
Alpha-Synuclein Aggregation Pathway in Parkinson’s Disease: Current Status and Novel Therapeutic Approaches


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Parkinson’s Disease
2.1. Genetic Factors Triggering PD
2.2. Environmental Factors Triggering PD
3. Alpha-Synuclein
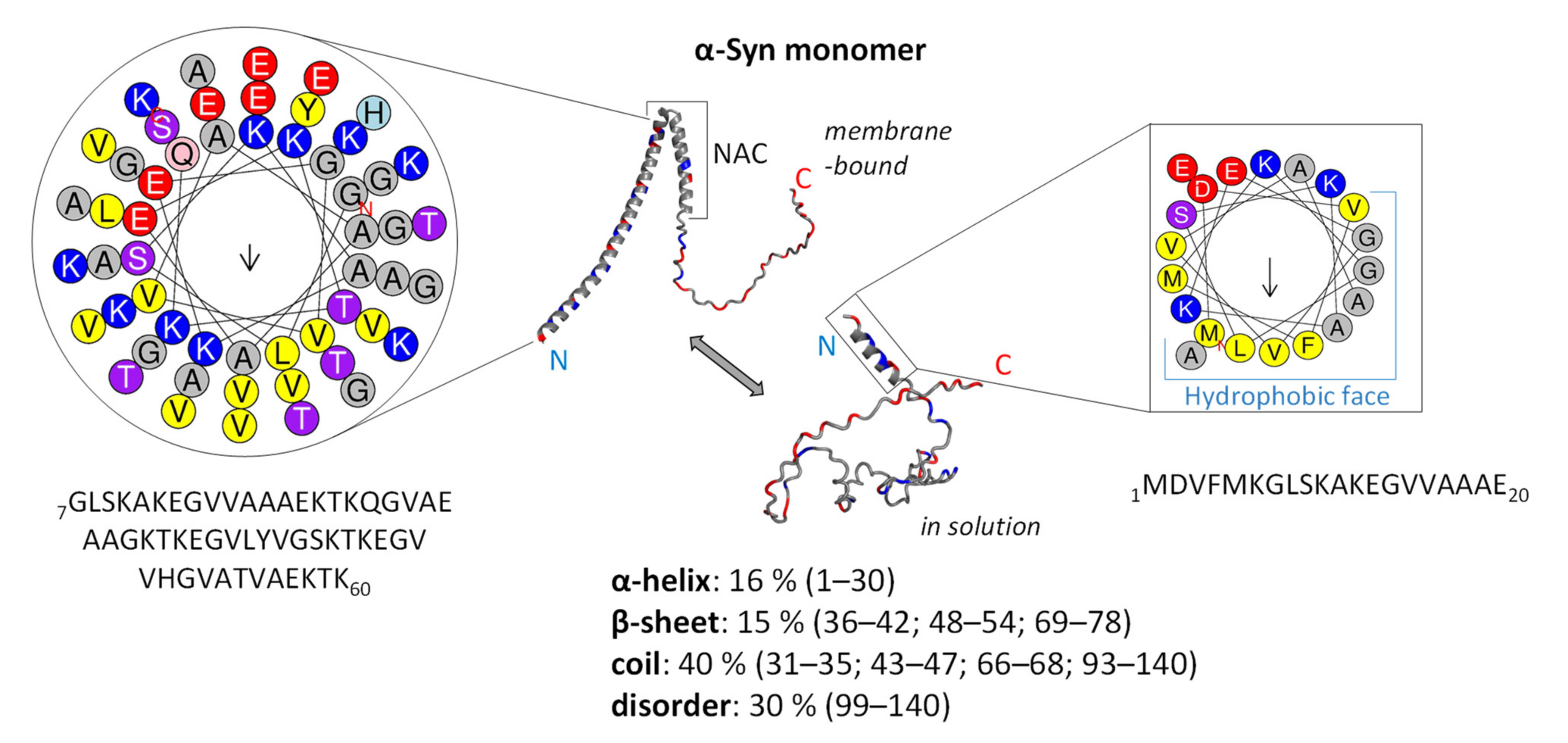
3.1. Structure and Function of Native α-Syn Monomer
3.2. Neurotoxicity of α-Syn
4. Therapies and Strategies against PD Related to α-Syn
4.1. Current PD Treatments
4.2. α-Syn-Targeted Therapies
4.2.1. Reduction of α-Syn Expression and Synthesis
4.2.2. Direct Inhibition of α-Syn Aggregation by Small Molecules
4.2.3. Direct Inhibition of α-Syn Aggregation by Short Peptides and Peptidomimetics
4.2.4. Clearance and Degradation of Toxic α-Syn Aggregates
4.2.5. Capturing Toxic α-Syn Aggregates and Blocking Transcellular Spreading
5. Conclusions and Further Perspectives
- Which species in the α-Syn aggregation pathway (monomer, oligomers, preformed fibrils) is the best target?
- Could selected disaggregators be used in combination with other ongoing approaches (gene silencing, immunotherapy, clearance stimulation) to achieve better efficiency in disease progress modification?
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Giorgetti, S.; Greco, C.; Tortora, P.; Aprile, F.A. Targeting amyloid aggregation: An overview of strategies and mechanisms. Int. J. Mol. Sci. 2018, 19, 2677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, J.; Perera, G.; Bhadbhade, M.; Halliday, G.M.; Dzamko, N. Autophagy activation promotes clearance of alpha-synuclein inclusions in fibril-seeded human neural cells. J. Biol. Chem. 2019, 294, 14241–14256. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Gao, G.; Duan, C.; Yang, H. Progress of immunotherapy of anti-α-synuclein in Parkinson’s disease. Biomed. Pharm. 2019, 115, 108843. [Google Scholar] [CrossRef] [PubMed]
- Zharikov, A.; Bai, Q.; De Miranda, B.R.; Van Laar, A.; Greenamyre, J.T.; Burton, E.A. Long-term RNAi knockdown of α-synuclein in the adult rat substantia nigra without neurodegeneration. Neurobiol. Dis. 2019, 125, 146–153. [Google Scholar] [CrossRef] [PubMed]
- Kouli, A.; Torsney, K.M.; Kuan, W.L. Parkinson’s disease: Etiology, neuropathology, and pathogenesis. In Parkinson’s Disease: Pathogenesis and Clinical Aspects; Stoker, T.B., Greenland, J.C., Eds.; Exon Publications: Brisbane, Australia, 2018; pp. 3–26. [Google Scholar]
- Meade, R.M.; Fairlie, D.P.; Mason, J.M. Alpha-synuclein structure and Parkinson’s disease—Lessons and emerging principles. Mol. Neurodegener. 2019, 14, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dorsey, E.R.; Elbaz, A.; Nicjols, E.; Abbasi, N.; Abd-Allah, F.; Abdelalim, A.; Adsur, J.C.; Ansha, M.G.; Brayne, C.; Choi, J.Y.J.; et al. Global, regional, and national burden of Parkinson’s disease, 1990–2016: A systematic analysis for the global burden of disease study. Lancet Neurol. 2018, 17, 939–953. [Google Scholar] [CrossRef] [Green Version]
- Ball, N.; Teo, W.P.; Chandra, S.; Chapman, J. Parkinson’s disease and the environment. Front. Neurol. 2019, 10, 218. [Google Scholar] [CrossRef] [Green Version]
- European Parkinson Disease Association. The European PD Standards of Care Consensus Statement. 2011. Available online: https://www.epda.eu.com/latest/resources/the-european-parkinsons-disease-standards-of-care-consensus-statement/ (accessed on 9 April 2022).
- Ou, Z.; Pan, J.; Tang, S.; Duan, D.; Yu, D.; Nong, H.; Wang, Z. Global trends in the incidence, prevalence, and years lived with disability of Parkinson’s disease in 204 countries/territories from 1990 to 2019. Front. Public Health 2021, 9, 776847. [Google Scholar] [CrossRef]
- Abbott, R.D.; Petrovitch, H.; White, L.R.; Masaki, K.H.; Tanner, C.M.; Curb, J.D.; Grandinetti, A.; Blanchette, P.L.; Popper, J.S.; Ross, G.W. Frequency of bowel movements and the future risk of Parkinson’s disease. Neurology 2001, 57, 456–462. [Google Scholar] [CrossRef]
- Jost, W.H. Gastrointestinal dysfunction in Parkinson’s disease. J. Neurol. Sci. 2010, 289, 69–73. [Google Scholar] [CrossRef]
- Schaeffer, E.; Kluge, A.; Böttner, M.; Zunke, F.; Cossais, F.; Berg, D.; Arnold, P. Alpha synuclein connects the gut-brain axis in Parkinson’s disease patients—A view on clinical aspects. Cellular pathology and analytical methodology. Front. Cell Dev. Biol. 2020, 8, 573696. [Google Scholar] [CrossRef]
- Kalia, L.V.; Lang, A.E. Parkinson’s disease. Lancet 2015, 386, 896–912. [Google Scholar] [CrossRef]
- Armstrong, M.J.; Okun, M.S. Diagnosis and treatment of Parkinson’s disease: A review. JAMA 2020, 323, 548–560. [Google Scholar] [CrossRef]
- He, S.; Zhong, S.; Liu, G.; Yang, J. Alpha-synuclein: The interplay of pathology, neuroinflammation, and environmental factors in Parkinson’s disease. Neurodegener. Dis. 2020, 20, 55–64. [Google Scholar] [CrossRef]
- Ganguly, U.; Singh, S.; Pal, S.; Prasad, S.; Agrawal, B.K.; Saini, R.V.; Chakrabarti, S. Alpha-synuclein as a biomarker of Parkinson’s disease: Good, but not good enough. Front. Aging Neurosci. 2021, 13, 702639. [Google Scholar] [CrossRef]
- Yang, W.; Hamilton, J.L.; Kopil, C.; Beck, J.C.; Tanner, C.M.; Albin, R.L.; Dorsey, R.; Dahodwala, N.; Cintina, I.; Hogan, P.; et al. Current and projected future economic burden of Parkinson’s disease in the US. NPJ Parkinsons Dis. 2020, 6, 15. [Google Scholar] [CrossRef]
- World Health Organisation, World Federation of Neurology. Atlas. In Country Resources for Neurological Disorders; World Health Organisation: Geneva, Switzerland, 2017. [Google Scholar]
- Muddapu, V.R.; Chakravarthy, V.S. Influence of energy deficiency on the subcellular processes of Substantia Nigra Pars Compacta cell for understanding Parkinsonian neurodegeneration. Sci. Rep. 2021, 11, 1754. [Google Scholar] [CrossRef]
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. Alpha-synuclein in Lewy bodies. Nature 1997, 388, 839–840. [Google Scholar] [CrossRef]
- Parmar, M.; Grealish, S.; Henchcliffe, C. The future of stem cell therapies for Parkinson’s disease. Nat. Rev. Neurosci. 2020, 21, 103–115. [Google Scholar] [CrossRef]
- Twohig, D.; Nielsen, H.M. α-synuclein in the pathophysiology of Alzheimer’s disease. Mol. Neurodegener. 2019, 14, 23. [Google Scholar] [CrossRef] [Green Version]
- Brás, I.C.; Outeiro, T.F. Alpha-synuclein: Mechanisms of release and pathology progression in synucleinopathies. Cells 2021, 10, 375. [Google Scholar] [CrossRef]
- Grosso Jasutkar, H.; Oh, S.E.; Mouradian, M.M. Therapeutics in the pipeline targeting α-synuclein for Parkinson’s disease. Pharmacol. Rev. 2022, 74, 207–237. [Google Scholar] [CrossRef]
- Polymeropoulos, M.H.; Lavedan, C.; Leroy, E.; Ide, S.E.; Dehejia, A.; Dutra, A.; Pike, B.; Root, H.; Rubenstein, J.; Boyer, R.; et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 1997, 276, 2045–2047. [Google Scholar] [CrossRef] [Green Version]
- Maraganore, D.M.; De Andrade, M.; Elbaz, A.; Farrer, M.J.; Ioannidis, J.P.; Krüger, R.; Walter, A.R.; Schneider, N.K.; Lesnick, T.G.; Lincoln, S.J.; et al. Genetic Epidemiology of Parkinson’s Disease (GEO-PD) Consortium. Collaborative analysis of α-synuclein gene promoter variability and Parkinson’s disease. JAMA 2006, 296, 661–670. [Google Scholar] [CrossRef] [Green Version]
- Soldner, F.; Stelzer, Y.; Shivalila, C.S.; Abraham, B.J.; Latourelle, J.C.; Barrasa, M.I.; Goldmann, J.; Myers, R.H.; Young, R.A.; Jaenisch, R. Parkinson-associated risk variant in distal enhancer of α-synuclein modulates target gene expression. Nature 2016, 533, 95–99. [Google Scholar] [CrossRef] [Green Version]
- Singleton, A.B.; Farrer, M.; Johnson, J.; Singleton, A.; Hague, S.; Kachergus, J.; Hulihan, M.; Peuralinna, T.; Dutra, A.; Nussbaum, R.; et al. Alpha-synuclein locus triplication causes Parkinson’s disease. Science 2003, 302, 841. [Google Scholar] [CrossRef] [Green Version]
- Chartier-Harlin, M.C.; Kachergus, J.; Roumier, C.; Mouroux, V.; Douay, X.; Lincoln, S.; Levecque, C.; Larvor, L.; Andrieux, J.; Hulihan, M.; et al. Alpha-synuclein locus duplication as a cause of familial Parkinson’s disease. Lancet 2004, 364, 1167–1169. [Google Scholar] [CrossRef]
- Wong, Y.C.; Krainc, D. Alpha-synuclein toxicity in neurodegeneration: Mechanism and therapeutic strategies. Nat. Med. 2017, 23, 1–13. [Google Scholar] [CrossRef]
- Trinh, J.; Zeldenrust, F.M.J.; Huan, J.; Kasten, M.; Schaake, S.; Petkovic, S.; Madoev, H.; Grünewald, A.; Almuammar, S.; König, I.R.; et al. Genotype-phenotype relations for the Parkinson’s disease genes SNCA, LRRK2, VPS35: MDS Gene systematic review. Mov. Disord. 2018, 3, 1857–1870. [Google Scholar] [CrossRef]
- Sardi, S.P.; Viel, C.; Clarke, J.; Treleaven, C.M.; Richards, A.M.; Park, H.; Olszewski, M.A.; Dodge, J.C.; Marshall, J.; Makino, E.; et al. Glucosylceramide synthase inhibition alleviates aberrations in synucleinopathy models. Proc. Natl. Acad. Sci. USA 2017, 114, 2699–2704. [Google Scholar] [CrossRef] [Green Version]
- Grova, N.; Schroeder, H.; Olivier, J.-L.; Turner, J.D. Epigenetic and neurological impairments associated with early life exposure to persistent organic pollutants. Int. J. Genom. 2019, 2019, 2085496. [Google Scholar] [CrossRef] [Green Version]
- Jowaed, A.; Schmitt, I.; Kaut, O.; Wüllner, U. Methylation regulates alpha-synuclein expression and is decreased in Parkinson’s disease patients’ brains. J. Neurosci. 2010, 30, 6355–6359. [Google Scholar] [CrossRef] [PubMed]
- Ascherio, A.; Schwarzschild, M.A. The epidemiology of Parkinson’s disease: Risk factors and prevention. Lancet Neurol. 2016, 15, 1257–1272. [Google Scholar] [CrossRef]
- Wassouf, Z.; Schulze-Hentrich, J.M. Alpha-synuclein at the nexus of genes and environment: The impact of environmental enrichment and stress on brain health and disease. J. Neurochem. 2019, 150, 591–604. [Google Scholar] [CrossRef] [PubMed]
- Chin-Chan, M.; Navarro-Yepes, J.; Quintanilla-Vega, B. Environmental pollutants as risk factors for neurodegenerative disorders: Alzheimer’s and Parkinson’s diseases. Front. Cell Neurosci. 2015, 9, 124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narayan, S.; Liew, Z.; Bronsteinb, J.M.; Ritz, B. Occupational pesticide use and Parkinson’s disease in the Parkinson environment gene (PEG). Study Environ. Int. 2017, 107, 266–273. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, C.; Almeida, C.; Tenreiro, S.; Quintas, A. Neuroprotection or neurotoxicity of illicit drugs on Parkinson’s disease. Life 2020, 10, 86. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, H.; Abushouk, A.I.; Gabr, M.; Negida, A.; Abdel-Daim, M.M. Parkinson’s disease and pesticides: A meta analysis of disease connection and genetic alterations. Biomed. Pharmacother. 2017, 90, 638–649. [Google Scholar] [CrossRef]
- Islam, M.D.; Azim, F.; Saju, H.; Zargaran, A.; Shirzad, M.; Kamal, M.; Fatema, K.; Rehman, S.; Momith Azad, M.A.; Ebrahimi-Barough, S. Pesticides and Parkinson’s disease: Current and future perspective. J. Chem. Neuroanat. 2021, 115, 101966. [Google Scholar] [CrossRef]
- Manning-Bog, A.B.; McCormack, A.L.; Li, J.; Uversky, V.N.; Fink, A.L.; Di Monte, D.A. The herbicide paraquat causes up-regulation and aggregation of alpha-synuclein in mice: Paraquat and alpha-synuclein. J. Biol. Chem. 2002, 277, 1641–1644. [Google Scholar] [CrossRef] [Green Version]
- Sherer, T.B.; Kim, J.H.; Betarbet, R.; Greenamyre, J.T. Subcutaneous rotenone exposure causes highly selective dopaminergic degeneration and alpha-synuclein aggregation. Exp. Neurol. 2003, 179, 9–16. [Google Scholar] [CrossRef]
- Uversky, V.N.; Li, J.; Fink, A.L. Pesticides directly accelerate the rate of alpha-synuclein fibril formation: A possible factor in Parkinson’s disease. FEBS Lett. 2001, 500, 105–108. [Google Scholar] [CrossRef] [Green Version]
- Kaidery, N.A.; Tarannum, S.; Thomas, B. Epigenetic landscape of Parkinson’s disease: Emerging role in disease mechanisms and therapeutic modalities. Neurotherapeutics 2013, 10, 698–708. [Google Scholar] [CrossRef] [Green Version]
- Ritz, B.; Lee, P.C.; Hansen, J.; FuchLassen, C.; Ketzel, M.; Sørensen, M.; Raaschou-Nielsen, O. Traffic-related air pollution and Parkinson’s disease in Denmark: A case-control study. Environ. Health Perspect. 2016, 124, 351–356. [Google Scholar] [CrossRef] [Green Version]
- Calderón-Garcidueñas, L.; Solt, A.C.; Henríquez-Roldán, C.; Torres-Jardón, R.; Nuse, B.; Herritt, L.; Villarreal-Calderón, R.; Osnaya, N.; Stone, I.; García, R.; et al. Long-term air pollution exposure is associated with neuroinflammation, an altered innate immune response, disruption of the blood-brain barrier, ultrafine particulate deposition, and accumulation of amyloid β-42 and α-synuclein in children and young adults. Toxicol. Pathol. 2008, 36, 289–310. [Google Scholar] [CrossRef]
- Calderón-Garcidueñas, L. Parkinson’s disease and air pollution: Does what we breathe matter? Nat. Rev. Neurol. 2021, 17, 467–468. [Google Scholar] [CrossRef]
- Kasdagli, M.-I.; Katsouyanni, K.; Dimakopoulou, K.; Samoli, E. Air pollution and Parkinson’s disease: A systematic review and meta-analysis up to 2018. Int. J. Hyg. Environ. Health. 2019, 222, 402–409. [Google Scholar] [CrossRef]
- Bjorklund, G.; Stejskal, V.; Urbina, M.A.; Dadar, M.; Chirumbolo, S.; Mutter, J. Metals and Parkinson’s disease: Mechanisms and biochemical processes. Curr. Med. Chem. 2018, 25, 2198–2214. [Google Scholar] [CrossRef]
- Breydo, L.; Wu, J.W.; Uversky, V.N. α-Synuclein misfolding and Parkinson’s disease. Biochim. Biophys. Acta Mol. Basis Dis. 2012, 1822, 261–285. [Google Scholar] [CrossRef] [Green Version]
- Santner, A.; Uversky, V.N. Metalloproteomics and metal toxicology of alpha-synuclein. Metallomics 2010, 2, 378–392. [Google Scholar] [CrossRef]
- Bisaglia, M.; Bubacco, L. Copper ions and Parkinson’s disease: Why is homeostasis so relevant? Biomolecules 2020, 10, 195. [Google Scholar] [CrossRef] [Green Version]
- Paik, S.R.; Shin, H.J.; Lee, J.H.; Chang, C.S.; Kim, J. Copper (II)-induced self-oligomerization of alpha-synuclein. Biochem. J. 1999, 340, 821–828. [Google Scholar] [CrossRef]
- Wei, X.; Cai, M.; Land, J.L. The function of the metals in regulating epigenetics during Parkinson’s disease. Front. Genet. 2021, 11, 616083. [Google Scholar] [CrossRef]
- Weisskopf, M.G.; Weuve, J.; Nie, H.; Saint-Hilaire, M.-H.; Sudarsky, L.; Simon, D.K.; Hersh, B.; Schwartz, J.; Wright, R.O.; Hu, H. Association of cumulative lead exposure with Parkinson’s disease. Environ. Health Perspect. 2010, 118, 1609–1613. [Google Scholar] [CrossRef]
- Jankovic, J.; Tan, E.K. Parkinson’s disease: Etiopathogenesis and treatment. J. Neurol. Neurosurg. Psychiatry 2020, 91, 795–808. [Google Scholar] [CrossRef]
- Kingwell, K. Zeroing in on neurodegenerative α-synuclein. Nat. Rev. Drug. Discov. 2017, 16, 371–373. [Google Scholar] [CrossRef]
- Ulmer, T.S.; Bax, A.; Cole, N.B.; Nussbaum, R.L. Structure and dynamics of micelle-bound human α-synuclein. J. Biol. Chem. 2005, 280, 9595–9603. [Google Scholar] [CrossRef] [Green Version]
- Georgieva, E.R.; Ramlall, T.F.; Borbat, P.P.; Freed, J.H.; Eliezer, D. Membrane-bound α-synuclein forms an extended helix: Long-distance pulsed ESR measurements using vesicles, bicelles, and rodlike micelles. J. Amer. Chem. Soc. 2008, 130, 12856–12857. [Google Scholar] [CrossRef] [Green Version]
- Kim, T.D.; Paik, S.R.; Yang, C.H. Structural and functional implications of C-terminal regions of alpha-synuclein. Biochemistry 2002, 41, 13782–13790. [Google Scholar] [CrossRef]
- Piovesan, D.; Walsh, I.; Minervini, G.; Tosatto, S.C.E. FELLS: Fast estimator of latent local structure. Bioinformatics 2017, 33, 1889–1891. [Google Scholar] [CrossRef] [PubMed]
- Gautier, R.; Douguet, D.; Antonny, B.; Drin, G. HELIQUEST: A web server to screen sequences with specific alpha-helical properties. Bioinformatics 2008, 24, 2101–2102. [Google Scholar] [CrossRef] [PubMed]
- Mirdita, M.; Schütze, K.; Moriwaki, Y.; Heo, L.; Ovchinnikov, S.; Steinegger, M. ColabFold-Making protein folding accessible to all. bioRxiv 2021. [Google Scholar] [CrossRef]
- Habchi, J.; Tompa, P.; Longhi, S.; Uversky, V.N. Introducing protein intrinsic disorder. Chem. Rev. 2014, 114, 6561–6588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bondos, S.E.; Dunker, A.K.; Uversky, V.N. Intrinsically disordered proteins play diverse roles in cell signaling. Cell Commun. Signal. 2022, 20, 20. [Google Scholar] [CrossRef]
- Zhang, S.; Liu, Y.Q.; Jia, C.; Lim, Y.J.; Feng, G.; Xu, E.; Long, H.; Kimura, Y.; Tao, Y.; Zhao, C.; et al. Mechanistic basis for receptor-mediated pathological α-synuclein fibril cell-to-cell transmission in Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2021, 118, e2011196118. [Google Scholar] [CrossRef]
- Bertoncini, C.W.; Jung, Y.S.; Fernandez, C.O.; Hoyer, W.; Griesinger, C.; Jovin, T.M.; Zweckstetter, M. Release of long-range tertiary interactions potentiates aggregation of natively unstructured alpha-synuclein. Proc. Natl. Acad. Sci. USA 2005, 102, 1430–1435. [Google Scholar] [CrossRef] [Green Version]
- Nuber, S.; Rajsombath, M.; Minakaki, G.; Winkler, J.; Müller, C.P.; Ericsson, M.; Caldarone, B.; Dettmer, U.; Selkoe, D.J. Abrogating native α-synuclein tetramers in mice causes a L-DOPA-responsive motor syndrome closely resembling Parkinson’s disease. Neuron 2018, 100, 75–90. [Google Scholar] [CrossRef] [Green Version]
- Bartels, T.; Choi, J.G.; Selkoe, D.J. Alpha-Synuclein occurs physiologically as a helically folded tetramer that resists aggregation. Nature 2011, 477, U107–U123. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Perovic, I.; Chittuluru, J.; Kaganovich, A.; Nguyen, L.T.; Liao, J.; Auclair, J.R.; Johnson, D.; Landeru, A.; Simorellis, A.K.; et al. A soluble alpha-synuclein construct forms a dynamic tetramer. Proc. Natl. Acad. Sci. USA 2011, 108, 17797–17802. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Das, U.; Scott, D.A.; Tang, Y.; McLean, P.J.; Roy, S. α-Synuclein multimers cluster synaptic vesicles and attenuate recycling. Curr. Biol. 2014, 24, 2319–2326. [Google Scholar] [CrossRef] [Green Version]
- Bendor, J.T.; Logan, T.P.; Edwards, R.H. The function of alpha-synuclein. Neuron 2013, 79, 1044–1066. [Google Scholar] [CrossRef] [Green Version]
- Goers, J.; Manning-Bog, A.B.; McCormack, A.L.; Millett, I.S.; Doniach, S.; Di Monte, D.A.; Uversky, V.N.; Fink, A.L. Nuclear localization of alpha-synuclein and its interaction with histones. Biochemistry 2003, 42, 8465–8471. [Google Scholar] [CrossRef]
- Takahashi, M.; Kanuka, H.; Fujiwara, H.; Koyama, A.; Hasegawa, M.; Miura, M.; Iwatsubo, T. Phosphorylation of α-synuclein characteristic of synucleinopathy lesions is recapitulated in α-synuclein transgenic Drosophila. Neurosci. Lett. 2003, 336, 155–158. [Google Scholar] [CrossRef]
- Siddiqui, A.; Chinta, S.J.; Mallajosyula, J.K.; Rajagopolan, S.; Hanson, I.; Rane, A.; Melov, S.; Andersen, J.K. Selective binding of nuclear alpha-synuclein to the PGC1alpha promoter under conditions of oxidative stress may contribute to losses in mitochondrial function: Implications for Parkinson’s disease. Free Radic. Biol. Med. 2012, 53, 993–1003. [Google Scholar] [CrossRef] [Green Version]
- Ma, K.L.; Song, L.K.; Yuan, Y.H.; Zhang, Y.; Yang, J.L.; Zhu, P.; Chen, N.H. Alpha-Synuclein is prone to interaction with the GC-box-like sequence in vitro. Cell Mol. Neurobiol. 2014, 34, 603–609. [Google Scholar] [CrossRef]
- Pinho, R.; Paiva, I.; Jercic, K.G.; Fonseca-Ornelas, L.; Gerhardt, E.; Fahlbusch, C.; Garcia-Esparcia, P.; Kerimoglu, C.; Pavlou, M.A.S.; Villar-Piqué, A.; et al. Nuclear localization and phosphorylation modulate pathological effects of alpha-synuclein. Hum. Mol. Genet. 2019, 28, 31–50. [Google Scholar] [CrossRef]
- Bernal-Conde, L.D.; Ramos-Acevedo, R.; Reyes-Hernández, M.A.; Balbuena-Olvera, A.J.; Morales-Moreno, I.D.; Argüero-Sánchez, R.; Schüle, B.; Guerra-Crespo, M. Alpha-synuclein physiology and pathology: A perspective on cellular structures and organelles. Front. Neurosci. 2020, 13, 1399. [Google Scholar] [CrossRef] [Green Version]
- Burré, J.; Sharma, M.; Sudhof, T.C. Cell biology and pathophysiology of α-synuclein. Cold Spring Harb. Perspect. Med. 2018, 8, a024091. [Google Scholar] [CrossRef]
- Burré, J.; Sharma, M.; Tsetsenis, T.; Buchman, V.; Etherton, M.R.; Sudhof, T.C. Alpha-synuclein promotes SNARE-complex assembly in vivo and in vitro. Science 2010, 329, 1663–1667. [Google Scholar] [CrossRef] [Green Version]
- Campioni, S.; Riek, R. Membrane remodelling activity of α-synuclein. J. Neurol. Neuromed. 2016, 1, 23–27. [Google Scholar]
- Sun, J.; Wang, L.; Bao, H.; Premi, S.; Das, U.; Chapman, E.R.; Roy, S. Functional cooperation of alpha-synuclein and VAMP2 in synaptic vesicle recycling. Proc. Natl. Acad. Sci. USA 2019, 116, 11113–11115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeWitt, D.C.; Rhoades, E. Alpha-synuclein can inhibit SNARE-mediated vesicle fusion through direct interactions with lipid bilayers. Biochemistry 2013, 52, 2385–2387. [Google Scholar] [CrossRef] [PubMed]
- Burré, J.; Sharma, M.; Südhof, T.C. α-Synuclein assembles into higher-order multimers upon membrane binding to promote SNARE complex formation. Proc. Natl. Acad. Sci. USA 2014, 111, E4274–E4283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Payton, J.E.; Perrin, R.J.; Woods, W.S.; George, J.M. Structural determinants of PLD2 inhibition by α-synuclein. J. Mol. Biol. 2004, 337, 1001–1009. [Google Scholar] [CrossRef]
- Fields, C.R.; Bengoa-Vergniory, N.; Wade-Martins, R. Targeting alpha-synuclein as a therapy for Parkinson’s disease. Front. Mol. Neurosci. 2019, 12, 299. [Google Scholar] [CrossRef] [Green Version]
- Dalfo, E.; Ferrer, I. α-synuclein binding to rab3a in multiple system atrophy. Neurosci. Lett. 2005, 380, 170–175. [Google Scholar] [CrossRef]
- Paciotti, S.; Bellomo, G.; Gatticchi, L.; Parnetti, L. Are we ready for detecting α-synuclein prone to aggregation in patients? The case of “protein-misfolding cyclic amplification” and “real-time quaking-induced conversion” as diagnostic tools. Front. Neurol. 2018, 9, 415. [Google Scholar] [CrossRef] [Green Version]
- Del Tredici, K.; Braak, H. Sporadic Parkinson’s disease: Development and distribution of alpha-synuclein pathology. Neuropathol. Appl. Neurobiol. 2016, 42, 33–50. [Google Scholar] [CrossRef]
- Kuo, Y.-M.; Nwankwo, E.I.; Nussbaum, R.L.; Rogers, J.; Maccecchini, M.L. Translational inhibition of α-synuclein by posiphen normalizes distal colon motility in transgenic Parkinson mice. Am. J. Neurodegener. Dis. 2019, 8, 1–15. [Google Scholar]
- Karpowicz, R.J.; Trojanowski, J.Q.; Lee, V.M. Transmission of alpha-synuclein seeds in neurodegenerative disease: Recent developments. Lab. Investig. 2019, 99, 971–981. [Google Scholar] [CrossRef]
- Böttner, M.; Fricke, T.; Müller, M.; Barrenschee, M.; Deuschl, G.; Schneider, S.A.; Egberts, J.-H.; Becker, T.; Fritscher-Ravens, A.; Ellrichmann, M.; et al. Alpha-synuclein is associated with the synaptic vesicle apparatus in the human and rat enteric nervous system. Brain Res. 2015, 1614, 51–59. [Google Scholar] [CrossRef]
- Bu, L.-L.; Huang, K.-X.; Zheng, D.-Z.; Lin, D.-Y.; Chen, Y.; Jing, X.-N.; Liang, Y.-R.; Tao, E.-X. Alpha-synuclein accumulation and its phosphorylation in the enteric nervous system of patients without neurodegeneration: An explorative study. Front. Aging Neurosci. 2020, 12, 575481. [Google Scholar] [CrossRef]
- Martins, M.; Rosa, A.; Guedes, L.C.; Fonseca, B.V.; Gotovac, K.; Violante, S.; Mestre, T.; Coelho, M.; Rosa, M.M.; Martin, E.R.; et al. Convergence of miRNA expression profiling, alpha-synuclein interacton and GWAS in Parkinson’s disease. PLoS ONE 2011, 6, e25443. [Google Scholar] [CrossRef]
- Alam, P.; Bousset, L.; Melki, R.; Otzen, D.E. Alpha-synuclein oligomers and fibrils: A spectrum of species, a spectrum of toxicities. J. Neurochem. 2019, 150, 522–534. [Google Scholar] [CrossRef] [Green Version]
- Burré, J. The synaptic function of alpha-synuclein. J. Parkinson’s Dis. 2015, 5, 699–713. [Google Scholar] [CrossRef] [Green Version]
- Shahmoradian, S.H.; Lewis, A.J.; Genoud, C.; Hench, J.; Moors, T.E.; Navarro, P.P.; Castaño-Díez, D.; Schweighauser, G.; Graff-Meyer, A.; Goldie, K.N.; et al. Lewy pathology in Parkinson’s disease consists of crowded organelles and lipid membranes. Nat. Neurosci. 2019, 22, 1099–1109. [Google Scholar] [CrossRef] [Green Version]
- Fusco, G.; Chen, S.W.; Williamson, P.T.; Cascella, R.; Perni, M.; Jarvis, J.A.; Cecchi, C.; Vendruscolo, M.; Chiti, F.; Cremades, N.; et al. Structural basis of membrane disruption and cellular toxicity by α-synuclein oligomers. Science 2017, 358, 1440–1443. [Google Scholar] [CrossRef] [Green Version]
- Santos, J.; Gracia, P.; Navarro, S.; Peña-Díaz, S.; Pujols, J.; Cremades, N.; Pallarès, I.; Ventura, S. α-Helical peptidic scaffolds to target α-synuclein toxic species with nanomolar affinity. Nat. Commun. 2021, 12, 3752. [Google Scholar] [CrossRef]
- Uversky, V.N.; Cooper, E.M.; Bower, K.S.; Li, J.; Fink, A.L. Accelerated alpha-synuclein fibrillation in crowded milieu. FEBS Lett. 2002, 515, 99–103. [Google Scholar] [CrossRef] [Green Version]
- Beyer, K. Alpha-synuclein structure, posttranslational modification and alternative splicing as aggregation enhancers. Acta Neuropathol. 2006, 112, 237–251. [Google Scholar] [CrossRef]
- Schmidt, S.; Vogt Waisenhorn, D.M.; Wurs, W. Chapter 5—“Parkinson’s disease—A role of non-enzymatic posttranslational modifications in disease onset and progression?”. Mol. Aspect Med. 2022, 101096. [Google Scholar] [CrossRef]
- Fujiwara, H.; Hasegawa, M.; Dohmae, N.; Kawashima, A.; Masliah, E.; Goldberg, M.S.; Shen, J.; Takio, K.; Iwatsubo, T. Alpha-Synuclein is phosphorylated in synucleinopathy lesions. Nat. Cell Biol. 2002, 4, 160–164. [Google Scholar] [CrossRef]
- Beyer, K.; Ariza, A. α-Synuclein posttranslational modification and alternative splicing as a trigger for neurodegeneration. Mol. Neurobiol. 2013, 47, 509–524. [Google Scholar] [CrossRef]
- Burai, R.; Ait-Bouziad, N.; Chiki, A.; Lashuel, H.A. Elucidating the role of site-specific nitration of α-synuclein in the pathogenesis of Parkinson’s disease via protein semisynthesis and mutagenesis. J. Am. Chem. Soc. 2015, 137, 5041–5052. [Google Scholar] [CrossRef]
- Zhang, J.; Li, X.; Jia-Da, L. The roles of post-translational modifications on α-synuclein in the pathogenesis of Parkinson’s Diseases. Front. Neurosci. 2019, 13, 381. [Google Scholar] [CrossRef] [Green Version]
- Burmann, B.M.; Gerez, J.A.; Matečko-Burmann, I.; Campioni, S.; Kumari, P.; Ghosh, D.; Mazur, A.; Aspholm, E.E.; Šulskis, D.; Wawrzyniuk, M.; et al. Regulation of α-synuclein by chaperones in mammalian cells. Nature 2020, 577, 127–132. [Google Scholar] [CrossRef]
- Rott, R.; Szargel, R.; Shani, V.; Hamza, H.; Savyon, M.; Abd Elghani, F.; Bandopadhyay, R.; Engelender, S. SUMOylation and ubiquitination reciprocally regulate α-synuclein degradation and pathological aggregation. Proc. Natl. Acad. Sci. USA 2017, 114, 13176–13181. [Google Scholar] [CrossRef] [Green Version]
- Krumova, P.; Meulmeester, E.; Garrido, M.; Tirard, M.; Hsiao, H.H.; Bossis, G.; Urlaub, H.; Zweckstetter, M.; Kügler, S.; Melchior, F.; et al. Sumoylation inhibits alpha-synuclein aggregation and toxicity. J. Cell Biol. 2011, 194, 49–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abeywardana, T.; Pratt, M.R. Extent of inhibition of α-synuclein aggregation in vitro by SUMOylation is conjugation site- and SUMO isoform-selective. Biochemistry 2015, 54, 959–961. [Google Scholar] [CrossRef] [PubMed]
- Marotta, N.P.; Lin, Y.H.; Lewis, Y.E.; Ambroso, M.R.; Zaro, B.W.; Roth, M.T.; Arnold, D.B.; Langen, R.; Pratt, M.R. O-GlcNAc modification blocks the aggregation and toxicity of the protein α-synuclein associated with Parkinson’s disease. Nat. Chem. 2015, 11, 913–920. [Google Scholar] [CrossRef] [PubMed]
- Celej, M.S.; Sarroukh, R.; Goormaghtigh, E.; Fidelio, G.D.; Ruysschaert, J.M.; Raussens, V. Toxic prefibrillar α-synuclein amyloid oligomers adopt a distinctive antiparallel β-sheet structure. Biochem. J. 2012, 443, 719–726. [Google Scholar] [CrossRef]
- Madine, J.; Doig, A.J.; Middleton, D.A. Design of an N-methylated peptide inhibitor of alpha-synuclein aggregation guided by solid-state NMR. J. Am. Chem. Soc. 2008, 130, 7873–7881. [Google Scholar] [CrossRef] [Green Version]
- Mirecka, E.A.; Shaykhalishahi, H.; Gauhar, A.; Akgul, S.; Lecher, J.; Willbold, D.; Stoldt, M.; Hoyer, W. Sequestration of a beta-hairpin for control of alpha-synuclein aggregation. Angew. Chem. 2014, 53, 4227–4230. [Google Scholar] [CrossRef]
- Zhao, K.; Li, Y.; Liu, Z.; Long, H.; Zhao, C.; Luo, F.; Sun, Y.; Tao, Y.; Su, X.-D.; Li, D.; et al. Parkinson’s disease associated mutation E46K of α-synuclein triggers the formation of a distinct fibril structure. Nat. Commun. 2020, 11, 2643. [Google Scholar] [CrossRef]
- Guerrero-Ferreira, R.; Kovacik, L.; Ni, D.; Stahlberg, H. New insights on the structure of alpha-synuclein fibrils using cryo-electron microscopy. Curr. Opin. Neurobiol. 2020, 61, 89–95. [Google Scholar] [CrossRef]
- Tuttle, M.D.; Comellas, G.; Nieuwkoop, A.J.; Covell, D.J.; Berthold, D.A.; Kloepper, K.D.; Courtney, J.M.; Kim, J.K.; Barclay, A.M.; Kendall, A.; et al. Solid-state NMR structure of a pathogenic fibril of full-length human alpha-synuclein. Nat. Struct. Mol. Biol. 2016, 23, 409–415. [Google Scholar] [CrossRef]
- Yang, X.; Wang, B.; Hoop, C.L.; Williams, J.K.; Baum, J. NMR unveils an N-terminal interaction interface on acetylated-α-synuclein monomers for recruitment to fibrils. Proc. Natl. Acad. Sci. USA 2021, 118, e2017452118. [Google Scholar] [CrossRef]
- Zhao, K.; Lim, Y.J.; Liu, Z.; Long, H.; Sun, Y.; Hu, J.J.; Zhao, C.; Tao, Y.; Zhang, X.; Li, D.; et al. Parkinson’s disease-related phosphorylation at Tyr39 rearranges α-synuclein amyloid fibril structure revealed by cryo-EM. Proc. Natl. Acad. Sci. USA 2020, 117, 20305–20315. [Google Scholar] [CrossRef]
- Guerrero-Ferreira, R.; Taylor, N.M.; Mona, D.; Ringler, P.; Lauer, M.E.; Riek, R.; Britschgi, M.; Stahlberg, H. Cryo-EM structure of alpha-synuclein fibrils. eLife 2018, 7, e36402. [Google Scholar] [CrossRef]
- Li, B.; Ge, P.; Murray, K.A.; Sheth, P.; Zhang, M.; Nair, G.; Sawaya, M.R.; Shin, W.S.; Boyer, D.R.; Ye, S.; et al. Cryo-EM of full-length α-synuclein reveals fibril polymorphs with a common structural kernel. Nat. Commun. 2018, 9, 10. [Google Scholar] [CrossRef]
- Li, Y.W.; Zhao, C.; Luo, F.; Liu, Z.; Gui, X.; Luo, Z.; Zhang, X.; Li, D.; Liu, C.; Li, X. Amyloid fibril structure of α-synuclein determined by cryo-electron microscopy. Cell Res. 2018, 28, 897–903. [Google Scholar] [CrossRef] [Green Version]
- Terada, M.; Suzuki, G.; Nonaka, T.; Kametani, F.; Tamaoka, A.; Hasegawa, M. The effect of truncation on prion-like properties of α-synuclein. J. Biol. Chem. 2018, 293, 13910–13920. [Google Scholar] [CrossRef] [Green Version]
- Bavinton, C.E.; Sternke-Hoffmann, R.; Knipe, P.C.; Yamashita, T.; Hamilton, A.D.; Luo, J.; Thompson, S. Rationally designed helical peptidomimetics disrupt alpha-synuclein fibrillation. Chem. Commun. 2022, 58, 5132–5135. [Google Scholar] [CrossRef]
- Conway, K.A.; Rochet, J.C.; Bieganski, R.M.; Lansbury, P.T., Jr. Kinetic stabilization of the α-synuclein protofibril by a dopamine-α-synuclein adduct. Science 2001, 294, 1346–1349. [Google Scholar] [CrossRef]
- Padmaraju, V.; Bhaskar, J.J.; Prasada Rao, U.J.; Salimath, P.V.; Rao, K.S. Role of advanced glycation on aggregation and DNA binding properties of alpha-synuclein. J. Alzheimer’s Dis. 2011, 24 (Suppl. 2), 211–221. [Google Scholar] [CrossRef] [Green Version]
- Ma, K.L.; Song, L.K.; Yuan, Y.H.; Zhang, Y.; Han, N.; Gao, K.; Chen, N.H. The nuclear accumulation of alpha-synuclein is mediated by importin alpha and promotes neurotoxicity by accelerating the cell cycle. Neuropharmacology 2014, 82, 132–142. [Google Scholar] [CrossRef]
- Santos, J.; Pallarès, I.; Ventura, S. Is a cure for Parkinson’s disease hiding inside us? Trends Biochem. Sci 2022, S0968-0004(22)00025-1. [Google Scholar] [CrossRef]
- Angelova, P.R.; Ludtmann, M.H.; Horrocks, M.H.; Negoda, A.; Cremades, N.; Klenerman, D.; Dobson, C.M.; Wood, N.W.; Pavlov, E.V.; Gandhi, S.; et al. Ca2+ is a key factor in α-synuclein-induced neurotoxicity. J. Cell Sci. 2016, 129, 1792–1801. [Google Scholar] [CrossRef] [Green Version]
- Deas, E.; Cremades, N.; Angelova, P.R.; Ludtmann, M.H.; Yao, Z.; Chen, S.; Horrocks, M.H.; Banushi, B.; Little, D.; Devine, M.J.; et al. Alpha-synuclein oligomers interact with metal ions to induce oxidative stress and neuronal death in Parkinson’s disease. Antioxid. Redox Signal. 2016, 24, 376–391. [Google Scholar] [CrossRef] [Green Version]
- Ysselstein, D.; Joshi, M.; Mishra, V.; Griggs, A.M.; Asiago, J.M.; McCabe, G.P.; Stanciu, L.A.; Post, C.B.; Rochet, J.C. Effects of impaired membrane interactions on α-synuclein aggregation and neurotoxicity. Neurobiol. Dis. 2015, 79, 150–163. [Google Scholar] [CrossRef] [Green Version]
- Westphal, C.H.; Chandra, S.S. Monomeric synucleins generate membrane curvature. J. Biol. Chem. 2013, 288, 1829–1840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, G.; Imura, S.; Hosokawa, M.; Katsumata, R.; Nonaka, T.; Hisanaga, S.I.; Saeki, Y.; Hasegawa, M. α-synuclein strains that cause distinct pathologies differentially inhibit proteasome. eLife 2020, 9, e56825. [Google Scholar] [CrossRef] [PubMed]
- Holmes, B.B.; DeVos, S.L.; Kfoury, N.; Li, M.; Jacks, R.; Yanamandra, K.; Ouidja, M.O.; Brodsky, F.M.; Marasa, J.; Bagchi, D.P.; et al. Heparan sulfate proteoglycans mediate internalization and propagation of specific proteopathic seeds. Proc. Natl. Acad. Sci. USA 2013, 110, E3138–E3147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, C.; Ho, D.-H.; Suk, J.-E.; You, S.; Michael, S.; Kang, J.; Lee, S.J.; Masliah, E.; Hwang, D.; Lee, H.-J.; et al. Neuron-released oligomeric alpha-synuclein is an endogenous agonist of TLR2 for paracrine activation of microglia. Nat. Commun. 2013, 4, 1562. [Google Scholar] [CrossRef] [Green Version]
- Danzer, K.M.; Haasen, D.; Karow, A.R.; Moussaud, S.; Habeck, M.; Giese, A.; Kretzschmar, H.; Hengerer, B.; Kostka, M. Different species of alpha-synuclein oligomers induce calcium influx and seeding. J. Neurosci. 2007, 27, 9220–9232. [Google Scholar] [CrossRef]
- Su, R.; Zhou, T. Alpha-synuclein induced immune cells activation and associated therapy in Parkinson’s disease. Front. Aging Neurosci. 2021, 13, 769506. [Google Scholar] [CrossRef]
- Rostami, J.; Fotaki, G.; Sirois, J.; Mzezewa, R.; Bergström, J.; Essand, M.; Healy, L.; Erlandsson, S. Astrocytes have the capacity to act as antigen-presenting cells in the Parkinson’s disease brain. J. Neuroinflam. 2020, 17, 119. [Google Scholar] [CrossRef]
- Kwon, S.; Iba, M.; Masliah, E.; Kim, C. Targeting microglial and neuronal Toll-like receptor 2 in synucleinopathies. Exp. Neurobiol. 2019, 28, 547–553. [Google Scholar] [CrossRef] [Green Version]
- Kim, C.; Kwon, S.; Iba, M.; Spencer, B.; Rockenstein, E.; Mante, M.; Adame, A.; Shin, S.J.; Fields, J.A.; Robert, A.; et al. Effects of innate immune receptor stimulation on extracellular α-synuclein uptake and degradation by brain resident cells. Exp. Mol. Med. 2021, 53, 281–290. [Google Scholar] [CrossRef]
- Beach, T.G.; Serrano, G.E.; Kremer, T.; Canamero, M.; Dziadek, S.; Sade, H.; Derkinderen, P.; Corbillé, A.G.; Letournel, F.; Munoz, D.G.; et al. Immunohistochemical method and histopathology judging for the systemic synuclein sampling study (S4). J. Neuropathol. Exp. Neurol. 2018, 77, 793–802. [Google Scholar] [CrossRef]
- Stolzenberg, E.; Berry, D.; Yang, D.; Lee, E.Y.; Kroemer, A.; Kaufman, S.; Wong, G.C.L.; Oppenheim, J.J.; Sen, S.; Fishbein, T.; et al. A role for neuronal alpha-synuclein in gastrointestinal immunity. J. Innate Immun. 2019, 9, 456–463. [Google Scholar] [CrossRef]
- Tan, E.-K.; Chao, Y.-X.; West, A.; Chan, L.-L.; Poewe, W.; Jankovic, J. Parkinson disease and the immune system—Associations, mechanisms and therapeutics. Nat. Rev. Neurol. 2020, 16, 303–318. [Google Scholar] [CrossRef]
- Challis, C.; Hori, A.; Sampson, T.R.; Yoo, B.B.; Challis, R.C.; Hamilton, A.M.; Mazmanian, S.K.; Volpicelli-Daley, L.A.; Gradinaru, V. Gut-seeded alpha-synuclein fibrils promote gut dysfunction and brain pathology specifically in aged mice. Nat. Neurosci. 2020, 23, 327–336. [Google Scholar] [CrossRef]
- Kim, S.; Kwon, S.H.; Kam, T.I.; Panicker, N.; Karuppagounder, S.S.; Lee, S.; Lee, J.H.; Kim, W.R.; Kook, M.; Foss, C.A.; et al. Transneuronal propagation of pathologic α-synuclein from the gut to the brain models Parkinson’s disease. Neuron 2019, 103, 627–641. [Google Scholar] [CrossRef]
- Breen, D.P.; Halliday, G.M.; Lang, A.E. Gut-brain axis and the spread of α-synuclein pathology: Vagal highway or dead end? Mov. Disord. 2019, 34, 307–316. [Google Scholar] [CrossRef]
- Leclair-Visonneau, L.; Neunlist, M.; Derkinderen, P.; Lebouvier, T. The gut in Parkinson’s disease: Bottom-up, top-down, or neither? Neurogestroenerol. Motil. 2020, 32, e13777. [Google Scholar] [CrossRef]
- Du, T.; Wang, L.; Liu, W.; Zhu, G.; Chen, Y.; Zhang, J. Biomarkers and the role of α-synuclein in Parkinson’s disease. Front. Aging Neurosci. 2021, 13, 137. [Google Scholar] [CrossRef]
- Fayyad, M.; Salim, S.; Majbour, N.; Erskine, D.; Stoops, E.; Mollenhauer, B.; El-Agnaf, O.M. Parkinson’s disease biomarkers based on α-synuclein. J. Neurochem. 2019, 150, 626–636. [Google Scholar] [CrossRef]
- Alcalay, R.N.; Kehoe, C.; Shorr, E.; Battista, R.; Hall, A.; Simuni, T.; Marder, K.; Wills, A.-M.; Naito, A.; Beck, J.C.; et al. Genetic testing for Parkinson disease: Current practice, knowledge, and attitudes among US and Canadian movement disorders specialists. Genet. Med. 2020, 22, 574–580. [Google Scholar] [CrossRef] [Green Version]
- Musacchio, J.M. Enzymes involved in the biosynthesis and degradation of catecholamines. In Biochemistry of Biogenic Amines; Iverson, L., Ed.; Springer: Berlin/Heidelberg, Germany, 2013; pp. 1–35. [Google Scholar]
- Zahoor, I.; Shafi, A.; Haq, E. Pharmacological treatment of Parkinson’s disease. In Parkinson’s Disease: Pathogenesis and Clinical Aspects; Stoker, T.B., Greenland, J.C., Eds.; Exon Publications: Brisbane, Australia, 2018; pp. 129–144. [Google Scholar]
- Parkinson’s Disease Toolkit. Available online: http://www.toolkit.parkinson.org/content/first-line-meds-and-dosing (accessed on 6 April 2022).
- Nyholm, D.; Klangemo, K.; Johansson, A. Levodopa/carbidopa intestinal gel infusion long-term therapy in advanced Parkinson’s disease. Eur. J. Neurol. 2012, 19, 1079–1085. [Google Scholar] [CrossRef]
- Amjad, F.; Bhatti, D.; Davis, T.L.; Oguh, O.; Pahwa, R.; Kukreja, P.; Zamudio, J.; Metman, L.V. Current practices for outpatient initiation of levodopa-carbidopa intestinal gel for management of advanced Parkinson’s disease in the United States. Adv. Ther. 2019, 36, 2233–2246. [Google Scholar] [CrossRef] [Green Version]
- Bonam, S.R.; Tranchant, C.; Muller, S. Autophagy-lysosomal pathway as potential therapeutic target in Parkinson’s disease. Cells 2021, 10, 3547. [Google Scholar] [CrossRef]
- Athauda, D.; Gulyani, S.; Karnati, H.K.; Li, Y.; Tweedie, D.; Mustapic, M.; Chawla, S.; Chowdhury, K.; Skene, S.S.; Greig, N.H.; et al. Utility of neuronal-derived exosomes to examine molecular mechanisms that affect motor function in patients with Parkinson disease. JAMA Neurol. 2019, 76, 420. [Google Scholar] [CrossRef]
- Glotfelty, E.J.; Olson, L.; Karlsson, T.E.; Li, Y.; Greig, N.H. Glucagon-like peptide-1 (GLP-1)-based receptor agonists as a treatment for Parkinson’s disease. Expert Opin. Invest Drugs 2020, 29, 595–602. [Google Scholar] [CrossRef] [PubMed]
- Barker, R.A. Designing stem-cell-based dopamine cell replacement trials for Parkinson’s disease. Nat. Med. 2019, 25, 1045–1053. [Google Scholar] [CrossRef] [PubMed]
- Christin, C.W.; Bankiewicz, K.S.; Van Laar, A.D.; Richardson, R.M.; Ravina, B.; Kells, A.P.; Boot, B.; Martin, A.J.; Nutt, J.; Thompson, M.E.; et al. Magnetic resonance imaging guided phase 1 trial of putaminal AADC gene therapy for Parkinson’s disease. Ann. Neurol. 2019, 85, 704–714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez, R.; Garitaonandia, I.; Semechkin, A.; Kern, R. Derivation of neural stem cells from human parthenogenetic stem cells. Methods Mol. Biol. 2019, 1919, 43–57. [Google Scholar] [PubMed]
- Jankovic, J.; Okun, M.S.; Kordower, J.H. Stem cells: Scientific and ethical quandaries of a personalized approach to Parkinson’s disease. Mov. Disord. 2020, 35, 1312–1314. [Google Scholar] [CrossRef] [PubMed]
- Teil, M.; Arotcarena, M.-L.; Faggiani, E.; Laferriere, F.; Bezard, E.; Dehay, B. Targeting α-synuclein for PD therapeutics: A pursuit on all fronts. Biomolecules 2020, 10, 391. [Google Scholar] [CrossRef] [Green Version]
- Dansithong, W.; Paul, S.; Scoles, D.R.; Pulst, S.M.; Huynh, D.P. Generation of SNCA cell models using zinc finger nuclease (ZFN) technology for efficient high throughput drug screening. PLoS ONE 2015, 10, e0136930. [Google Scholar] [CrossRef]
- Kantor, B.; Tagliafierro, L.; Gu, J.; Zamora, M.E.; Ilich, E.; Grenier, C.; Huang, Z.Y.; Murphy, S.; Chiba-Falek, O. Downregulation of SNCA expression by targeted editing of DNA methylation: A potential strategy for precision therapy in PD. Mol. Ther. 2018, 26, 2638–2649. [Google Scholar] [CrossRef] [Green Version]
- Mittal, S.; Bjørnevik, K.; Im, D.S.; Flierl, A.; Dong, X.; Locascio, J.J.; Abo, K.M.; Long, E.; Jin, M.; Xu, B.; et al. B2-Adrenoreceptor is a regulator of the α-synuclein gene driving risk of Parkinson’s disease. Science 2017, 357, 891–898. [Google Scholar] [CrossRef] [Green Version]
- Gronich, N.; Abernethy, D.R.; Auriel, E.; Lavi, I.; Rennert, G.; Saliba, W. β2- adrenoceptor agonists and antagonists and risk of Parkinson’s disease. Mov. Disord. 2018, 33, 1465–1471. [Google Scholar] [CrossRef]
- Nielsen, S.S.; Gross, A.; Camacho-Soto, A.; Willis, A.W.; Racette, B.A. B2-adrenoreceptor medications and risk of Parkinson’s disease. Ann. Neurol. 2018, 84, 683–693. [Google Scholar] [CrossRef]
- Magistrelli, L.; Comi, C. Beta2-adrenoceptor agonists in Parkinson’s disease and other synucleinopathies. J. Neuroimmune. Pharmacol. 2020, 15, 74–81. [Google Scholar] [CrossRef]
- Hayashita-Kinoh, H.; Yamada, M.; Yokota, T.; Mizuno, Y.; Mochizuki, H. Down-regulation of α-synuclein expression can rescue dopaminergic cells from cell death in the substantia nigra of Parkinson’s disease rat model. Biochem. Biophys. Res. Commun. 2006, 341, 1088–1095. [Google Scholar] [CrossRef]
- Doxakis, E. Post-transcriptional regulation of α-synuclein expression by mir-7 and mir-153. J. Biol. Chem. 2010, 285, 12726–12734. [Google Scholar] [CrossRef] [Green Version]
- Zharikov, A.D.; Cannon, J.R.; Tapias, V.; Bai, Q.; Horowitz, M.P.; Shah, V.; El Ayadi, A.; Hastings, T.G.; Greenamyre, J.T.; Burton, E.A. shRNA targeting α-synuclein prevents neurodegeneration in a Parkinson’s disease model. J. Clin. Investig. 2015, 125, 2721–2735. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, M.; Suzuki, M.; Fukuoka, M.; Fujikake, N.; Watanabe, S.; Murata, M.; Wada, K.; Nagai, Y.; Hohjoh, H. Normalization of overexpressed α-synuclein causing Parkinson’s disease by a moderate gene silencing with RNA interference. Mol. Ther. Nucleic Acids. 2015, 4, e241. [Google Scholar] [CrossRef]
- Alarócn-Arís, D.; Recasens, A.; Galofre, M.; Carballo-Carbajal, I.; Zacchi, N.; Ruiz-Bronchal, E.; Pavia-Collado, R.; Chica, R.; Ferres-Coy, A.; Santos, M.; et al. Selective α-synuclein knockdown in monoamine neurons by intranasal oligonucleotide delivery: Potential therapy for Parkinson’s disease. Mol. Ther. 2018, 26, 550–567. [Google Scholar] [CrossRef] [Green Version]
- Di Fusco, D.; Dinallo, V.; Marafini, I.; Figliuzzi, M.M.; Romano, B.; Monteleone, G. Antisense oligonucleotide: Basic concepts and therapeutic application in inflammatory bowel disease. Front. Pharmacol. 2019, 10, 305. [Google Scholar] [CrossRef] [Green Version]
- Dhuri, K.; Bechtold, C.; Quijano, E.; Pham, H.; Gupta, A.; Vikram, A.; Bahal, R. Antisense oligonucleotides: An emerging area in drug discovery and development. J. Clin. Med. 2020, 9, 2004. [Google Scholar] [CrossRef]
- Cole, T.A.; Zhao, H.; Collier, T.J.; Sandoval, I.; Sortwell, C.E.; Steece-Collier, K.; Daley, B.F.; Booms, A.; Lipton, J.; Welch, M.; et al. α-Synuclein antisense oligonucleotides as a disease-modifying therapy for Parkinson’s disease. JCI Insight 2021, 6, e135633. [Google Scholar] [CrossRef]
- Gorbatyuk, O.S.; Li, S.; Nash, K.; Gorbatyuk, M.; Lewin, A.S.; Sullivan, L.F.; Mandel, R.J.; Chen, W.; Meyers, C.; Manfredsson, F.P.; et al. In vivo RNAi-mediated alpha-synuclein silencing induces nigrostriatal degeneration. Mol. Ther. 2010, 18, 1450–1457. [Google Scholar] [CrossRef]
- Khodr, C.E.; Becerra, A.; Han, Y.; Bohn, M.C. Targeting alpha-synuclein with a microRNA-embedded silencing vector in the rat substantia nigra: Positive and negative effects. Brain Res. 2014, 1550, 47–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benskey, M.J.; Sellnow, R.C.; Sandoval, I.M.; Sortwell, C.E.; Lipton, J.W.; Manfredsson, F.P. Silencing alpha-synuclein in mature nigral neurons results in rapid neuroinflammation and subsequent toxicity. Front. Mol. Neurosci. 2018, 11, 36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Yang, Y.; Hong, W.; Huang, M.; Wu, M.; Zhao, X. Applications of genome editing technology in the targeted therapy of human diseases: Mechanisms, advances and prospects. Signal. Transduct. Target. Ther. 2020, 5, 1. [Google Scholar] [CrossRef] [PubMed]
- Reinhardt, P.; Schmid, B.; Burbulla, L.F.; Schöndorf, D.C.; Wagner, L.; Glatza, M.; Höing, S.; Hargus, G.; Heck, S.A.; Dhingra, A.; et al. Genetic correction of a LRRK2 mutation in human iPSCs links parkinsonian neurodegeneration to ERK-dependent changes in gene expression. Cell Stem Cell 2013, 12, 354–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Disney, M.D.; Suresh, B.M.; Benhamou, R.I.; Childs-Disney, J.L. Progress toward the development of the small molecule equivalent of small interfering RNA. Curr. Opin. Chem. Biol. 2020, 56, 63–71. [Google Scholar] [CrossRef]
- Meyer, S.M.; Williams, C.C.; Akahori, Y.; Tanaka, T.; Aikawa, H.; Tong, Y.; Childs-Disney, J.L.; Disney, M.D. Small molecule recognition of disease-relevant RNA structures. Chem. Soc. Rev. 2020, 49, 7167–7199. [Google Scholar] [CrossRef]
- Zhou, Z.D.; Tan, E.-K. Iron regulatory protein (IRP)-iron responsive element (IRE) signaling pathway in human neurodegenerative diseases. Mol. Neurodegener. 2017, 12, 75. [Google Scholar] [CrossRef]
- Mikkilineni, S.; Cantuti-Castelvetri, I.; Cahill, C.M.; Balliedier, A.; Greig, N.H.; Rogers, J.T. The anticholinesterase phenserine and its enantiomer posiphen as 5’untranslated-region-directed translation blockers of the Parkinson’s alpha synuclein expression. Parkinsons Dis. 2012, 2012, 142372. [Google Scholar] [CrossRef] [Green Version]
- Lahiri, D.K.; Chen, D.; Maloney, B.; Holloway, H.W.; Yu, Q.-S.; Utsuki, T.; Giordano, T.; Sambamurti, K.; Greig, N.H. The experimental Alzheimer’s disease drug posiphen [(1)-phenserine] lowers amyloid-β peptide levels in cell culture and mice. J. Pharmacol. Exp. Ther. 2007, 320, 386–396. [Google Scholar] [CrossRef] [Green Version]
- Pujols, J.; Peña-Díaz, S.; Lázaro, D.F.; Peccati, F.; Pinheiro, F.; González, D.; Carija, A.; Navarro, S.; Conde-Giménez, M.; García, J.; et al. Small molecule inhibits alpha-synuclein aggregation, disrupts amyloid fibrils, and prevents degeneration of dopaminergic neurons. Proc. Natl. Acad. Sci. USA 2018, 115, 10481–10486. [Google Scholar] [CrossRef] [Green Version]
- Sardi, S.P.; Cedarbaum, J.M.; Brundin, P. Targeted therapies for Parkinson’s disease: From genetics to the clinic. Mov. Disord. 2018, 33, 684–696. [Google Scholar] [CrossRef] [Green Version]
- Shihabuddin, L.S.; Brundin, P.; Greenamyre, J.T.; Stephenson, D.; Sardi, S.P. New frontiers in Parkinson’s disease: From genetics to the clinic. J. Neurosci. 2018, 38, 9375–9382. [Google Scholar] [CrossRef] [Green Version]
- Caruana, M.; Hogen, T.; Levin, J.; Hillmer, A.; Giese, A.; Vassallo, N. Inhibition and disaggregation of alpha-synuclein oligomers by natural polyphenolic compounds. FEBS Lett. 2011, 585, 1113–1120. [Google Scholar] [CrossRef] [Green Version]
- Toni, M.; Massimino, M.L.; De Mario, A.; Angiulli, E.; Spisni, E. Metal dyshomeostasis and their pathological role in prion and prion-like diseases: The basis for a nutritional approach. Front. Neurosci. 2017, 11, 3. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, B.; Lapidus, L.J. Curcumin prevents aggregation in α-synuclein by increasing reconfiguration rate. J. Biol. Chem. 2012, 287, 9193–9199. [Google Scholar] [CrossRef] [Green Version]
- Ahsan, N.; Mishra, S.; Jain, M.K.; Surolia, A.; Gupta, S. Curcumin pyrazole and its derivative (N-(3-nitrophenylpyrazole) curcumin inhibit aggregation, disrupt fibrils and modulate toxicity of wild type and mutant α-synuclein. Sci. Rep. 2015, 5, 9862. [Google Scholar] [CrossRef] [Green Version]
- Macedo, D.; Tavares, L.; McDougall, G.J.; Vicente Miranda, H.; Stewart, D.; Ferreira, R.B.; Santos, C.N. (Poly)phenols protect from α-synuclein toxicity by reducing oxidative stress and promoting autophagy. Hum. Mol. Genet. 2015, 24, 1717–1732. [Google Scholar] [CrossRef] [Green Version]
- Freyssin, A.; Page, G.; Fauconneau, B.; RiouxBilan, A. Natural polyphenols effects on protein aggregates in Alzheimer’s and Parkinson’s prion-like diseases. Neural Regen. Res. 2018, 13, 955–961. [Google Scholar]
- Masuda, M.; Suzuki, N.; Taniguchi, S.; Oikawa, T.; Nonaka, T.; Iwatsubo, T.; Hasegawa, M. Small molecule inhibitors of α-synuclein filament assembly. Biochemistry 2006, 45, 6085–6094. [Google Scholar] [CrossRef]
- Dominguez-Meijide, A.; Vasili, E.; Konig, A.; Cima-Omori, M.S.; Ibanez de Opakua, A.; Leonov, A.; Ryazanov, S.; Zweckstetter, M.; Griesinger, C.; Outeiro, T.F. Effects of pharmacological modulators of alpha-synuclein and tau aggregation and internalization. Sci. Rep. 2020, 10, 1282. [Google Scholar] [CrossRef]
- Meng, X.; Munishkina, L.A.; Fink, A.L.; Uversky, V.N. Molecular mechanisms underlying the flavonoid-induced inhibition of alpha-synuclein fibrillation. Biochemistry 2009, 48, 8206–8224. [Google Scholar] [CrossRef]
- Ono, K.; Tsuji, M.; Yamasaki, T.R.; Pasinetti, G.M. Anti-aggregation effects of phenolic compounds on α-synuclein. Molecules 2020, 25, 2444. [Google Scholar] [CrossRef]
- Kazakova, O.; Giniyatullina, G.; Babkov, D.; Wimmer, Z. From marine metabolites to the drugs of the future: Squalamine, trodusquemine, their steroid and triterpene analogues. Int. J. Mol. Sci. 2022, 23, 1075. [Google Scholar] [CrossRef]
- Limbocker, R.; Staats, R.; Chia, S.; Ruggeri, F.S.; Mannini, B.; Xu, C.K.; Perni, M.; Cascella, R.; Bigi, A.; Sasser, L.R.; et al. Squalamine and its derivatives modulate the aggregation of amyloid-β and α-synuclein and suppress the toxicity of their oligomers. Front. Neurosci. 2021, 15, 680026. [Google Scholar] [CrossRef]
- Perni, M.; Galvagnion, C.; Maltsev, A.; Meisl, G.; Muller, M.B.; Challa, P.K.; Kirkegaard, J.B.; Flagmeier, P.; Cohen, S.I.; Cascella, R.; et al. A natural product inhibits the initiation of alpha-synuclein aggregation and suppresses its toxicity. Proc. Natl. Acad. Sci. USA 2017, 114, E1009–E1017. [Google Scholar] [CrossRef] [Green Version]
- Perni, M.; Flagmeier, P.; Limbocker, R.; Cascella, R.; Aprile, F.A.; Galvagnion, C.; Heller, G.T.; Meisl, G.; Chen, S.W.; Kumita, J.R.; et al. Multistep Inhibition of alpha-synuclein aggregation and toxicity in vitro and in vivo by trodusquemine. ACS Chem. Biol. 2018, 13, 2308–2319. [Google Scholar] [CrossRef]
- Moree, B.; Yin, G.; Lázaro, D.F.; Munari, F.; Strohäker, T.; Giller, K.; Becker, S.; Outeiro, T.F.; Zweckstetter, M.; Salafsky, J. Small molecules detected by second-harmonic generation modulate the conformation of monomeric α-synuclein and reduce its aggregation in cells. J. Biol. Chem. 2015, 290, 27582–27593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandez, C.O.; Hoyer, W.; Zweckstetter, M.; Jares-Erijman, E.A.; Subramaniam, V.; Griesinger, C.; Jovin, T.M. NMR of alpha-synuclein-polyamine complexes elucidates the mechanism and kinetics of induced aggregation. EMBO J. 2004, 23, 2039–2046. [Google Scholar] [CrossRef] [PubMed]
- Wagner, J.; Ryazanov, S.; Leonov, A.; Levin, J.; Shi, S.; Schmidt, F.; Prix, C.; Pan-Montojo, F.; Bertsch, U.; Mitteregger-Kretzschmar, G.; et al. Anle138b: A novel oligomer modulator for disease-modifying therapy of neurodegenerative diseases such as prion and Parkinson’s disease. Acta Neuropathol. 2013, 125, 795–813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levin, J.; Schmidt, F.; Boehm, C.; Prix, C.; Botzel, K.; Ryazanov, S.; Leonov, A.; Griesinger, C.; Giese, A. The oligomer modulator Anle138b inhibits disease progression in a Parkinson mouse model even with treatment started after disease onset. Acta Neuropathol. 2014, 127, 779–780. [Google Scholar] [CrossRef] [Green Version]
- Pujols, J.; Peña-Díaz, S.; Conde-Giménez, M.; Pinheiro, F.; Navarro, S.; Sancho, J.; Ventura, S. High-throughput screening methodology to identify alpha-synuclein aggregation inhibitors. Int. J. Mol. Sci. 2017, 18, 478. [Google Scholar] [CrossRef] [Green Version]
- Peña-Díaz, S.; Pujols, J.; Vasili, E.; Pinheiro, F.; Santos, J.; Manglano-Artuñedo, Z.; Outeiro, T.F.; Ventura, S. The small aromatic compound SynuClean-D inhibits the aggregation and seeded polymerization of multiple α-synuclein strains. J. Biol. Chem. 2022, 298, 101902. [Google Scholar] [CrossRef]
- Schrader, T.; Bitan, G.; Klärner, F.G. Molecular tweezers for lysine and arginine–powerful inhibitors of pathologic protein aggregation. Chem. Commun. 2016, 52, 11318–11334. [Google Scholar] [CrossRef] [Green Version]
- Attar, A.; Chan, W.T.; Klarner, F.G.; Schrader, T.; Bitan, G. Safety and pharmacological characterization of the molecular tweezer CLR01—A broad-spectrum inhibitor of amyloid proteins’ toxicity. BMC Pharm. Toxicol. 2014, 15, 23. [Google Scholar] [CrossRef] [Green Version]
- Hadrovic, I.; Rebmann, P.; Klarner, F.G.; Bitan, G.; Schrader, T. Molecular lysine tweezers counteract aberrant protein aggregation. Front. Chem. 2019, 7, 657. [Google Scholar] [CrossRef]
- Prabhudesai, S.; Sinha, S.; Attar, A.; Kotagiri, A.; Fitzmaurice, A.G.; Lakshmanan, R.; Ivanova, M.I.; Loo, J.A.; Klarner, F.G.; Schrader, T.; et al. A novel “molecular tweezer” inhibitor of alpha-synuclein neurotoxicity in vitro and in vivo. Neurotherapeutics 2012, 9, 464–476. [Google Scholar] [CrossRef] [Green Version]
- Kurnik, M.; Sahin, C.; Andersen, C.B.; Lorenzen, N.; Giehm, L.; Mohammad-Beigi, H.; Jessen, C.M.; Pedersen, J.S.; Christiansen, G.; Petersen, S.V.; et al. Potent alpha-synuclein aggregation inhibitors, identified by high-throughput screening, mainly target the monomeric state. Cell Chem. Biol. 2018, 25, 1389–1402. [Google Scholar] [CrossRef]
- Uversky, V.N. Intrinsically disordered proteins and their (disordered) proteomes in neurodegenerative disorders. Front. Aging Neurosci. 2015, 7, 18. [Google Scholar] [CrossRef] [Green Version]
- Schweighauser, M.; Shi, Y.; Tarutani, A.; Kametani, F.; Murzin, A.G.; Ghetti, B.; Matsubara, T.; Tomita, T.; Ando, T.; Hasegawa, K.; et al. Structures of alpha-synuclein filaments from multiple system atrophy. Nature 2020, 585, 464–469. [Google Scholar] [CrossRef]
- Guerrero-Ferreira, R.; Taylor, N.M.; Arteni, A.A.; Kumari, P.; Mona, D.; Ringler, P.; Britschgi, M.; Lauer, M.E.; Makky, A.; Verasdonck, J.; et al. Two new polymorphic structures of human full-length alpha-synuclein fibrils solved by cryo-electron microscopy. eLife 2019, 8, e48907. [Google Scholar] [CrossRef]
- Shahnawaz, M.; Mukherjee, A.; Pritzkow, S.; Mendez, N.; Rabadia, P.; Liu, X.; Hu, B.; Schmeichel, A.; Singer, W.; Wu, G.; et al. Discriminating alpha-synuclein strains in Parkinson’s disease and multiple system atrophy. Nature 2020, 578, 273–277. [Google Scholar] [CrossRef]
- Ryan, P.; Patel, B.; Makwana, V.; Jadhav, H.R.; Kiefel, M.; Davey, A.; Reekie, T.A.; Rudrawar, S.; Kassiou, M. Peptides, peptidomimetics, and carbohydrate–peptide conjugates as amyloidogenic aggregation inhibitors for Alzheimer’s disease. ACS Chem. Neurosci. 2018, 9, 1530–1551. [Google Scholar] [CrossRef]
- Azzarito, V.; Long, K.; Murphy, N.S.; Wilson, A.J. Inhibition of α-helix-mediated protein-protein interactions using designed molecules. Nat. Chem. 2013, 5, 161–173. [Google Scholar] [CrossRef]
- Sonti, R.; Gopi, H.N.; Muddegowda, U.; Ragothama, S.; Balaram, P. A designed three-stranded β-sheet in an α/β hybrid peptide. Chemistry 2013, 19, 5955–5965. [Google Scholar] [CrossRef]
- Wrasidlo, W.; Tsigelny, I.F.; Price, D.L.; Dutta, G.; Rockenstein, E.; Schwarz, T.C.; Ledolter, K.; Bonhaus, D.; Paulino, A.; Eleuteri, S.; et al. A de novo compound targeting α-synuclein improves deficits in models of Parkinson’s disease. Brain 2016, 139, 3217–3236. [Google Scholar] [CrossRef] [Green Version]
- Price, D.L.; Koike, M.A.; Khan, A.; Wrasidlo, W.; Rockenstein, E.; Masliah, E.; Bonhaus, D. The small molecule alpha-synuclein misfolding inhibitor, NPT200-11, produces multiple benefits in an animal model of Parkinson’s disease. Sci. Rep. 2018, 8, 16165. [Google Scholar] [CrossRef] [Green Version]
- Levenson, J.M.; Schroeter, S.; Carroll, J.C.; Cullen, V.; Asp, E.; Proschitsky, M.; Gannon, K.S. NPT088 reduces both amyloid-β and tau pathologies in transgenic mice. Alzheimers Dement. TRCI 2016, 2, 141–155. [Google Scholar] [CrossRef] [Green Version]
- Gordon, D.J.; Tappe, R.; Meredith, S.C. Design and characterization of a membrane permeable N-methyl amino acid-containing peptide that inhibits Abeta1-40 fibrillogenesis. J. Pept. Res. 2002, 60, 34–55. [Google Scholar]
- Adessi, C.; Frossard, M.-J.; Boissard, C.; Fraga, S.; Bieler, S.; Ruckle, T.; Vilbois, F.; Robinson, S.M.; Mutter, M.; Banks, W.A.; et al. Pharmacological profiles of peptide drug candidates for the treatment of Alzheimer’s disease. J. Biol. Chem. 2003, 278, 13905–13911. [Google Scholar] [CrossRef] [Green Version]
- Stott, K. Peptides Containing N-substituted L-amino Acids for Preventing Beta-Strand Association. 2001. Available online: https://patents.google.com/patent/WO2001007473A1/en (accessed on 14 April 2022).
- Ruzza, P.; Gazziero, M.; De Marchi, M.; Massalongo, G.; Marchiani, A.; Autiero, I.; Tessari, I.; Bubacco, L.; Calderan, A.P. Peptides as modulators of α-synuclein aggregation. Protein Pept. Lett. 2015, 22, 354–361. [Google Scholar] [CrossRef]
- Szegő, E.M.; Boß, F.; Komnig, D.; Gärtner, C.; Höfs, L.; Shaykhalishahi, H.; Wördehoff, M.M.; Saridaki, T.; Schulz, J.B.; Hoyer, W.; et al. A β-Wrapin targeting the N-terminus of α-synuclein monomers reduces fibril-induced aggregation in neurons. Front. Neurosci. 2021, 15, 696440. [Google Scholar] [CrossRef]
- Rodriguez, J.A.; Ivanova, M.I.; Sawaya, M.R.; Cascio, D.; Reyes, F.E.; Shi, D.; Sangwan, S.; Guenther, E.L.; Johnson, L.M.; Zhang, M.; et al. Structure of the toxic core of α-synuclein from invisible crystals. Nature 2015, 525, 486–490. [Google Scholar] [CrossRef] [Green Version]
- Sangwan, S.; Sahay, S.; Murray, K.A.; Morgan, S.; Guenther, E.L.; Jiang, L.; Williams, C.K.; Vinters, H.V.; Goedert, M.; Eisenberg, D.S. Inhibition of synucleinopathic seeding by rationally designed inhibitors. eLife 2020, 9, e46775. [Google Scholar] [CrossRef]
- Giacomelli, C.; Daniele, S.; Martini, C. Potential biomarkers and novel pharmacological targets in protein aggregation-related neurodegenerative diseases. Biochem. Pharmacol. 2017, 131, 1–15. [Google Scholar] [CrossRef]
- Lie, P.P.; Nixon, R.A. Lysosome trafficking and signaling in health and neurodegenerative diseases. Neurobiol. Dis. 2019, 122, 94–105. [Google Scholar] [CrossRef]
- Bonam, S.R.; Wang, F.; Muller, S. Lysosomes as a therapeutic target. Nat. Rev. Drug Discove. 2019, 18, 923–948. [Google Scholar] [CrossRef] [Green Version]
- Assêncio, F.R. Alpha-synuclein as therapeutic target in Parkinson’s disease. Neuroforum 2019, 25, 129–136. [Google Scholar] [CrossRef]
- Fakhree, M.A.A.; Konings, I.B.M.; Kole, J.; Cambi, A.; Blum, C.; Claessens, M.M.A.E. The localization of alpha-synuclein in the endocytic pathway. Neuroscience 2021, 457, 186–195. [Google Scholar] [CrossRef] [PubMed]
- Cuervo, A.M.; Dice, J.F.; Fredenburg, R.; Lansbury, P.T.; Sulzer, D. Impaired degradation of mutant α-synuclein by chaperone-mediated autophagy. Science 2004, 305, 1292–1295. [Google Scholar] [CrossRef] [PubMed]
- Mazzulli, J.R.; Zunke, F.; Isacson, O.; Studer, L.; Krainc, D. α-synuclein-induced lysosomal dysfunction occurs through disruptions in protein trafficking in human midbrain synucleinopathy models. Proc. Natl. Acad. Sci. USA 2016, 113, 1931–1936. [Google Scholar] [CrossRef] [Green Version]
- Scheidt, T.; Łapińska, U.; Kumita, J.R.; Whiten, D.R.; Klenerman, D.; Wilson, M.R.; Chen, S.A.I.; Linse, S.; Vendruscolo, M.; Dopson, C.M.; et al. Secondary nucleation and elongation occur at different sites on Alzheimer’s amyloid—Aggregates. Sci. Adv. 2019, 5, eaau3112. [Google Scholar] [CrossRef] [Green Version]
- Shin, Y.; Klucken, J.; Patterson, C.; Hyman, B.T.; McLean, P.J. The co-chaperone carboxyl terminus of Hsp70-interacting protein (CHIP) mediates alpha-synuclein degradation decisions between proteasomal and lysosomal pathways. J. Biol. Chem. 2005, 280, 23727–23734. [Google Scholar] [CrossRef] [Green Version]
- Luk, K.C.; Mills, I.P.; Trojanowski, J.Q.; Lee, V.M. Interactions between Hsp70 and the hydrophobic core of alpha-synuclein inhibit fibril assembly. Biochemistry 2008, 47, 12614–12625. [Google Scholar] [CrossRef] [Green Version]
- Danzer, K.M.; Ruf, W.P.; Putcha, P.; Joyner, D.; Hashimoto, T.; Glabe, C.; Hyman, B.T.; McLean, P.J. Heat-shock protein 70 modulates toxic extracellular alpha-synuclein oligomers and rescues trans-synaptic toxicity. FASEB J. 2011, 25, 326–336. [Google Scholar] [CrossRef] [Green Version]
- Kalia, L.V.; Kalia, S.K.; Chau, H.; Lozano, A.M.; Hyman, B.T.; McLean, P.J. Ubiquitinylation of alpha-synuclein by carboxyl terminus Hsp70-interacting protein (CHIP) is regulated by Bcl-2-associated athanogene 5 (BAG5). PLoS ONE 2011, 6, e14695. [Google Scholar] [CrossRef]
- Hu, S.; Tan, J.; Qin, L.; Lv, L.; Yan, W.; Zhang, H.; Tang, B.S.; Wang, C. Molecular chaperones and Parkinson’s disease. Neurobiol. Dis. 2021, 160, 105527. [Google Scholar] [CrossRef]
- Klucken, J.; Shin, Y.; Masliah, E.; Hyman, B.; Mclean, P. Hsp70 reduces alpha-synuclein aggregation and toxicity. J. Biol. Chem. 2004, 279, 25497–25502. [Google Scholar] [CrossRef] [Green Version]
- Jia, C.; Ma, X.; Liu, Z.; Gu, J.; Zhang, X.; Li, D.; Zhang, S. Different heat shock proteins bind α-synuclein with distinct mechanisms and synergistically prevent its amyloid aggregation. Front. Neurosci. 2019, 13, 1124. [Google Scholar] [CrossRef] [Green Version]
- Jones, D.R.; Moussaud, S.; McLean, P. Targeting heat shock proteins to modulate α-synuclein toxicity. Ther. Adv. Neurol. Disor. 2014, 7, 33–51. [Google Scholar] [CrossRef] [Green Version]
- Cullen, V.; Lindfors, M.; Ng, J.; Paetau, A.; Swinton, E.; Kolodziej, P.; Boston, H.; Saftig, P.; Woulfe, J.; Feany, M.B.; et al. Cathepsin D expression level affects alpha-synuclein processing, aggregation, and toxicity in vivo. Mol. Brain. 2009, 2, 5. [Google Scholar] [CrossRef] [Green Version]
- Colacurcio, D.J.; Nixon, R.A. Disorders of lysosomal acidification—The emerging role of v-ATPase in aging and neurodegenerative disease. Ageing Res. Rev. 2016, 32, 75–88. [Google Scholar] [CrossRef] [Green Version]
- Stefanis, L.; Emmanouilidou, E.; Pantazopoulou, M.; Kirik, D.; Vekrellis, K.; Tofaris, G.K. How is alpha-synuclein cleared from the cell? J. Neurochem. 2019, 150, 577–590. [Google Scholar] [CrossRef]
- Masliah, E.; Rockenstein, E.; Mante, M.; Crews, L.; Spencer, B.; Adame, A.; Patrick, C.; Trejo, M.; Ubhi, K.; Rohn, T.T.; et al. Passive immunization reduces behavioral and neuropathological deficits in an alpha-synuclein transgenic model of Lewy body disease. PLoS ONE 2011, 6, e19338. [Google Scholar] [CrossRef] [Green Version]
- Schenk, D.B.; Koller, M.; Ness, D.K.; Griffith, S.G.; Grundman, M.; Zago, W.; Soto, J.; Atiee, G.; Ostrowitzki, S.; Kinney, G.G. First-in-human assessment of PRX002, an anti-α-synuclein monoclonal antibody, in healthy volunteers. Mov. Disord. 2017, 32, 211–218. [Google Scholar] [CrossRef]
- Games, D.; Valera, E.; Spencer, B.; Rockenstein, E.; Mante, M.; Adame, A.; Patrick, C.; Ubhi, K.; Nuber, S.; Sacayon, P.; et al. Reducing C-terminal-truncated alphasynuclein by immunotherapy attenuates neurodegeneration and propagation in Parkinson’s disease-like models. J. Neurosci. 2014, 34, 9441–9454. [Google Scholar] [CrossRef]
- Weihofen, A.; Liu, Y.; Arndt, J.W.; Huy, C.; Quan, C.; Smith, B.A.; Baeriswyl, J.-L.; Cavegn, N.; Senn, L.; Su, L.; et al. Development of an aggregate-selective, human-derived α-synuclein antibody BIIB054 that ameliorates disease phenotypes in Parkinson’s disease models. Neurobiol. Dis. 2019, 124, 276–288. [Google Scholar] [CrossRef]
- Messer, A.; Butler, D.C. Optimizing intracellular antibodies (intrabodies/nanobodies) to treat neurodegenerative disorders. Neurobiol. Dis. 2019, 134, 104619. [Google Scholar] [CrossRef] [PubMed]
- Lynch, S.M.; Zhou, C.; Messer, A. An scFv intrabody against the nonamyloid component of alpha-synuclein reduces intracellular aggregation and toxicity. J. Mol. Biol. 2008, 377, 136–147. [Google Scholar] [CrossRef] [Green Version]
- El Turk, F.; De Genst, E.; Guilliams, T.; Fauvet, B.; Hejjaoui, M.; Di Trani, J.; Chiki, A.; Mittermaier, A.; Vendruscolo, M.; Lashuel, H.A.; et al. Exploring the role of post-translational modifications in regulating alpha-synuclein interactions by studying the effects of phosphorylation on nanobody binding. Protein Sci. 2018, 27, 1262–1274. [Google Scholar] [CrossRef] [Green Version]
- Nimmo, J.T.; Verma, A.; Dodart, J.-C.; Wang, C.Y.; Savistchenko, J.; Melki, R.; Carare, R.O.; Nicoll, J.A.R. Novel antibodies detect additional α-synuclein pathology in synucleinopathies: Potential development for immunotherapy. Alzheimers Res. Ther. 2020, 12, 159. [Google Scholar] [CrossRef]
- Volc, D.; Poewe, W.; Kutzelnigg, A.; Lührs, P.; Thun-Hohenstein, C.; Schneeberger, A.; Galabova, G.; Majbour, N.; Vaikath, N.; El-Agnaf, O.; et al. Safety and immunogenicity of the α-synuclein active immunotherapeutic PD01A in patients with Parkinson’s disease: A randomised, single-blinded, phase 1 trial. Lancet Neurol. 2020, 19, 591–600. [Google Scholar] [CrossRef]
- Mandler, M.; Valera, E.; Rockenstein, E.; Weninger, H.; Patrick, C.; Adame, A.; Santic, R.; Meindl, S.; Vigl, B.; Smrzka, O.; et al. Next-generation active immunization approach for synucleinopathies: Implications for Parkinson’s disease clinical trials. Acta Neuropathol. 2014, 127, 861–879. [Google Scholar] [CrossRef] [Green Version]
- Rockenstein, E.; Ostroff, G.; Dikengil, F.; Rus, F.; Mante, M.; Florio, J.; Adame, A.; Trinth, I.; Kim, C.; Overak, C. Combined active humoral and cellular immunization approaches for the treatment of synucleinopathies. J. Neurosci. 2018, 38, 1000–1014. [Google Scholar] [CrossRef]
- Mao, X.; Ou, M.T.; Karuppagounder, S.S.; Kam, T.I.; Yin, X.; Xiong, Y.; Ge, P.; Umanah, G.E.; Brahmachari, S.; Shin, J.H.; et al. Pathological α-synuclein transmission initiated by binding lymphocyte-activation gene 3. Science 2016, 353, 6307. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Yun, S.P.; Lee, S.; Umanah, G.E.; Bandaru, V.V.R.; Yin, X.; Rhee, P.; Karuppagounder, S.S.; Kwon, S.H.; Lee, H.; et al. GBA1 deficiency negatively affects physiological α-synuclein tetramers and related multimers. Proc. Natl. Acad. Sci. USA 2018, 115, 798–803. [Google Scholar] [CrossRef] [Green Version]
- Parkinson’s Disease: Challenges, Progress, and Promise. NIH Publication No.15-5595. 2016. Available online: https://www.ninds.nih.gov/Disorders/All-Disorders/Parkinsons-Disease-Challenges-Progress-and-Promise (accessed on 9 April 2022).



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vidović, M.; Rikalovic, M.G. Alpha-Synuclein Aggregation Pathway in Parkinson’s Disease: Current Status and Novel Therapeutic Approaches. Cells 2022, 11, 1732. https://doi.org/10.3390/cells11111732
Vidović M, Rikalovic MG. Alpha-Synuclein Aggregation Pathway in Parkinson’s Disease: Current Status and Novel Therapeutic Approaches. Cells. 2022; 11(11):1732. https://doi.org/10.3390/cells11111732
Chicago/Turabian StyleVidović, Marija, and Milena G. Rikalovic. 2022. "Alpha-Synuclein Aggregation Pathway in Parkinson’s Disease: Current Status and Novel Therapeutic Approaches" Cells 11, no. 11: 1732. https://doi.org/10.3390/cells11111732