
STING Agonists/Antagonists: Their Potential as Therapeutics and Future Developments


Abstract
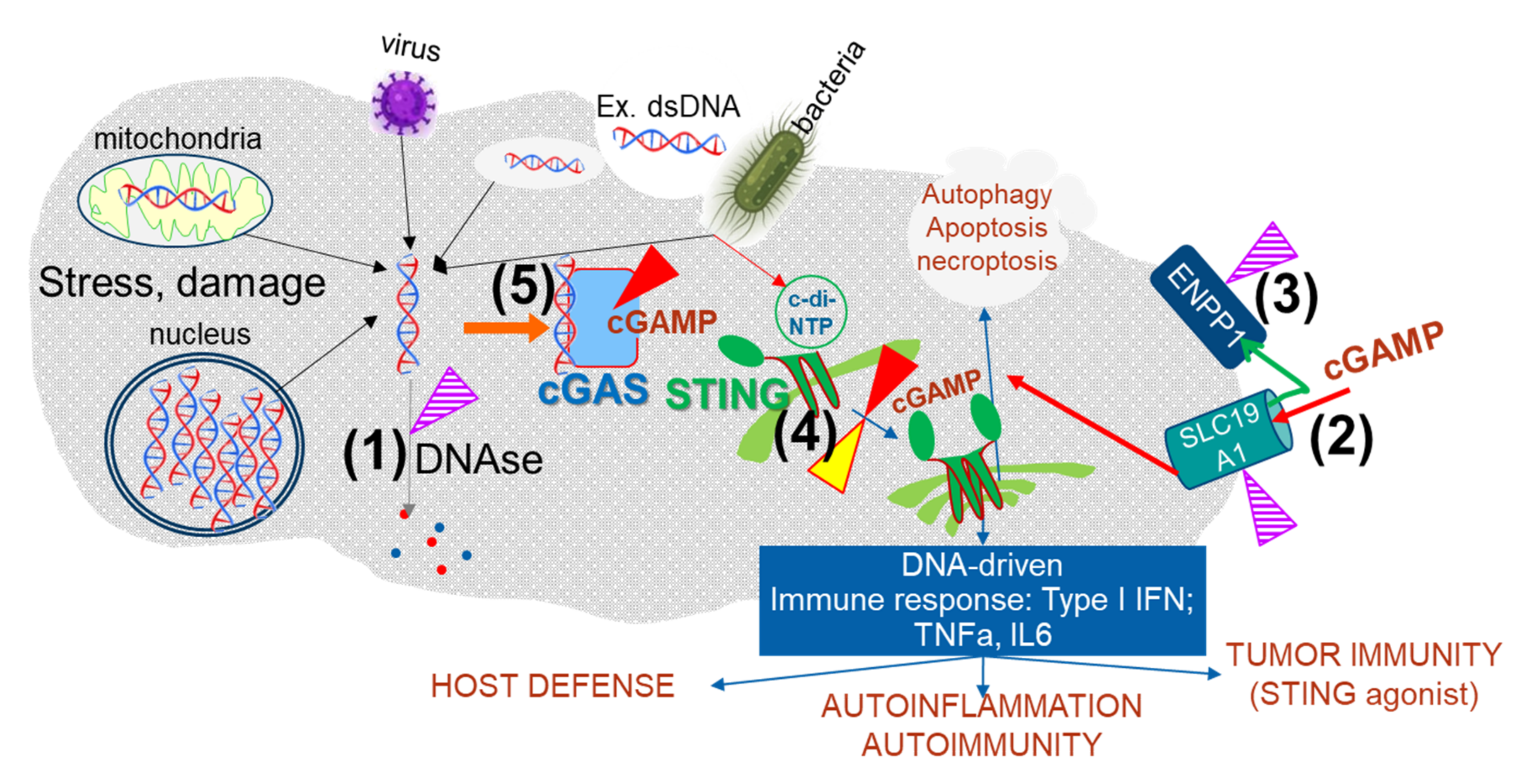
:1. The cGAS/STING, a Nucleic Acid Sensing Pathway, Plays a Role in Many Diseases
2. How to “Control” the cGAS STING Pathway
3. STING Agonists in Cancer
4. STING Antagonists in Inflammatory Diseases

{kind=link}
| STING Agonist | Publication | Characteristic | Mode of Action |
|---|---|---|---|
Amidobenz-imidazole (diABZI)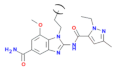 | [65] | An agonist with nM affinity to STING once it forms a dimer (diABZ). The monomer has weak activity to STING: A monomer summarizing the basic chemical properties of the amidobenzimidazole is depicted | diABZI bind in the C-terminal domain of STING in the open conformation, e.g., like-cGAMP. The compound shows activity in preclinical tumor models. Recently, a derivative of diABZI showed efficacy in an animal model for SARS-CoV-2 virus infection [66] |
MSA-2 | [69] | The monomer of the compound called MSA-2 forms a pH-dependent dimer that shows high affinity to STING. (20–100× fold increased affinity compared to monomer). | The MSA-2 is a weak acid, and it is preferentially taken up in an acidic tumor environment, where it can form a dimer, showing low systemic effects. |
| STING Antagonist | Publication | Characteristic | Mode of Action |
H-151 | [70] | H-151 is a small covalent inhibitor of STING, depicted shows both mouse and human activity. Another class of small covalent inhibitors (C-176), with a different structure from H-151, shows a preferential effect at mouse STING | The compound H-151 binds to the stalk region of STING, preventing the dimerization (multimerization) required for the activation of STING. Both H-151 and C-176 have good in vivo activity, showing positive effects in the Trex1 KO mouse model |
SN-11 | [74] | SN-011 is a novel STING inhibitor that targets the cyclic dinucleotide binding pocket and shows good efficacy in vivo. It has been suggested to prevent cGAMP binding to STING and, therefore, prevent its activation | SN-011 depicted in the left panel has been shown to work in vivo as a potent inhibitor of the cGAS/STING pathway. The SN-011 shows similar efficacy as the covalent compound H-151 in the Trex1 KO mouse model |
5. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Barber, G.N. Innate immune DNA sensing pathways: STING, AIMII and the regulation of interferon production and inflammatory responses. Curr. Opin. Immunol. 2011, 23, 10–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishikawa, H.; Barber, G.N. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 2008, 455, 674–678. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Wu, J.; Du, F.; Chen, X.; Chen, Z.J. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 2013, 339, 786–791. [Google Scholar] [CrossRef] [Green Version]
- Cai, X.; Chiu, Y.H.; Chen, Z.J. The cGAS-cGAMP-STING pathway of cytosolic DNA sensing and signaling. Mol. Cell 2014, 54, 289–296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishikawa, H.; Ma, Z.; Barber, G.N. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature 2009, 461, 788–792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Q.; Sun, L.; Chen, Z.J. Regulation and function of the cGAS-STING pathway of cytosolic DNA sensing. Nat. Immunol. 2016, 17, 1142–1149. [Google Scholar] [CrossRef]
- Ablasser, A.; Goldeck, M.; Cavlar, T.; Deimling, T.; Witte, G.; Rohl, I.; Hopfner, K.P.; Ludwig, J.; Hornung, V. cGAS produces a 2′–5′-linked cyclic dinucleotide second messenger that activates STING. Nature 2013, 498, 380–384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crow, Y.J.; Stetson, D.B. The type I interferonopathies: 10 years on. Nat. Rev. Immunol. 2021, 2021, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Crow, Y.J.; Manel, N. Aicardi-Goutieres syndrome and the type I interferonopathies. Nat. Rev. Immunol. 2015, 15, 429–440. [Google Scholar] [CrossRef] [PubMed]
- Fiehn, C. Familial Chilblain Lupus—What Can We Learn from Type I Interferonopathies? Curr. Rheumatol. Rep. 2017, 19, 61. [Google Scholar] [CrossRef] [PubMed]
- Rice, G.I.; Rodero, M.P.; Crow, Y.J. Human disease phenotypes associated with mutations in TREX1. J. Clin. Immunol. 2015, 35, 235–243. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Jesus, A.A.; Marrero, B.; Yang, D.; Ramsey, S.E.; Sanchez, G.A.M.; Tenbrock, K.; Wittkowski, H.; Jones, O.Y.; Kuehn, H.S.; et al. Activated STING in a vascular and pulmonary syndrome. N. Engl. J. Med. 2014, 371, 507–518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gray, E.E.; Winship, D.; Snyder, J.M.; Child, S.J.; Geballe, A.P.; Stetson, D.B. The AIM2-like Receptors Are Dispensable for the Interferon Response to Intracellular DNA. Immunity 2016, 45, 255–266. [Google Scholar] [CrossRef] [Green Version]
- Motwani, M.; Pesiridis, S.; Fitzgerald, K.A. DNA sensing by the cGAS-STING pathway in health and disease. Nat. Rev. Genet. 2019, 20, 657–674. [Google Scholar] [CrossRef]
- Harley, I.T.; Kaufman, K.M.; Langefeld, C.D.; Harley, J.B.; Kelly, J.A. Genetic susceptibility to SLE: New insights from fine mapping and genome-wide association studies. Nat. Rev. Genet. 2009, 10, 285–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huijser, E.; Bodewes, I.L.A.; Lourens, M.S.; van Helden-Meeuwsen, C.G.; van den Bosch, T.P.P.; Grashof, D.G.B.; van de Werken, H.J.G.; Lopes, A.P.; van Roon, J.A.G.; van Daele, P.L.A.; et al. Hyperresponsive cytosolic DNA-sensing pathway in monocytes from primary Sjogren’s syndrome. Rheumatology 2022, 2022, keac016. [Google Scholar] [CrossRef]
- Skopelja-Gardner, S.; An, J.; Tai, J.; Tanaka, L.; Sun, X.; Hermanson, P.; Baum, R.; Kawasumi, M.; Green, R.; Gale, M., Jr.; et al. The early local and systemic Type I interferon responses to ultraviolet B light exposure are cGAS dependent. Sci. Rep. 2020, 10, 7908. [Google Scholar] [CrossRef]
- Wang, J.; Li, R.; Lin, H.; Qiu, Q.; Lao, M.; Zeng, S.; Wang, C.; Xu, S.; Zou, Y.; Shi, M.; et al. Accumulation of cytosolic dsDNA contributes to fibroblast-like synoviocytes-mediated rheumatoid arthritis synovial inflammation. Int. Immunopharmacol. 2019, 76, 105791. [Google Scholar] [CrossRef]
- Willemsen, J.; Neuhoff, M.T.; Hoyler, T.; Noir, E.; Tessier, C.; Sarret, S.; Thorsen, T.N.; Littlewood-Evans, A.; Zhang, J.; Hasan, M.; et al. TNF leads to mtDNA release and cGAS/STING-dependent interferon responses that support inflammatory arthritis. Cell Rep. 2021, 37, 109977. [Google Scholar] [CrossRef]
- Aden, K.; Tran, F.; Ito, G.; Sheibani-Tezerji, R.; Lipinski, S.; Kuiper, J.W.; Tschurtschenthaler, M.; Saveljeva, S.; Bhattacharyya, J.; Hasler, R.; et al. ATG16L1 orchestrates interleukin-22 signaling in the intestinal epithelium via cGAS-STING. J. Exp. Med. 2018, 215, 2868–2886. [Google Scholar] [CrossRef] [Green Version]
- Ahn, J.; Son, S.; Oliveira, S.C.; Barber, G.N. STING-Dependent Signaling Underlies IL-10 Controlled Inflammatory Colitis. Cell Rep. 2017, 21, 3873–3884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canesso, M.C.C.; Lemos, L.; Neves, T.C.; Marim, F.M.; Castro, T.B.R.; Veloso, E.S.; Queiroz, C.P.; Ahn, J.; Santiago, H.C.; Martins, F.S.; et al. The cytosolic sensor STING is required for intestinal homeostasis and control of inflammation. Mucosal. Immunol. 2018, 11, 820–834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, G.R.; Blomquist, C.M.; Henare, K.L.; Jirik, F.R. Stimulator of interferon genes (STING) activation exacerbates experimental colitis in mice. Sci. Rep. 2019, 9, 14281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Q.; Man, S.M.; Gurung, P.; Liu, Z.; Vogel, P.; Lamkanfi, M.; Kanneganti, T.D. Cutting edge: STING mediates protection against colorectal tumorigenesis by governing the magnitude of intestinal inflammation. J. Immunol. 2014, 193, 4779–4782. [Google Scholar] [CrossRef]
- Marinho, F.V.; Benmerzoug, S.; Oliveira, S.C.; Ryffel, B.; Quesniaux, V.F.J. The Emerging Roles of STING in Bacterial Infections. Trends Microbiol. 2017, 25, 906–918. [Google Scholar] [CrossRef]
- Hu, Q.; Ren, H.; Li, G.; Wang, D.; Zhou, Q.; Wu, J.; Zheng, J.; Huang, J.; Slade, D.A.; Wu, X.; et al. STING-mediated intestinal barrier dysfunction contributes to lethal sepsis. EBioMedicine 2019, 41, 497–508. [Google Scholar] [CrossRef] [Green Version]
- Benmerzoug, S.; Rose, S.; Bounab, B.; Gosset, D.; Duneau, L.; Chenuet, P.; Mollet, L.; Le Bert, M.; Lambers, C.; Geleff, S.; et al. STING-dependent sensing of self-DNA drives silica-induced lung inflammation. Nat. Commun. 2018, 9, 5226. [Google Scholar] [CrossRef]
- Benmerzoug, S.; Bounab, B.; Rose, S.; Gosset, D.; Biet, F.; Cochard, T.; Xavier, A.; Rouxel, N.; Fauconnier, L.; Horsnell, W.G.C.; et al. Sterile Lung Inflammation Induced by Silica Exacerbates Mycobacterium tuberculosis Infection via STING-Dependent Type 2 Immunity. Cell Rep. 2019, 27, 2649–2664. [Google Scholar] [CrossRef] [Green Version]
- Benmerzoug, S.; Ryffel, B.; Togbe, D.; Quesniaux, V.F.J. Self-DNA Sensing in Lung Inflammatory Diseases. Trends Immunol. 2019, 40, 719–734. [Google Scholar] [CrossRef]
- Allison, S.J. STING activation by cytoplasmic mtDNA triggers renal inflammation and fibrosis. Nat. Rev. Nephrol. 2019, 15, 661. [Google Scholar] [CrossRef]
- Bennion, B.G.; Ingle, H.; Ai, T.L.; Miner, C.A.; Platt, D.J.; Smith, A.M.; Baldridge, M.T.; Miner, J.J. A Human Gain-of-Function STING Mutation Causes Immunodeficiency and Gammaherpesvirus-Induced Pulmonary Fibrosis in Mice. J. Virol. 2019, 93, e01806-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iracheta-Vellve, A.; Petrasek, J.; Gyongyosi, B.; Satishchandran, A.; Lowe, P.; Kodys, K.; Catalano, D.; Calenda, C.D.; Kurt-Jones, E.A.; Fitzgerald, K.A.; et al. Endoplasmic Reticulum Stress-induced Hepatocellular Death Pathways Mediate Liver Injury and Fibrosis via Stimulator of Interferon Genes. J. Biol. Chem. 2016, 291, 26794–26805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, S.C.; Han, R.; Hou, S.S.; Yi, H.Q.; Chi, S.J.; Zhang, A.H. Juglanin alleviates bleomycin-induced lung injury by suppressing inflammation and fibrosis via targeting sting signaling. Biomed. Pharm. 2020, 127, 110119. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Rao, H.; Zhao, J.; Wee, A.; Li, X.; Fei, R.; Huang, R.; Wu, C.; Liu, F.; Wei, L. STING expression in monocyte-derived macrophages is associated with the progression of liver inflammation and fibrosis in patients with nonalcoholic fatty liver disease. Lab. Investig. 2020, 100, 542–552. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, W.; Wang, Y. STING is an essential regulator of heart inflammation and fibrosis in mice with pathological cardiac hypertrophy via endoplasmic reticulum (ER) stress. Biomed. Pharm. 2020, 125, 110022. [Google Scholar] [CrossRef]
- Platt, D.J.; Lawrence, D.; Rodgers, R.; Schriefer, L.; Qian, W.; Miner, C.A.; Menos, A.M.; Kennedy, E.A.; Peterson, S.T.; Stinson, W.A.; et al. Transferrable protection by gut microbes against STING-associated lung disease. Cell Rep. 2021, 35, 109113. [Google Scholar] [CrossRef]
- Gluck, S.; Guey, B.; Gulen, M.F.; Wolter, K.; Kang, T.W.; Schmacke, N.A.; Bridgeman, A.; Rehwinkel, J.; Zender, L.; Ablasser, A. Innate immune sensing of cytosolic chromatin fragments through cGAS promotes senescence. Nat. Cell Biol. 2017, 19, 1061–1070. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Wang, H.; Ren, J.; Chen, Q.; Chen, Z.J. cGAS is essential for cellular senescence. Proc. Natl. Acad. Sci. USA 2017, 114, E4612–E4620. [Google Scholar] [CrossRef] [Green Version]
- Dou, Z.; Ghosh, K.; Vizioli, M.G.; Zhu, J.; Sen, P.; Wangensteen, K.J.; Simithy, J.; Lan, Y.; Lin, Y.; Zhou, Z.; et al. Cytoplasmic chromatin triggers inflammation in senescence and cancer. Nature 2017, 550, 402–406. [Google Scholar] [CrossRef] [Green Version]
- Sliter, D.A.; Martinez, J.; Hao, L.; Chen, X.; Sun, N.; Fischer, T.D.; Burman, J.L.; Li, Y.; Zhang, Z.; Narendra, D.P.; et al. Parkin and PINK1 mitigate STING-induced inflammation. Nature 2018, 561, 258–262. [Google Scholar] [CrossRef]
- Hou, Y.; Wei, Y.; Lautrup, S.; Yang, B.; Wang, Y.; Cordonnier, S.; Mattson, M.P.; Croteau, D.L.; Bohr, V.A. NAD+ supplementation reduces neuroinflammation and cell senescence in a transgenic mouse model of Alzheimer’s disease via cGAS-STING. Proc. Natl. Acad. Sci. USA 2021, 118, e2011226118. [Google Scholar] [CrossRef]
- McCauley, M.E.; O’Rourke, J.G.; Yanez, A.; Markman, J.L.; Ho, R.; Wang, X.; Chen, S.; Lall, D.; Jin, M.; Muhammad, A.; et al. C9orf72 in myeloid cells suppresses STING-induced inflammation. Nature 2020, 585, 96–101. [Google Scholar] [CrossRef]
- Maekawa, H.; Inoue, T.; Ouchi, H.; Jao, T.M.; Inoue, R.; Nishi, H.; Fujii, R.; Ishidate, F.; Tanaka, T.; Tanaka, Y.; et al. Mitochondrial Damage Causes Inflammation via cGAS-STING Signaling in Acute Kidney Injury. Cell Rep. 2019, 29, 1261–1273.e6. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Raman, A.; Coffey, N.J.; Sheng, X.; Wahba, J.; Seasock, M.J.; Ma, Z.; Beckerman, P.; Laczko, D.; Palmer, M.B.; et al. The key role of NLRP3 and STING in APOL1-associated podocytopathy. J. Clin. Investig. 2021, 131, e136329. [Google Scholar] [CrossRef] [PubMed]
- Teijaro, J.R.; Ng, C.; Lee, A.M.; Sullivan, B.M.; Sheehan, K.C.; Welch, M.; Schreiber, R.D.; de la Torre, J.C.; Oldstone, M.B. Persistent LCMV infection is controlled by blockade of type I interferon signaling. Science 2013, 340, 207–211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, E.B.; Yamada, D.H.; Elsaesser, H.; Herskovitz, J.; Deng, J.; Cheng, G.; Aronow, B.J.; Karp, C.L.; Brooks, D.G. Blockade of chronic type I interferon signaling to control persistent LCMV infection. Science 2013, 340, 202–207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carozza, J.A.; Brown, J.A.; Bohnert, V.; Fernandez, D.; AlSaif, Y.; Mardjuki, R.E.; Smith, M.; Li, L. Structure-Aided Development of Small-Molecule Inhibitors of ENPP1, the Extracellular Phosphodiesterase of the Immunotransmitter cGAMP. Cell Chem. Biol. 2020, 27, 1347–1358.e5. [Google Scholar] [CrossRef] [PubMed]
- Luteijn, R.D.; Zaver, S.A.; Gowen, B.G.; Wyman, S.K.; Garelis, N.E.; Onia, L.; McWhirter, S.M.; Katibah, G.E.; Corn, J.E.; Woodward, J.J.; et al. SLC19A1 transports immunoreactive cyclic dinucleotides. Nature 2019, 573, 434–438. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, C.; Cordova, A.F.; Hess, G.T.; Bassik, M.C.; Li, L. SLC19A1 Is an Importer of the Immunotransmitter cGAMP. Mol. Cell 2019, 75, 372–381.e5. [Google Scholar] [CrossRef] [PubMed]
- Cordova, A.F.; Ritchie, C.; Bohnert, V.; Li, L. Human SLC46A2 Is the Dominant cGAMP Importer in Extracellular cGAMP-Sensing Macrophages and Monocytes. ACS Cent. Sci. 2021, 7, 1073–1088. [Google Scholar] [CrossRef]
- Zhou, C.; Chen, X.; Planells-Cases, R.; Chu, J.; Wang, L.; Cao, L.; Li, Z.; Lopez-Cayuqueo, K.I.; Xie, Y.; Ye, S.; et al. Transfer of cGAMP into Bystander Cells via LRRC8 Volume-Regulated Anion Channels Augments STING-Mediated Interferon Responses and Anti-viral Immunity. Immunity 2020, 52, 767–781.e6. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Fei, M.; Zhang, G.; Liang, W.C.; Lin, W.; Wu, Y.; Piskol, R.; Ridgway, J.; McNamara, E.; Huang, H.; et al. Blockade of the Phagocytic Receptor MerTK on Tumor-Associated Macrophages Enhances P2X7R-Dependent STING Activation by Tumor-Derived cGAMP. Immunity 2020, 52, 357–373.e9. [Google Scholar] [CrossRef] [PubMed]
- Ablasser, A.; Schmid-Burgk, J.L.; Hemmerling, I.; Horvath, G.L.; Schmidt, T.; Latz, E.; Hornung, V. Cell intrinsic immunity spreads to bystander cells via the intercellular transfer of cGAMP. Nature 2013, 503, 530–534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jameson, M.B.; Thompson, P.I.; Baguley, B.C.; Evans, B.D.; Harvey, V.J.; Porter, D.J.; McCrystal, M.R.; Small, M.; Bellenger, K.; Gumbrell, L.; et al. Clinical aspects of a phase I trial of 5,6-dimethylxanthenone-4-acetic acid (DMXAA), a novel antivascular agent. Br. J. Cancer 2003, 88, 1844–1850. [Google Scholar] [CrossRef]
- Kim, S.; Li, L.; Maliga, Z.; Yin, Q.; Wu, H.; Mitchison, T.J. Anticancer flavonoids are mouse-selective STING agonists. ACS Chem. Biol. 2013, 8, 1396–1401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, P.; Ascano, M.; Zillinger, T.; Wang, W.; Dai, P.; Serganov, A.A.; Gaffney, B.L.; Shuman, S.; Jones, R.A.; Deng, L.; et al. Structure-function analysis of STING activation by c[G(2′,5′)pA(3′,5′)p] and targeting by antiviral DMXAA. Cell 2013, 154, 748–762. [Google Scholar] [CrossRef] [Green Version]
- Sivick, K.E.; Desbien, A.L.; Glickman, L.H.; Reiner, G.L.; Corrales, L.; Surh, N.H.; Hudson, T.E.; Vu, U.T.; Francica, B.J.; Banda, T.; et al. Magnitude of Therapeutic STING Activation Determines CD8+ T Cell-Mediated Anti-tumor Immunity. Cell Rep. 2018, 25, 3074–3085.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, C.; Song, Z.; Shen, A.; Chen, T.; Zhang, A. Small molecules targeting the innate immune cGASSTINGTBK1 signaling pathway. Acta Pharm. Sin. B 2020, 10, 2272–2298. [Google Scholar] [CrossRef] [PubMed]
- Amouzegar, A.; Chelvanambi, M.; Filderman, J.N.; Storkus, W.J.; Luke, J.J. STING Agonists as Cancer Therapeutics. Cancers 2021, 13, 2695. [Google Scholar] [CrossRef] [PubMed]
- Decout, A.; Katz, J.D.; Venkatraman, S.; Ablasser, A. The cGAS-STING pathway as a therapeutic target in inflammatory diseases. Nat. Rev. Immunol. 2021, 21, 548–569. [Google Scholar] [CrossRef]
- Hong, Z.; Mei, J.; Guo, H.; Zhu, J.; Wang, C. Intervention of cGASSTING signaling in sterile inflammatory diseases. J. Mol. Cell Biol. 2022, 2022, mjac005. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Tian, S.; Liang, J.; Fan, J.; Lai, J.; Chen, Q. Therapeutic Development by Targeting the cGAS-STING Pathway in Autoimmune Disease and Cancer. Front. Pharmacol. 2021, 12, 779425. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Lu, X.; Qin, N.; Qiao, Y.; Xing, S.; Liu, W.; Feng, F.; Liu, Z.; Sun, H. STING, a promising target for small molecular immune modulator: A review. Eur. J. Med. Chem. 2021, 211, 113113. [Google Scholar] [CrossRef] [PubMed]
- Motedayen Aval, L.; Pease, J.E.; Sharma, R.; Pinato, D.J. Challenges and Opportunities in the Clinical Development of STING Agonists for Cancer Immunotherapy. J. Clin. Med. 2020, 9, 3323. [Google Scholar] [CrossRef] [PubMed]
- Ramanjulu, J.M.; Pesiridis, G.S.; Yang, J.; Concha, N.; Singhaus, R.; Zhang, S.Y.; Tran, J.L.; Moore, P.; Lehmann, S.; Eberl, H.C.; et al. Design of amidobenzimidazole STING receptor agonists with systemic activity. Nature 2018, 564, 439–443. [Google Scholar] [CrossRef] [PubMed]
- Humphries, F.; Liraz, S.G.; Jiang, Z.; Wilson, R.; Landis, P.; Ng, S.; Parsi, K.M.; Maehr, R.; Cruz, J.; Morales, A.; et al. A diamidobenzimidazole STING agonist protects against SARS-CoV-2 infection. Sci. Immunol. 2021, 6, eabi9002. [Google Scholar] [CrossRef]
- Li, M.; Ferretti, M.; Ying, B.; Descamps, H.; Lee, E.; Dittmar, M.; Lee, J.S.; Whig, K.; Kamalia, B.; Dohnalova, L.; et al. Pharmacological activation of STING blocks SARS-CoV-2 infection. Sci. Immunol. 2021, 6, eabi9007. [Google Scholar] [CrossRef]
- Zhu, Q.; Zhang, Y.; Wang, L.; Yao, X.; Wu, D.; Cheng, J.; Pan, X.; Liu, H.; Yan, Z.; Gao, L. Inhibition of coronavirus infection by a synthetic STING agonist in primary human airway system. Antivir. Res. 2021, 187, 105015. [Google Scholar] [CrossRef]
- Pan, B.S.; Perera, S.A.; Piesvaux, J.A.; Presland, J.P.; Schroeder, G.K.; Cumming, J.N.; Trotter, B.W.; Altman, M.D.; Buevich, A.V.; Cash, B.; et al. An orally available non-nucleotide STING agonist with antitumor activity. Science 2020, 369, eaba6098. [Google Scholar] [CrossRef] [PubMed]
- Haag, S.M.; Gulen, M.F.; Reymond, L.; Gibelin, A.; Abrami, L.; Decout, A.; Heymann, M.; van der Goot, F.G.; Turcatti, G.; Behrendt, R.; et al. Targeting STING with covalent small-molecule inhibitors. Nature 2018, 559, 269–273. [Google Scholar] [CrossRef] [PubMed]
- Gray, E.E.; Treuting, P.M.; Woodward, J.J.; Stetson, D.B. Cutting Edge: cGAS is required for Lethal Autoimmune Disease in the Trex1-Deficient Mouse Model of Aicardi-Goutieres Syndrome. J. Immunol. 2015, 195, 1939–1943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grieves, J.L.; Fye, J.M.; Harvey, S.; Grayson, J.M.; Hollis, T.; Perrino, F.W. Exonuclease TREX1 degrades double-stranded DNA to prevent spontaneous lupus-like inflammatory disease. Proc. Natl. Acad. Sci. USA 2015, 112, 5117–5122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, N.; Wei, J.; Xu, S.; Du, H.; Huang, M.; Zhang, S.; Ye, W.; Sun, L.; Chen, Q. cGAS activation causes lupus-like autoimmune disorders in a TREX1 mutant mouse model. J. Autoimmun. 2019, 100, 84–94. [Google Scholar] [CrossRef] [PubMed]
- Hong, Z.; Mei, J.; Li, C.; Bai, G.; Maimaiti, M.; Hu, H.; Yu, W.; Sun, L.; Zhang, L.; Cheng, D.; et al. STING inhibitors target the cyclic dinucleotide binding pocket. Proc. Natl. Acad. Sci. USA 2021, 118, e2105465118. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guerini, D. STING Agonists/Antagonists: Their Potential as Therapeutics and Future Developments. Cells 2022, 11, 1159. https://doi.org/10.3390/cells11071159
Guerini D. STING Agonists/Antagonists: Their Potential as Therapeutics and Future Developments. Cells. 2022; 11(7):1159. https://doi.org/10.3390/cells11071159
Chicago/Turabian StyleGuerini, Danilo. 2022. "STING Agonists/Antagonists: Their Potential as Therapeutics and Future Developments" Cells 11, no. 7: 1159. https://doi.org/10.3390/cells11071159
APA StyleGuerini, D. (2022). STING Agonists/Antagonists: Their Potential as Therapeutics and Future Developments. Cells, 11(7), 1159. https://doi.org/10.3390/cells11071159