
Hydrogen Sulfide Produced by Gut Bacteria May Induce Parkinson’s Disease

{kind=link}
Abstract
:1. Introduction
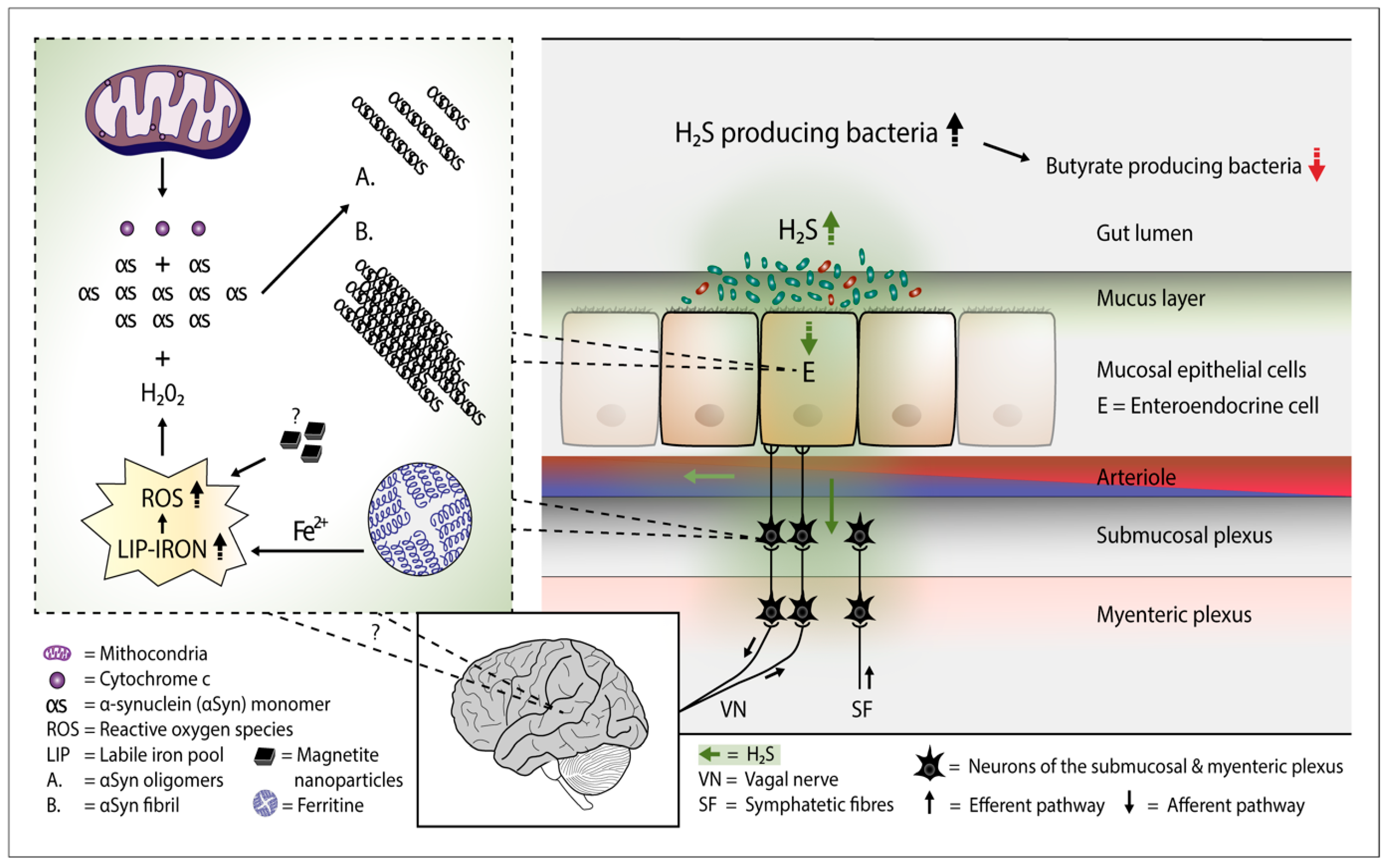
2. H2S Releases Cytochrome c from the Mitochondria—Start for Alpha-Synuclein Aggregation
3. Overgrowth of H2S Producing Gut Bacteria—Putative Consequences
4. H2S and aSyn Containing Gut Cells
5. H2S Producing Colonic Bacteria and PD
6. H2S Producing Small Intestinal Bacteria and PD
7. Viral Infections, PD, and H2S Producing Gut Bacteria
8. Bacterially Produced H2S and Risk Factors for PD
9. Conclusions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wang, R. Physiological implications of hydrogen sulfide: A whiff exploration that blossomed. Physiol. Rev. 2012, 92, 791–896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linden, D.R.; Sha, L.; Mazzone, A.; Stoltz, G.J.; Bernard, C.E.; Furne, J.K.; Levitt, M.D.; Farrugia, G.; Szurszewski, J.H. Production of the gaseous signal molecule hydrogen sulfide in mouse tissue. J. Neurochem. 2008, 106, 1577–1585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furne, J.; Saeed, A.; Levitt, M.D. Whole tissue hydrogen sulfide concentrations are orders of magnitude lower than presently accepted values. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2008, 295, R479–R1485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karunya, R.; Jayaprakash, K.S.; Gaikwad, R.; Sajeesh, P.; Ramshad, K.; Muraleedharan, K.M.; Dixit, M.; Thangaraj, P.R.; Sen, A.K. Rapid measurement of hydrogen sulfide in human blood plasma using a microfluid method. Sci. Rep. 2019, 9, 3258. [Google Scholar] [CrossRef] [PubMed]
- Buret, A.G.; Allain, T.; Motta, J.-P.; Wallace, J.L. Effects of hydrogen sulfide on the microbiome: From toxicity to therapy. Antioxid. Redox Sign. 2022, 36, 211–219. [Google Scholar] [CrossRef] [PubMed]
- Haouzi, P.; Sonobe, T.; Judenherc-Haouzi, A. Hydrogen sulfide intoxication induced brain injury and methylene blue. Neurobiol. Dis. 2020, 133, 104474. [Google Scholar] [CrossRef] [PubMed]
- Klingerman, C.M.; Trushin, N.; Prokopczyk, B.; Haouzi, P. H2S concentration in the arterial blood during H2S administration in relation to its toxicity and effects on breathing. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2013, 305, R630–R638. [Google Scholar] [CrossRef]
- Bianco, C.L.; Savitsky, A.; Feelisch, M.; Cortese-Krott, M.M. Investigations on the role of hemoglobin in sulfide metabolism by intact human red blood cells. Biochem. Pharmacol. 2018, 149, 163–174. [Google Scholar] [CrossRef] [PubMed]
- Jensen, B.; Fago, A. Reactions of ferric hemoglobin and myoglobin with hydrogen sulfide under physiologic conditions. J. Inorg. Biochem. 2018, 182, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Zaorska, E.; Tomasova, L.; Loszelewski, D.; Ostaszewski, R.; Ufnal, M. Hydrogen sulfide in pharmacotherapy, beyond the hydrogen sulfide-donors. Biomolecules 2020, 10, 323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dorman, D.C.; Moulin, F.J.-M.; McManus, B.E.; Mahle, K.C.; James, A.; Struve, M.F. Cytochrome oxidase inhibition induced by acute hydrogen sulfide inhalation: Correlation with tissue sulfide concentrations in the rat brin, liver, lung, and nasal epithelium. Toxicol. Sci. 2002, 65, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Murros, K.E. Sulfate reducing gut bacteria and Parkinson’s disease. Eur. J. Neurol. 2021, 28, e21. [Google Scholar] [CrossRef] [PubMed]
- Murros, K.E.; Huynh, V.A.; Takala, T.M.; Saris, P.E.J. Desulfovibrio bacteria are associated with Parkinson’s disease. Front. Cell. Infect. Microbiol. 2021, 11, 652617. [Google Scholar] [CrossRef] [PubMed]
- Garrido, C.; Galluzzi, L.; Brunet, M.; Puig, P.E.; Didelot, C.; Kroemer, G. Mechanism of cytochrome c release from mitochondria. Cell Death Diff. 2006, 13, 1423–1433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baskar, R.; Li, L.; Moore, P.K. Hydrogen sulfide-induces DNA damage and changes in apoptotic gene expression in human lung fibroblast cells. Faseb J. 2007, 21, 247–255. [Google Scholar] [CrossRef] [PubMed]
- Calenic, B.; Yaegaki, K.; Murata, T.; Imai, T.; Aoyama, I.; Sato, T.; Ii, H. Oral malodorous compound triggers mitochondrial-dependent apoptosis and causes genomic DNA damage in human gingival epithelial cells. J. Periodontal Res. 2010, 45, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, C.; Yaegaki, K.; Calenic, B.; Ishkitiev, N.; Imai, T.; Ii, H.; Aoyama, I.; Kobayashi, H.; Izumi, Y.; Haapasalo, M. Hydrogen sulfide causes apoptosis in human pulp stem cells. J. Endod. 2011, 37, 479–484. [Google Scholar] [CrossRef] [PubMed]
- Clayton, R.; Clark, J.B.; Sharpe, M. Cytochrome c release from rat brain mitocondria is proportional to the mitochondrial funtional deficit: Implications for apoptosis and neurodegenerative disease. J. Neurochem. 2005, 92, 840–849. [Google Scholar] [CrossRef]
- Tomasková, N.; Varhac, T.; Lysáková, V.; Musatov, A.; Sedlák, E. Peroxidase activity of cytochrome c in its compact state dependes on dynamics of the heme region. Biochim. Biophys. Acta Proteins Proteom. 2018, 1866, 1073–1083. [Google Scholar] [CrossRef] [PubMed]
- Belikova, N.A.; Vladimirov, Y.A.; Osipov, A.N.; Kapralov, A.A.; Tyurin, V.A.; Potapovich, M.V.; Basova, L.V.; Peterson, J.; Kurnikov, I.V.; Kagan, V.E. Peroxidase activity and structural transitions of cytochrome c bound to cardiolipin-contiainig membranes. Biochemistry 2006, 45, 4998–5009. [Google Scholar] [CrossRef] [Green Version]
- Bayir, H.; Kapralov, A.A.; Jiang, J.; Huang, Z.; Tyurina, Y.Y.; Tyurin, V.A.; Zhao, Q.; Belikova, N.A.; Vlasova, I.I.; Maeda, A.; et al. Peroxidase mechanism of lipid-dependent cross-linking of synuclein with cytochrome c. J. Biol. Chem. 2008, 284, 15951–15969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hashimoto, M.; Takeda, A.; Hsu, L.J.; Takenouchi, T.; Masliah, E. Role of cytochrome c as astimulator of α-synuclein aggregation in Lewy-body disease. J. Biol. Chem. 1999, 274, 28849–28852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, A.; Ganini, D.; Mason, R.P. Role of cytochrome c in α-synuclein radical formation: Implications of α-synuclein in neuronal death in Maneb- and paraquat-induced model of Parkinson’s disease. Mol. Neurodegener. 2016, 11, 70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cassanelli, S.; Moulis, J. Sulfide is an efficient iron releasing agent for mammalian ferritins. Biochim. Biophys. Acta 2001, 1547, 174–182. [Google Scholar] [CrossRef]
- Hälldin, J.; Land, T. Sulfide increases labile iron pool in RD4 cells. Biometals 2008, 21, 127–131. [Google Scholar] [CrossRef] [PubMed]
- Truong, D.H.; Eghbal, M.A.; Hindmarsh, W.; Roth, S.R.; O’Brien, P.J. Molecular mechanisms of hydrogen sulfide toxicity. Drug Metab. Rev. 2006, 38, 733–744. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Chen, X.; Huang, S.; Li, G.; Mo, M.; Zhang, L.; Chen, C.; Guo, W.; Zhou, M.; Wu, Z.; et al. Iron promotes α-synuclein aggregation and transmission by inhibiting TFEB-mediated autophagosome-lysosome fusion. J. Neurochem. 2018, 145, 34–50. [Google Scholar] [CrossRef] [PubMed]
- Cabantchik, Z.I. Labile iron in cells and body fluids: Physiology, pathology, and pharmacology. Front. Pharmacol. 2014, 5, 45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murros, K.; Wasiljeff, J.; Macias-Sánchez, E.; Faivre, D.; Soinne, L.; Valtonen, J.; Pohja, M.; Saari, P.; Pesonen, L.J.; Salminen, J.M. Magnetic nanoparticles in human cervical skin. Front. Med. 2019, 6, 123. [Google Scholar] [CrossRef] [PubMed]
- Könczöl, M.; Ebeling, S.; Goldenberg, E.; Treude, F.; Gminski, R.; Gieré, R.; Grobéty, B.; Rothen-Rutishauser, B.; Merfort, I.; Merch-Sundermann, V. Cytotoxicity and genotoxicity of size-fractionde iron oxide (magnetite) in A549 human lung epithelial cells: Role of ROS, JNK, and NF-κB. Chem. Res. Toxicol. 2011, 24, 1460–1475. [Google Scholar] [CrossRef] [PubMed]
- Mathai, J.C.; Missner, A.; Kügler, P.; Saparov, S.M.; Zeidel, M.L.; Lee, J.K.; Pohl, P. No facilitator required fro membrane transport of hydrogen sulfide. PNAS 2009, 106, 16633–16638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, S.B.; Lin, H.C. Hydrogen sulfide in physiology and diseases of the digestive tract. Microorganism 2015, 3, 866–889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, X.; Carlström, M.; Borniquel, S.; Jädert, C.; Kevill, C.G.; Lundberg, J. Microbial regulation of host hydrogen sulfide bioavailability and metabolism. Free Radic. Biol. Med. 2013, 60, 195–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hertel, J.; Harms, A.C.; Heinken, A.; Baldini, F.; Thinnes, C.C.; Glaab, E.; Vasco, D.A.; Pietzner, M.; Stewart, I.D.; Wareham, N.J.; et al. Integrated analyses on microbiome and longitudinal metabolome data reveal microbial-host interactions on sulfur metabolism in Parkinson’s disease. Cell Rep. 2019, 29, 1767–1777.e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makletsova, M.G.; Rikhireva, G.T.; Poleshuk, V.V.; Grjakalov, K.V.; Timerbaeva, S.L.; Fedorova, T.N. The effects of antioxidants on in vivo and in vitro methemoglobin formation in erythorcytes of patients with Parkinson’s disease. Biomed. Khim. 2016, 62, 193–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greco, V.; Neri, C.; Pieragostino, D.; Spalloni, A.; Persichilli, S.; Gastaldi, M.; Mercuri, N.B.; Longone, P.; Urbani, A. Investigating different forms of hydrogen sulfide in cerebrospinal fulid of various neurological disorders. Metabolites 2021, 11, 152. [Google Scholar] [CrossRef] [PubMed]
- Lagouette, E.; Mimoun, S.; Andriamihaja, M.; Chaumontet, C.; Blachier, F.; Bouillaud, F. Oxidation of hydrogen sulfide remains a priority in mammalian cells and causes reverse electron transfer in colonocytes. Biochim. Biophys. Acta 2010, 1797, 1500–1511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhatia, D.; Grozdanov, V.; Ruf, W.P.; Kassubek, J.; Ludolph, A.C.; Weishaupt, J.H.; Danzer, K.M. T-cell dysregulation is associated with disease severity in Parkinson’s disease. J. Neuroinflamm. 2021, 18, 250. [Google Scholar] [CrossRef] [PubMed]
- Mirandola, P.; Gobbi, G.; Sponzilli, I.; Pambianco, M.; Malinverno, C.; Cacchioli, A.; De Panfilis, G.; Vitale, M. Exogenous hydrogen sulfide induces functional inhibition and cell desth of cytotoxic lymphocytes subsets. J. Cellul. Physiol. 2007, 213, 826–833. [Google Scholar] [CrossRef]
- Hyndman, D.; Liu, S.; Miner, J.N. Urate handling in the human body. Curr. Rheumatol. Rep. 2016, 18, 34. [Google Scholar] [CrossRef] [Green Version]
- Annanmäki, T.; Muuronen, A.; Murros, K. Low plasma uric acid in Parkinson’s disease. Mov. Disord. 2007, 22, 1133–1137. [Google Scholar] [CrossRef] [PubMed]
- Pardue, S.; Kolluru, G.K.K.; Shen, X.; Lewis, S.; Saffle, C.; Kelley, E.; Kevil, C. Hydrogen sulfide stimulates xanthine oxidoreductase conversion to nitrite reductase and formation of NO. Redox Biol. 2020, 34, 101447. [Google Scholar] [CrossRef] [PubMed]
- Chandra, R.; Hiniker, A.; Kuo, Y.-M.; Nussbaum, R.L.; Liddle, R.A. α-Synuclein in gut endocrine cells and its implications for Parkinsons’s disease. JCI Insight 2017, 2, e92295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liddle, R.A. Interactions of gut endocrine cells with epithelium and neurons. Compr. Physiol. 2019, 8, 1019–1030. [Google Scholar] [CrossRef]
- Bu, L.-L.; Huang, K.-X.; Zheng, D.-Z.; Lin, D.-Y.; Chen, Y.; Jing, X.-N.; Liang, Y.-R.; Tao, E.-X. Alpha-synuclein accumulation and its phosphorylation in the enteric nervous system of patients without neurodegeneration: An explorative study. Front. Aging Neurosci. 2020, 12, 575481. [Google Scholar] [CrossRef] [PubMed]
- El-Agnaf, O.M.A.; Salem, S.A.; Paleologou, K.E.; Curran, M.D.; Gibson, M.L.; Court, J.A.; Schlossmacher, M.G.; Allsop, D. Detection of oligomeric forms of alpha-synucelin protein in human plasma as a potential biomarker for Parkinson’s disease. Faseb J. 2006, 20, 419–425. [Google Scholar] [CrossRef] [PubMed]
- Holmqvist, S.; Chutna, O.; Bousset, L.; Aldrin-Kirk, P.; Li, W.; Björklund, T.; Wang, Z.-Y.; Roybon, L.; Melki, R.; Li, J.-Y. Direct evidence of Parkinson pathology spread from the gastrointestinal tract to the brain in rats. Acta Neuropathol. 2014, 128, 805–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Den Berge, N.; Ferreira, N.; Gram, H.; Mikkelsen, T.W.; Alstrup, A.K.O.; Casadei, N.; Tsung-Pin, P.; Nyengaard, J.R.; Tamgüney, G.; Jensen, P.H.; et al. Evidence of bidirectional and trans-synaptic parasymphathetic and symphathetic propagation of alpha-synuclein in rats. Acta Neuropathol. 2019, 138, 535–550. [Google Scholar] [CrossRef] [Green Version]
- Dordević, D.; Janciková, S.; Vitezová, M.; Kushkevych, I. Hydrogen sulfide toxicity in the gut environment: Meta-analysis of sulfate-reducing and lactic acid bacteria in inflammatory processes. J. Adv. Res. 2021, 27, 55–69. [Google Scholar] [CrossRef] [PubMed]
- Gibson, G.R.; Macfarlane, S.; Macfarlane, G.T. Metabolic interactions involving sulphate-reducing and methanogenic bacteria in the human large intestine. FEMS Microbiol. 1993, 12, 117–125. [Google Scholar] [CrossRef]
- Carbonero, F.; Benefiel, A.C.; Alizadeh-Ghamsari, A.H.; Gaskins, H.R. Microbial pathways in colonic sulfur metabolism and links with health and disease. Front. Physiol. 2012, 3, 448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braccia, J.; Jiang, X.; Pop, M.; Hall, A.B. The capacity to produce hydrogen sulfide (H2S) via cysteine degradation is ubiquitos in the human gut microbiome. Front. Microbiol. 2021, 12, 3193. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.-K.; Wang, J.-H.; Lei, W.-Y.; Chen, C.-L.; Chang, C.-Y.; Liou, L.-S. Helicobacter pylori infection is associated with an increased risk of Parkinson’s disease: A population-based retrospective cohort study. Park. Rel. Dis. 2018, 47, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Kang, X.; Ploner, A.; Ludvigsson, J.F.; Williams, D.M.; Larsson, H.; Pedersen, N.L.; Wirdefeldt, P.K. Clostridium difficile infection and risk of Parkinson’s disaease: A Swedish population-based cohort study. Eur. J. Neurol. 2020, 27, 2134–2141. [Google Scholar] [CrossRef] [PubMed]
- Romano, S.; Savva, G.M.; Bedarf, J.R.; Charles, I.G.; Hildebrand, F.; Narbad, A. Meta-analysis of the Parkinson’s disease gut microbiota suggest alterations linked to intestinal inflammation. Npj. Park. Dis. 2021, 7, 27. [Google Scholar] [CrossRef] [PubMed]
- Wallen, Z.D.; Appah, M.; Dean, M.N.; Sesler, C.L.; Factor, S.A.; Molho, E.; Zabetian, C.P.; Standaert, D.G.; Payami, H. Characterizing dysbiosis of gut microbiome in PD: Evidence for overabundance of opportunistic pathogens. Npj. Park. Dis. 2020, 6, 11. [Google Scholar] [CrossRef] [PubMed]
- Blachier, F.; Davila, A.-M.; Mimoun, S.; Benetti, P.-H.; Atanasiu, C.; Andriamihaja, M.; Benamouzig, R.; Boillaud, F.; Tomé, D. Luminal sulfide and large intestine mucosa: Friend or foe? Amino Acids 2009, 39, 335–347. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-H.; Chen, C.-C.; Chiang, H.-L.; Liou, J.-M.; Chang, C.-M.; Lu, T.-P.; Chuang, E.Y.; Tai, Y.-C.; Cheng, C.; Lin, H.-Y.; et al. Altered microbiota and inflammatory cytokine responses in patients with Parkinson’s disease. J. Neuroinflamm. 2019, 16, 129. [Google Scholar] [CrossRef] [PubMed]
- Petrov, V.A.; Saltykova, I.V.; Zhukova, I.A.; Alifirova, V.M.; Zhukova, N.G.; Dorofeeva, Y.B.; Tyakht, A.V.; Kovarsky, B.A.; Alekseev, D.G.; Kostryukova, E.S.; et al. Analysis of gut microbiota in patients with Parkinson’s disease. Bull. Exp. Biol. Med. 2017, 162, 734–737. [Google Scholar] [CrossRef] [PubMed]
- Jin, M.; Liu, F.; Wang, K.; Wang, L.; Liang, S.; Tao, H.; Zhu, B.; Alkasir, R. Analysis of the gut microflora in patients with Parkinson’s disease. Front. Neurosci. 2019, 13, 1184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Babidge, W.; Millard, S.; Roediger, W. Sulfides impair short chain fatty acid β-oxidation at acyl-CoA dehydrogenase level in colonocytes: Implications for ulcerative colitis. Mol. Cell. Biochem. 1998, 181, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Landry, A.P.; Moon, S.; Kim, H.; Yadav, P.K.; Guha, A.; Cho, U.-S.; Banerjee, R. A catalytic trisulfide in human sulfide quinone oxidoreductase catalyzes Coenzyme A persulfide synthesis and inhibits butyrate oxidation. Cell. Chem. Biol. 2019, 26, 1515–1525.e4. [Google Scholar] [CrossRef] [PubMed]
- Clark, R.L.; Connors, B.M.; Stevenson, D.M.; Hromada, S.E.; Hamilton, J.J.; Amador-Noguez, D.; Venturelli, O.S. Design of synthetic human gut microbiome assembly and butyrate production. Nature Comm. 2021, 12, 3254. [Google Scholar] [CrossRef] [PubMed]
- de la Cuesta-Zuluaga, J.; Mueller, N.T.; Alvarez-Quintero, R.; Velásguez-Mejia, E.; Sierra, J.A.; Corrales-Agudelo, V.; Carmona, J.A.; Abad, J.M.; Escobar, J.S. Higher fecal short-chain fatty acid levels are associated with gut microbiome dysbiosis, obesity, hypertension and cardiometbolic disease risk factors. Nutritients 2019, 11, 51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.-J.; Liang, C.-Y.; Yang, L.-Q.; Ren, S.-M.; Xia, Y.-M.; Cui, L.; Li, X.-F.; Gao, B.-L. Association of Parkinson’s disease with microbes and microbiological therapy. Front. Cell. Infect. Microbiol. 2021, 11, 619354. [Google Scholar] [CrossRef] [PubMed]
- Vascellari, S.; Palmas, V.; Melis, M.; Pisanu, S.; Cusano, R.; Uva, P.; Perra, D.; Madau, V.; Sarchioto, M.; Oppo, V.; et al. Gut microbiota and metabolome alterations associated with Parkinson’s disease. Msystems 2020, 5, e00561-20. [Google Scholar] [CrossRef] [PubMed]
- Washio, J.; Sakuma, Y.; Shimada, Y.; Takahashi, N. Hydrogen-sulfide production from various substrates by oral Veillonella and effects of lactate on the production. J. Med. Microbiol. 2009, 54, 889–895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, A.; Zheng, W.; He, Y.; Tang, W.; Wei, X.; He, R.; Huang, W.; Su, Y.; Huang, Y.; Zhou, H.; et al. Gut microbiota in patients with Parkinson’s disease in southern China. Park. Rel. Dis. 2018, 53, 82–88. [Google Scholar] [CrossRef] [PubMed]
- Cirstea, M.S.; Yu, A.C.; Golz, E.; Sundvick, K.; Kliger, D.; Radisavljevic, N.; Foulger, L.M.; Mackenzie, M.; Huan, T.; Finlay, B.; et al. Microbiota composition and metabolism are associated with gut function in Parkinson’s disease. Mov. Disord. 2020, 35, 1208–1217. [Google Scholar] [CrossRef] [PubMed]
- van der Marck, M.; Dicke, H.C.; Uc, E.Y.; Kentin, Z.H.A.; Borm, G.F.; Bloem, B.R.; Overeem, S.; Munneke, M. Body mass index in Parkinson’s disease: A meta-analysis. Park. Rel. Dis. 2012, 18, 263–267. [Google Scholar] [CrossRef] [PubMed]
- Waters, J.L.; Ley, R. The human gut bacteria Christensenellaceae are widespread, heritable, and associated with health. MMC Biol. 2019, 17, 83. [Google Scholar] [CrossRef] [PubMed]
- Geerlings, S.Y.; Kostopoulos, I.; de Vos, W.M.; Belzer, C. Akkermansia muciniphila in he human gastrointrestinal tract: When, where, and how? Microorganisms 2018, 6, 75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Derrien, M.; Vaughan, E.E.; Plugge, C.M.; de Vos, W.M. Akkermansia muciniphila gen. nov., sp. nov., a human intestinal mucin-degrading bacterium. Int. J. Syst. Evol. Microbiol. 2004, 54, 1469–1476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nava, G.M.; Carbonero, F.; Croix, J.A.; Greenberg, E.; Gaskins, H.R. Abundance and diversity of mucosa-associated hydrogenotrophic microbes in the healthy human colon. ISME J. 2012, 6, 57–80. [Google Scholar] [CrossRef] [PubMed]
- Earley, H.; Lennon, G.; Balfe, A.; Coffey, J.C.; Winter, D.C.; O’Connell, P.R. The abundance of Akkermansia muciniphila and its relationship in health and ulceratice colitis. Sci. Rep. 2019, 9, 15683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baldini, F.; Hertel, J.; Sandt, E.; Thinnes, C.C.; Neuberger-Castillo, L.; Pavelka, L.; Betsou, F.; Krüger, R.; Thiele, I.; on behalf of the NCER-PD Consortium. Parkinson’s disease-associated alterations of the gut microbiome predict disease-relevant changes in metabolic functions. BMC Biol. 2020, 18, 62. [Google Scholar] [CrossRef] [PubMed]
- Keshavarzian, A.; Green, S.J.; Engen, P.A.; Voigt, R.M.; Naqib, A.; Forsyth, C.B.; Mutlu, E.; Shannon, K.M. Colonic bacterial composition in Parkinson’s disease. Mov. Disord. 2015, 30, 1351–1360. [Google Scholar] [CrossRef] [PubMed]
- Sicard, J.F.; Le Bihan, G.; Vogeleer, P.; Jacques, M.; Harel, J. Interactions of intestinal bacteria with components of the intestinal mucus. Front. Cell Infect. Microbiol. 2017, 7, 387. [Google Scholar] [CrossRef]
- Forsyth, C.B.; Shannon, K.M.; Kordower, J.H.; Voigt, R.M.; Shaikh, M.; Jaglin, J.A.; Estes, J.D.; Dodiya, H.B.; Keshavarzian, A. Increased intestinal permeability correlates with sigmoid mucosa alpha-synuclein staining and endotoxin exposure markers in early Parkinosn’s disease. PLoS ONE 2011, 6, e28032. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Wu, X.; Hu, X.; Wang, T.; Liang, S.; Duan, Y.; Jin, F.; Qin, B. Structural changes of gut microbiota in Parkinson’s disease and its correlations with clinical features. Sci. China Life Sci. 2017, 60, 1223–1233. [Google Scholar] [CrossRef]
- Li, X.; Feng, X.; Jiang, Z.; Jiang, Z. Association of small intestinal bacterial overgrowth with Parkinson’s disease: A systematic review and meta-analysis. Gut Pathog. 2021, 13, 25. [Google Scholar] [CrossRef]
- Leite, G.G.S.; Weitsman, S.; Parodi, G.; Celly, S.; Sedighi, R.; Sanchez, M.; Morales, W.; Villanueva-Millan, M.J.; Barlow, G.M.; Mathur, R.; et al. Mapping the segmental microbiomes in the human small bowel in comparison with stool: A REIMAGINE study. Digest. Dis. Sci. 2020, 65, 2595–2604. [Google Scholar] [CrossRef] [Green Version]
- Leite, G.; Morales, W.; Weitsman, S.; Celly, S.; Parodi, G.; Mathur, R.; Barlow, G.M.; Sedighi, R.; Millan, M.J.V.; Rezaie, A.; et al. The duodenal microbiome is altered in small intestinal bacterial overgrowth. PloS ONE 2020, 15, e02344906. [Google Scholar] [CrossRef]
- Chen, C.-K.; Wu, Y.-T.; Chang, Y.-C. Periodontal inflammatory disease is associated with the risk of Parkinson’s disease: A population-based retrospective matched-cohort study. PeerJ 2017, 10, e3647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, M.; Wei, Y.; Hu, W.; Nie, Y.; Wu, X.; Lu, R. The subgingival micrbiome of periodontal pockets with different probing depths in chronic and aggressive periodontitis: A pilot study. Front. Cell. Infect. Microbiol. 2018, 8, 124. [Google Scholar] [CrossRef] [PubMed]
- Kushkevych, I.; Coufalová, M.; Vitezová, M.; Ritmann, S.K.-M.R. Sulfate-reducing bacteria of the oral cavity and their relation with periodontitis-recent advances. J. Clin. Med. 2020, 9, 2347. [Google Scholar] [CrossRef]
- Pereira, P.A.B.; Aho, V.T.E.; Paulin, L.; Pekkonen, E.; Auvinen, P.; Scheperjans, F. Oral and nasal microbiota in Parkinson’s disease. Park. Rel. Dis. 2017, 38, 61–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harris, M.A.; Tsui, J.K.; Marion, S.A.; Shen, H.; Teschke, K. Asoociation of Parkinson’s disease with infections and occupational exposure to possible vectors. Mov. Disord 2012, 27, 1111–1117. [Google Scholar] [CrossRef] [PubMed]
- Cocoros, N.M.; Svensson, E.; Szépligeti, S.K.; Vestergaard, S.V.; Szentkúti, P.; Thomsen, R.W.; Borghammer, P.; Sorensen, H.T.; Henderson, V.W. Long-term risk of Parkinson’s disease following influenza and other infections. JAMA Neurol. 2021, 78, 1461–1470. [Google Scholar] [CrossRef]
- Yildiz, S.; Mazel-Sanchez, B.; Kandasamy, M.; Manicassamy, B.; Schmolke, M. Influenza A virus infection impacts systemic microbiota dynamics and causes quantitative enteric dysbiosis. Microbiome 2018, 6, 9. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Li, F.; Wei, H.; Lian, Z.-X.; Sun, R.; Tian, Z. Respiratory influenza virus infection induces intestinal immune injury via microbiota-mediated Th17 cell-dependent inflammation. J. Exp. Med. 2014, 211, 2397–2410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deriu, E.; Boxx, G.M.; He, X.; Pan, C.; Benavidez, S.D.; Cen, L.; Rozengurt, N.; Shi, W.; Cheng, G. Influenza virus affects interstinal microbiota and secondary Salmonella infection in the gut through type 1 interferons. PloS Pathog. 2016, 15, e1005572. [Google Scholar] [CrossRef]
- Sencio, V.; Barthelemy, A.; Tavares, L.P.; Machado, M.G.; Soulard, D.; Cuinat, C.; Queiroz-Junior, C.M.; Noordine, M.-L.; Salomé-Desnoulez, S.; Deryuter, L.; et al. Gut dysbiosis during influenza contributes to pulmonary pneumococcal superinfection through altered short-chain fatty acid production. Cell Rep. 2020, 30, 2934–2947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, S.; Chen, Y.; Wu, Z.; Chen, Y.; Gao, H.; Lv, L.; Guo, F.; Zhang, X.; Luo, R.; Huang, C.; et al. Alterations of the gut microbiota in patients with Coronavirus disease 2019 or H1N1 influenza. Clin. Infect. Dis. 2020, 71, 2669–2678. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.Y.-Y.; Kang, K.-H.; Chen, S.L.-S.; Chiu, S.Y.-H.; Yen, A.M.-F.; Fann, J.C.-T.; Su, C.-W.; Liu, H.-C.; Lee, C.-Z.; Fu, W.-M.; et al. Hepatitis C virus infection: A risk for Parkinson’s disease. J. Viral Hepat. 2015, 22, 784–791. [Google Scholar] [CrossRef] [PubMed]
- El-Mowafy, M.; Elgaml, A.; El-Mesery, M.; Sultan, S.; Ahmed, T.A.E.; Gomaa, A.I.; Aly, M.; Mottawea, W. Changes of gut-microbiota-liver axis in hepatitis C virus infections. Biology 2021, 10, 55. [Google Scholar] [CrossRef] [PubMed]
- Scheperjans, F.; Aho, V.; Pereira, P.A.B.; Koskinen, K.; Paulin, L.; Pekkonen, E.; Haapaniemi, E.; Kaakkola, S.; Eerola-Rautio, J.; Pohja, M.; et al. Gut microbiota are related to Parkinson’s disease and clinicla phenotype. Mov. Disord. 2015, 30, 350–358. [Google Scholar] [CrossRef] [PubMed]
- Unger, M.M.; Spiegel, J.; Dillmann, K.-U.; Grundmann, D.; Philippelt, H.; Bürmann, J.; Fassbender, K.; Scwiertz, A.; Schäfer, K.-H. Short chain fatty acids and gut microbiota differ between patients witn Parkinson’n disease and age-matched controls. Park. Rel. Dis. 2016, 32, 66–72. [Google Scholar] [CrossRef] [PubMed]
- Vascellari, S.; Melis, M.; Palmas, V.; Serra, A.; Perra, D.; Santoru, M.L.; Oppo, V.; Uva, P.; Atzori, L.; Morelli, M.; et al. Clinical phenotypes of Parkinson’s disease associate with distinct gut microbiota and metabolome enterotypes. Biomolecules 2021, 11, 144. [Google Scholar] [CrossRef] [PubMed]
- GBD 2016 Parkinson’s Disease Collaborators. Global, regional, and national burden of Parkinson’s disease, 1990–2016: A systematic analysis for the global burden of disease study 2016. Lancet Neurol. 2018, 17, 939–953. [Google Scholar] [CrossRef] [Green Version]
- Xu, C.; Zhu, H.; Qiu, P. Aging progression of human microbiota. BMC Microbiol. 2019, 19, 236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gillies, G.; Pienaar, I.S.; Vohra, S.; Qamhawi, Z. Sex differences in Parkinson’s disease. Front. Neuroendocrin. 2014, 35, 370–384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bourque, M.; Dluzen, D.E.; Di Paolo, T. Neuroprotective actions of sex steroids in Parkinson’s disease. Front Neuroendocrinol. 2009, 30, 142–157. [Google Scholar] [CrossRef] [PubMed]
- Gatto, N.M.; Deapen, D.; Stoyanoff, S.; Pinder, R.; Bordelon, T.; Ritz, B. Lifetime exposure to estrogens and Parkinson’s disease in California teachers. Park. Rel. Dis. 2014, 20, 1149–1156. [Google Scholar] [CrossRef] [PubMed]
- Bagetta, C.; Chiappetta, O.; Amantea, D.; Iannone, M.; Rotiroti, D.; Costa, A.; Nappi, G.; Corasaniti, M.T. Estradiol reduces cytochrome c translocation and minimizes hippocampal damage caused by transient global ischemia in rat. Neurosci. Lett. 2004, 368, 87–91. [Google Scholar] [CrossRef] [PubMed]
- Morkuniene, R.; Arandarcikaite, O.; Borutaite, V. Estradiol prevents release of cytochrome c form mitochondria and inhibits ischemia-induced apoptosis in perfused heart. Exp. Gerontol. 2006, 41, 704–708. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Rüb, U.; Gai, W.P.; Del Tredici, K. Idiopathic Parkinson’s disease: Possible routes by which vulnerable neuronal types may be subject to neuroinvasion by an unknown pathogen. J. Neural. Transm. 2003, 110, 517–536. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Murros, K.E. Hydrogen Sulfide Produced by Gut Bacteria May Induce Parkinson’s Disease. Cells 2022, 11, 978. https://doi.org/10.3390/cells11060978
Murros KE. Hydrogen Sulfide Produced by Gut Bacteria May Induce Parkinson’s Disease. Cells. 2022; 11(6):978. https://doi.org/10.3390/cells11060978
Chicago/Turabian StyleMurros, Kari Erik. 2022. "Hydrogen Sulfide Produced by Gut Bacteria May Induce Parkinson’s Disease" Cells 11, no. 6: 978. https://doi.org/10.3390/cells11060978
APA StyleMurros, K. E. (2022). Hydrogen Sulfide Produced by Gut Bacteria May Induce Parkinson’s Disease. Cells, 11(6), 978. https://doi.org/10.3390/cells11060978