
Integrative Computational Modeling of Cardiomyocyte Calcium Handling and Cardiac Arrhythmias: Current Status and Future Challenges



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Cardiomyocyte Calcium Handling: The Complex Hub of Excitation and Contraction
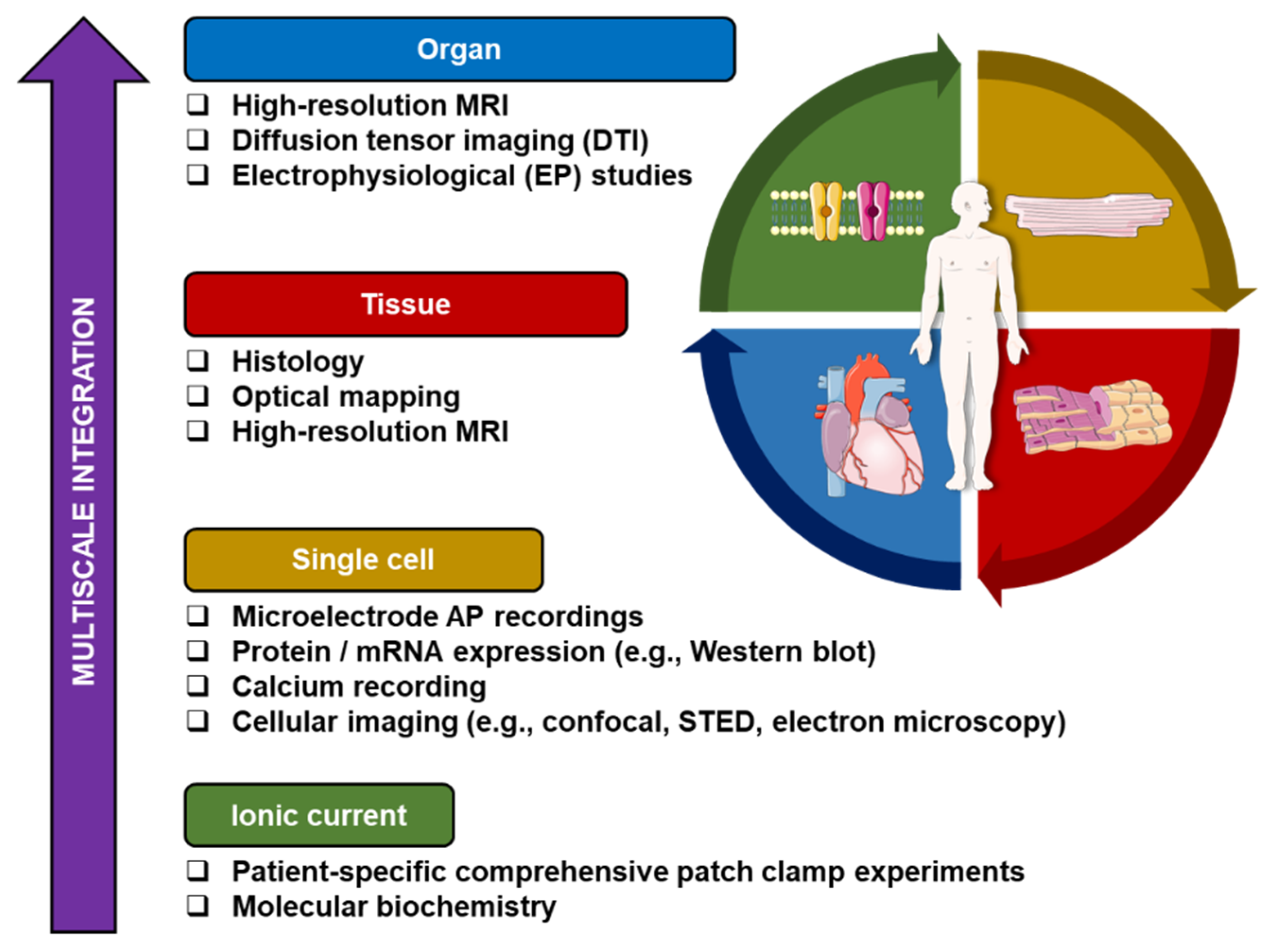
2. Integrative Experimentation: Is It Necessary?
3. Why Do We Need Integrative Computational Modeling?
4. All Models Are Wrong, but Some Are Useful
4.1. Temperature Dependence
4.2. Interspecies Diversity
4.3. Intra- and Inter-Individual Heterogeneities
4.4. Model-Dependent Effects
5. The Future Outlook of Integrative In Silico Modeling
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sutanto, H.; Lyon, A.; Lumens, J.; Schotten, U.; Dobrev, D.; Heijman, J. Cardiomyocyte calcium handling in health and disease: Insights from in vitro and in silico studies. Prog. Biophys. Mol. Biol. 2020, 157, 54–75. [Google Scholar] [CrossRef]
- Lakatta, E.G.; Maltsev, V.A.; Vinogradova, T.M. A coupled SYSTEM of intracellular Ca2+ clocks and surface membrane voltage clocks controls the timekeeping mechanism of the heart’s pacemaker. Circ. Res. 2010, 106, 659–673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lang, D.; Glukhov, A.V. Cellular and Molecular Mechanisms of Functional Hierarchy of Pacemaker Clusters in the Sinoatrial Node: New Insights into Sick Sinus Syndrome. J. Cardiovasc. Dev. Dis. 2021, 8, 43. [Google Scholar] [CrossRef] [PubMed]
- Capel, R.A.; Terrar, D.A. The importance of Ca2+-dependent mechanisms for the initiation of the heartbeat. Front. Physiol. 2015, 6, 80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venetucci, L.A.; Trafford, A.W.; O’Neill, S.C.; Eisner, D.A. The sarcoplasmic reticulum and arrhythmogenic calcium release. Cardiovasc. Res. 2008, 77, 285–292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandez-Tenorio, M.; Niggli, E. Stabilization of Ca2+ signaling in cardiac muscle by stimulation of SERCA. J. Mol. Cell. Cardiol. 2018, 119, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Lyon, A.R.; Bannister, M.L.; Collins, T.; Pearce, E.; Sepehripour, A.H.; Dubb, S.S.; Garcia, E.; O’Gara, P.; Liang, L.; Kohlbrenner, E.; et al. SERCA2a gene transfer decreases sarcoplasmic reticulum calcium leak and reduces ventricular arrhythmias in a model of chronic heart failure. Circ. Arrhythm. Electrophysiol. 2011, 4, 362–372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prunier, F.; Kawase, Y.; Gianni, D.; Scapin, C.; Danik, S.B.; Ellinor, P.T.; Hajjar, R.J.; Del Monte, F. Prevention of ventricular arrhythmias with sarcoplasmic reticulum Ca2+ ATPase pump overexpression in a porcine model of ischemia reperfusion. Circulation 2008, 118, 614–624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beavers, D.L.; Wang, W.; Ather, S.; Voigt, N.; Garbino, A.; Dixit, S.S.; Landstrom, A.P.; Li, N.; Wang, Q.L.; Olivotto, I.; et al. Mutation E169K in Junctophilin-2 Causes Atrial Fibrillation Due to Impaired RyR2 Stabilization. J. Am. Coll. Cardiol. 2013, 62, 2010–2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hove-Madsen, L.; Llach, A.; Bayes-Genis, A.; Roura, S.; Rodriguez Font, E.; Aris, A.; Cinca, J. Atrial fibrillation is associated with increased spontaneous calcium release from the sarcoplasmic reticulum in human atrial myocytes. Circulation 2004, 110, 1358–1363. [Google Scholar] [CrossRef] [Green Version]
- Molina, C.E.; Abu-Taha, I.H.; Wang, Q.; Rosello-Diez, E.; Kamler, M.; Nattel, S.; Ravens, U.; Wehrens, X.H.T.; Hove-Madsen, L.; Heijman, J.; et al. Profibrotic, Electrical, and Calcium-Handling Remodeling of the Atria in Heart Failure Patients with and without Atrial Fibrillation. Front. Physiol. 2018, 9, 1383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neef, S.; Dybkova, N.; Sossalla, S.; Ort, K.R.; Fluschnik, N.; Neumann, K.; Seipelt, R.; Schondube, F.A.; Hasenfuss, G.; Maier, L.S. CaMKII-dependent diastolic SR Ca2+ leak and elevated diastolic Ca2+ levels in right atrial myocardium of patients with atrial fibrillation. Circ. Res. 2010, 106, 1134–1144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voigt, N.; Li, N.; Wang, Q.; Wang, W.; Trafford, A.W.; Abu-Taha, I.; Sun, Q.; Wieland, T.; Ravens, U.; Nattel, S.; et al. Enhanced sarcoplasmic reticulum Ca2+ leak and increased Na+-Ca2+ exchanger function underlie delayed afterdepolarizations in patients with chronic atrial fibrillation. Circulation 2012, 125, 2059–2070. [Google Scholar] [CrossRef] [Green Version]
- Christ, T.; Rozmaritsa, N.; Engel, A.; Berk, E.; Knaut, M.; Metzner, K.; Canteras, M.; Ravens, U.; Kaumann, A. Arrhythmias, elicited by catecholamines and serotonin, vanish in human chronic atrial fibrillation. Proc. Natl. Acad. Sci. USA 2014, 111, 11193–11198. [Google Scholar] [CrossRef] [Green Version]
- Greiser, M.; Kerfant, B.G.; Williams, G.S.; Voigt, N.; Harks, E.; Dibb, K.M.; Giese, A.; Meszaros, J.; Verheule, S.; Ravens, U.; et al. Tachycardia-induced silencing of subcellular Ca2+ signaling in atrial myocytes. J. Clin. Investig. 2014, 124, 4759–4772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greiser, M.; Schotten, U. Dynamic remodeling of intracellular Ca2+ signaling during atrial fibrillation. J. Mol. Cell. Cardiol. 2013, 58, 134–142. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, A.D.; Joca, H.C.; Boyman, L.; Greiser, M. Calcium Signaling Silencing in Atrial Fibrillation: Implications for Atrial Sodium Homeostasis. Int. J. Mol. Sci. 2021, 22, 513. [Google Scholar] [CrossRef] [PubMed]
- van Gorp, P.R.R.; Trines, S.A.; Pijnappels, D.A.; de Vries, A.A.F. Multicellular In vitro Models of Cardiac Arrhythmias: Focus on Atrial Fibrillation. Front. Cardiovasc. Med. 2020, 7, 43. [Google Scholar] [CrossRef] [PubMed]
- Harlaar, N.; Dekker, S.O.; Zhang, J.; Snabel, R.R.; Veldkamp, M.W.; Verkerk, A.O.; Fabres, C.C.; Schwach, V.; Lerink, L.J.S.; Rivaud, M.R.; et al. Conditional immortalization of human atrial myocytes for the generation of in vitro models of atrial fibrillation. Nat. Biomed. Eng. 2022, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Libbus, I.; Wan, X.; Rosenbaum, D.S. Electrotonic load triggers remodeling of repolarizing current Ito in ventricle. Am. J. Physiol. Heart Circ. Physiol. 2004, 286, H1901–H1909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Decker, K.F.; Heijman, J.; Silva, J.R.; Hund, T.J.; Rudy, Y. Properties and ionic mechanisms of action potential adaptation, restitution, and accommodation in canine epicardium. Am. J. Physiol. Heart Circ. Physiol. 2009, 296, H1017–H1026. [Google Scholar] [CrossRef] [Green Version]
- Salama, G.; Hwang, S.M. Simultaneous optical mapping of intracellular free calcium and action potentials from Langendorff perfused hearts. Curr. Protoc. Cytom. 2009, 49, 12–17. [Google Scholar] [CrossRef] [Green Version]
- Jaimes, R., 3rd; McCullough, D.; Siegel, B.; Swift, L.; Hiebert, J.; McLnerney, D.; Posnack, N.G. Lights, camera, path splitter: A new approach for truly simultaneous dual optical mapping of the heart with a single camera. BMC Biomed. Eng. 2019, 1, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Lee, P.; Calvo, C.J.; Alfonso-Almazan, J.M.; Quintanilla, J.G.; Chorro, F.J.; Yan, P.; Loew, L.M.; Filgueiras-Rama, D.; Millet, J. Low-Cost Optical Mapping Systems for Panoramic Imaging of Complex Arrhythmias and Drug-Action in Translational Heart Models. Sci. Rep. 2017, 7, 43217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, J.S.; Small, D.M.; Nishimura, N. In Vivo Calcium Imaging of Cardiomyocytes in the Beating Mouse Heart with Multiphoton Microscopy. Front. Physiol. 2018, 9, 969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, P.; Taghavi, F.; Yan, P.; Ewart, P.; Ashley, E.A.; Loew, L.M.; Kohl, P.; Bollensdorff, C.; Woods, C.E. In situ optical mapping of voltage and calcium in the heart. PLoS ONE 2012, 7, e42562. [Google Scholar] [CrossRef]
- Kim, J.; Park, S.; Lee, C.; Kim, J.Y.; Kim, C. Organic Nanostructures for Photoacoustic Imaging. Chemnanomat 2016, 2, 156–166. [Google Scholar] [CrossRef]
- Heijman, J.; Guichard, J.B.; Dobrev, D.; Nattel, S. Translational Challenges in Atrial Fibrillation. Circ. Res. 2018, 122, 752–773. [Google Scholar] [CrossRef] [PubMed]
- Mirams, G.R.; Arthurs, C.J.; Bernabeu, M.O.; Bordas, R.; Cooper, J.; Corrias, A.; Davit, Y.; Dunn, S.J.; Fletcher, A.G.; Harvey, D.G.; et al. Chaste: An open source C++ library for computational physiology and biology. PLoS Comput. Biol. 2013, 9, e1002970. [Google Scholar] [CrossRef] [Green Version]
- Plank, G.; Loewe, A.; Neic, A.; Augustin, C.; Huang, Y.L.; Gsell, M.A.F.; Karabelas, E.; Nothstein, M.; Prassl, A.J.; Sanchez, J.; et al. The openCARP simulation environment for cardiac electrophysiology. Comput. Methods Progr. Biomed. 2021, 208, 106223. [Google Scholar] [CrossRef]
- Clerx, M.; Collins, P.; de Lange, E.; Volders, P.G. Myokit: A simple interface to cardiac cellular electrophysiology. Prog. Biophys. Mol. Biol. 2016, 120, 100–114. [Google Scholar] [CrossRef]
- Sutanto, H.; van Sloun, B.; Schonleitner, P.; van Zandvoort, M.; Antoons, G.; Heijman, J. The Subcellular Distribution of Ryanodine Receptors and L-Type Ca2+ Channels Modulates Ca2+-Transient Properties and Spontaneous Ca2+-Release Events in Atrial Cardiomyocytes. Front. Physiol. 2018, 9, 1108. [Google Scholar] [CrossRef] [PubMed]
- Heijman, J.; Muna, A.P.; Veleva, T.; Molina, C.E.; Sutanto, H.; Tekook, M.A.; Wang, Q.; Abu-Taha, I.; Gorka, M.; Kunzel, S.; et al. Atrial Myocyte NLRP3/CaMKII Nexus Forms a Substrate for Post-Operative Atrial Fibrillation. Circ. Res. 2020, 127, 1036–1055. [Google Scholar] [CrossRef]
- Amuzescu, B.; Airini, R.; Epureanu, F.B.; Mann, S.A.; Knott, T.; Radu, B.M. Evolution of mathematical models of cardiomyocyte electrophysiology. Math. Biosci. 2021, 334, 108567. [Google Scholar] [CrossRef]
- Clancy, C.E.; Zhu, Z.I.; Rudy, Y. Pharmacogenetics and anti-arrhythmic drug therapy: A theoretical investigation. Am. J. Physiol. Heart Circ. Physiol. 2007, 292, H66–H75. [Google Scholar] [CrossRef] [PubMed]
- Dutta, S.; Chang, K.C.; Beattie, K.A.; Sheng, J.; Tran, P.N.; Wu, W.W.; Wu, M.; Strauss, D.G.; Colatsky, T.; Li, Z. Optimization of an In silico Cardiac Cell Model for Proarrhythmia Risk Assessment. Front. Physiol. 2017, 8, 616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaeger, K.H.; Wall, S.; Tveito, A. Computational prediction of drug response in short QT syndrome type 1 based on measurements of compound effect in stem cell-derived cardiomyocytes. PLoS Comput. Biol. 2021, 17, e1008089. [Google Scholar] [CrossRef]
- Sutanto, H.; Laudy, L.; Clerx, M.; Dobrev, D.; Crijns, H.; Heijman, J. Maastricht antiarrhythmic drug evaluator (MANTA): A computational tool for better understanding of antiarrhythmic drugs. Pharmacol. Res. 2019, 148, 104444. [Google Scholar] [CrossRef]
- Sutanto, H.; Heijman, J. Beta-adrenergic receptor stimulation modulates the cellular proarrhythmic effects of chloroquine and azithromycin. Front. Physiol. 2020, 11, 587709. [Google Scholar] [CrossRef]
- Benson, A.P.; Stevenson-Cocks, H.J.; Whittaker, D.G.; White, E.; Colman, M.A. Multi-scale approaches for the simulation of cardiac electrophysiology: II—Tissue-level structure and function. Methods 2021, 185, 60–81. [Google Scholar] [CrossRef] [PubMed]
- Sutanto, H.; Cluitmans, M.J.M.; Dobrev, D.; Volders, P.G.A.; Bebarova, M.; Heijman, J. Acute effects of alcohol on cardiac electrophysiology and arrhythmogenesis: Insights from multiscale in silico analyses. J. Mol. Cell. Cardiol. 2020, 146, 69–83. [Google Scholar] [CrossRef] [PubMed]
- Mackiewicz, U.; Lewartowski, B. Temperature dependent contribution of Ca2+ transporters to relaxation in cardiac myocytes: Important role of sarcolemmal Ca2+-ATPase. J. Physiol. Pharmacol. 2006, 57, 3–15. [Google Scholar]
- Fu, Y.; Zhang, G.Q.; Hao, X.M.; Wu, C.H.; Chai, Z.; Wang, S.Q. Temperature dependence and thermodynamic properties of Ca2+ sparks in rat cardiomyocytes. Biophys. J. 2005, 89, 2533–2541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Severi, S.; Corsi, C.; Cerbai, E. From in vivo plasma composition to in vitro cardiac electrophysiology and in silico virtual heart: The extracellular calcium enigma. Philos. Trans. A Math. Phys. Eng. Sci. 2009, 367, 2203–2223. [Google Scholar] [CrossRef] [PubMed]
- O’Hara, T.; Rudy, Y. Quantitative comparison of cardiac ventricular myocyte electrophysiology and response to drugs in human and nonhuman species. Am. J. Physiol. Heart Circ. Physiol. 2012, 302, H1023–H1030. [Google Scholar] [CrossRef] [Green Version]
- Gaur, N.; Qi, X.Y.; Benoist, D.; Bernus, O.; Coronel, R.; Nattel, S.; Vigmond, E.J. A computational model of pig ventricular cardiomyocyte electrophysiology and calcium handling: Translation from pig to human electrophysiology. PLoS Comput. Biol. 2021, 17, e1009137. [Google Scholar] [CrossRef]
- Clauss, S.; Bleyer, C.; Schuttler, D.; Tomsits, P.; Renner, S.; Klymiuk, N.; Wakili, R.; Massberg, S.; Wolf, E.; Kaab, S. Animal models of arrhythmia: Classic electrophysiology to genetically modified large animals. Nat. Rev. Cardiol. 2019, 16, 457–475. [Google Scholar] [CrossRef]
- Bondarenko, V.E.; Szigeti, G.P.; Bett, G.C.; Kim, S.J.; Rasmusson, R.L. Computer model of action potential of mouse ventricular myocytes. Am. J. Physiol. Heart Circ. Physiol. 2004, 287, H1378–H1403. [Google Scholar] [CrossRef]
- Niederer, S.A.; Smith, N.P. A mathematical model of the slow force response to stretch in rat ventricular myocytes. Biophys. J. 2007, 92, 4030–4044. [Google Scholar] [CrossRef] [Green Version]
- Noble, D.; Varghese, A.; Kohl, P.; Noble, P. Improved guinea-pig ventricular cell model incorporating a diadic space, IKr and IKs, and length- and tension-dependent processes. Can. J. Cardiol. 1998, 14, 123–134. [Google Scholar]
- Mahajan, A.; Shiferaw, Y.; Sato, D.; Baher, A.; Olcese, R.; Xie, L.H.; Yang, M.J.; Chen, P.S.; Restrepo, J.G.; Karma, A.; et al. A rabbit ventricular action potential model replicating cardiac dynamics at rapid heart rates. Biophys. J. 2008, 94, 392–410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Hara, T.; Virag, L.; Varro, A.; Rudy, Y. Simulation of the undiseased human cardiac ventricular action potential: Model formulation and experimental validation. PLoS Comput. Biol. 2011, 7, e1002061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whittaker, D.G.; Clerx, M.; Lei, C.L.; Christini, D.J.; Mirams, G.R. Calibration of ionic and cellular cardiac electrophysiology models. Wiley Interdiscip. Rev. Syst. Biol. Med. 2020, 12, e1482. [Google Scholar] [CrossRef] [Green Version]
- Ahrens-Nicklas, R.C.; Christini, D.J. Anthropomorphizing the mouse cardiac action potential via a novel dynamic clamp method. Biophys. J. 2009, 97, 2684–2692. [Google Scholar] [CrossRef] [Green Version]
- Heijman, J.; Ghezelbash, S.; Dobrev, D. Investigational antiarrhythmic agents: Promising drugs in early clinical development. Expert Opin. Investig. Drugs 2017, 26, 897–907. [Google Scholar] [CrossRef]
- Muszkiewicz, A.; Britton, O.J.; Gemmell, P.; Passini, E.; Sanchez, C.; Zhou, X.; Carusi, A.; Quinn, T.A.; Burrage, K.; Bueno-Orovio, A.; et al. Variability in cardiac electrophysiology: Using experimentally-calibrated populations of models to move beyond the single virtual physiological human paradigm. Prog. Biophys. Mol. Biol. 2016, 120, 115–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gourdie, R.G. The Cardiac Gap Junction has Discrete Functions in Electrotonic and Ephaptic Coupling. Anat. Rec. 2019, 302, 93–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Britton, O.J.; Abi-Gerges, N.; Page, G.; Ghetti, A.; Miller, P.E.; Rodriguez, B. Quantitative comparison of effects of dofetilide, sotalol, quinidine, and verapamil between human ex vivo trabeculae and in silico ventricular models incorporating inter-individual action potential variability. Front. Physiol. 2017, 8, 597. [Google Scholar] [CrossRef]
- Sutanto, H. Individual Contributions of Cardiac Ion Channels on Atrial Repolarization and Reentrant Waves: A Multiscale In-Silico Study. J. Cardiovasc. Dev. Dis. 2022, 9, 28. [Google Scholar] [CrossRef]
- Bartolucci, C.; Passini, E.; Hyttinen, J.; Paci, M.; Severi, S. Simulation of the Effects of Extracellular Calcium Changes Leads to a Novel Computational Model of Human Ventricular Action Potential with a Revised Calcium Handling. Front. Physiol. 2020, 11, 314. [Google Scholar] [CrossRef]
- Grandi, E.; Pasqualini, F.S.; Bers, D.M. A novel computational model of the human ventricular action potential and Ca transient. J. Mol. Cell. Cardiol. 2010, 48, 112–121. [Google Scholar] [CrossRef] [Green Version]
- Heijman, J.; Volders, P.G.; Westra, R.L.; Rudy, Y. Local control of beta-adrenergic stimulation: Effects on ventricular myocyte electrophysiology and Ca2+-transient. J. Mol. Cell. Cardiol. 2011, 50, 863–871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Passini, E.; Minchole, A.; Coppini, R.; Cerbai, E.; Rodriguez, B.; Severi, S.; Bueno-Orovio, A. Mechanisms of pro-arrhythmic abnormalities in ventricular repolarisation and anti-arrhythmic therapies in human hypertrophic cardiomyopathy. J. Mol. Cell. Cardiol. 2016, 96, 72–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomek, J.; Bueno-Orovio, A.; Passini, E.; Zhou, X.; Minchole, A.; Britton, O.; Bartolucci, C.; Severi, S.; Shrier, A.; Virag, L.; et al. Development, calibration, and validation of a novel human ventricular myocyte model in health, disease, and drug block. Elife 2019, 8, e48890. [Google Scholar] [CrossRef] [PubMed]
- ten Tusscher, K.H.; Panfilov, A.V. Alternans and spiral breakup in a human ventricular tissue model. Am. J. Physiol. Heart Circ. Physiol. 2006, 291, H1088–H1100. [Google Scholar] [CrossRef] [PubMed]
- Winslow, R.L.; Rice, J.; Jafri, S.; Marban, E.; O’Rourke, B. Mechanisms of altered excitation-contraction coupling in canine tachycardia-induced heart failure, II: Model studies. Circ. Res. 1999, 84, 571–586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayourian, J.; Sobie, E.A.; Costa, K.D. An Introduction to Computational Modeling of Cardiac Electrophysiology and Arrhythmogenicity. Methods Mol. Biol. 2018, 1816, 17–35. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Perez, A.; Sebastian, R.; Ferrero, J.M. Three-dimensional cardiac computational modelling: Methods, features and applications. Biomed. Eng. Online 2015, 14, 35. [Google Scholar] [CrossRef] [Green Version]
- Galappaththige, S.K.; Pathmanathan, P.; Bishop, M.J.; Gray, R.A. Effect of Heart Structure on Ventricular Fibrillation in the Rabbit: A Simulation Study. Front. Physiol. 2019, 10, 564. [Google Scholar] [CrossRef] [PubMed]
- Colman, M.A. Arrhythmia mechanisms and spontaneous calcium release: Bi-directional coupling between re-entrant and focal excitation. PLoS Comput. Biol. 2019, 15, e1007260. [Google Scholar] [CrossRef] [Green Version]
- Boyle, P.M.; Zghaib, T.; Zahid, S.; Ali, R.L.; Deng, D.; Franceschi, W.H.; Hakim, J.B.; Murphy, M.J.; Prakosa, A.; Zimmerman, S.L.; et al. Computationally guided personalized targeted ablation of persistent atrial fibrillation. Nat. Biomed. Eng. 2019, 3, 870–879. [Google Scholar] [CrossRef] [PubMed]
- Prakosa, A.; Arevalo, H.J.; Deng, D.; Boyle, P.M.; Nikolov, P.P.; Ashikaga, H.; Blauer, J.J.E.; Ghafoori, E.; Park, C.J.; Blake, R.C., 3rd; et al. Personalized virtual-heart technology for guiding the ablation of infarct-related ventricular tachycardia. Nat. Biomed. Eng. 2018, 2, 732–740. [Google Scholar] [CrossRef]
- Corral-Acero, J.; Margara, F.; Marciniak, M.; Rodero, C.; Loncaric, F.; Feng, Y.; Gilbert, A.; Fernandes, J.F.; Bukhari, H.A.; Wajdan, A.; et al. The ‘Digital Twin’ to enable the vision of precision cardiology. Eur. Heart J. 2020, 41, 4556–4564. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sutanto, H.; Heijman, J. Integrative Computational Modeling of Cardiomyocyte Calcium Handling and Cardiac Arrhythmias: Current Status and Future Challenges. Cells 2022, 11, 1090. https://doi.org/10.3390/cells11071090
Sutanto H, Heijman J. Integrative Computational Modeling of Cardiomyocyte Calcium Handling and Cardiac Arrhythmias: Current Status and Future Challenges. Cells. 2022; 11(7):1090. https://doi.org/10.3390/cells11071090
Chicago/Turabian StyleSutanto, Henry, and Jordi Heijman. 2022. "Integrative Computational Modeling of Cardiomyocyte Calcium Handling and Cardiac Arrhythmias: Current Status and Future Challenges" Cells 11, no. 7: 1090. https://doi.org/10.3390/cells11071090
APA StyleSutanto, H., & Heijman, J. (2022). Integrative Computational Modeling of Cardiomyocyte Calcium Handling and Cardiac Arrhythmias: Current Status and Future Challenges. Cells, 11(7), 1090. https://doi.org/10.3390/cells11071090