-S.; Cho, D.-H. Primary cilia mediate mitochondrial stress responses to promote dopamine neuron survival in a Parkinson’s disease model. Cell Death Dis. 2019, 10, 952. [Google Scholar] [CrossRef]
- Lee, J.; Park, K.C.; Sul, H.J.; Hong, H.J.; Kim, K.-H.; Kero, J.; Shong, M. Loss of primary cilia promotes mitochondria-dependent apoptosis in thyroid cancer. Sci. Rep. 2021, 11, 4181. [Google Scholar] [CrossRef]
- Atkinson, K.F.; Sherpa, R.T.; Nauli, S.M. The Role of the primary cilium in sensing extracellular pH. Cells 2019, 8, 704. [Google Scholar] [CrossRef] [Green Version]
- Collins, I.; Wann, A. Regulation of the extracellular matrix by ciliary machinery. Cells 2020, 9, 278. [Google Scholar] [CrossRef] [Green Version]
- Waters, A.M.; Beales, P.L. Ciliopathies: An expanding disease spectrum. Pediatr. Nephrol. 2011, 26, 1039–1056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baker, K.; Beales, P.L. Making sense of cilia in disease: The human ciliopathies. Am. J. Med. Genet. Part C Semin. Med. Genet. 2009, 151C, 281–295. [Google Scholar] [CrossRef] [PubMed]
- Marshall, J.D.; Maffei, P.; Collin, G.B.; Naggert, J.K. Alstrom syndrome: Genetics and clinical overview. Curr. Genom. 2011, 12, 225–235. [Google Scholar] [CrossRef] [Green Version]
- Kamal, R.; Dahiya, P.; Kaur, S.; Bhardwaj, R.; Chaudhary, K. Ellis-van Creveld syndrome: A rare clinical entity. J. Oral Maxillofac. Pathol. JOMFP 2013, 17, 132–135. [Google Scholar] [CrossRef] [PubMed]
- Keppler-Noreuil, K.M.; Adam, M.P.; Welch, J.; Muilenburg, A.; Willing, M.C. Clinical insights gained from eight new cases and review of reported cases with Jeune syndrome (asphyxiating thoracic dystrophy). Am. J. Med. Genet. Part A 2011, 155, 1021–1032. [Google Scholar] [CrossRef]
- Devi, A.R.R.; Naushad, S.M.; Lingappa, L. Clinical and molecular diagnosis of joubert syndrome and related disorders. Pediatr. Neurol. 2020, 106, 43–49. [Google Scholar] [CrossRef]
- Tsang, S.H.; Sharma, T. Leber congenital amaurosis. Adv. Exp. Med. Biol. 2018, 1085, 131–137. [Google Scholar] [CrossRef]
- Schaefer, E.; Durand, M.; Stoetzel, C.; Doray, B.; Viville, B.; Hellé, S.; Danse, J.-M.; Hamel, C.; Bitoun, P.; Goldenberg, A.; et al. Molecular diagnosis reveals genetic heterogeneity for the overlapping MKKS and BBS phenotypes. Eur. J. Med. Genet. 2011, 54, 157–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartill, V.; Szymanska, K.; Sharif, S.M.; Wheway, G.; Johnson, C.A. Meckel-Gruber syndrome: An update on diagnosis, clinical management, and research advances. Front. Pediatr. 2017, 5, 244. [Google Scholar] [CrossRef] [Green Version]
- Luo, F.; Tao, Y. Nephronophthisis: A review of genotype–phenotype correlation. Nephrology 2018, 23, 904–911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsang, S.H.; Aycinena, A.R.P.; Sharma, T. Ciliopathy: Senior-Løken syndrome. Adv. Exp. Med. Biol. 2018, 1085, 175–178. [Google Scholar] [CrossRef] [PubMed]
- Walczak-Sztulpa, J.; Eggenschwiler, J.; Osborn, D.; Brown, D.A.; Emma, F.; Klingenberg, C.; Hennekam, R.C.; Torre, G.; Garshasbi, M.; Tzschach, A.; et al. Cranioectodermal dysplasia, sensenbrenner syndrome, is a ciliopathy caused by mutations in the IFT122 gene. Am. J. Hum. Genet. 2010, 86, 949–956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naki, M.M.; Gür, D.; Zemheri, E.; Tekcan, C.; Kanadikirik, F.; Has, R.; Kanadıkırık, F. Short rib-polydactyly syndrome. Arch. Gynecol. Obstet. 2004, 272, 173–175. [Google Scholar] [CrossRef] [PubMed]
- Villalobos, E.; Criollo, A.; Schiattarella, G.; Altamirano, F.; French, K.M.; May, H.; Jiang, N.; Nguyen, N.U.N.; Romero, D.; Roa, J.C.; et al. Fibroblast primary cilia are required for cardiac fibrosis. Circulation 2019, 139, 2342–2357. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Chalothorn, D.; Faber, J.E. Collateral vessels have unique endothelial and smooth muscle cell phenotypes. Int. J. Mol. Sci. 2019, 20, 3608. [Google Scholar] [CrossRef] [Green Version]
- Toomer, K.A.; Yu, M.; Fulmer, D.; Guo, L.; Moore, K.S.; Moore, R.; Drayton, K.D.; Glover, J.; Peterson, N.; Ramos-Ortiz, S.; et al. Primary cilia defects causing mitral valve prolapse. Sci. Transl. Med. 2019, 11, 11. [Google Scholar] [CrossRef] [Green Version]
- Kaur, S.; McGlashan, S.R.; Ward, M.-L. Evidence of primary cilia in the developing rat heart. Cilia 2018, 7, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, K.; Carson, J.; Lo, C. Genetics of congenital heart disease. Biomolecules 2019, 9, 879. [Google Scholar] [CrossRef] [Green Version]
- Fulmer, D.; Toomer, K.; Guo, L.; Moore, K.; Glover, J.; Moore, R.; Stairley, R.; Lobo, G.; Zuo, X.; Dang, Y.; et al. Defects in the Exocyst-Cilia machinery cause bicuspid aortic valve disease and aortic stenosis. Circulation 2019, 140, 1331–1341. [Google Scholar] [CrossRef]
- Delling, F.N.; Vasan, R.S. Epidemiology and pathophysiology of mitral valve prolapse: New insights into disease progression, genetics, and molecular basis. Circulation 2014, 129, 2158–2170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Durst, R.; Sauls, K.; Peal, D.S.; de Vlaming, A.; Toomer, K.; Leyne, M.; Salani, M.; Talkowski, M.; Brand, H.; Perrocheau, M.; et al. Mutations in DCHS1 cause mitral valve prolapse. Nature 2015, 525, 109–113. [Google Scholar] [CrossRef]
- Toomer, K.A.; Fulmer, D.; Guo, L.; Drohan, A.; Peterson, N.; Swanson, P.; Brooks, B.; Mukherjee, R.; Body, S.; Lipschutz, J.H.; et al. A role for primary cilia in aortic valve development and disease. Dev. Dyn. 2017, 246, 625–634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leask, A. Getting to the heart of the matter: New insights into cardiac fibrosis. Circ. Res. 2015, 116, 1269–1276. [Google Scholar] [CrossRef] [Green Version]
- Levine, R.A.; Jerosch-Herold, M.; Hajjar, R.J. Mitral Valve Prolapse: A Disease of Valve and Ventricle. J. Am. Coll. Cardiol. 2018, 72, 835–837. [Google Scholar] [CrossRef] [PubMed]
- Kitkungvan, D.; Nabi, F.; Kim, R.J.; Bonow, R.O.; Khan, A.; Xu, J.; Little, S.H.; Quinones, M.A.; Lawrie, G.M.; Zoghbi, W.A.; et al. Myocardial fibrosis in patients with primary mitral regurgitation with and without prolapse. J. Am. Coll. Cardiol. 2018, 72, 823–834. [Google Scholar] [CrossRef] [PubMed]
- Dobaczewski, M.; de Haan, J.J.; Frangogiannis, N.G. The Extracellular matrix modulates fibroblast phenotype and function in the infarcted myocardium. J. Cardiovasc. Transl. Res. 2012, 5, 837–847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeisberg, E.M.; Tarnavski, O.; Zeisberg, M.; Dorfman, A.L.; McMullen, J.R.; Gustafsson, E.; Chandraker, A.; Yuan, X.; Pu, W.T.; Roberts, A.B.; et al. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat. Med. 2007, 13, 952–961. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Adam, M.; Matkar, P.N.; Bugyei-Twum, A.; Desjardins, J.-F.; Chen, H.H.; Nguyen, H.; Bazinet, H.; Michels, D.; Liu, Z.; et al. Endothelial-specific Loss of IFT88 promotes endothelial-to-mesenchymal transition and exacerbates bleomycin-induced pulmonary fibrosis. Sci. Rep. 2020, 10, 4466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Egorova, A.D.; Khedoe, P.P.; Goumans, M.-J.T.; Yoder, B.K.; Nauli, S.M.; Dijke, P.T.; Poelmann, R.E.; Hierck, B.P. Lack of primary cilia primes shear-induced endothelial-to-mesenchymal transition. Circ. Res. 2011, 108, 1093–1101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spasic, M.; Jacobs, C.R. Primary cilia: Cell and molecular mechanosensors directing whole tissue function. Semin. Cell Dev. Biol. 2017, 71, 42–52. [Google Scholar] [CrossRef] [PubMed]
- Luu, V.Z.; Chowdhury, B.; Al-Omran, M.; Hess, D.A.; Verma, S. Role of endothelial primary cilia as fluid mechanosensors on vascular health. Atherosclerosis 2018, 275, 196–204. [Google Scholar] [CrossRef] [PubMed]
- Nauli, S.M.; Kawanabe, Y.; Kaminski, J.J.; Pearce, W.J.; Ingber, D.E.; Zhou, J. Endothelial cilia are fluid shear sensors that regulate calcium signaling and nitric oxide production through polycystin-1. Circulation 2008, 117, 1161–1171. [Google Scholar] [CrossRef] [Green Version]
- Pala, R.; Jamal, M.; Alshammari, Q.; Nauli, S.M. The roles of primary cilia in cardiovascular diseases. Cells 2018, 7, 233. [Google Scholar] [CrossRef] [Green Version]
- Cheng, C.; Tempel, D.; Van Haperen, R.; Van Der Baan, A.; Grosveld, F.; Daemen, M.; Krams, R.; De Crom, R. Atherosclerotic lesion size and vulnerability are determined by patterns of fluid shear stress. Circulation 2006, 113, 2744–2753. [Google Scholar] [CrossRef] [Green Version]
- Van der Heiden, K.; Hierck, B.; Krams, R.; de Crom, R.; Cheng, C.; Baiker, M.; Pourquie, M.J.; Alkemade, F.E.; DeRuiter, M.C.; Gittenberger-de Groot, A.C.; et al. Endothelial primary cilia in areas of disturbed flow are at the base of atherosclerosis. Atherosclerosis 2008, 196, 542–550. [Google Scholar] [CrossRef] [PubMed]
- Espinha, L.C.; Hoey, D.; Fernandes, P.R.; Rodrigues, H.; Jacobs, C.R. Oscillatory fluid flow influences primary cilia and microtubule mechanics. Cytoskeleton 2014, 71, 435–445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dinsmore, C.; Reiter, J.F. Endothelial primary cilia inhibit atherosclerosis. EMBO Rep. 2016, 17, 156–166. [Google Scholar] [CrossRef] [Green Version]
- Patch, C.; Charlton, J.; Roderick, P.J.; Gulliford, M.C. Use of antihypertensive medications and mortality of patients with autosomal dominant polycystic kidney disease: A population-based study. Am. J. Kidney Dis. 2011, 57, 856–862. [Google Scholar] [CrossRef]
- Abdul-Majeed, S.; Nauli, S.M. Dopamine receptor type 5 in the primary cilia has dual chemo- and mechano-sensory roles. Hypertension 2011, 58, 325–331. [Google Scholar] [CrossRef]
- Zeng, C.; Jose, P.A. Dopamine receptors. Hypertension 2011, 57, 11–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frey, N.; Olson, E.N. Cardiac hypertrophy: The good, the bad, and the ugly. Annu. Rev. Physiol. 2003, 65, 45–79. [Google Scholar] [CrossRef] [PubMed]
- Takeda, N.; Manabe, I.; Uchino, Y.; Eguchi, K.; Matsumoto, S.; Nishimura, S.; Shindo, T.; Sano, M.; Otsu, K.; Snider, P.; et al. Cardiac fibroblasts are essential for the adaptive response of the murine heart to pressure overload. J. Clin. Investig. 2010, 120, 254–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roman, M.J.; Ganau, A.; Saba, P.S.; Pini, R.; Pickering, T.G.; Devereux, R.B. Impact of arterial stiffening on left ventricular structure. Hypertension 2000, 36, 489–494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Volz, A.-K.; Frei, A.; Kretschmer, V.; de Jesus Domingues, A.M.; Ketting, R.F.; Ueffing, M.; Boldt, K.; Krämer-Albers, E.-M.; May-Simera, H.L. Bardet-Biedl syndrome proteins modulate the release of bioactive extracellular vesicles. Nat. Commun. 2021, 12, 5671. [Google Scholar] [CrossRef] [PubMed]
- Mohieldin, A.M.; Pala, R.; Sherpa, R.T.; Alanazi, M.; Alanazi, A.; Shamloo, K.; Ahsan, A.; AbouAlaiwi, W.A.; Moresco, J.J.; Yates, J.R., 3rd; et al. Proteomic identification reveals the role of ciliary extracellular-like vesicle in cardiovascular function. Adv. Sci. 2020, 7, 1903140. [Google Scholar] [CrossRef] [PubMed]
- Menting, M.E.; Cuypers, J.A.; Opić, P.; Utens, E.M.; Witsenburg, M.; van den Bosch, A.E.; van Domburg, R.T.; Meijboom, F.J.; Boersma, E.; Bogers, A.J.; et al. The unnatural history of the ventricular septal defect: Outcome up to 40 years after surgical closure. J. Am. Coll. Cardiol. 2015, 65, 1941–1951. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).